Balancing STAT Activity as a Therapeutic Strategy
by
 and
and
Kelsey L. Polak
1,†, 
Noah M. Chernosky
1,†,
Jacob M. Smigiel
1,
Ilaria Tamagno
1 and
Mark W. Jackson
1,2,* 1
Department of Pathology, Case Western Reserve University, School of Medicine, Cleveland, OH 44106, USA
2
Case Comprehensive Cancer Center, Case Western Reserve University, School of Medicine, Cleveland, OH 44106, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2019, 11(11), 1716; https://doi.org/10.3390/cancers11111716
Submission received: 17 September 2019
/
Revised: 23 October 2019
/
Accepted: 31 October 2019
/
Published: 3 November 2019
(This article belongs to the Special Issue Targeting STAT3 and STAT5 in Cancer)
Abstract
:Driven by dysregulated IL-6 family member cytokine signaling in the tumor microenvironment (TME), aberrant signal transducer and activator of transcription (STAT3) and (STAT5) activation have been identified as key contributors to tumorigenesis. Following transformation, persistent STAT3 activation drives the emergence of mesenchymal/cancer-stem cell (CSC) properties, important determinants of metastatic potential and therapy failure. Moreover, STAT3 signaling within tumor-associated macrophages and neutrophils drives secretion of factors that facilitate metastasis and suppress immune cell function. Persistent STAT5 activation is responsible for cancer cell maintenance through suppression of apoptosis and tumor suppressor signaling. Furthermore, STAT5-mediated CD4+/CD25+ regulatory T cells (Tregs) have been implicated in suppression of immunosurveillance. We discuss these roles for STAT3 and STAT5, and weigh the attractiveness of different modes of targeting each cancer therapy. Moreover, we discuss how anti-tumorigenic STATs, including STAT1 and STAT2, may be leveraged to suppress the pro-tumorigenic functions of STAT3/STAT5 signaling.
1. Introduction
A complex milieu of both cellular and non-cellular components creates a heterogeneous tumor and microenvironment [1,2,3,4,5,6,7,8]. As a tumor becomes more heterogeneous, the risk of metastasis and therapy failure increases [9,10,11,12]. Cancer cells, pericytes, immune cells, fibroblasts, and endothelial cells are just some of the cellular components of the tumor [13,14]. A highly dysregulated network of cytokines and growth factors, emanating from cancer and stromal tumor microenvironment (TME) cells, contributes to the evolution of cancer cells, and often, suppression of immune cell function. Many of these cytokines and growth factors result in the phosphorylation and activation of signal transducers and activators of transcription (STAT) proteins, which can drive cell-specific changes in gene expression. STAT3 and STAT5 activity is often elevated in aggressive subtypes of cancer and serve as prognostic indicators [15,16,17,18,19,20,21,22,23,24]. Here, we discuss the impact of microenvironmental signals on STAT3/STAT5 activation during cancer development and progression. Elevated STAT3 activity promotes epithelial-mesenchymal transition (EMT) and a stem cell program in cancer cells, while also suppressing the function of immune cells within the tumor, all of which are important steps that underlie metastasis and therapy failure [25,26,27,28,29,30]. Likewise, persistent STAT5 activity induces (i) transformation, (ii) proliferation, and (iii) anti-apoptotic signals that contribute to hematological malignancies, while also suppressing anti-tumor immunity by expanding CD4+/CD25+ regulatory T cells (Tregs) [28,31,32,33,34,35,36]. We will discuss options for targeting STAT3/5 in cancer, either directly or indirectly through the inhibition of upstream kinases, receptors and/or ligands. In addition, we will discuss how STAT1/2 activity can counter the more aggressive phenotypes induced by STAT3/5. We propose that balancing STAT-activated cytokine signaling in the TME may serve as an effective therapeutic strategy.
2. Activating STAT3 and STAT5
The STAT family of transcription factors is comprised of seven different members, STAT1, -2, -3, -4, -5a, -5b, and -6. Hereafter, when discussing the overlapping functions of STAT5A and STAT5B, we will refer to them as “STAT5”. STAT proteins transduce signals from the cell membrane to the nucleus, bypassing the need for second messengers [37,38,39]. STATs are phosphorylated by Janus Kinases (JAK1, 2, 3) or Tyrosine Kinase 2 (Tyk2), which are recruited to ligand-activated receptors, including cytokine, growth factor, or g-protein associated receptors. Most important of the STAT3/5 activators are the IL-6 family members, which include IL-6, IL-11, ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), oncostatin-m (OSM), cardiotrophin 1 (CT-1), cardiotrophin-like cytokine (CLC), and IL-27. IL-6 family cytokines form a heterodimeric complex consisting of gp130 and a cytokine specific subunit (IL-6Rα, IL-11Rα, CNTFRα, gp130, LIFR, and OSMR), recruiting and activating the JAKs or Tyk2. Once phosphorylated, STATs dimerize and translocate to the nucleus, where they bind to short stretches of DNA and act as transcription factors to induce the expression of genes implicated in cell proliferation, survival, differentiation, motility, apoptosis, and metabolism (Figure 1) [37,40,41,42,43].
The crucial role STATs play in these normal physiological processes was first demonstrated in STAT-deficient mouse models. The generation of tissue-specific STAT3 knockout models have identified STAT3 as a key component in a wide variety of processes, including, but not limited to: T-cell proliferation, suppression of apoptosis, epidermal regeneration during wound healing, macrophage and neutrophil anti-inflammatory responses, and mammary gland involution [44,45,46]. Deletion of STAT5 in mice demonstrated high incidence of perinatal lethality, prevented the appropriate development of B-cells and T-cells, and inhibited the function of hematopoietic stem cells [47]. Under normal conditions, JAK/STAT activation is tightly regulated by protein inhibitors of activated STATs (PIAS), tyrosine phosphatases, and suppressors of cytokine signaling (SOCS) proteins that inhibit JAK catalytic activity [48,49]. In cancer however, STAT3 and STAT5 activity becomes dysregulated, resulting in elevated STAT3/5-driven responses in tumor, stromal, and immune cells.
Advances in sequencing technologies have allowed scientists to investigate the frequency of mutations in the JAK/STAT signaling pathway, resulting in the identification of mutations that constitutively activate STAT3, STAT5, and JAK2. The majority of STAT mutations occur in the SH2 and C-terminal domains of STAT3 and STAT5B and associate with leukemias and lymphomas [50,51]. STAT3 is the most frequently mutated member of the STAT family, with high incidence of mutation in T-cell large granular lymphocytic leukemias and NK lymphoproliferative disorders [52,53]. STAT5B mutations were similarly identified in these diseases, but with lower frequency. In addition, sequencing of STAT3 exons in diffuse large B-cell lymphoma patients identified a missense point mutation (M206K) in the coiled-coil domain that affected SH2 domain function and drove robust proliferation [54,55]. Furthermore, JAK2 V617F, a constitutively active JAK2 mutant, phosphorylates both STAT3 and STAT5 proteins and has high frequency in patients with hematopoietic stem cell diseases, such as myeloproliferative diseases, essential thrombocythemia, polycythemia vera (PV), and idiopathic myelofibrosis (IMF) [56]. In addition, JAK2 V617F mutations have transformation potential in in vivo bone marrow transplantation assays and induce persistent activation of STAT5 and a PV phenotype [57]. While persistent activation of both STAT3 and STAT5 has been implicated in the transformation process, the greater impact of dysregulated STAT3/5 appears to be their influence on the induction of aggressive cancer cell properties and immunosuppression. The impacts of both cell-intrinsic reprogramming and immune dysfunction are discussed below.
3. IL-6 Family Cytokine Dysregulation in the TME
A number of IL-6 family members have long been recognized for their involvement in the pathogenesis of aggressive cancers [19,77,78,79,80,81]. For example, IL-6 levels serve as a prognostic biomarker and predictor of therapeutic response in pancreatic ductal adenocarcinoma (PDAC), bladder, gastric, lung adenocarcinoma, colorectal, cervical, liver, and breast cancers: all of which have a high incidence of metastasis and resistance [77,82,83,84,85,86]. This is due to the pro-tumor effects of IL-6 in tumor cells and stromal components. In tumor cells, IL-6 can drive EMT, therapy resistance, and invasive characteristics [87,88,89]. Concurrently, IL-6 can shift the anti-tumor immune responses towards immunosuppression via recruitment of myeloid-derived suppressor cells (MDSCs) and expansion of FoxP3+ Tregs [90,91,92]. Furthermore, IL-6 secretion by certain stromal components (cancer-associated fibroblasts, macrophages, and neutrophils) drives EMT in associated tumor populations, which is supported by immunohistochemical staining at the invasive edge of patient breast tumors, where levels of phosphorylated STAT3 and IL-6 is elevated [93,94].
Interestingly, IL-6 activation of STAT3 functions in a positive feed-forward loop to drive the secretion of new IL-6 into the TME, which then interacts with IL-6R/gp130 to further activate JAK/STAT signaling [93,95]. OSM also potently activates a feed-forward loop, resulting in the de novo production of additional OSM and OSMR by tumor and immune cells [96]. Interestingly, OSM was first identified to play a tumor suppressive role and inhibited the proliferation of melanoma cell models [97]. However, later studies correlated OSM-OSMR signaling with robust STAT3 and STAT5 activation and more aggressive tumor phenotypes [98,99,100,101,102,103,104,105,106]. Like IL-6, OSM feed-forward signaling is observed in aggressive cancers with limited-therapeutic options, including glioblastoma (GBM), non-small cell lung carcinoma (NSCLC), PDAC, and triple negative breast cancer (TNBC) [107,108,109,110,111]. Elevated signaling through the OSM/OSMR axis induces high levels of an ‘inflammatory module’, which includes IL-6, CCL2, IL-1, CXCL1, CXCL9, CXCL10, and CXCL11—all of which have been implicated in migration, invasion, therapy failure, and dedifferentiation to a cancer stem cell (CSC) program. Importantly, neutralization of OSM by treatment with an Fc-tagged soluble OSMR suppresses this inflammatory module suggesting that the OSM/OSMR feed-forward loop may be critical for the long-term maintenance of inflammatory signaling [112,113].
IL-11 similarly engages in an autocrine feed-forward mechanism to drive persistent JAK2/STAT3 activation, which can promote resistance to platinum-based therapies [114]. Furthermore, IL-11 and LIF both contribute to tumorigenesis by enhancing tumor cell survival through STAT3-mediated activation of anti-apoptotic proteins, Bcl-2 and Survivin, and the inhibition of tumor suppressor p53 [114,115,116]. IL-11 and LIF also contribute to cancer progression by driving EMT through STAT3 and Akt/mTOR signaling, thereby conferring an enhanced migratory capacity [81,117,118]. The impact of CT-1, CLC, CNTF, and IL-27 on cancer progression is currently understudied. While IL-6 family cytokines are important mediators of STAT3 activation, other cancer-associated receptors can also activate STAT3. STAT3-driven tumor progression can also be achieved by epidermal growth factor receptors (both wild-type EGFR and EGFRvIII), fibroblast growth factor receptors (FGFR), insulin-like growth factor receptor (IGFR), hepatocyte growth factor (HGFR, also known as MET), platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), v-src, and Bcr-Abl [119,120,121,122,123,124,125,126,127]. Interestingly, acetylation of STAT3 also induces tumor progression through enhanced pro-tumorigenic IL-17A secretion [128,129]. These examples demonstrate the abundance of mechanisms through which STAT3 can become activated.
4. STAT3 Activation of a Mesenchymal/CSC Program in Cancer Cells
The majority of cancers have constitutive activation of STAT3 [130,131,132]. STAT3 was first termed oncogenic when its persistent activation was discovered in v-src transformed mouse embryonic fibroblasts [133]. Subsequent studies demonstrated that expression of a constitutively active STAT3 isoform (STAT3-C) can drive transformation of pre-malignant human mammary epithelial cells (HMEC) and MCF-10A cells to malignant breast cancer [134]. In addition, Ras-induced transformation in bladder and breast carcinoma models exhibit mitochondrial accumulation of STAT3 and more robust cellular glycolysis, a characteristic of cancer cells [135]. In PDAC, STAT3 is required for both the development of pre-malignant pancreatic interepithelial neoplasias (PanINs), as well as the progression of PanINs to PDAC [136]. While STAT3 has been strongly implicated as a driver of oncogenesis, evidence suggests that persistent cytokine activation of STAT3 in pre-malignant lesions, engages a tumor suppressive senescence response. In non-transformed HMEC models, OSM engages senescence through a direct STAT3 interaction with mothers against decapentaplegic-3 (SMAD3). However, downstream constitutive expression of c-Myc could overcome OSM-induced senescence and drive EMT and invasion [137]. Beyond the genetic events that occur as a normal cell becomes transformed, cancer progression relies heavily on an evolving microenvironment, which impacts both the cancer cells and the immune system, ultimately influencing patient outcomes. In cancer cells, increased STAT3 activity induces EMT-driving transcription factors, such as ZEB1, SNAIL, and Twist, which initiate the repression of epithelial markers and expression of mesenchymal markers (N-cadherin, Vimentin) [138,139,140,141]. In addition, STAT3 induces the expression of matrix metalloproteinases (MMP), which can also contribute to the loss of cell-cell contacts [142,143,144].
While EMT is important in normal physiology to allow cells to migrate during development and wound healing, inappropriate EMT and the loss of cell-cell interactions contributes to aggressive cancer cell behaviors, including metastasis and resistance to therapy. In TNBC, the loss of E-cadherin or gain of Vimentin, N-Cadherin, or Snail expression, typically at the tumor’s edge, correlates with poor clinical outcome [145,146,147,148,149]. Likewise, the presence and abundance of circulating tumor cells (CTC) that express mesenchymal markers have been referred to as the “silent predictors of metastasis” because they correlate with tumor cell dissemination [150]. Moreover, mesenchymal CTC can be used to track a patient’s response to therapy, with increasing numbers correlating with increased risk of relapse [151,152,153,154,155].
Likewise, the emergence of CSC properties, which are often defined experimentally as using tumorsphere- or tumor-initiating assays, frequently occur concomitantly with a mesenchymal phenotype [156,157,158,159,160,161,162,163]. STAT3 has been identified as an essential driver of the de novo reprogramming to a CSC state through activation of Sox2, Nanog, and Oct4 [164,165,166,167,168,169,170]. Further evidence linking EMT and a CSC phenotype is provided by the observation that a subset of CTC, responsible for initiating metastasis termed the metastasis-initiating cells, express high levels of the STAT3-driven CSC marker CD44 [151,152,153,171,172,173,174]
The importance of mesenchymal/CSC reprogramming in metastasis and therapy failure continues to emerge. For example, single-cell analysis identified a predominantly mesenchymal/CSC program in early-stage TNBC micro-metastases, in contrast to late-stage metastases [154]. The findings are consistent with the idea that mesenchymal/CSC initiate metastatic outgrowth at a secondary site, followed by differentiation. Single cell RNA sequencing (scRNA-seq) confirmed that EMT in primary tumors proceeds through distinct, hybrid states, ranging from completely epithelial to completely mesenchymal [155]. The epithelial-mesenchymal hybrids, which harbor the greatest level of phenotypic plasticity, are more efficient at intravasating, surviving in circulation, extravasating to the lungs, and forming metastases [175]. Acute exposure to Adriamycin or Taxanes drives the adaptive emergence of therapy-resistant, CD44HIGH CSC in both breast tumor explants and breast cancer cell lines [176]. scRNA-seq determined that chemo-resistant cells activate an EMT program, which was not evident before treatment [177]. In mouse models, the ability to undergo EMT is important for therapeutic resistance [178,179]. These findings suggest that potent mesenchymal/CSC programming has significant consequences that allow cancer cells to adapt to chemotherapy and survive, ultimately contributing to therapy failure.
Indeed, STAT3 has also been implicated in acquired therapy resistance in many cancers including NSCLC, GBM, PDAC, melanoma, and breast cancer [107,180,181,182,183,184,185]. Elevated STAT3 phosphorylation in ovarian cancer is associated with paclitaxel resistance and increased tumor cell invasion post-therapy, consistent with the elevated expression of mesenchymal/CSC genes and increased tumor initiating potential [176,186,187]. In Trastuzumab-resistant HER2+ breast cancer, STAT3 feed-forward loops generate a TME rich with STAT3-activating cytokines that promote and maintain the mesenchymal/CSC phenotypes. Inhibition of the STAT3 feed-forward activation diminished tumor growth and metastasis and resensitized cells to therapy [188]. In cancer cells driven by diverse receptor tyrosine kinases (RTKs) (EGFR, HER2, ALK, and MET), MEK inhibitors drive feedback activation of STAT3 through FGFR and JAKs, resulting in therapy failure [189]. Likewise, in NSCLC cells resistant to molecularly-targeted therapies (EGFR-TKI, ALK inhibitor Crizotinib, and MEK inhibitor Selumetinib), OSM/JAK1/STAT3–signaling protects cells from targeted drug-induced apoptosis [190]. JAK/STAT3 signaling also interacts with numerous other growth, proliferative, and survival pathways, such as PI3K/Akt, MAPK, NF-kB, Notch, Wnt/β-Catenin, and TGF-β, among others [191,192,193,194,195,196]. Tamoxifen-resistant breast cancers have elevated STAT3 and Notch4 expression associated with metastasis and tumorigenicity. Interestingly, inhibition of Notch4 was able to reduce the phosphorylation of STAT3 and suppress metastasis, suggesting STAT3 and Notch4 cooperate to promote therapy resistance [197]. In addition, a STAT3/NF-kB complex promotes cisplatin resistance in malignant mesothelioma, and targeted inhibition of this complex inhibits the growth of refractory tumors [198]. Such evidence suggests STAT3 may not be solely responsible for therapy failure. Instead, STAT3-mediated cross-talk with additional signaling effectors may coordinate resistance [140,199,200,201]. Nonetheless, the common theme appears to be a STAT3-activated, mesenchymal/CSC program.
5. A STAT3-Generated Pro-Metastatic Immune Microenvironment
An important and rapidly emerging area of research is the impact of the immune microenvironment on metastasis and therapy failure. Beyond its impact on tumor cells, STAT3 has an important role in restricting immune cell functions and producing immunosuppressive factors. STAT3-induced cytokines are important mediators of the crosstalk between tumor cells, tumor-associated macrophages (TAMs), and tumor-associated neutrophils (TANs), which are responsible for generating a pro-metastatic and pro-angiogenic TME [202]. In the TME, macrophages and neutrophils are exposed to a host of STAT3-induced cytokines, such as IL-4, IL-6, IL-10, IL-13, VEGFA, and TGF-β that drive their polarization to a pro-tumorigenic M2 (macrophages) or N2 (neutrophils) state [203,204]. Conversely, robust STAT3 signaling activity in M2-TAMs and N2-TANs correlates with the production of factors able to drive cancer cell EMT (OSM, IL-6, TGF-β) and angiogenesis (VEGF, TGF-β, PDGF, and FGF) [205,206,207,208].
Importantly, because of their localization at the invasive edge of tumors, the contribution of M2-TAMs and N2-TANs to metastasis is increasingly being recognized. For example, TAMs have been demonstrated to form simultaneous physical contacts with tumor cells and endothelial cells that result in the formation of invadopodia, which assist cancer cells in transendothelial migration and escape into the circulatory system. These sites of cancer cell intravasation are called “tumor microenvironment of metastasis (TMEM)” and have been validated as prognostic markers of metastasis. Chemotherapy increases TMEM in breast cancer patients, thereby potentially facilitating metastasis [209,210]. STAT3 inhibition in TAM populations re-sensitizes breast cancer cells to paclitaxel, further suggesting tumor and TAM crosstalk is an essential component in STAT3-driven therapy resistance [205,211]. TANs, on the other hand, were found to function as circulatory escorts of CTC and promote their proliferation, survival, and seeding of secondary sites [212,213,214]. Importantly, OSM and IL-6 were two of the 4 most frequent cytokines secreted by neutrophils found clustered with CTC. These cytokines interact with OSMR and IL-6R, which are expressed on cancer cells [212]. The neutrophil/CTC cross-talk promotes the cell cycle progression in CTC, thereby expanding their metastatic potential.
Following the classical events of the metastatic cascade, after a tumor cell intravasates into circulation, it extravasates to establish secondary sites. An emerging concept in the metastatic cascade is the role of myeloid cells- such as basophils, neutrophils, eosinophils, monocytes, and macrophages- in the establishment of a pre-metastatic niche [215]. Although the exact TME components required for colonization of secondary sites remain a mystery, the activation of a STAT3-sphingosine 1-phosphate receptor-1 (S1PR1) axis in tumor cells, and secretion of IL-6 and IL-10, was observed to persistently activate STAT3 at distant, pre-metastatic sites. At these pre-metastatic sites, persistent STAT3 phosphorylation was associated with myeloid cell migration from the primary tumor to the secondary site. Subsequent targeting of STAT3 in myeloid cells disrupted metastatic tumor outgrowth, suggesting STAT3 plays an integral role in priming distant metastatic sites for tumor cell outgrowth [216].
6. STAT5 in Cancer Cells
As previously mentioned, cells produce two different STAT5 proteins, STAT5A and STAT5B, which share greater than 90% sequence homology [217]. However, evidence suggests STAT5A and STAT5B play different functional roles in normal and cancer cell systems. Genetic deletion of STAT5 in pure mouse backgrounds are embryonic lethal, due to the essential roles of STAT5 in erythropoiesis and iron metabolism [218,219,220]. While the distinct roles of STAT5A and STAT5B remain understudied, data from mammary gland-specific knockouts suggest that STAT5A is required for lactogenesis in the mammary gland, while STAT5B is imperative for mammary gland differentiation and development [221]. The function of STAT5A and STAT5B appear to be cell-specific. They can have either synergistic or opposing effects, such as in memory B-cell differentiation, which may be due to (i) the formation of STAT5A/STAT5B homo- or hetero-dimers, and/or (ii) differences in nuclear shuttling mechanisms [222,223,224]. Furthermore, genetic tuning models depleting STAT5A and/or STAT5B have demonstrated a critical role for STAT5 in the accumulation and development of innate lymphoid cells, such as NK cells [225,226,227].
Given its role in lymphoid cell development and differentiation, it is not surprising that STAT5 activity contributes to hematologic malignancies. Deletion of STAT5 prevents transformation by the Abl oncogene, thereby preventing leukemia development [228]. Genetic and pharmacologic inhibition of STAT5 activity decreases expression of apoptosis inhibitors MCL1 and BCL2 and inhibits leukemogenesis of BCR-ABL1+ acute lymphoblastic leukemia (ALL), both in cell lines and newly diagnosed and relapsed/TKI-resistant ALL patients [229]. Likewise, a new, STAT5 inhibitor suppressed the proliferation of human acute myeloid leukemia (AML) cell lines and primary FLT3-ITD+ AML patient cells [74]. Combined inhibition of STAT3 and STAT5 by shRNAs also suppressed growth in chronic myeloid leukemia, suggesting that combinatorial suppression of STAT3 and STAT5 may be efficacious in treating hematological malignancies [230].
STAT5 activation has also been implicated in the progression of solid tumor malignancies. Deletion of STAT5 in the mammary gland, hepatocytes, and prostate cells delays the development of mammary, liver, and prostate cancer [32,231,232]. Like STAT3, experimental evidence implicates STAT5B as a driver of tumorigenesis, as it can drive EMT and increased invasiveness in hepatocellular carcinoma (HCC) [233]. Moreover, in mammary epithelial cells, thymocytes, and epithelial prostate cancer cells, persistent activation of STAT5 is sufficient to drive transformation [234,235,236]. Furthermore, the function of STAT5 in solid tumors extends beyond oncogenesis as evidence has emerged that STAT5 signaling can induce a metastatic cascade. For example, STAT5 inhibition in colorectal cancer induces G1 cell cycle arrest and reduces cancer cell migration, demonstrating the role of STAT5 in proliferation and metastasis [237]. Additional studies demonstrate that STAT5A/B signaling in prostate cancer and squamous cell carcinoma of the head and neck drive EMT programming, which results in enhanced cell migration, invasion, and formation of metastases [238,239].
An increasing number of reports demonstrate that STAT5 drives tumorigenesis and cancer progression through cooperation with other intracellular signaling cascades and activation of additional feed-forward loops. STAT5 activates transcription of AKT1 and PI3K, and, in turn, Akt1 phosphorylates STAT5 to induce cell survival [240,241]. Furthermore, STAT5-dependent Akt restores cyclin D expression, which promotes proliferation [241]. Once cells aberrantly proliferate, apoptosis suppressors Bcl-xL and Bcl-2 are activated via persistent STAT5 signaling, driving tumor cell survival [16,234,242,243,244]. BCL-XL expression is also enhanced via formation of a transcription factor complex comprised of phosphorylated STAT5 and nuclear EGFRvIII. This transcription factor complex binds to the BCL-XL promoter to induce its transcription and promote anti-apoptotic signaling [245]. Similar to STAT3, STAT5 translocates to tumor cell mitochondria, suggesting an interaction with the mitochondrial genome to promote aerobic glycolysis (the Warburg Effect), a defining characteristic of cancer cells [246]. Collectively, this data demonstrates that STAT5 mediates crosstalk between cancer cell survival, and proliferation, and metabolism signaling pathways.
Surprisingly, STAT5 in certain model systems has been demonstrated to function in a tumor suppressive manner. Human breast cancers infrequently (~7%) show signs of STAT5 activation (compared to 40% of STAT3 activation). This elevated STAT5 activity trends with more differentiated and lower grade tumors, suggesting that STAT5 does not induce the aggressive cancer cell program initiated by STAT3, at least in breast cancer [247]. STAT5 expression in these models stabilizes E-cadherin surface marker expression and reverses the undifferentiated mesenchymal phenotype [248]. In normal human fibroblasts, aberrant activation of STAT5A induces a senescence response concurrent with accumulation of p53 and DNA damaged foci. Furthermore, knockdown DNA-repair kinase, ATM, and tumor suppressor, retinoblastoma protein did not eliminate damaged foci, providing evidence for the persistence of DNA damage in pre-malignant lesions [249]. Interestingly in HCC models, liver specific STAT5 knockout results in tumor formation through the enhanced activation of TGFβ/STAT3 signaling [20]. Physiologically, STAT proteins have been identified as drivers of erythropoiesis. STAT5A/B double knockout mice in a mixed genetic background (Sv129 x C57Bl/6) results in mild hematopoietic phenotypes, due to compensatory activation and enhanced DNA-binding of STAT1/3 [218].
7. STAT5 in Treg-Associated Immunosuppression
In addition to its impact on cancer cells, activated STAT5 dampens anti-tumor immune function. This immunosuppressive function is largely driven by CD4+/CD25+ Tregs, a subset of T cells that contribute to tumor progression and metastasis [250] and correlates with poor patient prognosis [251,252]. Sustained STAT5 phosphorylation in progenitor T cells induces the differentiation to a Treg population that, in turn, significantly diminishes the function of cytotoxic and helper T cells [253,254,255]. Experimental depletion of Tregs from the TME results in enhanced infiltration of mature CD4+ and CD8+ T cells into the tumor, leading to tumor rejection [256,257]. Furthermore, STAT5-mediated Treg expansion increases IL-10, IL-4, and IL-13, which skew TAMs to an M2 immunosuppressive phenotype. M2 macrophages are immunosuppressive because they release elevated levels of IL-10 and transforming growth factor-β (TGF-β) and restrict secretion of immune stimulatory cytokines via NF-kB repression [258,259]. STAT5-induced Tregs and M2 macrophage populations secrete VEGF-A, which, along with TGF-β, promotes angiogenesis [255,260,261].
Tumor clearance mechanisms are also suppressed by Treg expansion through impediment of B cell development and maturation [262]. STAT5-activated Tregs result in a decrease in follicular helper T (Tfh) cell populations via Blimp-1, which severely hampers germinal center formation in lymph nodes [263]. This reduction in germinal centers diminishes the number of B cells that can be recruited and primed to aid in an anti-tumor immune response. Interestingly, the differentiation and self-renewal of memory B cells are respectively influenced by STAT5A-mediated repression and STAT5B-mediated induction of BCL-6. Immunosuppression driven by the expansion of Tregs, relies on STAT5-mediated alteration in T-cell metabolism. Mature effector T cells preferentially undergo glycolysis and require a de novo fatty acid synthesis reliant on acetyl-coA carboxylase 1 whereas Tregs undergo lipid-oxidation and readily synthesize fatty acids because of a structural reconfiguration of mitochondrial cristae [264,265,266]. Accumulation of intracellular lipids impairs autophagy, providing a mechanism for Treg-mediated immunosuppression [267,268]. Just as STAT5 function in the mitochondria impacts tumor cell functions, as described earlier, mitochondrial STAT5 activation drives metabolic shifts in the immune compartment, inducing an expansion of Tregs.
However, while the suppression of Tregs increases tumor immunity, provided STAT5 remains functional, the overall impact of suppressing STAT5 signaling in the remaining immune cells of the TME remains open to debate. Mouse models deficient in STAT5 have depleted CD8+ T cell, NK cell, and Treg populations, suggesting STAT5 plays an integral role in the proper development of multiple immune cell types [269]. STAT5 signaling contributes to the differentiation of naive CD4+ T cells into CD8+ T cells, Th1, Th2, Th9, ThGM, and Tregs, while inhibition of STAT5 is required for the generation of Th17 and Tfh cells [270,271]. Importantly, STAT5 is heavily involved in the development, survival, and lytic function of NK cells. Knock-out or suppression of STAT5 in NK populations sparked the secretion of VEGF-A, a growth factor that supports tumor-associated angiogenesis in melanoma and leukemia models [269,272]. Taken together, these findings suggest that targeting STAT5 may threaten the integrity of anti-tumor immune functions and drive worse outcome in patients [269,272].
8. Targeting STAT Activity
Given the roles of STAT3 and STAT5 in tumor progression and immunosuppression discussed above (Figure 2), multiple methods of inhibiting their activity are being pursued. Successful inhibition of STAT3 would prevent the acquisition of, and potentially revert, a mesenchymal/CSC program, making cancer cells less invasive and more sensitive to therapy. Moreover, STAT3 inhibition would help activate anti-tumor immunity by reducing immunosuppressive factors and increasing the infiltration of immune cells into the TME. Likewise, suppressing STAT5 in cancer cells, particularly leukemia, halts proliferation and induces apoptosis, suggesting that STAT5 may be a valuable therapeutic target [74]. However, due to STAT5′s controversial roles in tumor progression and immune cell maturation and differentiation, further studies are required to elucidate the effects of targeting STAT5 in cancer patients. [273]. Generally speaking, the majority of small molecule inhibitors are designed to have high affinity for the catalytic domain of an enzyme; the ATP binding site for example, which STAT proteins lack. Therefore, direct inhibition of STAT3 relies on disruption of binding motifs necessary for downstream signal transduction. A series of STAT3 phospho-ester SH-2 domain inhibitors have been developed, with the intent to inhibit dimerization of activated STAT3 (PY*L, S3I-2001, OPB-31121, etc.). While these approaches are promising, the compounds continue to be refined [274,275,276,277]. An alternative approach would involve targeting the upstream activator of STAT3, which could include neutralizing antibodies for specific IL-6 family cytokines, competitive antibodies hindering cytokine-receptor interactions, and JAK inhibitors.
One of the first IL-6 monoclonal antibodies generated was Tocilizumab, which inhibits IL-6 signaling by preventing IL-6 binding to both the soluble and transmembrane forms of IL-6R [278,279]. Pre-clinical studies demonstrated strong anti-tumor cell activity in multiple myeloma and therapeutic efficacy in Castelman’s disease, rheumatoid arthritis, and cytokine release syndrome, however, Tocilizumab is not currently FDA-approved for cancer treatments [280,281,282,283]. Clinical trials using Tocilizumab in combination with other monoclonal antibodies, chemotherapies, and immunotherapies to treat cancers such as HER2+ breast cancer, B-cell Non-Hodgkin Lymphoma, metastatic NSCLC, and recurrent metastatic colorectal adenocarcinoma are ongoing [284]. In addition to Tocilizumab, Siltuximab is an IL-6 specific neutralizing antibody that has emerged as another promising therapy. Pre-clinical studies support Siltuximab use in KRAS-mutant lung cancer, particularly in tumors with elevated stromal production of IL-6 [285]. Similar to Tocilizumab, Siltuximab is undergoing clinical trials for patients with malignant solid tumors in cancers with elevated stromal presence, such as ovarian, pancreatic, colorectal, head and neck, and lung neoplasms [286]. Furthermore, phase II studies of combination therapy with Siltuximab have demonstrated anti-tumor effects for patients diagnosed with metastatic prostate cancer, suggesting clinical efficacy of targeting the IL-6-STAT signaling axis with combination therapies [287].
Our lab has focused on OSM, given its potent activity at inducing numerous inflammatory cytokines that promote mesenchymal/CSC reprogramming in cancer cells and generate a pro-tumor immune microenvironment [106,288,289,290]. Neither OSM nor OSMR protein are abundantly expressed in normal tissues in the absence of inflammation, in contrast to JAK1, JAK2, and STAT3 (key OSM effectors) [109]. This finding is supported by the observation that knocking out OSM or OSMR from mice results in only mild phenotypes [291]. Moreover, OSMR has characteristics unique from other IL-6 family co-receptors, resulting in distinct signaling and biological effects [43,292,293]. For example, OSMR strongly recruits SHC, resulting in the hyper-activation of the MAPK signaling cascade [293]. Other gp130 co-receptors fail to do this, and rely solely on the gp130-mediated SHP-2 recruitment for MAPK activation, which is less robust than the gp130/OSMR heterodimer [43]. Therefore, suppressing the OSM signaling axis may have benefits in aggressive subtypes of cancer. Anti-OSM antibody (GSK315234) and OSMR-fusion protein (OR-FC) were first examined in pre-clinical studies in chronic inflammatory disease models, such as rheumatoid arthritis and inflammatory heart disease [294,295]. Antibodies suppressing OSM signaling ameliorate these pathological conditions, suggesting that OSM is a driver of these chronic hyper-inflamed states. More recently, OSM signaling was identified as a driver of inflammatory bowel disease (IBD), especially in patients who fail to respond to anti-tumor necrosis factor-α (TNF-α) antibodies. Neutralization of OSM in IBD models suppresses a cadre of inflammatory cytokines and reduces colitis severity, further supporting the OSM feed-forward loop as a critical mediator in the long-term maintenance of inflammatory signaling, a state common in the TME [112].
Another recent study investigating the effects of an anti-OSM antibody in a murine model of lupus nephritis demonstrated that OSM-driven EMT and extracellular matrix secretion, leading to renal fibrosis, could be suppressed, concomitant with the suppression of JAK/STAT3 activation [296]. More recently, a clinical grade OSM neutralizing antibody was used to treat pre-clinical models of squamous cell carcinoma. Again, OSM neutralizing antibodies suppressed the STAT3 feed forward signaling, resulting in reduced invasion and metastasis [96,297]. Disrupting cell surface receptor activation of STAT3 by inhibiting OSMR signaling was recently described in a study of an OSMR/gp130 antagonist (SN79), which prevents STAT3 phosphorylation in astrogliosis [298]. While these studies demonstrate the potential of OSM and OSMR inhibition as a therapeutic strategy, a number of ligand/receptors activate STAT3 and STAT5, as described above. Therefore, additional studies will be needed to define whether single ligand-receptor inhibitors can sufficiently impact STAT3 activation, thus suppressing tumor growth.
Currently, JAK inhibitors are the most promising inhibitors of STAT-driven phenotypes. Commonly used JAK 1/2 inhibitor, Ruxolitinib, has been shown to robustly block both STAT3 and STAT5 activation. Yet, while JAK inhibitors can reduce STAT3 activation, there is conflicting data on the impact of JAK inhibitors. Some studies find that JAK inhibition suppresses tumor growth [93], while other studies find that they ultimately enhance metastasis, likely because they also suppress the positive influence of JAK activity on other STAT proteins (including STAT5 and STAT1/2, as discussed below) [299]. In addition, side effects of JAK inhibitors may be more pronounced, as JAKs activate other pathways, such as MAPK and PI3K in normal cells as well as cancer [43].
9. Balancing Opposing STAT-Activated Cytokine Signaling as a Therapeutic Strategy
Though STAT proteins are grouped together based on their structural similarities and common functions as transcription factors, individual STAT proteins can have diverse functions. We have focused extensively on STAT3 and STAT5 and their described roles in cancer progression, however this is not the universal effect among all STATs. For example, Interferon-β (IFNβ) induces the phosphorylation and activation of STAT1/2, which form a complex with IRF9 to create the transcription factor complex ISGF3. IFNβ/P-ISGF3 signaling induces interferon-stimulated genes (ISG), mesenchymal-epithelial transition (MET), and the differentiation of CSC into a less aggressive epithelial, non-CSC state with reduced migratory potential and reduced tumorsphere forming capabilities [111,300,301]. Importantly, the IFNβ and OSM/STAT3 signaling pathways strongly oppose one another. OSM represses transcription of IFNβ, thereby eliminating autocrine and paracrine IFNβ-mediated activation of P-ISGF3 and repressing ISG expression in both cancer cells and immune cells [301].
In addition to the impact of IFNβ on cancer cells, increasing rationale supports developing methods for the delivery of P-ISGF3 activators (or Type I IFN-agonists more generally) directly to the TME. First, favorable responses to frontline chemotherapy correlate with robust IFN signaling in both mouse and human studies [111,302,303,304]. Elevated IFN signaling in the tumor correlates with immunologically “hot” tumors harboring elevated numbers of tumor infiltrating lymphocytes (TILs), activated immune surveillance, increased tumor antigen cross presentation, and diminished numbers of immunosuppressive cells including MDSCs and Tregs [111,302,305,306,307]. Second, loss of Type I IFN signaling correlates with metastasis and decreased survival. Restoration of Type I IFN signaling significantly decreases metastasis and improves survival outcome [303,304]. Third, in contrast to STAT3 activators (which promote a pro-tumorigenic M2 state), addition of type I IFN inhibits macrophage polarization to an M2 state [308,309]. Type I IFNs also induce the differentiation of neutrophils into anti-tumor N1s [310]. Fourth, administration of IFNβ prior to surgical resection significantly improves response rates to immunotherapies such as anti-PD1/anti-PDL-1 [311]. Therefore, we propose balancing pro-tumor STAT3 activation with anti-tumor STAT1/STAT2 activation as a novel therapeutic approach. This STAT3/STAT1 balancing would (i) reprogram mesenchymal/CSC to a non-CSC state, making them more susceptible to chemotherapy and (ii) enhance anti-tumor immunity, thereby facilitating immune cell-mediated tumor cell killing. Yet, while IFN treatment is currently approved to treat hematological malignancies and some solid tumors (melanoma), the high doses of IFNs needed to inhibit cancer cell proliferation or induce cell death result in side-effects that limit its effectiveness [311,312,313]. We suggest tumor targeting antibodies (or nanobodies) linked to IFNβ. Generation of an oncogenic cytokine or receptor antibody conjugated to an IFNβ first demonstrated success in limiting resistance to EGFR inhibitors in breast cancer [314]. The designed therapy sought to re-activate innate and adaptive immune components, while simultaneously targeting the oncogenic receptor EGFR [303]. IFNβ-conjugated antibodies show immense promise. They would limit toxicity by using tumor-associated receptors to target IFNs to the TME and suppress STAT3 activation, while simultaneously, activating STAT1/2 in both tumor cells and immune cells [315].
10. Conclusions
As discussed throughout this review, STAT3 and STAT5 have emerged as essential components involved in regulating tumor progression. Cytokines and cytokine receptors of the IL-6 family are some of the most widely recognized STAT activators and are abundantly expressed on cancer cells as well as tumor-infiltrating immune cells. The role of STAT3 in promoting molecular programs in cancer cells that induce tumor metastasis and therapy resistance mechanisms continue to emerge, as does the impact of STAT3 as a suppressor of immune cell function in the TME. These findings suggest that specifically suppressing STAT3 activation would be beneficial to patients. Targeting STAT5 in hematological malignancies is gaining traction, and the clinical successes of JAK and tyrosine kinase inhibitors in disrupting STAT3 and STAT5 activation, provide strong support for the development of direct STAT3 and STAT5 inhibitors, summarized in Table 1. Specific suppression of STAT5 in immune-suppressive Tregs could also prove beneficial, but STAT5 is essential for many other immune cells as well. Thus, systemic suppression of STAT5 activity could undermine tumor immunity and promote tumor progression, as recently reported for STAT5 knock-out from NK cells [272]. However, reducing the aberrant activation of STAT5 without complete ablation may have therapeutic efficacy upon combination with other vulnerabilities. We conclude that targeting STAT3, either directly by disrupting STAT3-homodimer formation or indirectly by suppressing the activation of receptors responsible for persistent STAT3 phosphorylation, would reverse the cellular programs driving metastasis and therapy failure. Furthermore, by activating STAT1/2 within TME cells, the programs that prevent metastasis and enhance therapeutic efficacy could be re-engaged. This STAT balancing would improve outcomes for patients, particularly those with aggressive cancers that may currently have limited therapeutic options.
Funding
This research is supported by T32 (CA059366) to Kelsey Polak.
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e920. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra254. [Google Scholar] [CrossRef]
- Natrajan, R.; Sailem, H.; Mardakheh, F.K.; Arias Garcia, M.; Tape, C.J.; Dowsett, M.; Bakal, C.; Yuan, Y. Microenvironmental Heterogeneity Parallels Breast Cancer Progression: A Histology-Genomic Integration Analysis. PLoS Med. 2016, 13, e1001961. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e1236. [Google Scholar] [CrossRef]
- Rybinski, B.; Yun, K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016, 7, 72322–72342. [Google Scholar] [CrossRef] [Green Version]
- Mroz, E.A.; Tward, A.D.; Pickering, C.R.; Myers, J.N.; Ferris, R.L.; Rocco, J.W. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 2013, 119, 3034–3042. [Google Scholar] [CrossRef]
- Caswell, D.R.; Swanton, C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 2017, 15, 133. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef]
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol. Cancer 2019, 18, 67. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323. [Google Scholar] [CrossRef]
- Wagner, K.U.; Rui, H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J. Mammary Gland. Biol. Neoplasia 2008, 13, 93–103. [Google Scholar] [CrossRef]
- Ahonen, T.J.; Xie, J.; LeBaron, M.J.; Zhu, J.; Nurmi, M.; Alanen, K.; Rui, H.; Nevalainen, M.T. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J. Biol. Chem. 2003, 278, 27287–27292. [Google Scholar] [CrossRef]
- Tan, S.H.; Dagvadorj, A.; Shen, F.; Gu, L.; Liao, Z.; Abdulghani, J.; Zhang, Y.; Gelmann, E.P.; Zellweger, T.; Culig, Z.; et al. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res. 2008, 68, 236–248. [Google Scholar] [CrossRef]
- Leong, P.L.; Xi, S.; Drenning, S.D.; Dyer, K.F.; Wentzel, A.L.; Lerner, E.C.; Smithgall, T.E.; Grandis, J.R. Differential function of STAT5 isoforms in head and neck cancer growth control. Oncogene 2002, 21, 2846–2853. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Hosui, A.; Kimura, A.; Yamaji, D.; Zhu, B.M.; Na, R.; Hennighausen, L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation. J. Exp. Med. 2009, 206, 819–831. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.M.; Jin, X.; Lin, H.J.; Huang, M.; Liu, R.; Reynolds, R.K.; Lin, J. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 2001, 20, 7925–7934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Wu, D.; Zhao, L.; Huang, L.; Shen, G.; Huang, J.; Chai, Y. Prognostic role of STAT3 in solid tumors: A systematic review and meta-analysis. Oncotarget 2016, 7, 19863–19883. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.L.; Li, Z.W.; Lou, C.J.; Pang, D.; Zhang, Y.Q. Phospho-STAT5 expression is associated with poor prognosis of human colonic adenocarcinoma. Pathol. Oncol. Res. 2011, 17, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Qi, J.; Lin, S.Q.; Zhang, C.Y.; Liu, F.Y.; Xie, W.D.; Li, X. STAT3 targets ERR-alpha to promote epithelial-mesenchymal transition, migration and invasion in triple negative breast cancer cells. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.X.; Huang, F.J.; Aldape, K.D.; Kang, S.H.; Liu, M.; Gershenwald, J.E.; Xie, K.; Sawaya, R.; Huang, S. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006, 66, 3188–3196. [Google Scholar] [CrossRef] [PubMed]
- Abdulghani, J.; Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Zellweger, T.; Alanen, K.; Mirtti, T.; et al. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 2008, 172, 1717–1728. [Google Scholar] [CrossRef]
- Klupp, F.; Diers, J.; Kahlert, C.; Neumann, L.; Halama, N.; Franz, C.; Schmidt, T.; Lasitschka, F.; Warth, A.; Weitz, J.; et al. Expressional STAT3/STAT5 Ratio is an Independent Prognostic Marker in Colon Carcinoma. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S1548–S1555. [Google Scholar] [CrossRef]
- Real, P.J.; Sierra, A.; De Juan, A.; Segovia, J.C.; Lopez-Vega, J.M.; Fernandez-Luna, J.L. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 2002, 21, 7611–7618. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef]
- Baskiewicz-Masiuk, M.; Machalinski, B. The role of the STAT5 proteins in the proliferation and apoptosis of the CML and AML cells. Eur. J. Haematol. 2004, 72, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Morcinek, J.C.; Weisser, C.; Geissinger, E.; Schartl, M.; Wellbrock, C. Activation of STAT5 triggers proliferation and contributes to anti-apoptotic signalling mediated by the oncogenic Xmrk kinase. Oncogene 2002, 21, 1668–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmud, S.A.; Manlove, L.S.; Farrar, M.A. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT 2013, 2, e23154. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Toker, A.; Liu, Z.Q.; Ohashi, P.S. Turning the Tide Against Regulatory T Cells. Front. Oncol. 2019, 9, 279. [Google Scholar] [CrossRef]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: A systematic review and meta-analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef]
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef]
- Clevenger, C.V. Roles and Regulation of Stat Family Transcription Factors in Human Breast Cancer. Am. J. Pathol. 2004, 165, 1449–1460. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Kovarik, P.; Meinke, A. GAS elements: A few nucleotides with a major impact on cytokine-induced gene expression. J. Interferon Cytokine Res. 1997, 17, 121–134. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raz, R.; Lee, C.K.; Cannizzaro, L.A.; d’Eustachio, P.; Levy, D.E. Essential role of STAT3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirito, K.; Osawa, M.; Morita, H.; Shimizu, R.; Yamamoto, M.; Oda, A.; Fujita, H.; Tanaka, M.; Nakajima, K.; Miura, Y.; et al. A functional role of Stat3 in in vivo megakaryopoiesis. Blood 2002, 99, 3220–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Cui, Y.; Watford, W.T.; Bream, J.H.; Yamaoka, K.; Hissong, B.D.; Li, D.; Durum, S.K.; Jiang, Q.; Bhandoola, A.; et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Song, D.; Li, H.; Yang, Y.; Ma, X.; Deng, S.; Ren, C.; Shu, X. Negative regulators of STAT3 signaling pathway in cancers. Cancer Manag. Res. 2019, 11, 4957–4969. [Google Scholar] [CrossRef]
- Kershaw, N.J.; Murphy, J.M.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. Regulation of Janus kinases by SOCS proteins. Biochem. Soc. Trans. 2013, 41, 1042–1047. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Muller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [Green Version]
- Orlova, A.; Wingelhofer, B.; Neubauer, H.A.; Maurer, B.; Berger-Becvar, A.; Keseru, G.M.; Gunning, P.T.; Valent, P.; Moriggl, R. Emerging therapeutic targets in myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas. Expert Opin. Targets 2018, 22, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Shahmarvand, N.; Nagy, A.; Shahryari, J.; Ohgami, R.S. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018, 109, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Kee, W.H.; Seow, K.T.; Fung, W.; Cao, X. The coiled-coil domain of Stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol. Cell Biol. 2000, 20, 7132–7139. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Witzig, T.E.; Gupta, M. A novel missense (M206K) STAT3 mutation in diffuse large B cell lymphoma deregulates STAT3 signaling. PLoS ONE 2013, 8, e67851. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Zhang, L.; Snow, J.W.; Jones, D.M.; Smith, A.M.; El Kasmi, K.C.; Liu, F.; Goldsmith, M.A.; Link, D.C.; Murray, P.J.; et al. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood 2006, 108, 3682–3690. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Mutze, J.; Roth, J.; Gerstberger, R.; Hubschle, T. Nuclear translocation of the transcription factor STAT5 in the rat brain after systemic leptin administration. Neurosci. Lett. 2007, 417, 286–291. [Google Scholar] [CrossRef]
- Ribeiro, D.; Melao, A.; van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018, 2, 2199–2213. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, J.B.; Uyttenhove, C.; Lejeune, D.; Mui, A.; Groner, B.; Renauld, J.C. STAT5 activation is required for interleukin-9-dependent growth and transformation of lymphoid cells. Cancer Res. 2000, 60, 3971–3977. [Google Scholar] [PubMed]
- Rochman, Y.; Kashyap, M.; Robinson, G.W.; Sakamoto, K.; Gomez-Rodriguez, J.; Wagner, K.U.; Leonard, W.J. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7-induced signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 19455–19460. [Google Scholar] [CrossRef] [PubMed]
- Okutani, Y.; Kitanaka, A.; Tanaka, T.; Kamano, H.; Ohnishi, H.; Kubota, Y.; Ishida, T.; Takahara, J. Src directly tyrosine-phosphorylates STAT5 on its activation site and is involved in erythropoietin-induced signaling pathway. Oncogene 2001, 20, 6643–6650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirito, K.; Watanabe, T.; Sawada, K.; Endo, H.; Ozawa, K.; Komatsu, N. Thrombopoietin regulates Bcl-xL gene expression through Stat5 and phosphatidylinositol 3-kinase activation pathways. J. Biol. Chem. 2002, 277, 8329–8337. [Google Scholar] [CrossRef]
- Scheeren, F.A.; Diehl, S.A.; Smit, L.A.; Beaumont, T.; Naspetti, M.; Bende, R.J.; Blom, B.; Karube, K.; Ohshima, K.; van Noesel, C.J.; et al. IL-21 is expressed in Hodgkin lymphoma and activates STAT5: Evidence that activated STAT5 is required for Hodgkin lymphomagenesis. Blood 2008, 111, 4706–4715. [Google Scholar] [CrossRef]
- Maier, E.; Mittermeir, M.; Ess, S.; Neuper, T.; Schmiedlechner, A.; Duschl, A.; Horejs-Hoeck, J. Prerequisites for Functional Interleukin 31 Signaling and Its Feedback Regulation by Suppressor of Cytokine Signaling 3 (SOCS3). J. Biol. Chem. 2015, 290, 24747–24759. [Google Scholar] [CrossRef] [Green Version]
- Toniolo, P.A.; Liu, S.; Yeh, J.E.; Moraes-Vieira, P.M.; Walker, S.R.; Vafaizadeh, V.; Barbuto, J.A.; Frank, D.A. Inhibiting STAT5 by the BET bromodomain inhibitor JQ1 disrupts human dendritic cell maturation. J. Immunol. 2015, 194, 3180–3190. [Google Scholar] [CrossRef]
- Tripathi, P.; Kurtulus, S.; Wojciechowski, S.; Sholl, A.; Hoebe, K.; Morris, S.C.; Finkelman, F.D.; Grimes, H.L.; Hildeman, D.A. STAT5 is critical to maintain effector CD8+ T cell responses. J. Immunol. 2010, 185, 2116–2124. [Google Scholar] [CrossRef]
- Kieslinger, M.; Woldman, I.; Moriggl, R.; Hofmann, J.; Marine, J.C.; Ihle, J.N.; Beug, H.; Decker, T. Antiapoptotic activity of Stat5 required during terminal stages of myeloid differentiation. Genes Dev. 2000, 14, 232–244. [Google Scholar]
- Kuroda, E.; Ho, V.; Ruschmann, J.; Antignano, F.; Hamilton, M.; Rauh, M.J.; Antov, A.; Flavell, R.A.; Sly, L.M.; Krystal, G. SHIP represses the generation of IL-3-induced M2 macrophages by inhibiting IL-4 production from basophils. J. Immunol. 2009, 183, 3652–3660. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Flores Santa Cruz, D.; Sexl, V.; Gottgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016, 35, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.R.; Ulirsch, J.C.; Wilmes, S.; Unal, E.; Moraga, I.; Karakukcu, M.; Yuan, D.; Kazerounian, S.; Abdulhay, N.J.; King, D.S.; et al. Functional Selectivity in Cytokine Signaling Revealed Through a Pathogenic EPO Mutation. Cell 2017, 168, 1053–1064 e1015. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Page, B.D.; Khoury, H.; Laister, R.C.; Fletcher, S.; Vellozo, M.; Manzoli, A.; Yue, P.; Turkson, J.; Minden, M.D.; Gunning, P.T. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J. Med. Chem. 2012, 55, 1047–1055. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef]
- Unver, N.; McAllister, F. IL-6 family cytokines: Key inflammatory mediators as biomarkers and potential therapeutic targets. Cytokine Growth Factor Rev. 2018, 41, 10–17. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Putoczki, T.L.; Ernst, M. IL-11 signaling as a therapeutic target for cancer. Immunotherapy 2015, 7, 441–453. [Google Scholar] [CrossRef]
- Lu, J.; Ksendzovsky, A.; Yang, C.; Mehta, G.U.; Yong, R.L.; Weil, R.J.; Park, D.M.; Mushlin, H.M.; Fang, X.; Balgley, B.M.; et al. CNTF receptor subunit alpha as a marker for glioma tumor-initiating cells and tumor grade: Laboratory investigation. J. Neurosurg. 2012, 117, 1022–1031. [Google Scholar] [CrossRef]
- Li, X.; Yang, Q.; Yu, H.; Wu, L.; Zhao, Y.; Zhang, C.; Yue, X.; Liu, Z.; Wu, H.; Haffty, B.G.; et al. LIF promotes tumorigenesis and metastasis of breast cancer through the AKT-mTOR pathway. Oncotarget 2014, 5, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.C.; Lin, J.T.; Wu, C.Y.; Huang, S.P.; Lin, M.T.; Wu, A.S.; Huang, Y.J.; Wu, M.S. Serum interleukin-6 level but not genotype predicts survival after resection in stages II and III gastric carcinoma. Clin. Cancer Res. 2008, 14, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.P.; Yang, D.C.; Elliott, R.L.; Head, J.F. Reduction in serum IL-6 after vacination of breast cancer patients with tumour-associated antigens is related to estrogen receptor status. Cytokine 2000, 12, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-C.; Chang, Y.-F. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 2003, 83, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Tawara, K.; Oxford, J.T.; Jorcyk, C.L. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: Potential of anti-IL-6 therapies. Cancer Manag. Res. 2011, 3, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Komoda, H.; Tanaka, Y.; Honda, M.; Matsuo, Y.; Hazama, K.; Takao, T. Interleukin-6 levels in colorectal cancer tissues. World J. Surg. 1998, 22, 895–898. [Google Scholar] [CrossRef]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef]
- Gyamfi, J.; Lee, Y.H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- Jiang, M.; Chen, J.; Zhang, W.; Zhang, R.; Ye, Y.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. Interleukin-6 Trans-Signaling Pathway Promotes Immunosuppressive Myeloid-Derived Suppressor Cells via Suppression of Suppressor of Cytokine Signaling 3 in Breast Cancer. Front. Immunol. 2017, 8, 1840. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yu, S.; Li, A.; Xu, H.; Han, X.; Wu, K. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour Biol. 2017, 39, 1010428317712445. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin. Cancer Res. 2018, 24, 4820–4833. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Q.; Daly, L.; Bromberg, J. The IL-6 feed-forward loop: A driver of tumorigenesis. Semin. Immunol. 2014, 26, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, J.; Parvani, J.G.; Tamagno, I.; Polak, K.; Jackson, M.W. Breaking the oncostatin M feed-forward loop to suppress metastasis and therapy failure. J. Pathol. 2018, 245, 6–8. [Google Scholar] [CrossRef] [Green Version]
- Zarling, J.M.; Shoyab, M.; Marquardt, H.; Hanson, M.B.; Lioubin, M.N.; Todaro, G.J. Oncostatin M: A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9739–9743. [Google Scholar] [CrossRef]
- Auguste, P.; Guillet, C.; Fourcin, M.; Olivier, C.; Veziers, J.; Pouplard-Barthelaix, A.; Gascan, H. Signaling of type II oncostatin M receptor. J. Biol. Chem. 1997, 272, 15760–15764. [Google Scholar] [CrossRef]
- Mirmohammadsadegh, A.; Hassan, M.; Bardenheuer, W.; Marini, A.; Gustrau, A.; Nambiar, S.; Tannapfel, A.; Bojar, H.; Ruzicka, T.; Hengge, U.R. STAT5 phosphorylation in malignant melanoma is important for survival and is mediated through SRC and JAK1 kinases. J. Investig. Derm. 2006, 126, 2272–2280. [Google Scholar] [CrossRef]
- Lacreusette, A.; Nguyen, J.M.; Pandolfino, M.C.; Khammari, A.; Dreno, B.; Jacques, Y.; Godard, A.; Blanchard, F. Loss of oncostatin M receptor β in metastatic melanoma cells. Oncogene 2006, 26, 881–892. [Google Scholar] [CrossRef]
- Hassel, J.C.; Winnemoller, D.; Schartl, M.; Wellbrock, C. STAT5 contributes to antiapoptosis in melanoma. Melanoma Res. 2008, 18, 378–385. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Murray, J.I.; Watson, P.H. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014, 33, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Lapeire, L.; Hendrix, A.; Lambein, K.; Van Bockstal, M.; Braems, G.; Van Den Broecke, R.; Limame, R.; Mestdagh, P.; Vandesompele, J.; Vanhove, C.; et al. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin M and Jak/STAT3 signaling. Cancer Res. 2014, 74, 6806–6819. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Louwagie, J.; Carvalho, B.; Terhaar Sive Droste, J.S.; Park, H.L.; Chae, Y.K.; Yamashita, K.; Liu, J.; Ostrow, K.L.; Ling, S.; et al. Promoter DNA methylation of oncostatin m receptor-beta as a novel diagnostic and therapeutic marker in colon cancer. PLoS ONE 2009, 4, e6555. [Google Scholar] [CrossRef] [PubMed]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast cancer cells stimulate neutrophils to produce oncostatin M: Potential implications for tumor progression. Cancer Res. 2005, 65, 8896–8904. [Google Scholar] [CrossRef]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef] [Green Version]
- Jahani-Asl, A.; Yin, H.; Soleimani, V.D.; Haque, T.; Luchman, H.A.; Chang, N.C.; Sincennes, M.-C.; Puram, S.V.; Scott, A.M.; Lorimer, I.A.J.; et al. Control of glioblastoma tumorigenesis by feed-forward cytokine signaling. Nat. Neurosci. 2016, 19, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Shien, K.; Papadimitrakopoulou, V.A.; Ruder, D.; Behrens, C.; Shen, L.; Kalhor, N.; Song, J.; Lee, J.J.; Wang, J.; Tang, X.; et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol. Cancer 2017, 16, 2234–2245. [Google Scholar] [CrossRef]
- Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Potent EMT and CSC Phenotypes Are Induced By Oncostatin-M in Pancreatic Cancer. Mol. Cancer Res. 2017, 15, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Tawara, K.; Bolin, C.; Koncinsky, J.; Kadaba, S.; Covert, H.; Sutherland, C.; Bond, L.; Kronz, J.; Garbow, J.R.; Jorcyk, C.L. OSM potentiates preintravasation events, increases CTC counts, and promotes breast cancer metastasis to the lung. Breast Cancer Res. 2018, 20, 53. [Google Scholar] [CrossRef]
- Doherty, M.R.; Cheon, H.; Junk, D.J.; Vinayak, S.; Varadan, V.; Telli, M.L.; Ford, J.M.; Stark, G.R.; Jackson, M.W. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13792–13797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Gortz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Diveu, C.; Venereau, E.; Froger, J.; Ravon, E.; Grimaud, L.; Rousseau, F.; Chevalier, S.; Gascan, H. Molecular and functional characterization of a soluble form of oncostatin M/interleukin-31 shared receptor. J. Biol. Chem. 2006, 281, 36673–36682. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef]
- Yu, H.; Yue, X.; Zhao, Y.; Li, X.; Wu, L.; Zhang, C.; Liu, Z.; Lin, K.; Xu-Monette, Z.Y.; Young, K.H.; et al. LIF negatively regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nat. Commun. 2014, 5, 5218. [Google Scholar] [CrossRef]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, Y.; Zhang, C.; Li, J.; Liu, Z.; Liu, J.; Hu, W. Leukemia inhibitory factor promotes EMT through STAT3-dependent miR-21 induction. Oncotarget 2016, 7, 3777–3790. [Google Scholar] [CrossRef]
- Bohrer, L.R.; Chuntova, P.; Bade, L.K.; Beadnell, T.C.; Leon, R.P.; Brady, N.J.; Ryu, Y.; Goldberg, J.E.; Schmechel, S.C.; Koopmeiners, J.S.; et al. Activation of the FGFR-STAT3 pathway in breast cancer cells induces a hyaluronan-rich microenvironment that licenses tumor formation. Cancer Res. 2014, 74, 374–386. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Ojeda-Marquez, L.; Marrugal, A.; Yague, P.; Ponce-Aix, S.; Salinas, A.; Carnero, A.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. The FGFR4-388arg Variant Promotes Lung Cancer Progression by N-Cadherin Induction. Sci. Rep. 2018, 8, 2394. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Su, L.; Shan, J.; Zhu, C.; Liu, L.; Liu, C.; Xu, Y.; Yang, Z.; Bian, X.; Shao, J.; et al. IGF/STAT3/NANOG/Slug Signaling Axis Simultaneously Controls Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer. Stem Cells 2016, 34, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Ji, J.; Jiang, J.; Cai, Q.; Wang, C.; Shi, M.; Yu, Y.; Zhu, Z.; Zhang, J. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018, 9, 867. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Liao, P.N.; Ho, A.S.; Lim, K.H.; Chang, J.; Su, Y.W.; Chen, C.G.; Chiang, Y.W.; Yang, B.L.; Lin, H.C.; et al. STAT3 exacerbates survival of cancer stem-like tumorspheres in EGFR-positive colorectal cancers: RNAseq analysis and therapeutic screening. J. Biomed. Sci. 2018, 25, 60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Ando, T.; Kudo, Y.; Iizuka, S.; Tsunematsu, T.; Umehara, H.; Shrestha, M.; Matsuo, T.; Kubo, T.; Shimose, S.; Arihiro, K.; et al. Ameloblastin induces tumor suppressive phenotype and enhances chemosensitivity to doxorubicin via Src-Stat3 inactivation in osteosarcoma. Sci. Rep. 2017, 7, 40187. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Xu, Z.; Wang, J.; Zhu, Z.; Lin, G.; Jiang, L.; Lu, X.; Zou, C. Matrine inhibits BCR/ABL mediated ERK/MAPK pathway in human leukemia cells. Oncotarget 2017, 8, 108880–108889. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Yeung, C.M.; Jahani-Asl, A.; Lin, C.; de la Iglesia, N.; Konopka, G.; Jackson-Grusby, L.; Bonni, A. STAT3-iNOS Signaling Mediates EGFRvIII-Induced Glial Proliferation and Transformation. J. Neurosci. 2012, 32, 7806–7818. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Berger, H.; Chalons, P.; Vegan, F.; Humblin, E.; Boidot, R.; Rebe, C.; Derangere, V.; et al. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012, 1, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Dechow, T.N.; Pedranzini, L.; Leitch, A.; Leslie, K.; Gerald, W.L.; Linkov, I.; Bromberg, J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA 2004, 101, 10602–10607. [Google Scholar] [CrossRef] [Green Version]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Fukuda, A.; Wang, S.C.; Morris, J.P.t.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef]
- Bryson, B.L.; Junk, D.J.; Cipriano, R.; Jackson, M.W. STAT3-mediated SMAD3 activation underlies Oncostatin M-induced Senescence. Cell Cycle 2017, 16, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Rokavec, M.; Oner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.H.; Jeong, K.J.; Shin, S.C.; Kang, J.; Park, C.G.; Lee, H.Y. STAT3 mediates TGF-beta1-induced TWIST1 expression and prostate cancer invasion. Cancer Lett. 2013, 336, 167–173. [Google Scholar] [CrossRef]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.-w.; Ren, L.-l.; Wang, Y.-c.; Su, W.-y.; Wang, J.-l.; Cui, Y.; Wang, Z.-h.; et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.X.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Z.H.; Jia, Y.; Guo, F.J.; Chen, J.; Zhang, X.W.; Cui, M.H. Phosphorylation of STAT3 at Tyr705 regulates MMP-9 production in epithelial ovarian cancer. PLoS ONE 2017, 12, e0183622. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Tsai, P.H.; Kandaswami, C.C.; Lee, P.P.; Huang, C.J.; Hwang, J.J.; Lee, M.T. Matrix metalloproteinase-9 cooperates with transcription factor Snail to induce epithelial-mesenchymal transition. Cancer Sci. 2011, 102, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.; Evans, A.J.; Rakha, E.A.; Green, A.R.; Ellis, I.O. Influence of E-cadherin expression on the mammographic appearance of invasive nonlobular breast carcinoma detected at screening. Radiology 2009, 253, 51–55. [Google Scholar] [CrossRef]
- Rakha, E.A.; Abd El Rehim, D.; Pinder, S.E.; Lewis, S.A.; Ellis, I.O. E-cadherin expression in invasive non-lobular carcinoma of the breast and its prognostic significance. Histopathology 2005, 46, 685–693. [Google Scholar] [CrossRef]
- Rakha, E.A.; Patel, A.; Powe, D.G.; Benhasouna, A.; Green, A.R.; Lambros, M.B.; Reis-Filho, J.S.; Ellis, I.O. Clinical and biological significance of E-cadherin protein expression in invasive lobular carcinoma of the breast. Am. J. Surg. Pathol. 2010, 34, 1472–1479. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, X.; Shang, M.; Zhang, Y.; Xia, B.; Niu, M.; Liu, Y.; Pang, D. Dysregulated expression of Slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J. Surg. Oncol. 2013, 107, 188–194. [Google Scholar] [CrossRef]
- Markiewicz, A.; Welnicka-Jaskiewicz, M.; Seroczynska, B.; Skokowski, J.; Majewska, H.; Szade, J.; Zaczek, A.J. Epithelial-mesenchymal transition markers in lymph node metastases and primary breast tumors—Relation to dissemination and proliferation. Am. J. Transl. Res. 2014, 6, 793–808. [Google Scholar]
- Zhou, L.; Dicker, D.T.; Matthew, E.; El-Deiry, W.S.; Alpaugh, R.K. Circulating tumor cells: Silent predictors of metastasis. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, S.; Liu, Z.; Huang, J.; Pu, X.; Li, J.; Yang, D.; Deng, H.; Yang, N.; Xu, J. Classification of circulating tumor cells by epithelial-mesenchymal transition markers. PLoS ONE 2015, 10, e0123976. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Mitra, A.; Brownlee, Z.; Xia, X.; Bellister, S.; Overman, M.J.; Kopetz, S.; Ellis, L.M.; Meng, Q.H.; Li, S. Epithelial-mesenchymal transitioned circulating tumor cells capture for detecting tumor progression. Clin. Cancer Res. 2015, 21, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8, 8. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Hayashida, T.; Jinno, H.; Kitagawa, Y.; Kitajima, M. Cooperation of cancer stem cell properties and epithelial-mesenchymal transition in the establishment of breast cancer metastasis. J. Oncol. 2011, 2011, 591427. [Google Scholar] [CrossRef]
- Choi, S.; Yu, J.; Park, A.; Dubon, M.J.; Do, J.; Kim, Y.; Nam, D.; Noh, J.; Park, K.S. BMP-4 enhances epithelial mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci. Rep. 2019, 9, 11724. [Google Scholar] [CrossRef]
- Hung, J.J.; Kao, Y.S.; Huang, C.H.; Hsu, W.H. Overexpression of Aiolos promotes epithelial-mesenchymal transition and cancer stem cell-like properties in lung cancer cells. Sci. Rep. 2019, 9, 2991. [Google Scholar] [CrossRef] [PubMed]
- Reiman, J.M.; Knutson, K.L.; Radisky, D.C. Immune promotion of epithelial-mesenchymal transition and generation of breast cancer stem cells. Cancer Res. 2010, 70, 3005–3008. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Rezende, N.C.; Scotland, K.B.; Shaffer, S.M.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. Regulation of stem cell pluripotency and differentiation involves a mutual regulatory circuit of the NANOG, OCT4, and SOX2 pluripotency transcription factors with polycomb repressive complexes and stem cell microRNAs. Stem Cells Dev. 2009, 18, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal 2013, 25, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Bareiss, P.M.; Paczulla, A.; Wang, H.; Schairer, R.; Wiehr, S.; Kohlhofer, U.; Rothfuss, O.C.; Fischer, A.; Perner, S.; Staebler, A.; et al. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer Res. 2013, 73, 5544–5555. [Google Scholar] [CrossRef]
- Jiang, J.; Li, Z.; Yu, C.; Chen, M.; Tian, S.; Sun, C. MiR-1181 inhibits stem cell-like phenotypes and suppresses SOX2 and STAT3 in human pancreatic cancer. Cancer Lett. 2015, 356, 962–970. [Google Scholar] [CrossRef]
- Yin, X.; Zhang, B.H.; Zheng, S.S.; Gao, D.M.; Qiu, S.J.; Wu, W.Z.; Ren, Z.G. Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling. J. Hematol. Oncol. 2015, 8, 23. [Google Scholar] [CrossRef]
- Van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.T.; Lippitt, J.M.; Guzman-Ramirez, N.; Hamdy, F.C.; Eaton, C.L.; Thalmann, G.N.; Cecchini, M.G.; et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010, 70, 5163–5173. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893 e813. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, H.; Yin, X.; Long, L.; Chen, X.; Feng, F.; Liu, Y.; Zhao, P.; Xu, Y.; Li, M.; et al. IL-6R/STAT3/miR-204 feedback loop contributes to cisplatin resistance of epithelial ovarian cancer cells. Oncotarget 2017, 8, 39154–39166. [Google Scholar] [CrossRef]
- Ganguly, D.; Fan, M.; Yang, C.H.; Zbytek, B.; Finkelstein, D.; Roussel, M.F.; Pfeffer, L.M. The critical role that STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma. Oncotarget 2018, 9, 22095–22112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Cao, J.; Wu, J.; Sullivan, K.; Shen, J.; Ryu, B.; Xu, Z.; Wei, W.; Cui, R. Stat3-targeted therapies overcome the acquired resistance to vemurafenib in melanomas. J. Investig. Derm. 2013, 133, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Castellanos, J.A.; Lamichhane, P.; Messaggio, F.; Shi, C.; Dai, X.; Rai, P.; Chen, X.; VanSaun, M.N.; Merchant, N.B. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res. 2018, 78, 6235–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ren, D.; Wu, X.; Lin, X.; Ye, L.; Lin, C.; Wu, S.; Zhu, J.; Peng, X.; Song, L. miR-1266 Contributes to Pancreatic Cancer Progression and Chemoresistance by the STAT3 and NF-kappaB Signaling Pathways. Mol. Nucleic Acids 2018, 11, 142–158. [Google Scholar] [CrossRef]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M.; et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, F.; Cui, J.Y.; Chen, L.; Chen, Y.T.; Liu, B.W. CAFs enhance paclitaxel resistance by inducing EMT through the IL6/JAK2/STAT3 pathway. Oncol. Rep. 2018, 39, 2081–2090. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef]
- Simon, G.R.; Somaiah, N. A tabulated summary of targeted and biologic therapies for non-small-cell lung cancer. Clin. Lung Cancer 2014, 15, 21–51. [Google Scholar] [CrossRef]
- Abell, K.; Watson, C.J. The Jak/Stat pathway: A novel way to regulate PI3K activity. Cell Cycle 2005, 4, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.A.; Kagan, P.; Sultan, M.; Tachlytski, I.; Safran, M.; Ben-Ari, Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, S.; Oishi, K.; Yoshimatsu, T.; Nakafuku, M.; Masuyama, N.; Gotoh, Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 2004, 6, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, M.A.; Patel, A.K.; Nakamura, R.E.; Yi, H.; Surapaneni, K.; Hackam, A.S. The Wnt/beta-catenin pathway cross-talks with STAT3 signaling to regulate survival of retinal pigment epithelium cells. PLoS ONE 2012, 7, e46892. [Google Scholar] [CrossRef]
- Tang, L.Y.; Heller, M.; Meng, Z.; Yu, L.R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-beta (TGF-beta) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef]
- Bui, Q.T.; Im, J.H.; Jeong, S.B.; Kim, Y.M.; Lim, S.C.; Kim, B.; Kang, K.W. Essential role of Notch4/STAT3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human breast cancer. Cancer Lett. 2017, 390, 115–125. [Google Scholar] [CrossRef]
- Canino, C.; Luo, Y.; Marcato, P.; Blandino, G.; Pass, H.I.; Cioce, M. A STAT3-NFkB/DDIT3/CEBPbeta axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget 2015, 6, 12637–12653. [Google Scholar] [CrossRef]
- Alt-Holland, A.; Sowalsky, A.G.; Szwec-Levin, Y.; Shamis, Y.; Hatch, H.; Feig, L.A.; Garlick, J.A. Suppression of E-cadherin function drives the early stages of Ras-induced squamous cell carcinoma through upregulation of FAK and Src. J. Investig. Derm. 2011, 131, 2306–2315. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- D’Amico, S.; Shi, J.; Martin, B.L.; Crawford, H.C.; Petrenko, O.; Reich, N.C. STAT3 is a master regulator of epithelial identity and KRAS-driven tumorigenesis. Genes Dev. 2018, 32, 1175–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef]
- Yang, C.; He, L.; He, P.; Liu, Y.; Wang, W.; He, Y.; Du, Y.; Gao, F. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med. Oncol. 2015, 32, 352. [Google Scholar] [CrossRef]
- Webb, N.J.; Myers, C.R.; Watson, C.J.; Bottomley, M.J.; Brenchley, P.E. Activated human neutrophils express vascular endothelial growth factor (VEGF). Cytokine 1998, 10, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cong, X.; Gao, H.; Lan, X.; Li, Z.; Wang, W.; Song, S.; Wang, Y.; Li, C.; Zhang, H.; et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef]
- Pignatelli, J.; Bravo-Cordero, J.J.; Roh-Johnson, M.; Gandhi, S.J.; Wang, Y.; Chen, X.; Eddy, R.J.; Xue, A.; Singer, R.H.; Hodgson, L.; et al. Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/MenaINV-initiated invadopodium formation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Pignatelli, J.; Goswami, S.; Jones, J.G.; Rohan, T.E.; Pieri, E.; Chen, X.; Adler, E.; Cox, D.; Maleki, S.; Bresnick, A.; et al. Invasive breast carcinoma cells from patients exhibit MenaINV- and macrophage-dependent transendothelial migration. Sci. Signal 2014, 7, ra112. [Google Scholar] [CrossRef]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Ferri, L.E. Neutrophils Promote Liver Metastasis via Mac-1–Mediated Interactions with Circulating Tumor Cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Deng, J.; Liu, Y.; Lee, H.; Herrmann, A.; Zhang, W.; Zhang, C.; Shen, S.; Priceman, S.J.; Kujawski, M.; Pal, S.K.; et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 2012, 21, 642–654. [Google Scholar] [CrossRef]
- Kanai, T.; Seki, S.; Jenks, J.A.; Kohli, A.; Kawli, T.; Martin, D.P.; Snyder, M.; Bacchetta, R.; Nadeau, K.C. Identification of STAT5A and STAT5B target genes in human T cells. PLoS ONE 2014, 9, e86790. [Google Scholar] [CrossRef]
- Grebien, F.; Kerenyi, M.A.; Kovacic, B.; Kolbe, T.; Becker, V.; Dolznig, H.; Pfeffer, K.; Klingmuller, U.; Muller, M.; Beug, H.; et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood 2008, 111, 4511–4522. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.M.; McLaughlin, S.K.; Na, R.; Liu, J.; Cui, Y.; Martin, C.; Kimura, A.; Robinson, G.W.; Andrews, N.C.; Hennighausen, L. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood 2008, 112, 2071–2080. [Google Scholar] [CrossRef] [Green Version]
- Kerenyi, M.A.; Grebien, F.; Gehart, H.; Schifrer, M.; Artaker, M.; Kovacic, B.; Beug, H.; Moriggl, R.; Mullner, E.W. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood 2008, 112, 3878–3888. [Google Scholar] [CrossRef] [Green Version]
- Barash, I. Stat5 in the mammary gland: Controlling normal development and cancer. J. Cell Physiol. 2006, 209, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Cella, N.; Groner, B.; Hynes, N.E. Characterization of Stat5a and Stat5b homodimers and heterodimers and their association with the glucocortiocoid receptor in mammary cells. Mol. Cell Biol. 1998, 18, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Aoki, Y.; Yoshida, M.; Arai, K.; Watanabe, S. Stat5B shuttles between cytoplasm and nucleus in a cytokine-dependent and -independent manner. J. Immunol. 2002, 168, 4567–4575. [Google Scholar] [CrossRef] [PubMed]
- Meinke, A.; Barahmand-Pour, F.; Wohrl, S.; Stoiber, D.; Decker, T. Activation of different Stat5 isoforms contributes to cell-type-restricted signaling in response to interferons. Mol. Cell Biol. 1996, 16, 6937–6944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.V.; Sciumè, G.; Davis, F.P.; Iwata, S.; Zitti, B.; Robinson, G.W.; Hennighausen, L.; Kanno, Y.; O’Shea, J.J. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J. Exp. Med. 2017, 214, 2999–3014. [Google Scholar] [CrossRef]
- Maurer, B.; Farlik, M.; Sexl, V. It is a differentiation game: STAT5 in a new role. Cell Death Differ. 2017, 24, 953–954. [Google Scholar] [CrossRef] [Green Version]
- Heltemes-Harris, L.M.; Farrar, M.A. The role of STAT5 in lymphocyte development and transformation. Curr. Opin. Immunol. 2012, 24, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006, 107, 4898–4906. [Google Scholar] [CrossRef] [Green Version]
- Minieri, V.; De Dominici, M.; Porazzi, P.; Mariani, S.A.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; Porcu, P.; Nevalainen, M.T.; Calabretta, B. Targeting STAT5 or STAT5-Regulated Pathways Suppresses Leukemogenesis of Ph+ Acute Lymphoblastic Leukemia. Cancer Res. 2018, 78, 5793–5807. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Schneeweiss, M.; Eisenwort, G.; Berger, D.; Herrmann, H.; Blatt, K.; Greiner, G.; Byrgazov, K.; Hoermann, G.; Konopleva, M.; et al. Combined targeting of STAT3 and STAT5: A novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica 2017, 102, 1519–1529. [Google Scholar] [CrossRef]
- Kaltenecker, D.; Themanns, M.; Mueller, K.M.; Spirk, K.; Golob-Schwarzl, N.; Friedbichler, K.; Kenner, L.; Haybaeck, J.; Moriggl, R. STAT5 deficiency in hepatocytes reduces diethylnitrosamine-induced liver tumorigenesis in mice. Cytokine 2018. [Google Scholar] [CrossRef] [PubMed]
- Boutillon, F.; Pigat, N.; Sala, L.S.; Reyes-Gomez, E.; Moriggl, R.; Guidotti, J.E.; Goffin, V. STAT5a/b Deficiency Delays, but does not Prevent, Prolactin-Driven Prostate Tumorigenesis in Mice. Cancers 2019, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Man, K.; Poon, R.T.P.; Lo, C.M.; Yuen, A.P.; Ng, I.O.; Ng, K.T.; Leonard, W.; Fan, S.T. Signal Transducers and Activators of Transcription 5b Activation Enhances Hepatocellular Carcinoma Aggressiveness through Induction of Epithelial-Mesenchymal Transition. Cancer Res. 2006, 66, 9948–9956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.N.; Bu, W.; Hein, S.; Garcia, S.; Camacho, L.; Xue, L.; Qin, L.; Nagi, C.; Hilsenbeck, S.G.; Kapali, J.; et al. Hyperprolactinemia-inducing antipsychotics increase breast cancer risk by activating JAK-STAT5 in precancerous lesions. Breast Cancer Res. 2018, 20, 42. [Google Scholar] [CrossRef] [Green Version]
- Vafaizadeh, V.; Klemmt, P.; Brendel, C.; Weber, K.; Doebele, C.; Britt, K.; Grez, M.; Fehse, B.; Desrivieres, S.; Groner, B. Mammary epithelial reconstitution with gene-modified stem cells assigns roles to Stat5 in luminal alveolar cell fate decisions, differentiation, involution, and mammary tumor formation. Stem Cells 2010, 28, 928–938. [Google Scholar] [CrossRef]
- Bessette, K.; Lang, M.L.; Fava, R.A.; Grundy, M.; Heinen, J.; Horne, L.; Spolski, R.; Al-Shami, A.; Morse, H.C., 3rd; Leonard, W.J.; et al. A Stat5b transgene is capable of inducing CD8+ lymphoblastic lymphoma in the absence of normal TCR/MHC signaling. Blood 2008, 111, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Su, W.Y.; Liang, Q.C.; Zhang, Z.G.; Chen, H.M.; Du, W.; Chen, Y.X.; Fang, J.Y. Inhibition of STAT5 induces G1 cell cycle arrest and reduces tumor cell invasion in human colorectal cancer cells. Lab. Investig. 2009, 89, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Koppikar, P.; Lui, V.W.; Man, D.; Xi, S.; Chai, R.L.; Nelson, E.; Tobey, A.B.; Grandis, J.R. Constitutive activation of signal transducer and activator of transcription 5 contributes to tumor growth, epithelial-mesenchymal transition, and resistance to epidermal growth factor receptor targeting. Clin. Cancer Res. 2008, 14, 7682–7690. [Google Scholar] [CrossRef]
- Talati, P.G.; Gu, L.; Ellsworth, E.M.; Girondo, M.A.; Trerotola, M.; Hoang, D.T.; Leiby, B.; Dagvadorj, A.; McCue, P.A.; Lallas, C.D.; et al. Jak2-Stat5a/b Signaling Induces Epithelial-to-Mesenchymal Transition and Stem-Like Cell Properties in Prostate Cancer. Am. J. Pathol. 2015, 185, 2505–2522. [Google Scholar] [CrossRef] [Green Version]
- Creamer, B.A.; Sakamoto, K.; Schmidt, J.W.; Triplett, A.A.; Moriggl, R.; Wagner, K.U. Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol. Cell Biol. 2010, 30, 2957–2970. [Google Scholar] [CrossRef]
- Schmidt, J.W.; Wehde, B.L.; Sakamoto, K.; Triplett, A.A.; Anderson, S.M.; Tsichlis, P.N.; Leone, G.; Wagner, K.U. Stat5 regulates the phosphatidylinositol 3-kinase/Akt1 pathway during mammary gland development and tumorigenesis. Mol. Cell Biol. 2014, 34, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Tsuruyama, T.; Hiratsuka, T.; Jin, G.; Imai, Y.; Takeuchi, H.; Maruyama, Y.; Kanaya, K.; Ozeki, M.; Takakuwa, T.; Haga, H.; et al. Murine leukemia retrovirus integration induces the formation of transcription factor complexes on palindromic sequences in the signal transducer and activator of transcription factor 5a gene during the development of pre-B lymphomagenesis. Am. J. Pathol. 2011, 178, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Q.; Zhang, L.; Zhong, Y.; Fan, G.; Zhang, Z.; Wang, R.; Jin, M.; Qiu, Y.; Kong, D. Stellettin B induces apoptosis in human chronic myeloid leukemia cells via targeting PI3K and Stat5. Oncotarget 2017, 8, 28906–28921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagvadorj, A.; Collins, S.; Jomain, J.B.; Abdulghani, J.; Karras, J.; Zellweger, T.; Li, H.; Nurmi, M.; Alanen, K.; Mirtti, T.; et al. Autocrine prolactin promotes prostate cancer cell growth via Janus kinase-2-signal transducer and activator of transcription-5a/b signaling pathway. Endocrinology 2007, 148, 3089–3101. [Google Scholar] [CrossRef] [PubMed]
- Latha, K.; Li, M.; Chumbalkar, V.; Gururaj, A.; Hwang, Y.; Dakeng, S.; Sawaya, R.; Aldape, K.; Cavenee, W.K.; Bogler, O.; et al. Nuclear EGFRvIII-STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl-XL promoter. Int. J. Cancer 2013, 132, 509–520. [Google Scholar] [CrossRef]
- Chueh, F.-Y.; Leong, K.-F.; Yu, C.-L. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem. Biophys. Res. Commun. 2010, 402, 778–783. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.R.; Xiang, M.; Frank, D.A. Distinct roles of STAT3 and STAT5 in the pathogenesis and targeted therapy of breast cancer. Mol. Cell Endocrinol. 2014, 382, 616–621. [Google Scholar] [CrossRef]
- Sultan, A.S.; Xie, J.; LeBaron, M.J.; Ealley, E.L.; Nevalainen, M.T.; Rui, H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene 2005, 24, 746–760. [Google Scholar] [CrossRef]
- Mallette, F.A.; Gaumont-Leclerc, M.F.; Ferbeyre, G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007, 21, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Knol, A.C.; Nguyen, J.M.; Quereux, G.; Brocard, A.; Khammari, A.; Dreno, B. Prognostic value of tumor-infiltrating Foxp3+ T-cell subpopulations in metastatic melanoma. Exp. Derm. 2011, 20, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Sasada, T.; Kimura, M.; Yoshida, Y.; Kanai, M.; Takabayashi, A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: Possible involvement of regulatory T cells in disease progression. Cancer 2003, 98, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Li, J.; Tian, Y.; Mao, J.; Liu, X.; Feng, S.; Li, J.; Bian, Q.; Ji, H.; Zhang, L. Restoring Th17/Treg balance via modulation of STAT3 and STAT5 activation contributes to the amelioration of chronic obstructive pulmonary disease by Bufei Yishen formula. J. Ethnopharmacol. 2018, 217, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef]
- Li, X.; Kostareli, E.; Suffner, J.; Garbi, N.; Hammerling, G.J. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur. J. Immunol. 2010, 40, 3325–3335. [Google Scholar] [CrossRef]
- Fisher, S.A.; Aston, W.J.; Chee, J.; Khong, A.; Cleaver, A.L.; Solin, J.N.; Ma, S.; Lesterhuis, W.J.; Dick, I.; Holt, R.A.; et al. Transient Treg depletion enhances therapeutic anti-cancer vaccination. Immun. Inflamm. Dis. 2017, 5, 16–28. [Google Scholar] [CrossRef]
- Schmidt, A.; Zhang, X.M.; Joshi, R.N.; Iqbal, S.; Wahlund, C.; Gabrielsson, S.; Harris, R.A.; Tegner, J. Human macrophages induce CD4(+)Foxp3(+) regulatory T cells via binding and re-release of TGF-beta. Immunol. Cell Biol. 2016, 94, 747–762. [Google Scholar] [CrossRef]
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol. 2017, 316, 1–10. [Google Scholar] [CrossRef]
- Müller-Hermelink, N.; Braumüller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A.; et al. TNFR1 Signaling and IFN-γ Signaling Determine whether T Cells Induce Tumor Dormancy or Promote Multistage Carcinogenesis. Cancer Cell 2008, 13, 507–518. [Google Scholar] [CrossRef]
- Edwards, J.P.; Thornton, A.M.; Shevach, E.M. Release of active TGF-beta1 from the latent TGF-beta1/GARP complex on T regulatory cells is mediated by integrin beta8. J. Immunol. 2014, 193, 2843–2849. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Schafer, C.C.; Hough, K.P.; Tousif, S.; Duncan, S.R.; Kearney, J.F.; Ponnazhagan, S.; Hsu, H.C.; Deshane, J.S. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer through IL-7 and STAT5. J. Immunol. 2018, 201, 278–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurieva, R.I.; Podd, A.; Chen, Y.; Alekseev, A.M.; Yu, M.; Qi, X.; Huang, H.; Wen, R.; Wang, J.; Li, H.S.; et al. STAT5 protein negatively regulates T follicular helper (Tfh) cell generation and function. J. Biol. Chem. 2012, 287, 11234–11239. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bahre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.X.; Li, P.; Liu, D.; Jin, H.T.; He, J.; Ata Ur Rasheed, M.; Rochman, Y.; Wang, L.; Cui, K.; Liu, C.; et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 2012, 36, 586–599. [Google Scholar] [CrossRef]
- Owen, D.L.; Farrar, M.A. STAT5 and CD4 (+) T Cell Immunity. F1000Research 2017, 6, 32. [Google Scholar] [CrossRef]
- Grange, M.; Buferne, M.; Verdeil, G.; Leserman, L.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Activated STAT5 promotes long-lived cytotoxic CD8+ T cells that induce regression of autochthonous melanoma. Cancer Res. 2012, 72, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, D.; Putz, E.M.; Grundschober, E.; Prchal-Murphy, M.; Straka, E.; Kudweis, P.; Heller, G.; Bago-Horvath, Z.; Witalisz-Siepracka, A.; Cumaraswamy, A.A.; et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 2016, 6, 414–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Sopper, S.; Pircher, A.; Gastl, G.; Wolf, A.M. Treg(s) in Cancer: Friends or Foe? J. Cell Physiol. 2015, 230, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Kim, J.S.; Zhang, S.; Yuan, J.; Huang, M.; Glenn, M.; Haura, E.; Sebti, S.; Hamilton, A.D.; Jove, R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer 2004, 3, 261–269. [Google Scholar]
- Fontaine, F.; Overman, J.; Francois, M. Pharmacological manipulation of transcription factor protein-protein interactions: Opportunities and obstacles. Cell Regen. 2015, 4, 2. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [Green Version]
- Siddiquee, K.A.; Gunning, P.T.; Glenn, M.; Katt, W.P.; Zhang, S.; Schrock, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef]
- Sato, K.; Tsuchiya, M.; Saldanha, J.; Koishihara, Y.; Ohsugi, Y.; Kishimoto, T.; Bendig, M.M. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res. 1993, 53, 851–856. [Google Scholar]
- Mihara, M.; Kasutani, K.; Okazaki, M.; Nakamura, A.; Kawai, S.; Sugimoto, M.; Matsumoto, Y.; Ohsugi, Y. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int. Immunopharmacol. 2005, 5, 1731–1740. [Google Scholar] [CrossRef]
- Nishimoto, N.; Sasai, M.; Shima, Y.; Nakagawa, M.; Matsumoto, T.; Shirai, T.; Kishimoto, T.; Yoshizaki, K. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000, 95, 56–61. [Google Scholar] [CrossRef]
- Nishimoto, N.; Yoshizaki, K.; Miyasaka, N.; Yamamoto, K.; Kawai, S.; Takeuchi, T.; Hashimoto, J.; Azuma, J.; Kishimoto, T. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: A multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004, 50, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Intravenous Tocilizumab: A Review of Its Use in Adults with Rheumatoid Arthritis. BioDrugs 2013, 28, 75–106. [Google Scholar] [CrossRef] [PubMed]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Dambacher, J.; Beigel, F.; Seiderer, J.; Haller, D.; Goke, B.; Auernhammer, C.J.; Brand, S. Interleukin 31 mediates MAP kinase and STAT1/3 activation in intestinal epithelial cells and its expression is upregulated in inflammatory bowel disease. Gut 2007, 56, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Smith, M.A.; Doshi, P.; Sasser, K.; Fulp, W.; Altiok, S.; Haura, E.B. Antitumor efficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J. Thorac. Oncol. 2014, 9, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chen, B. Siltuximab (CNTO 328): A promising option for human malignancies. Drug Des. Devel. 2015, 9, 3455–3458. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; De Bono, J.S.; Flechon, A.; Heidenreich, A.; Voog, E.; Davis, N.B.; Qi, M.; Bandekar, R.; Vermeulen, J.T.; Cornfeld, M.; et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 85–93. [Google Scholar] [CrossRef]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Machida, T.; Sakaguchi, S.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Oncostatin M-induced blood-brain barrier impairment is due to prolonged activation of STAT3 signaling in vitro. J. Cell Biochem. 2018, 119, 9055–9063. [Google Scholar] [CrossRef]
- Jorcyk, C.L.; Holzer, R.G.; Ryan, R.E. Oncostatin M induces cell detachment and enhances the metastatic capacity of T-47D human breast carcinoma cells. Cytokine 2006, 33, 323–336. [Google Scholar] [CrossRef]
- Sterbova, S.; Karlsson, T.; Persson, E. Oncostatin M induces tumorigenic properties in non-transformed human prostate epithelial cells, in part through activation of signal transducer and activator of transcription 3 (STAT3). Biochem. Biophys. Res. Commun. 2018, 498, 769–774. [Google Scholar] [CrossRef]
- Richards, C.D. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm. 2013, 2013, 512103. [Google Scholar] [CrossRef] [PubMed]
- Hintzen, C.; Evers, C.; Lippok, B.E.; Volkmer, R.; Heinrich, P.C.; Radtke, S.; Hermanns, H.M. Box 2 region of the oncostatin M receptor determines specificity for recruitment of Janus kinases and STAT5 activation. J. Biol. Chem. 2008, 283, 19465–19477. [Google Scholar] [CrossRef]
- Hermanns, H.M.; Radtke, S.; Schaper, F.; Heinrich, P.C.; Behrmann, I. Non-redundant signal transduction of interleukin-6-type cytokines. The adapter protein Shc is specifically recruited to rhe oncostatin M receptor. J. Biol. Chem. 2000, 275, 40742–40748. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Bendit, M.; McAleer, D.; Liu, F.; Feeney, M.; Brett, S.; Zamuner, S.; Campanile, A.; Toso, J. Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti- oncostatin M monoclonal antibody in rheumatoid arthritis: Results from phase II randomized, placebo-controlled trials. Arthritis Res. 2013, 15, R132. [Google Scholar] [CrossRef] [PubMed]
- Poling, J.; Gajawada, P.; Richter, M.; Lorchner, H.; Polyakova, V.; Kostin, S.; Shin, J.; Boettger, T.; Walther, T.; Rees, W.; et al. Therapeutic targeting of the oncostatin M receptor-beta prevents inflammatory heart failure. Basic Res. Cardiol. 2014, 109, 396. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Du, Y.; Li, K.; Zhang, W.; Feng, X.; Hao, J.; Li, H.; Liu, S. Anti-OSM Antibody Inhibits Tubulointerstitial Lesion in a Murine Model of Lupus Nephritis. Mediat. Inflamm. 2017, 2017, 3038514. [Google Scholar] [CrossRef] [PubMed]
- Kucia-Tran, J.A.; Tulkki, V.; Scarpini, C.G.; Smith, S.; Wallberg, M.; Paez-Ribes, M.; Araujo, A.M.; Botthoff, J.; Feeney, M.; Hughes, K.; et al. Anti-oncostatin M antibody inhibits the pro-malignant effects of oncostatin M receptor overexpression in squamous cell carcinoma. J. Pathol. 2018, 244, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.J.; Turner, R.C.; Naser, Z.J.; McCurdy, C.R.; O’Callaghan, J.P.; Huber, J.D.; Matsumoto, R.R. SN79, a sigma receptor antagonist, attenuates methamphetamine-induced astrogliosis through a blockade of OSMR/gp130 signaling and STAT3 phosphorylation. Exp. Neurol. 2014, 254, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7, 12258. [Google Scholar] [CrossRef] [Green Version]
- Doherty, M.R.; Jackson, M.W. Using interferon-beta to combat cancer stem cell properties in triple negative breast cancer. Oncoscience 2018, 5, 169–171. [Google Scholar] [CrossRef]
- Doherty, M.R.; Parvani, J.G.; Tamagno, I.; Junk, D.J.; Bryson, B.L.; Cheon, H.J.; Stark, G.R.; Jackson, M.W. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Fu, M.L.; Weichselbaum, R.R.; Gajewski, T.F.; Guo, Y.; Fu, Y.X. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Marini, F.; Konopleva, M.; Schober, W.; Shi, Y.; Burks, J.; Clise-Dwyer, K.; Wang, R.Y.; Zhang, W.; Yuan, X.; et al. Mesenchymal Stem Cells Overexpressing IFN-beta Inhibit Breast Cancer Growth and Metastases through Stat3 Signaling in a Syngeneic Tumor Model. Cancer Microenviron. 2010, 3, 83–95. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef]
- Liu, S.; Lachapelle, J.; Leung, S.; Gao, D.; Foulkes, W.D.; Nielsen, T.O. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012, 14, R48. [Google Scholar] [CrossRef]
- Xie, C.; Liu, C.; Wu, B.; Lin, Y.; Ma, T.; Xiong, H.; Wang, Q.; Li, Z.; Ma, C.; Tu, Z. Effects of IRF1 and IFN-beta interaction on the M1 polarization of macrophages and its antitumor function. Int. J. Mol. Med. 2016, 38, 148–160. [Google Scholar] [CrossRef]
- Moreira-Teixeira, L.; Sousa, J.; McNab, F.W.; Torrado, E.; Cardoso, F.; Machado, H.; Castro, F.; Cardoso, V.; Gaifem, J.; Wu, X.; et al. Type I IFN Inhibits Alternative Macrophage Activation during Mycobacterium tuberculosis Infection and Leads to Enhanced Protection in the Absence of IFN-gamma Signaling. J. Immunol. 2016, 197, 4714–4726. [Google Scholar] [CrossRef]
- Pylaeva, E.; Lang, S.; Jablonska, J. The Essential Role of Type I Interferons in Differentiation and Activation of Tumor-Associated Neutrophils. Front. Immunol. 2016, 7, 629. [Google Scholar] [CrossRef] [Green Version]
- Brockwell, N.K.; Owen, K.L.; Zanker, D.; Spurling, A.; Rautela, J.; Duivenvoorden, H.M.; Baschuk, N.; Caramia, F.; Loi, S.; Darcy, P.K.; et al. Neoadjuvant Interferons: Critical for Effective PD-1-Based Immunotherapy in TNBC. Cancer Immunol. Res. 2017, 5, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Jin, S.; Yang, W.; Zhou, G.; Zhao, M.; Fang, S.; Zhang, Z.; Hu, J. Interferon-alpha enhances the antitumour activity of EGFR-targeted therapies by upregulating RIG-I in head and neck squamous cell carcinoma. Br. J. Cancer 2018, 118, 509–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef]
Figure 1.
STAT3/5 Signaling Cascades and Therapy Targets. Schematic representation of canonical activation of signal transducer and activator of transcription-3 (STAT3) and STAT5 by IL-6 family member cytokines. IL-6 family cytokines drive receptor heterodimerization and subsequent Janus Kinase (JAK activation). JAKs phosphorylate tyrosine residues along the cytoplasmic domain of the receptor dimer, which recruits STAT proteins and facilitates their binding to interferon-gamma activation site-like (GAS-like) elements and regulation of large sets of genes. Asterisks (*) denote that additional info can be found in Table 1.
Figure 1.
STAT3/5 Signaling Cascades and Therapy Targets. Schematic representation of canonical activation of signal transducer and activator of transcription-3 (STAT3) and STAT5 by IL-6 family member cytokines. IL-6 family cytokines drive receptor heterodimerization and subsequent Janus Kinase (JAK activation). JAKs phosphorylate tyrosine residues along the cytoplasmic domain of the receptor dimer, which recruits STAT proteins and facilitates their binding to interferon-gamma activation site-like (GAS-like) elements and regulation of large sets of genes. Asterisks (*) denote that additional info can be found in Table 1.

Figure 2.
Biological Impact of STAT3 and STAT5 Activation. Pre-malignant cell populations, in which apoptotic signaling and immunosurveillance are functional, exhibit low levels of phosphorylated STAT3 (pSTAT3) and STAT5 (pSTAT5). Elevated activity of pSTAT3 and/or pSTAT5 accompanies tumorigenesis, leading to the inhibition of apoptotic pathways and repression of immune cell recognition of a burgeoning tumor. pSTAT3 is utilized by malignant cell populations to drive epithelial-mesenchymal transition (EMT) and by tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs) to drive metastasis. pSTAT5 activity in progenitor T-cells drives expansion of a Treg population that then secretes factors that inhibit the function of CD4+ and CD8+ T-cells as well as B-cells.
Figure 2.
Biological Impact of STAT3 and STAT5 Activation. Pre-malignant cell populations, in which apoptotic signaling and immunosurveillance are functional, exhibit low levels of phosphorylated STAT3 (pSTAT3) and STAT5 (pSTAT5). Elevated activity of pSTAT3 and/or pSTAT5 accompanies tumorigenesis, leading to the inhibition of apoptotic pathways and repression of immune cell recognition of a burgeoning tumor. pSTAT3 is utilized by malignant cell populations to drive epithelial-mesenchymal transition (EMT) and by tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs) to drive metastasis. pSTAT5 activity in progenitor T-cells drives expansion of a Treg population that then secretes factors that inhibit the function of CD4+ and CD8+ T-cells as well as B-cells.


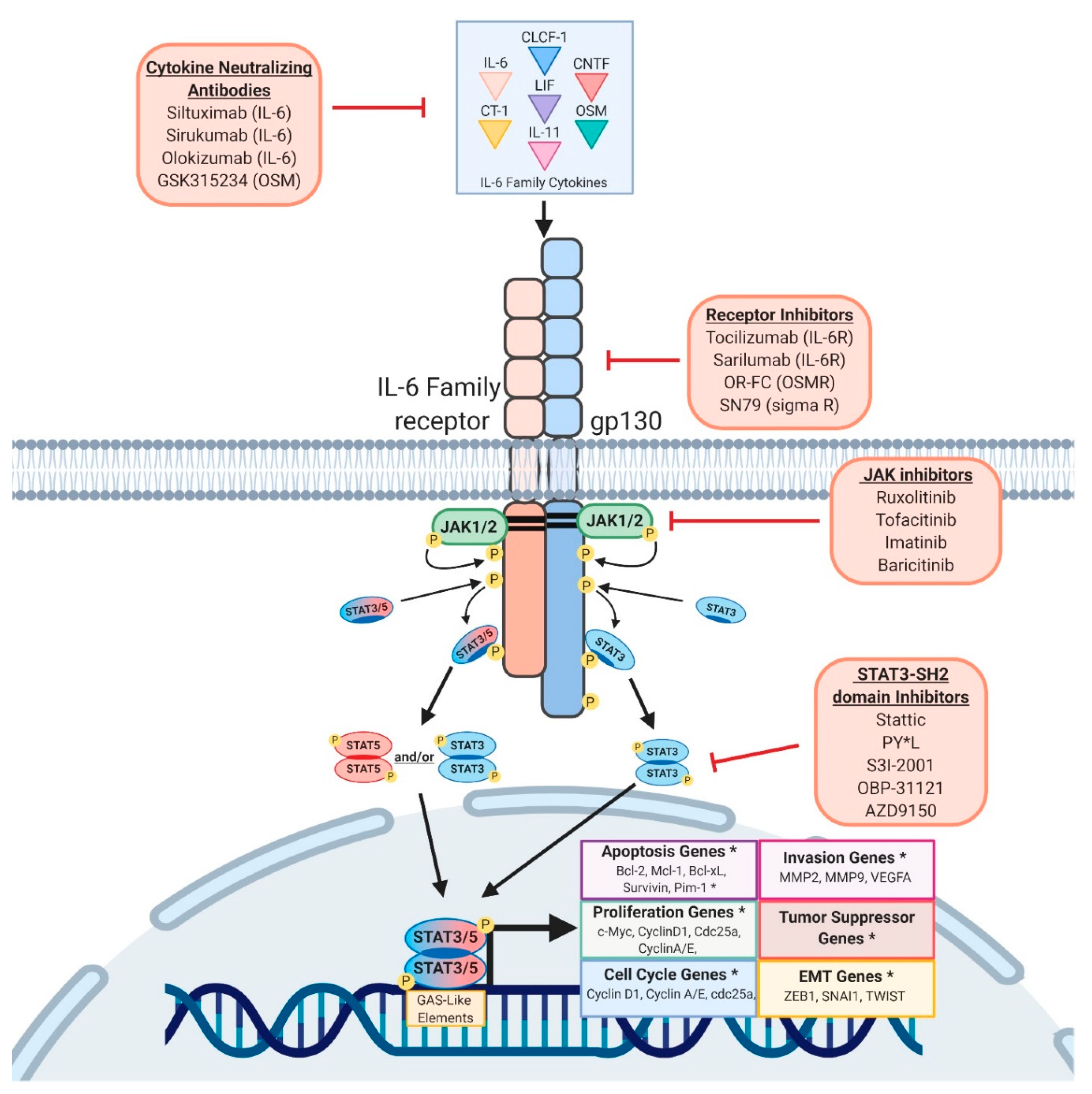{kind=link}
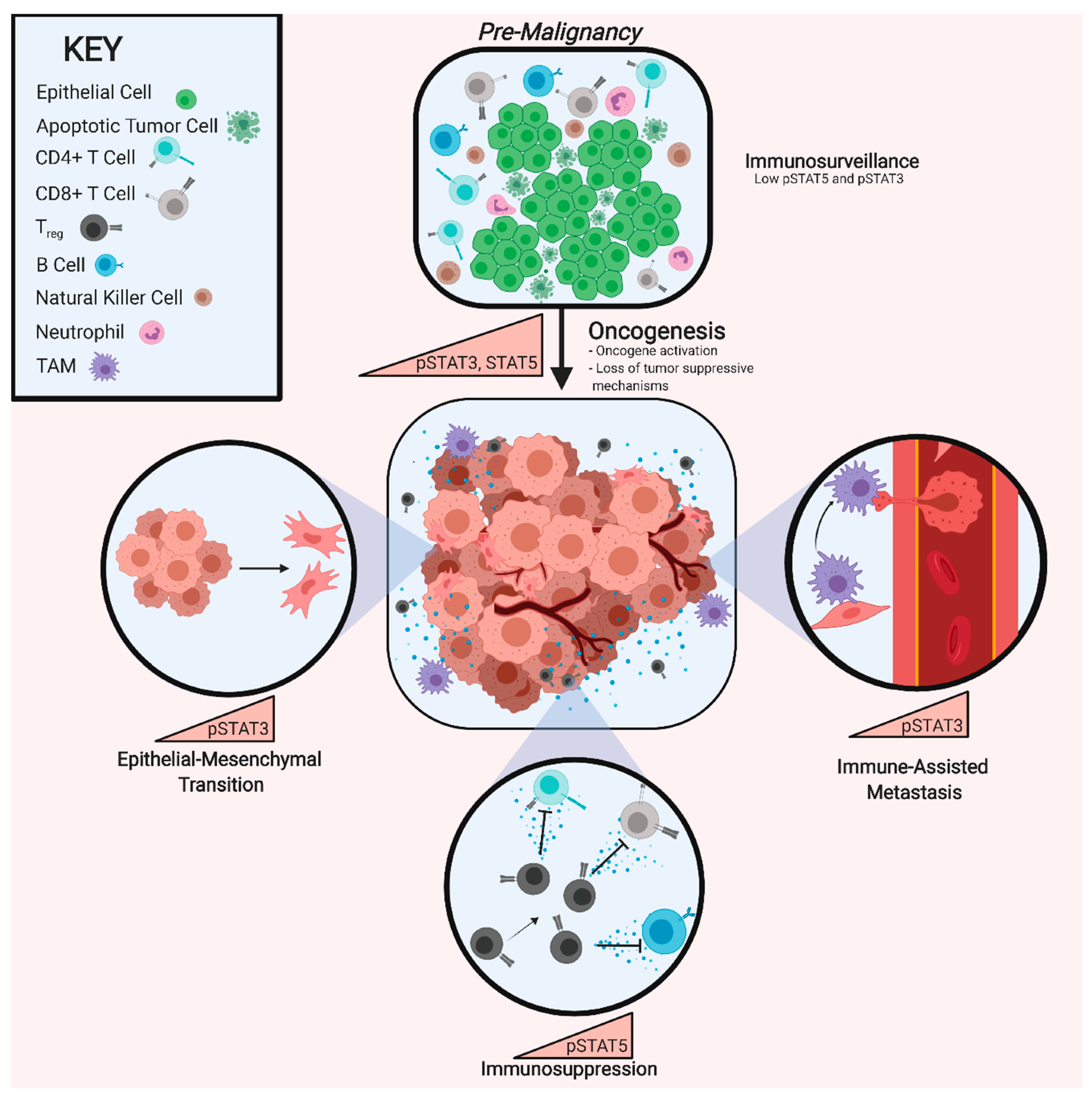{kind=link}
Table 1.
Summary of the Roles of STAT3 and STAT5 in Cancer and Strategies for their Inhibition.
| STAT-Family Protein | pYSTAT3/5-Activating Cytokines and Growth Factors | Normal Immune Function | Pro-Tumorigenic Genes Activated | Pro-Tumorigenic Effects | Therapies and Drugs in Development |
|---|---|---|---|---|---|
| STAT3 | Growth Factors: EGFs, FGFs, HGFs, Leptin (REF), PDGFs Lymphoid Cytokines: IL-4, IL-13 Myeloid Cytokines: G-CSF, IL-10 Ubiquitous Cytokines: CNTF, LIF, IL-6, OSM | Development of Mature Neutrophils [58] Neutrophil Mobilization from Bone Marrow [59] | Apoptosis: BCL2, BCL-XL, MCL1, PIM1 Cell Cycle: Cyclin A, Cyclin D1, CDC25A EMT: ZEB1, SNAI1, TWIST, CDH2, Vimentin Migration/Invasion: MMP2, MMP9, VEGFA Proliferation: cMYC, NOTCH4 Stem Cell: SOX2, NANOG, OCT4 Tumor Suppressor: p53 | Transformation Activation of Anti-Apoptotic Proteins EMT M2 Polarization N2 Polarization Enhanced Metastasis Therapy Resistance | Imatinib (JAK1/2) Ruxolitinib (JAK1/2) Tofacitinib (JAK1/2) Siltuximab (IL-6) Tocilizumab (IL-6R) GSK315234 (OSM) OS-FC (OSMR) PY*L (SH2 Domain) S3I-2001 (SH2 Domain) SN79 (Sigma Receptor) IFNβ |
| STAT5 | Growth Factors: EGFs, FGFs, FLT3L, Leptin [60], PDGFs, Prolactin, SCF Lymphoid Cytokines: IL-2, IL-4, IL-7 [61], IL-9 [62], IL-13, IL-15, Thymic Stromal Lymphopoietin [63] Myeloid Cytokines: Erythropoietin [64], G-CSF, GM-CSF, IL-3, IL-5, IL-10, Thrombopoietin [65] Ubiquitous Cytokines: IL-21 [66], IL-31 [67], OSM | Differentiation, Survival, and Lineage Expansion of NK and NKT Cells Dendritic Cell Function [68] T Cell Differentiation and Expansion Effector Memory CD8+ T Cell-Mediated Cancer Cell Clearance [69] Myeloid Cell Differentiation and Survival [70] | Apoptosis: BCL-2, BCL-XL, MCL1, PIM1, Survivin Cell Cycle: Cyclin D1, Cyclin E Immune Function: PRDM1, BCL-6 Migration/Invasion: MMP2, MMP9, VEGFA Proliferation: AKT1, cMYC, PI3K Tumor Suppressor: p53 | Transformation Activation of Anti-Apoptotic Proteins Treg Population Expansion M2 Polarization [71] Megakaryopoeisis [72] Erythropoeisis [73] | Imatinib (JAK1/2) Ruxolitinib (JAK1/2) Tofacitinib (JAK1/2) Siltuximab (IL-6) Tocilizumab (IL-6R) GSK315234 (OSM) OS-FC (OSMR) AC-4-130 (SH2 Domain) [74] BP-1-107 (SH2 Domain) [75] BP-1-108 (SH2 Domain) [75] Pomstafib-2 (SH2 Domain) [76] SF-1-087 (SH2 Domain) [75] SF-1-088 (SH2 Domain) [75] IFNβ |
Therapies included in the far right column are listed by their respective name followed by their biological target in parentheses. Italicized font denotes therapies that are not currently approved by the FDA as cancer therapies.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Polak, K.L.; Chernosky, N.M.; Smigiel, J.M.; Tamagno, I.; Jackson, M.W. Balancing STAT Activity as a Therapeutic Strategy. Cancers 2019, 11, 1716. https://doi.org/10.3390/cancers11111716
AMA Style
Polak KL, Chernosky NM, Smigiel JM, Tamagno I, Jackson MW. Balancing STAT Activity as a Therapeutic Strategy. Cancers. 2019; 11(11):1716. https://doi.org/10.3390/cancers11111716
Chicago/Turabian StylePolak, Kelsey L., Noah M. Chernosky, Jacob M. Smigiel, Ilaria Tamagno, and Mark W. Jackson. 2019. "Balancing STAT Activity as a Therapeutic Strategy" Cancers 11, no. 11: 1716. https://doi.org/10.3390/cancers11111716
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.