Optic Pathway Glioma in Type 1 Neurofibromatosis: Review of Its Pathogenesis, Diagnostic Assessment, and Treatment Recommendations

 ,
,  ,
,
Abstract
:1. Introduction
2. Optic Pathway Gliomas in NF1
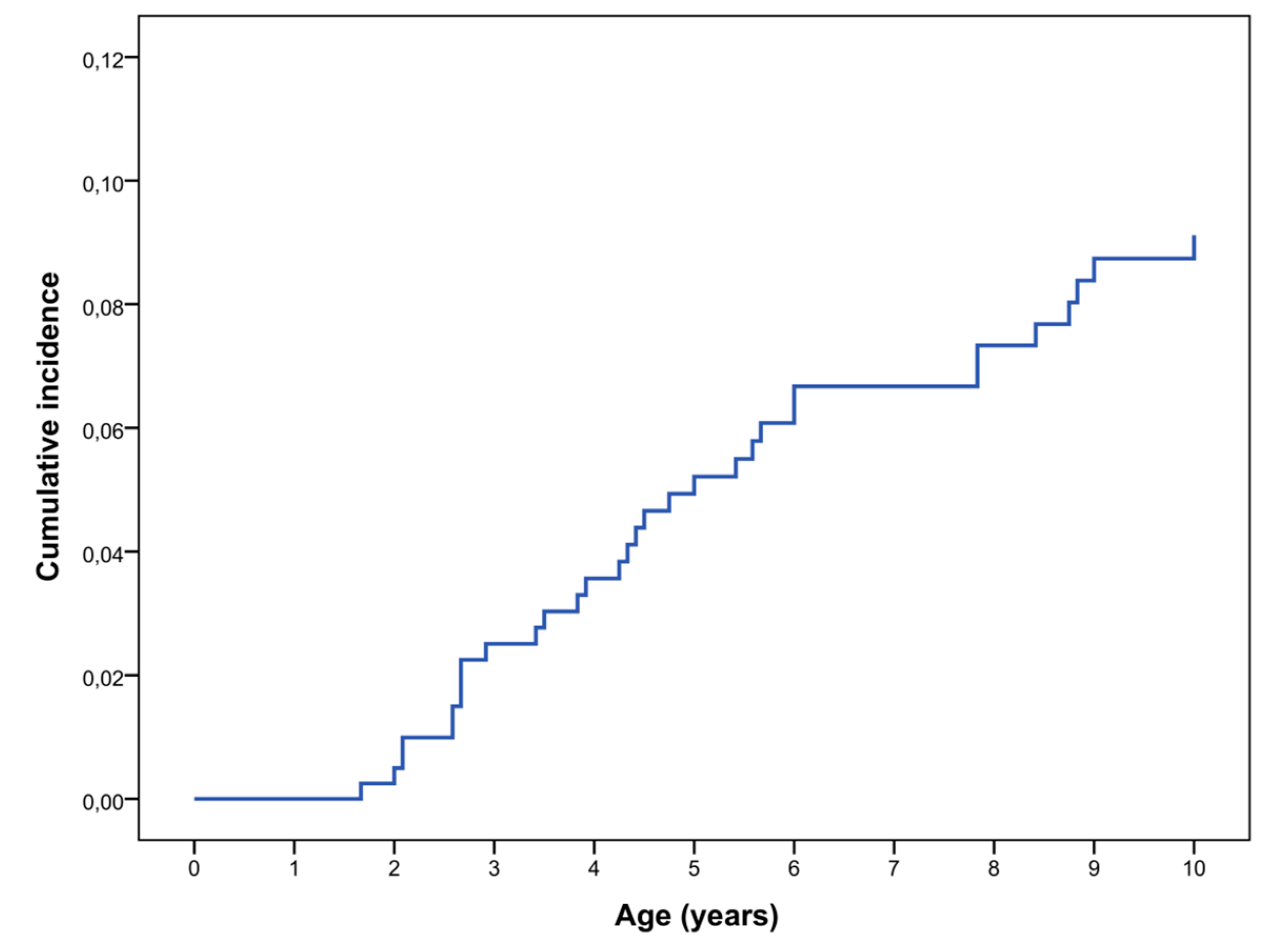
2.1. Prevalence, Clinical Features and Natural History of OPG
2.2. Genotype-phenotype Correlations in OPG
3. Screening and Diagnostic Protocol for OPG
3.1. The Role of Brain and Orbit MRI
3.2. Ophthalmological Assessment
3.2.1. Visual Function Assessment
Visual Acuity Testing
Visual Field Testing
Visual Evoked Potentials
Eye Movements Assessment
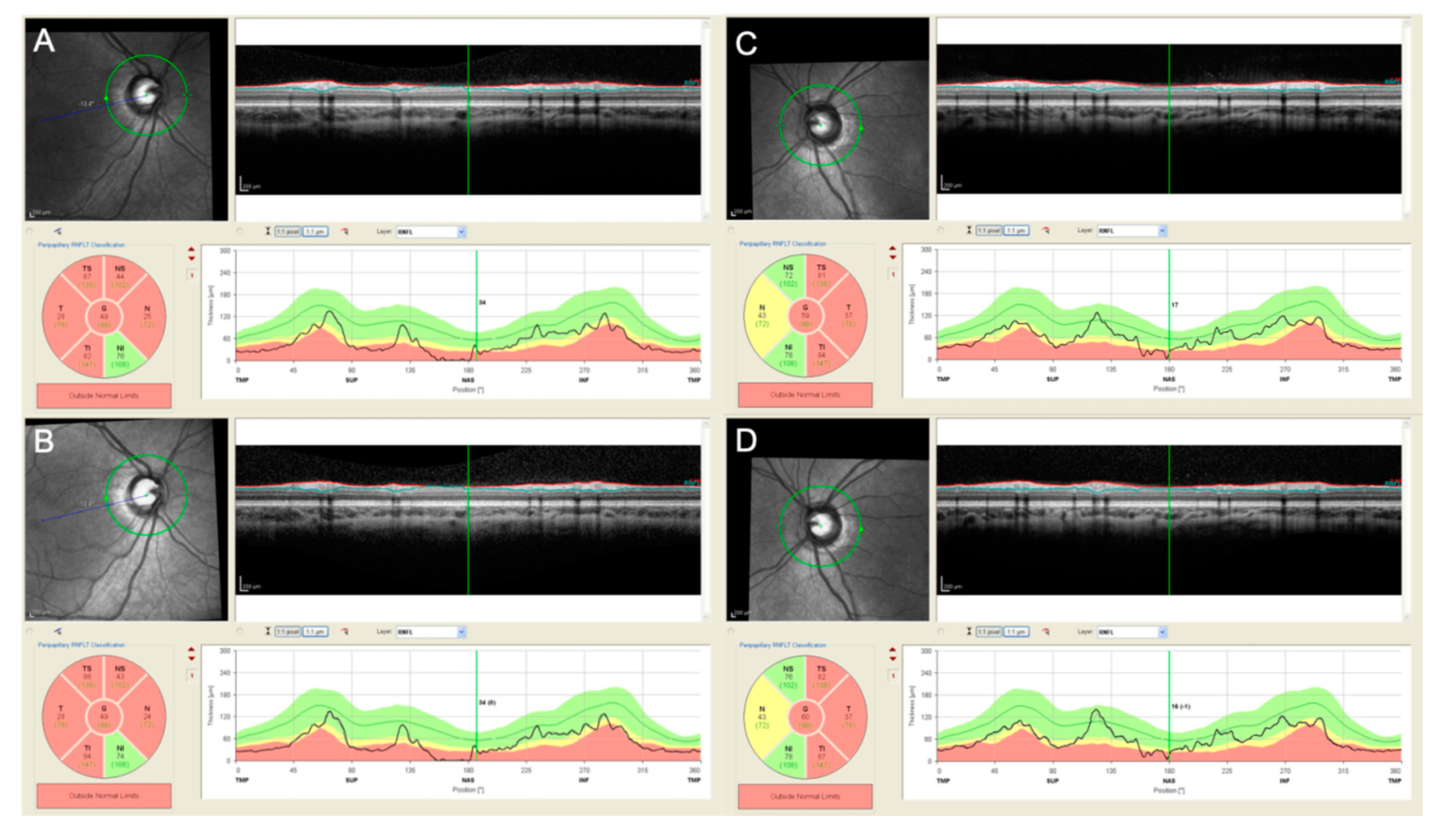
3.2.2. Optical Coherence Tomography (OCT)
3.2.3. Optic Disc Evaluation
4. Preclinical Models of OPG
5. Oncologic Treatment of Children with NF1-OPG
5.1. Standard Treatment
5.2. MEK Inhibitors
5.3. Other Potential Treatments for NF1-OPG
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clementi, M.; Barbujani, G.; Turolla, L.; Tenconi, R. Neurofibromatosis-1: A maximum likelihood estimation of mutation rate. Hum. Genet. 1990, 84, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.F.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841. [Google Scholar] [CrossRef]
- Wallis, D.; Li, K.; Lui, H.; Hu, K.; Chen, M.J.; Li, J.; Kang, J.; Das, S.; Korf, B.R.; Kesterson, R.A. Neurofibromin (NF1) genetic variant structure-function analyses using a full-length mouse cDNA. Hum. Mutat. 2018, 39, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.T.; Bearden, C.E.; Castellanos, F.X.; Castellanos, X.F.; Cutting, L.; Elgersma, Y.; Gioia, G.; Gutmann, D.H.; Lee, Y.S.; Legius, E.; et al. The Learning Disabilities Network (LeaDNet): Using neurofibromatosis type 1 (NF1) as a paradigm for translational research. Am. J. Med. Genet. A 2012, 158A, 2225–2232. [Google Scholar] [CrossRef] [PubMed]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Trevisson, E.; Cassina, M.; Opocher, E.; Vicenzi, V.; Lucchetta, M.; Parrozzani, R.; Miglionico, G.; Mardari, R.; Viscardi, E.; Midena, E.; et al. Natural history of optic pathway gliomas in a cohort of unselected patients affected by Neurofibromatosis 1. J. Neurooncol. 2017, 134, 279–287. [Google Scholar] [CrossRef]
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Sylvester, C.L.; Drohan, L.A.; Sergott, R.C. Optic-nerve gliomas, chiasmal gliomas and neurofibromatosis type 1. Curr. Opin. Ophthalmol. 2006, 17, 7–11. [Google Scholar] [CrossRef]
- Lewis, R.A.; Gerson, L.P.; Axelson, K.A.; Riccardi, V.M.; Whitford, R.P. von Recklinghausen neurofibromatosis: II. Incidence of optic gliomata. Ophthalmology 1984, 91, 929–935. [Google Scholar] [CrossRef]
- Listernick, R.; Charrow, J.; Greenwald, M.J.; Esterly, N.B. Optic gliomas in children with neurofibromatosis type 1. J. Pediatr. 1989, 114, 788–792. [Google Scholar] [CrossRef]
- Listernick, R.; Charrow, J.; Greenwald, M.; Mets, M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: A longitudinal study. J. Pediatr. 1994, 125, 63–66. [Google Scholar] [CrossRef]
- Listernick, R.; Darling, C.; Greenwald, M.; Strauss, L.; Charrow, J. Optic pathway tumors in children: The effect of neurofibromatosis type 1 on clinical manifestations and natural history. J. Pediatr. 1995, 127, 718–722. [Google Scholar] [CrossRef]
- Listernick, R.; Louis, D.N.; Packer, R.J.; Gutmann, D.H. Optic pathway gliomas in children with neurofibromatosis 1: Consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann. Neurol. 1997, 41, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Listernick, R.; Charrow, J.; Gutmann, D.H. Intracranial gliomas in neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 38–44. [Google Scholar] [CrossRef]
- Prada, C.E.; Hufnagel, R.B.; Hummel, T.R.; Lovell, A.M.; Hopkin, R.J.; Saal, H.M.; Schorry, E.K. The Use of Magnetic Resonance Imaging Screening for Optic Pathway Gliomas in Children with Neurofibromatosis Type 1. J. Pediatr. 2015, 167, 851–856.e851. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, G.; Lafforgue, M.P.; Lion-François, L.; Kemlin, I.; Rodriguez, D.; Castelnau, P.; Carneiro, M.; Meyer, P.; Rivier, F.; Barbarot, S.; et al. Systematic MRI in NF1 children under six years of age for the diagnosis of optic pathway gliomas. Study and outcome of a French cohort. Eur. J. Paediatr. Neurol. 2016, 20, 275–281. [Google Scholar] [CrossRef]
- Sellmer, L.; Farschtschi, S.; Marangoni, M.; Heran, M.K.S.; Birch, P.; Wenzel, R.; Mautner, V.F.; Friedman, J.M. Serial MRIs provide novel insight into natural history of optic pathway gliomas in patients with neurofibromatosis 1. Orphanet. J. Rare. Dis. 2018, 13, 62. [Google Scholar] [CrossRef]
- Miller, D.T.; Freedenberg, D.; Schorry, E.; Ullrich, N.J.; Viskochil, D.; Korf, B.R.; GENETICS, C.O.; Genomics, A.C.O.M.G.A. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics 2019, 143. [Google Scholar] [CrossRef]
- Fisher, M.J.; Loguidice, M.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Ullrich, N.J.; Packer, R.J.; Tabori, U.; Hoffman, R.O.; Ardern-Holmes, S.L.; et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: A multicenter retrospective analysis. Neuro Oncol. 2012, 14, 790–797. [Google Scholar] [CrossRef]
- Campen, C.J.; Gutmann, D.H. Optic Pathway Gliomas in Neurofibromatosis Type 1. J. Child. Neurol. 2018, 33, 73–81. [Google Scholar] [CrossRef]
- Mandiwanza, T.; Kaliaperumal, C.; Khalil, A.; Sattar, M.; Crimmins, D.; Caird, J. Suprasellar pilocytic astrocytoma: One national centre’s experience. Childs. Nerv. Syst. 2014, 30, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Blazo, M.A.; Lewis, R.A.; Chintagumpala, M.M.; Frazier, M.; McCluggage, C.; Plon, S.E. Outcomes of systematic screening for optic pathway tumors in children with Neurofibromatosis Type 1. Am. J. Med. Genet. A 2004, 127A, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, L.; Hagel, C.; Tatagiba, M.; Thomas, S.; Stavrou, D.; Ostertag, H.; von Deimling, A.; Mautner, V.F. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J. Neuropathol. Exp. Neurol. 2001, 60, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Diggs-Andrews, K.A.; Brown, J.A.; Gianino, S.M.; Rubin, J.B.; Wozniak, D.F.; Gutmann, D.H. Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann. Neurol. 2014, 75, 309–316. [Google Scholar] [CrossRef]
- Friedrich, R.E.; Nuding, M.A. Optic Pathway Glioma and Cerebral Focal Abnormal Signal Intensity in Patients with Neurofibromatosis Type 1: Characteristics, Treatment Choices and Follow-up in 134 Affected Individuals and a Brief Review of the Literature. Anticancer Res. 2016, 36, 4095–4121. [Google Scholar]
- Parkhurst, E.; Abboy, S. Optic Gliomas in Neurofibromatosis Type 1. J. Pediatr. Ophthalmol. Strabismus. 2016, 53, 334–338. [Google Scholar] [CrossRef]
- Listernick, R.; Ferner, R.E.; Liu, G.T.; Gutmann, D.H. Optic pathway gliomas in neurofibromatosis-1: Controversies and recommendations. Ann. Neurol. 2007, 61, 189–198. [Google Scholar] [CrossRef]
- Listernick, R.; Ferner, R.E.; Piersall, L.; Sharif, S.; Gutmann, D.H.; Charrow, J. Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology 2004, 63, 1944–1946. [Google Scholar] [CrossRef]
- Créange, A.; Zeller, J.; Rostaing-Rigattieri, S.; Brugières, P.; Degos, J.D.; Revuz, J.; Wolkenstein, P. Neurological complications of neurofibromatosis type 1 in adulthood. Brain 1999, 122 Pt 3, 473–481. [Google Scholar] [CrossRef]
- Piccirilli, M.; Lenzi, J.; Delfinis, C.; Trasimeni, G.; Salvati, M.; Raco, A. Spontaneous regression of optic pathways gliomas in three patients with neurofibromatosis type I and critical review of the literature. Childs. Nerv. Syst. 2006, 22, 1332–1337. [Google Scholar] [CrossRef]
- Brzowski, A.E.; Bazan, C.; Mumma, J.V.; Ryan, S.G. Spontaneous regression of optic glioma in a patient with neurofibromatosis. Neurology 1992, 42, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Parsa, C.F.; Hoyt, C.S.; Lesser, R.L.; Weinstein, J.M.; Strother, C.M.; Muci-Mendoza, R.; Ramella, M.; Manor, R.S.; Fletcher, W.A.; Repka, M.X.; et al. Spontaneous regression of optic gliomas: Thirteen cases documented by serial neuroimaging. Arch. Ophthalmol. 2001, 119, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.J.; Avery, R.A.; Allen, J.C.; Ardern-Holmes, S.L.; Bilaniuk, L.T.; Ferner, R.E.; Gutmann, D.H.; Listernick, R.; Martin, S.; Ullrich, N.J.; et al. Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology 2013, 81, S15–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, T.; Jaspan, T.; Milano, G.; Gregson, R.; Parker, T.; Ritzmann, T.; Benson, C.; Walker, D.; Group, P.S. Radiological classification of optic pathway gliomas: Experience of a modified functional classification system. Br. J. Radiol. 2008, 81, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Guillamo, J.S.; Créange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 2003, 126, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.J.; Loguidice, M.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Ullrich, N.J.; Packer, R.J.; Tabori, U.; Hoffman, R.O.; Ardern-Holmes, S.L.; et al. Gender as a disease modifier in neurofibromatosis type 1 optic pathway glioma. Ann. Neurol. 2014, 75, 799–800. [Google Scholar] [CrossRef]
- Balcer, L.J.; Liu, G.T.; Heller, G.; Bilaniuk, L.; Volpe, N.J.; Galetta, S.L.; Molloy, P.T.; Phillips, P.C.; Janss, A.J.; Vaughn, S.; et al. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: Relation to tumor location by magnetic resonance imaging. Am. J. Ophthalmol. 2001, 131, 442–445. [Google Scholar] [CrossRef]
- Zeid, J.L.; Charrow, J.; Sandu, M.; Goldman, S.; Listernick, R. Orbital optic nerve gliomas in children with neurofibromatosis type 1. J. AAPOS 2006, 10, 534–539. [Google Scholar] [CrossRef]
- Opocher, E.; Kremer, L.C.; Da Dalt, L.; van de Wetering, M.D.; Viscardi, E.; Caron, H.N.; Perilongo, G. Prognostic factors for progression of childhood optic pathway glioma: A systematic review. Eur. J. Cancer 2006, 42, 1807–1816. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef] [Green Version]
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet. Med. 2019, 21, 867–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Trevisson, E.; Morbidoni, V.; Forzan, M.; Daolio, C.; Fumini, V.; Parrozzani, R.; Cassina, M.; Midena, E.; Salviati, L.; Clementi, M. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol. Genet. Genom. Med. 2019, 7, e616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frayling, I.M.; Mautner, V.F.; van Minkelen, R.; Kallionpaa, R.A.; Aktaş, S.; Baralle, D.; Ben-Shachar, S.; Callaway, A.; Cox, H.; Eccles, D.M.; et al. Breast cancer risk in neurofibromatosis type 1 is a function of the type of. J. Med. Genet. 2019, 56, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Sharif, S.; Upadhyaya, M.; Ferner, R.; Majounie, E.; Shenton, A.; Baser, M.; Thakker, N.; Evans, D.G. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet. 2011, 48, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Bolcekova, A.; Nemethova, M.; Zatkova, A.; Hlinkova, K.; Pozgayova, S.; Hlavata, A.; Kadasi, L.; Durovcikova, D.; Gerinec, A.; Husakova, K.; et al. Clustering of mutations in the 5’ tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma 2013, 60, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Hutter, S.; Piro, R.M.; Waszak, S.M.; Kehrer-Sawatzki, H.; Friedrich, R.E.; Lassaletta, A.; Witt, O.; Korbel, J.O.; Lichter, P.; Schuhmann, M.U.; et al. No correlation between NF1 mutation position and risk of optic pathway glioma in 77 unrelated NF1 patients. Hum. Genet. 2016, 135, 469–475. [Google Scholar] [CrossRef]
- Anastasaki, C.; Morris, S.M.; Gao, F.; Gutmann, D.H. Children with 5’-end. Neurol. Genet. 2017, 3, e192. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Xiong, H.; Han, Y.; Li, C.; Mai, S.; Huang, Z.; Ai, X.; Guo, Z.; Zeng, F.; Guo, Q. Identification of Mutation Regions on. Front. Genet. 2018, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Gao, F.; Gutmann, D.H. Commentary: Identification of Mutation Regions on. Front. Genet. 2019, 10, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.; Listernick, R.; Charrow, J.; Piersall, L.; Gutmann, D.H. Optic pathway gliomas in neurofibromatosis type 1: The effect of presenting symptoms on outcome. Am. J. Med. Genet. A 2003, 122A, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Listernick, R.; Charrow, J. Knowledge without truth: Screening for complications of neurofibromatosis type 1 in childhood. Am. J. Med. Genet. A 2004, 127A, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Schneider, K.W.; Scott, H.S.; Plon, S.E.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 1. Clin. Cancer Res. 2017, 23, e46–e53. [Google Scholar] [CrossRef] [PubMed]
- Parrozzani, R.; Clementi, M.; Kotsafti, O.; Miglionico, G.; Trevisson, E.; Orlando, G.; Pilotto, E.; Midena, E. Optical coherence tomography in the diagnosis of optic pathway gliomas. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8112–8118. [Google Scholar] [CrossRef] [Green Version]
- Parrozzani, R.; Miglionico, G.; Leonardi, F.; Pulze, S.; Trevisson, E.; Clementi, M.; Opocher, E.; Licata, V.; Viscardi, E.; Pilotto, E.; et al. Correlation of peripapillary retinal nerve fibre layer thickness with visual acuity in paediatric patients affected by optic pathway glioma. Acta Ophthalmol. 2018, 96, e1004–e1009. [Google Scholar] [CrossRef] [Green Version]
- De Blank, P.M.K.; Fisher, M.J.; Liu, G.T.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Avery, R.A. Optic Pathway Gliomas in Neurofibromatosis Type 1: An Update: Surveillance, Treatment Indications, and Biomarkers of Vision. J. Neuroophthalmol. 2017, 37 (Suppl. 1), S23–S32. [Google Scholar] [CrossRef]
- Avery, R.A.; Bouffet, E.; Packer, R.J.; Reginald, A. Feasibility and comparison of visual acuity testing methods in children with neurofibromatosis type 1 and/or optic pathway gliomas. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1034–1038. [Google Scholar] [CrossRef] [Green Version]
- Anstice, N.S.; Thompson, B. The measurement of visual acuity in children: An evidence-based update. Clin. Exp. Optom. 2014, 97, 3–11. [Google Scholar] [CrossRef]
- Campagna, M.; Opocher, E.; Viscardi, E.; Calderone, M.; Severino, S.M.; Cermakova, I.; Perilongo, G. Optic pathway glioma: Long-term visual outcome in children without neurofibromatosis type-1. Pediatr. Blood Cancer 2010, 55, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.P.; Weiss, A.H. Detection of tumor progression in optic pathway glioma with and without neurofibromatosis type 1. Neuro Oncol. 2013, 15, 1560–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassiman, C.; Laenen, A.; Jacobs, S.; Demaerel, P.; Legius, E.; Casteels, I. Ophthalmological examination in neurofibromatosis type 1: A long-term retrospective analysis. Acta Ophthalmol. 2018, 96, e1044–e1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, D.L.; Beiser, A.S.; Warner, A.F.; Pratt, E.M.; Raye, K.N.; Lang, J.M. Monocular acuity norms for the Teller Acuity Cards between ages one month and four years. Investig. Ophthalmol. Vis. Sci. 1995, 36, 671–685. [Google Scholar]
- Hargadon, D.D.; Wood, J.; Twelker, J.D.; Harvey, E.M.; Dobson, V. Recognition acuity, grating acuity, contrast sensitivity, and visual fields in 6-year-old children. Arch. Ophthalmol. 2010, 128, 70–74. [Google Scholar] [CrossRef] [Green Version]
- Becker, R.; Hübsch, S.; Gräf, M.H.; Kaufmann, H. Examination of young children with Lea symbols. Br. J. Ophthalmol. 2002, 86, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.I.; Chandna, A.; Norcia, A.M.; Pettet, M.; Stone, D. The repeatability of best corrected acuity in normal and amblyopic children 4 to 12 years of age. Investig. Ophthalmol. Vis. Sci. 2006, 47, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Drover, J.R.; Felius, J.; Cheng, C.S.; Morale, S.E.; Wyatt, L.; Birch, E.E. Normative pediatric visual acuity using single surrounded HOTV optotypes on the Electronic Visual Acuity Tester following the Amblyopia Treatment Study protocol. J. AAPOS 2008, 12, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tarczy-Hornoch, K.; Cotter, S.A.; Wen, G.; Borchert, M.S.; Azen, S.P.; Varma, R.; Group, M.-E.P.E.D.S. Visual acuity norms in pre-school children: The Multi-Ethnic Pediatric Eye Disease Study. Optom. Vis. Sci. 2009, 86, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Listernick, R.; Charrow, J.; Gutmann, D.H. Comments on neurofibromatosis 1 and optic pathway tumors. Am. J. Med. Genet. 2001, 102, 105. [Google Scholar] [CrossRef]
- Beres, S.J. Optic Pathway Gliomas in Neurofibromatosis Type 1: Imaging and Monitoring. Int. Ophthalmol. Clin. 2018, 58, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Fried, I.; Tabori, U.; Tihan, T.; Reginald, A.; Bouffet, E. Optic pathway gliomas: A review. CNS Oncol. 2013, 2, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Avery, R.A.; Fisher, M.J.; Liu, G.T. Optic pathway gliomas. J. Neuroophthalmol. 2011, 31, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert-Boire, V.; Rosca, L.; Samson, Y.; Ospina, L.H.; Perreault, S. Clinical Presentation and Outcome of Patients with Optic Pathway Glioma. Pediatr. Neurol. 2017, 75, 55–60. [Google Scholar] [CrossRef]
- Dotan, G.; Qureshi, H.M.; Toledano-Alhadef, H.; Azem, N.; Yahalom, C. Prevalence of Strabismus Among Children With Neurofibromatosis Type 1 Disease With and Without Optic Pathway Glioma. J. Pediatr. Ophthalmol. Strabismus. 2019, 56, 19–22. [Google Scholar] [CrossRef]
- Yanni, S.E.; Wang, J.; Cheng, C.S.; Locke, K.I.; Wen, Y.; Birch, D.G.; Birch, E.E. Normative reference ranges for the retinal nerve fiber layer, macula, and retinal layer thicknesses in children. Am. J. Ophthalmol. 2013, 155, 354–360.e351. [Google Scholar] [CrossRef]
- Avery, R.A.; Mansoor, A.; Idrees, R.; Trimboli-Heidler, C.; Ishikawa, H.; Packer, R.J.; Linguraru, M.G. Optic pathway glioma volume predicts retinal axon degeneration in neurofibromatosis type 1. Neurology 2016, 87, 2403–2407. [Google Scholar] [CrossRef] [Green Version]
- Avery, R.A.; Cnaan, A.; Schuman, J.S.; Trimboli-Heidler, C.; Chen, C.L.; Packer, R.J.; Ishikawa, H. Longitudinal Change of Circumpapillary Retinal Nerve Fiber Layer Thickness in Children With Optic Pathway Gliomas. Am. J. Ophthalmol. 2015, 160, 944–952.e941. [Google Scholar] [CrossRef] [Green Version]
- Sahinoglu-Keskek, N.; Altan-Yaycioglu, R.; Canan, H.; Coban-Karatas, M.; Erbay, A.; Yazıcı, N.; Alkan, O. Measurements of Retinal Nerve Fiber Thickness and Ganglion Cell Complex in Neurofibromatosis Type 1, with and Without Optic Pathway Gliomas: A Case Series. Curr. Eye Res. 2018, 43, 424–427. [Google Scholar] [CrossRef]
- Gu, S.; Glaug, N.; Cnaan, A.; Packer, R.J.; Avery, R.A. Ganglion cell layer-inner plexiform layer thickness and vision loss in young children with optic pathway gliomas. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1402–1408. [Google Scholar] [CrossRef] [Green Version]
- Parrozzani, R.; Clementi, M.; Frizziero, L.; Miglionico, G.; Perrini, P.; Cavarzeran, F.; Kotsafti, O.; Comacchio, F.; Trevisson, E.; Convento, E.; et al. In Vivo Detection of Choroidal Abnormalities Related to NF1: Feasibility and Comparison With Standard NIH Diagnostic Criteria in Pediatric Patients. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6036–6042. [Google Scholar] [CrossRef] [PubMed]
- Alpfidan, I.; Sakarya, Y.; Goktas, S.; Ozcimen, M.; Sakarya, R. Ophthalmological assessment of children with neurofibromatosis type 1. Eur. J. Pediatr. 2014, 173, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrozzani, R.; Leonardi, F.; Frizziero, L.; Trevisson, E.; Clementi, M.; Pilotto, E.; Fusetti, S.; Miglionico, G.; Midena, E. Retinal Vascular and Neural Remodeling Secondary to Optic Nerve Axonal Degeneration: A Study Using OCT Angiography. Ophthalmol. Retina. 2018, 2, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Parrozzani, R.; Pilotto, E.; Clementi, M.; Frizziero, L.; Leonardi, F.; Convento, E.; Miglionico, G.; Pulze, S.; Perrini, P.; Trevisson, E.; et al. Retinal vascular abnormalities in a large cohort of patients affected by neurofibromatosis type 1: A study using optical coherence tomography angiography. Retina 2018, 38, 585–593. [Google Scholar] [CrossRef]
- Freret, M.E.; Gutmann, D.H. Insights into optic pathway glioma vision loss from mouse models of neurofibromatosis type 1. J. Neurosci. Res. 2019, 97, 45–56. [Google Scholar] [CrossRef]
- Jacks, T.; Shih, T.S.; Schmitt, E.M.; Bronson, R.T.; Bernards, A.; Weinberg, R.A. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994, 7, 353–361. [Google Scholar] [CrossRef]
- Bajenaru, M.L.; Hernandez, M.R.; Perry, A.; Zhu, Y.; Parada, L.F.; Garbow, J.R.; Gutmann, D.H. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003, 63, 8573–8577. [Google Scholar]
- Zhu, Y.; Harada, T.; Liu, L.; Lush, M.E.; Guignard, F.; Harada, C.; Burns, D.K.; Bajenaru, M.L.; Gutmann, D.H.; Parada, L.F. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development 2005, 132, 5577–5588. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, B.; Dasgupta, B.; Shin, J.E.; Emnett, R.J.; Hart-Mahon, E.K.; Elghazi, L.; Bernal-Mizrachi, E.; Gutmann, D.H. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 2007, 1, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Solga, A.C.; Toonen, J.A.; Pan, Y.; Cimino, P.J.; Ma, Y.; Castillon, G.A.; Gianino, S.M.; Ellisman, M.H.; Lee, D.Y.; Gutmann, D.H. The cell of origin dictates the temporal course of neurofibromatosis-1 (Nf1) low-grade glioma formation. Oncotarget 2017, 8, 47206–47215. [Google Scholar] [CrossRef]
- Hegedus, B.; Banerjee, D.; Yeh, T.H.; Rothermich, S.; Perry, A.; Rubin, J.B.; Garbow, J.R.; Gutmann, D.H. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008, 68, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Toonen, J.A.; Cimino, P.J.; Gianino, S.M.; Gutmann, D.H. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol. 2015, 17, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See, W.L.; Tan, I.L.; Mukherjee, J.; Nicolaides, T.; Pieper, R.O. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res. 2012, 72, 3350–3359. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Gianino, S.M.; Gutmann, D.H. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci. 2010, 30, 5579–5589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daginakatte, G.C.; Gutmann, D.H. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum. Mol. Genet. 2007, 16, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Pong, W.W.; Higer, S.B.; Gianino, S.M.; Emnett, R.J.; Gutmann, D.H. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann. Neurol. 2013, 73, 303–308. [Google Scholar] [CrossRef]
- Solga, A.C.; Pong, W.W.; Kim, K.Y.; Cimino, P.J.; Toonen, J.A.; Walker, J.; Wylie, T.; Magrini, V.; Griffith, M.; Griffith, O.L.; et al. RNA Sequencing of Tumor-Associated Microglia Reveals Ccl5 as a Stromal Chemokine Critical for Neurofibromatosis-1 Glioma Growth. Neoplasia 2015, 17, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Pan, Y.; Gutmann, D.H. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro Oncol. 2019. [Google Scholar] [CrossRef]
- Toonen, J.A.; Ma, Y.; Gutmann, D.H. Defining the temporal course of murine neurofibromatosis-1 optic gliomagenesis reveals a therapeutic window to attenuate retinal dysfunction. Neuro Oncol. 2017, 19, 808–819. [Google Scholar] [CrossRef]
- Isakson, S.H.; Rizzardi, A.E.; Coutts, A.W.; Carlson, D.F.; Kirstein, M.N.; Fisher, J.; Vitte, J.; Williams, K.B.; Pluhar, G.E.; Dahiya, S.; et al. Genetically engineered minipigs model the major clinical features of human neurofibromatosis type 1. Commun. Biol. 2018, 1, 158. [Google Scholar] [CrossRef] [Green Version]
- Wegscheid, M.L.; Anastasaki, C.; Gutmann, D.H. Human stem cell modeling in neurofibromatosis type 1 (NF1). Exp. Neurol. 2018, 299, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, F.; Ceccarelli, M.; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; Berzero, G.; et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Gnekow, A.K.; Kandels, D.; van Tilburg, C.; Azizi, A.A.; Opocher, E.; Stokland, T.; Driever, P.H.; Meeteren, A.Y.N.S.; Thomale, U.W.; Schuhmann, M.U.; et al. SIOP-E-BTG and GPOH Guidelines for Diagnosis and Treatment of Children and Adolescents with Low Grade Glioma. Klin. Padiatr. 2019, 231, 107–135. [Google Scholar] [CrossRef] [PubMed]
- Ater, J.L.; Xia, C.; Mazewski, C.M.; Booth, T.N.; Freyer, D.R.; Packer, R.J.; Sposto, R.; Vezina, G.; Pollack, I.F. Nonrandomized comparison of neurofibromatosis type 1 and non-neurofibromatosis type 1 children who received carboplatin and vincristine for progressive low-grade glioma: A report from the Children’s Oncology Group. Cancer 2016, 122, 1928–1936. [Google Scholar] [CrossRef]
- De Blank, P.M.; Berman, J.I.; Fisher, M.J. Systemic Chemotherapy and White Matter Integrity in Tracts Associated with Cognition Among Children with Neurofibromatosis Type 1. Pediatr. Blood Cancer 2016, 63, 818–824. [Google Scholar] [CrossRef]
- Liu, G.T.; Brodsky, M.C.; Phillips, P.C.; Belasco, J.; Janss, A.; Golden, J.C.; Bilaniuk, L.L.; Burson, G.T.; Duhaime, A.C.; Sutton, L.N. Optic radiation involvement in optic pathway gliomas in neurofibromatosis. Am. J. Ophthalmol. 2004, 137, 407–414. [Google Scholar] [CrossRef]
- Moreno, L.; Bautista, F.; Ashley, S.; Duncan, C.; Zacharoulis, S. Does chemotherapy affect the visual outcome in children with optic pathway glioma? A systematic review of the evidence. Eur. J. Cancer 2010, 46, 2253–2259. [Google Scholar] [CrossRef]
- Dalla Via, P.; Opocher, E.; Pinello, M.L.; Calderone, M.; Viscardi, E.; Clementi, M.; Battistella, P.A.; Laverda, A.M.; Da Dalt, L.; Perilongo, G. Visual outcome of a cohort of children with neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol. 2007, 9, 430–437. [Google Scholar] [CrossRef]
- Shofty, B.; Ben-Sira, L.; Freedman, S.; Yalon, M.; Dvir, R.; Weintraub, M.; Toledano, H.; Constantini, S.; Kesler, A. Visual outcome following chemotherapy for progressive optic pathway gliomas. Pediatr Blood Cancer 2011, 57, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Jakacki, R.; Goldman, S.; Hargrave, D.; Hawkins, C.; Shroff, M.; Hukin, J.; Bartels, U.; Foreman, N.; Kellie, S.; et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J. Clin. Oncol. 2012, 30, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, A.; Scheinemann, K.; Zelcer, S.M.; Hukin, J.; Wilson, B.A.; Jabado, N.; Carret, A.S.; Lafay-Cousin, L.; Larouche, V.; Hawkins, C.E.; et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. J. Clin. Oncol. 2016, 34, 3537–3543. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.J.; Jakacki, R.; Horn, M.; Rood, B.; Vezina, G.; MacDonald, T.; Fisher, M.J.; Cohen, B. Objective response of multiply recurrent low-grade gliomas to bevacizumab and irinotecan. Pediatr. Blood Cancer 2009, 52, 791–795. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- Duncan, K.E.; Chang, L.Y.; Patronas, M. MEK inhibitors: A new class of chemotherapeutic agents with ocular toxicity. Eye (Lond) 2015, 29, 1003–1012. [Google Scholar] [CrossRef]
- Maubon, L.; Hirji, N.; Petrarca, R.; Ursell, P. MEK inhibitors: A new class of chemotherapeutic agents with ocular toxicity. Eye (Lond) 2016, 30, 330. [Google Scholar] [CrossRef] [Green Version]
- Falsini, B.; Chiaretti, A.; Rizzo, D.; Piccardi, M.; Ruggiero, A.; Manni, L.; Soligo, M.; Dickmann, A.; Federici, M.; Salerni, A.; et al. Nerve growth factor improves visual loss in childhood optic gliomas: A randomized, double-blind, phase II clinical trial. Brain 2016, 139, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D.; Gutmann, D.H.; Rubin, J.B. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Test | Authors | Age (Years) | Mean Visual Acuity | logMAR Equivalent | Approximate Snellen Fraction |
|---|---|---|---|---|---|
| Teller acuity cards | Mayer et al. [64] | (1 month) | 1 cycle per degree | 1.5 | 6/180 |
| (6 months) | 6 cycles per degree | 0.70 | 6/30 | ||
| 4 | 25 cycles per degree | 0.07 | 6/7 | ||
| Hargdon et al. [65] | 5–6 | 24.5 cycles per degree | 0.10 | 6/7.5 | |
| Single Lea symbol | Becker et al. [66] | 2–6 | 0.88 Snellen decimal | 0.07 | 6/7 |
| Crowded Lea symbols | Becker et al. [66] | 2–6 | 0.74 Snellen decimal | 0.12 | 6/8 |
| Chen et al. [67] | 4.5–8.5 | 0.08 ± 0.09 | 6/7.5 | ||
| HOTV crowded | Drover et al. [68] | 3–4 | 0.08 | 6/7.5 | |
| Drover et al. [68] | 5 | 0.03 | 6/6 | ||
| Drover et al. [68] | 6 | −0.03 | 6/6 | ||
| Pan et al. [69] | 3–4 | 0.17 ± 0.13 | 6/9 | ||
| 4–6 | 0.08 ± 0.11 | 6/7.5 |
| RNFL Thickness | Cut-off | SE | SP | PPV | NPP |
|---|---|---|---|---|---|
| RNFL thickness (G) | |||||
| Most sensitive | 88 µm | 100.0% | 55.8% | 54.8% | 100.0% |
| Best balanced | 76 µm | 91.3% | 76.7% | 67.7% | 94.3% |
| RNFL thickness (T) | |||||
| Most sensitive | 59 µm | 100.0% | 60.5% | 57.5% | 100.0% |
| Best balanced | 49 µm | 87.0% | 76.7% | 66.7% | 91.7% |
| RNFL thickness (S) | |||||
| Most sensitive | 115 µm | 100.0% | 41.9% | 47.9% | 100.0% |
| Best balanced | 95 µm | 87.0% | 81.4% | 71.4% | 92.1% |
| RNFL thickness (N) | |||||
| Most sensitive | 111 µm | 100.0% | 2.3% | 35.4% | 100.0% |
| Best balanced | 54 µm | 78.3% | 72.1% | 60.0% | 86.1% |
| RNFL thickness (I) | |||||
| Most sensitive | 117 µm | 100.0% | 51.2% | 52.3% | 100.0% |
| Best balanced | 99 µm | 87.0% | 79.1% | 69.0% | 91.9% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassina, M.; Frizziero, L.; Opocher, E.; Parrozzani, R.; Sorrentino, U.; Viscardi, E.; Miglionico, G.; Midena, E.; Clementi, M.; Trevisson, E. Optic Pathway Glioma in Type 1 Neurofibromatosis: Review of Its Pathogenesis, Diagnostic Assessment, and Treatment Recommendations. Cancers 2019, 11, 1790. https://doi.org/10.3390/cancers11111790
Cassina M, Frizziero L, Opocher E, Parrozzani R, Sorrentino U, Viscardi E, Miglionico G, Midena E, Clementi M, Trevisson E. Optic Pathway Glioma in Type 1 Neurofibromatosis: Review of Its Pathogenesis, Diagnostic Assessment, and Treatment Recommendations. Cancers. 2019; 11(11):1790. https://doi.org/10.3390/cancers11111790
Chicago/Turabian StyleCassina, Matteo, Luisa Frizziero, Enrico Opocher, Raffaele Parrozzani, Ugo Sorrentino, Elisabetta Viscardi, Giacomo Miglionico, Edoardo Midena, Maurizio Clementi, and Eva Trevisson. 2019. "Optic Pathway Glioma in Type 1 Neurofibromatosis: Review of Its Pathogenesis, Diagnostic Assessment, and Treatment Recommendations" Cancers 11, no. 11: 1790. https://doi.org/10.3390/cancers11111790