Recreating Tumour Complexity in a Dish: Organoid Models to Study Liver Cancer Cells and their Extracellular Environment

 and
and
Abstract
:1. Introduction
2. Limitations of Current in Vitro Cancer Models
2.1. Two-Dimensional Cell Line Models
2.2. Three-Dimensional Liver Cancer Models
2.2.1. Scaffold-Based Models
2.2.2. Scaffold-Free Models
3. Emerging Organoid Models
3.1. Organoid Culture Systems
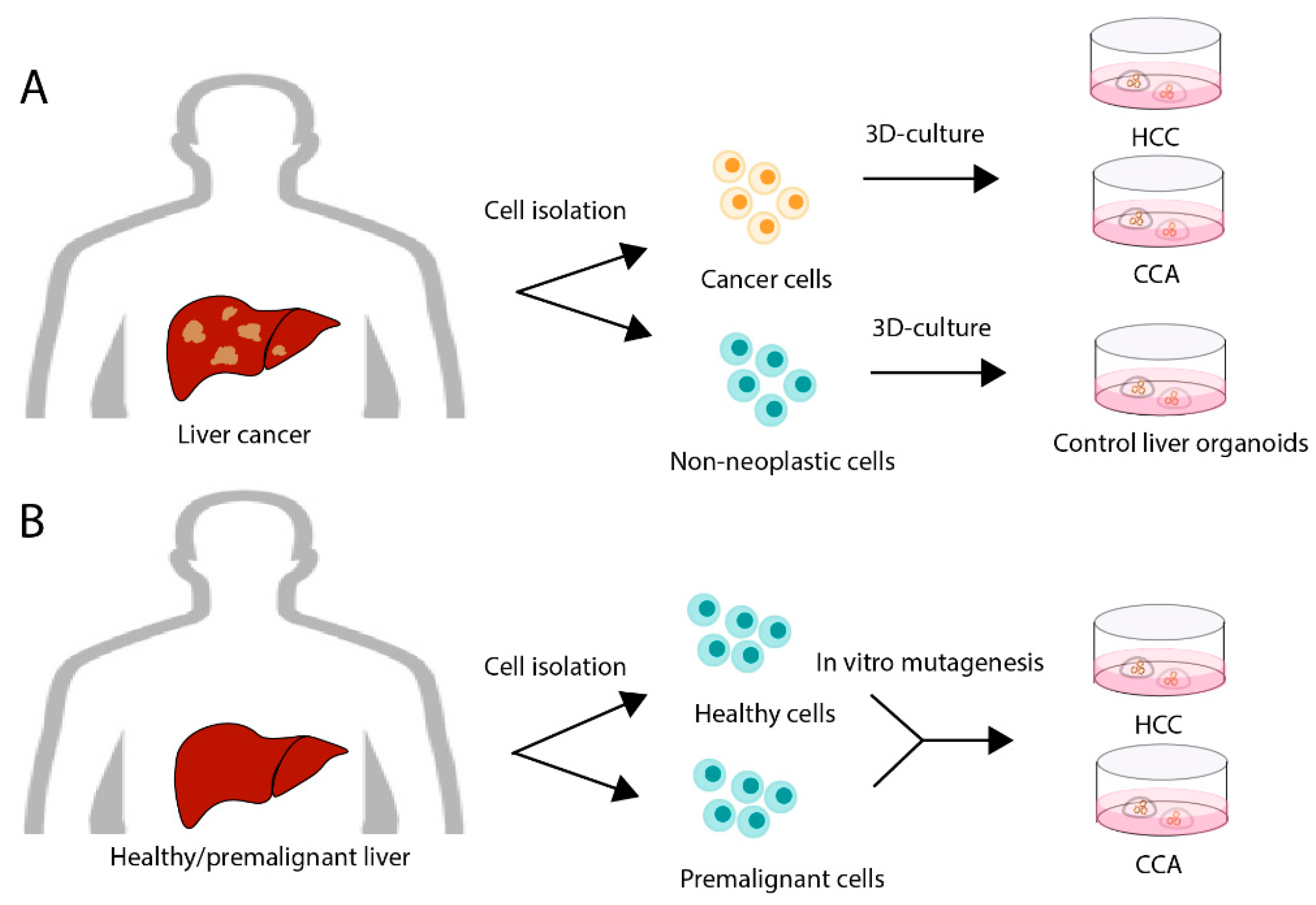
3.2. Primary Liver Cancer (PLC) Organoids
3.3. Current Limitations of Organoid Models
4. The Liver Cancer Extracellular Environment
4.1. Role of the Extracellular Environment in Hepatocellular Carcinoma
4.2. Role of the Extracellular Environment in Cholangiocarcinoma
5. Adapting Organoid Models to Study Liver Cancer Cell Interactions with the Extracellular Environment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PLC | Primary liver cancer |
| CCA | Cholangiocarcinoma |
| iCCA | Intrahepatic cholangiocarcinoma |
| pCCA | Perihilar cholangiocarcinoma |
| dCCA | Distal cholangiocarcinoma |
| CAF | Cancer-associated fibroblast |
| ECM | Extracellular matrix |
| PVA | Poly(vinyl alcohol) |
| RWV | Rotary wall vessel |
| LGR5 | Leucine-rich repeat-containing G-protein coupled receptor 5 |
| KRAS | Ki-ras |
| Pik3ca | Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha |
| FGFR2-AHCYL1 | Fibroblast growth factor receptor 2-adenosylhomocysteinase like 1 |
| PEG | Polyethylene glycol |
| MMP | Matrix metallopeptidases |
| TME | Tumour microenvironment |
| PVTT | Portal vein tumour thrombus |
| EMT | Epithelial-to-mesenchymal transition |
| TEE | Tumour extracellular environment |
| LOXL2 | Lysyl oxidase like 2 |
| YAP | Yes-associated protein |
| TAZ | Transcriptional co-activator with PDZ-binding motif |
| PDAC | Pancreatic ductal adenocarcinoma |
References
- Bosch, F.X.; Ribes, J.; Cléries, R.; Díaz, M. Epidemiology of hepatocellular carcinoma. Clin. Liver Dis. 2005, 9, 191–211. [Google Scholar] [CrossRef]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, e408. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Zheng, R.; Guo, Y.; Zhang, S.; Zou, X.; Wang, N.; Zhang, L.; Tang, J.; Chen, J.; Wei, K. Cancer survival in c hina, 2003–2005: A population-based study. Int. J. Cancer 2015, 136, 1921–1930. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, J.S.; Shi, J.F.; Li, N.; Wang, Y.T.; Qu, C.; Zhang, Y.; Dai, M. International trends in primary liver cancer incidence from 1973 to 2007. BMC Cancer 2015, 15, e94. [Google Scholar] [CrossRef]
- Valery, P.C.; Laversanne, M.; Clark, P.J.; Petrick, J.L.; McGlynn, K.A.; Bray, F. Projections of primary liver cancer to 2030 in 30 countries worldwide. Hepatology 2018, 67, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.M.; Bu, H.; Chen, J.; Dong, H.; Zhu, Y.Y.; Feng, L.H.; Chen, J.; Guideline, C. Practice guidelines for the pathological diagnosis of primary liver cancer: 2015 update. World J. Gastroenterol. 2016, 22, e9279. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, e653. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef]
- Dragani, T.A. Risk of hcc: Genetic heterogeneity and complex genetics. J. Hepatol. 2010, 52, 252–257. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Classification, diagnosis, and management of cholangiocarcinoma. Clin. Gastroenterol. Hepatol. 2013, 11, 13–21. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Mian, I.; Blechacz, B. Cancer review: Cholangiocarcinoma. J. Carcinog. 2015, 14, e1. [Google Scholar] [CrossRef]
- Walter, D.; Döring, C.; Feldhahn, M.; Battke, F.; Hartmann, S.; Winkelmann, R.; Schneider, M.; Bankov, K.; Schnitzbauer, A.; Zeuzem, S. Intratumoral heterogeneity of intrahepatic cholangiocarcinoma. Oncotarget 2017, 8, e14957. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Illades, D.; McMillin, M.; Quinn, M.; DeMorrow, S. Cholangiocarcinoma pathogenesis: Role of the tumor microenvironment. Transl. Gastrointest. Cancer 2012, 1, e71. [Google Scholar]
- Yaacoub, K.; Pedeux, R.; Tarte, K.; Guillaudeux, T. Role of the tumor microenvironment in regulating apoptosis and cancer progression. Cancer Lett. 2016, 378, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, M.; Melin, A.A.; Douaiher, J.; Lin, C.; Zhen, W.K.; Hussain, S.M.; Geschwind, J.F.H.; Doyle, M.B.M.; Abou-Alfa, G.K.; Are, C. A review and update of treatment options and controversies in the management of hepatocellular carcinoma. Ann. Surg. 2016, 263, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and future treatment of hepatocellular carcinoma: An updated comprehensive review. J. Clin. Transl. Hepatol. 2018, 6, e69. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Shiani, A.; Narayanan, S.; Pena, L.; Friedman, M. The role of diagnosis and treatment of underlying liver disease for the prognosis of primary liver cancer. Cancer Control 2017, 24, e1073274817729240. [Google Scholar] [CrossRef]
- Li, L.; Wang, H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016, 379, 191–197. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.L.; Schirmacher, P.; Vilgrain, V. Easl clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X. Future directions in the treatment of cholangiocarcinoma. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, e13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, Z.Y.; Hou, J.X. Hepatocellular carcinoma: Insight from animal models. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, e32. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Wagner, E.F. Mouse models for liver cancer. Mol. Oncol. 2013, 7, 206–223. [Google Scholar] [CrossRef]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, e23306. [Google Scholar] [CrossRef]
- Vicent, S.; Lieshout, R.; Saborowski, A.; Verstegen, M.M.A.; Raggi, C.; Recalcati, S.; Invernizzi, P.; van der Laan, L.J.W.; Alvaro, D.; Calvisi, D.F. Experimental models to unravel the molecular pathogenesis, cell of origin and stem cell properties of cholangiocarcinoma. Liver Int. 2019, 39, 79–97. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Hulkower, K.I.; Herber, R.L. Cell migration and invasion assays as tools for drug discovery. Pharmaceutics 2011, 3, 107–124. [Google Scholar] [CrossRef]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In vitro tumor models: Advantages, disadvantages, variables, and selecting the right platform. Front. Bioeng. Biotechnol. 2016, 4, e12. [Google Scholar] [CrossRef]
- Stock, K.; Estrada, M.F.; Vidic, S.; Gjerde, K.; Rudisch, A.; Santo, V.E.; Barbier, M.; Blom, S.; Arundkar, S.C.; Selvam, I. Capturing tumor complexity in vitro: Comparative analysis of 2d and 3d tumor models for drug discovery. Sci. Rep. 2016, 6, e28951. [Google Scholar] [CrossRef] [PubMed]
- Konstantopoulos, K.; Wu, P.H.; Wirtz, D. Dimensional control of cancer cell migration. Biophys. J. 2013, 104, e279. [Google Scholar] [CrossRef] [PubMed]
- Pisano, M.; Triacca, V.; Barbee, K.A.; Swartz, M.A. An in vitro model of the tumor–lymphatic microenvironment with simultaneous transendothelial and luminal flows reveals mechanisms of flow enhanced invasion. Integr. Biol. 2015, 7, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, S.; Truong, D.; Mouneimne, G.; Nikkhah, M. Microfluidic tumor–vascular model to study breast cancer cell invasion and intravasation. Adv. Healthcare Mater. 2018, 7, e1701257. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Varghese, S. Ex vivo tumor-on-a-chip platforms to study intercellular interactions within the tumor microenvironment. Adv. Healthcare Mater. 2019, 8, e1801198. [Google Scholar] [CrossRef]
- Wilding, J.L.; Bodmer, W.F. Cancer cell lines for drug discovery and development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef]
- Hirschfield, H.; Bian, C.B.; Higashi, T.; Nakagawa, S.; Zeleke, T.Z.; Nair, V.D.; Fuchs, B.C.; Hoshida, Y. In vitro modeling of hepatocellular carcinoma molecular subtypes for anti-cancer drug assessment. Exp. Mol. Med. 2018, 50, e419. [Google Scholar] [CrossRef]
- Varamo, C.; Peraldo-Neia, C.; Ostano, P.; Basiricò, M.; Raggi, C.; Bernabei, P.; Venesio, T.; Berrino, E.; Aglietta, M.; Leone, F. Establishment and characterization of a new intrahepatic cholangiocarcinoma cell line resistant to gemcitabine. Cancers 2019, 11, 519. [Google Scholar] [CrossRef]
- Lacoste, B.; Raymond, V.A.; Cassim, S.; Lapierre, P.; Bilodeau, M. Highly tumorigenic hepatocellular carcinoma cell line with cancer stem cell-like properties. PLoS ONE 2017, 12, e0171215. [Google Scholar] [CrossRef]
- Castven, D.; Becker, D.; Czauderna, C.; Wilhelm, D.; Andersen, J.B.; Strand, S.; Hartmann, M.; Heilmann-Heimbach, S.; Roth, W.; Hartmann, N. Application of patient-derived liver cancer cells for phenotypic characterization and therapeutic target identification. Int. J. Cancer 2019, 144, 2782–2794. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Piorońska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wölfl, S.; Meyer, C. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J. Exp. Clin. Cancer Res. 2018, 37, e211. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, e839. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension–how 3d culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Chiang, T.; Liu, C.H.; Chern, G.G.; Lin, T.T.; Gao, D.Y.; Chen, Y. Delivery of sirna using cxcr4-targeted nanoparticles modulates tumor microenvironment and achieves a potent antitumor response in liver cancer. Mol. Ther. 2015, 23, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.K.; Tennant, D.A.; McKeating, J.A. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: Current understanding and future directions. J. Hepatol. 2014, 61, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Oudin, M.J.; Weaver, V.M. Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2016; pp. 189–205. [Google Scholar]
- Théry, M.; Racine, V.; Piel, M.; Pépin, A.; Dimitrov, A.; Chen, Y.; Sibarita, J.B.; Bornens, M. Anisotropy of cell adhesive microenvironment governs cell internal organization and orientation of polarity. Proc. Natl. Acad. Sci. USA 2006, 103, 19771–19776. [Google Scholar] [CrossRef] [Green Version]
- Pathak, A.; Kumar, S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc. Natl. Acad. Sci. USA 2012, 109, 10334–10339. [Google Scholar] [CrossRef] [Green Version]
- Justice, B.A.; Badr, N.A.; Felder, R.A. 3D cell culture opens new dimensions in cell-based assays. Drug Discov. Today 2009, 14, 102–107. [Google Scholar] [CrossRef]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef]
- Moscato, S.; Ronca, F.; Campani, D.; Danti, S. Poly (vinyl alcohol)/gelatin hydrogels cultured with hepg2 cells as a 3D model of hepatocellular carcinoma: A morphological study. J. Funct. Biomater. 2015, 6, 16–32. [Google Scholar] [CrossRef]
- Fang, M.; Peng, C.W.; Liu, S.P.; Yuan, J.P.; Li, Y. In vitro invasive pattern of hepatocellular carcinoma cell line hcclm9 based on three-dimensional cell culture and quantum dots molecular imaging. J. Huazhong Univ. Sci. Technol. 2013, 33, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.; Kievit, F.M.; Florczyk, S.J.; Veiseh, O.; Wu, J.; Park, J.O.; Zhang, M. Chitosan-alginate scaffold culture system for hepatocellular carcinoma increases malignancy and drug resistance. Pharm. Res. 2010, 27, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Yip, D.; Cho, C.H. A multicellular 3d heterospheroid model of liver tumor and stromal cells in collagen gel for anti-cancer drug testing. Biochem. Biophys. Res. Commun. 2013, 433, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Takai, A.; Fako, V.; Dang, H.; Forgues, M.; Yu, Z.; Budhu, A.; Wang, X.W. Three-dimensional organotypic culture models of human hepatocellular carcinoma. Sci. Rep. 2016, 6, e21174. [Google Scholar] [CrossRef] [PubMed]
- Iwase, Y.; Nakayama, M.; Yamato, M.; Okano, T. A biomimicking tumor tissue model using hepatocellular carcinoma cell sheet in a collagen sandwich system. Anticancer Res. 2015, 35, 6481–6486. [Google Scholar]
- Parasramka, M.; Yan, I.K.; Wang, X.; Nguyen, P.; Matsuda, A.; Maji, S.; Foye, C.; Asmann, Y.; Patel, T. Bap1 dependent expression of long non-coding rna neat-1 contributes to sensitivity to gemcitabine in cholangiocarcinoma. Mol. Cancer 2017, 16, e22. [Google Scholar] [CrossRef]
- Campbell, D.J.W.; Dumur, C.I.; Lamour, N.F.; DeWitt, J.L.; Sirica, A.E. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol. Res. 2012, 42, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3d tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6, e19103. [Google Scholar] [CrossRef]
- Santo, V.E.; Rebelo, S.P.; Estrada, M.F.; Alves, P.M.; Boghaert, E.; Brito, C. Drug screening in 3d in vitro tumor models: Overcoming current pitfalls of efficacy read-outs. Biotechnol. J. 2017, 12. [Google Scholar] [CrossRef]
- Ong, S.M.; Zhao, Z.; Arooz, T.; Zhao, D.; Zhang, S.; Du, T.; Wasser, M.; van Noort, D.; Yu, H. Engineering a scaffold-free 3D tumor model for in vitro drug penetration studies. Biomaterials 2010, 31, 1180–1190. [Google Scholar] [CrossRef]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Dong, Y.; Xie, X.; Chen, J.; Gao, D.; Liu, Y.; Ren, Z.; Cui, J. Screening candidate metastasis-associated genes in three-dimensional hcc spheroids with different metastasis potential. Int. J. Clin. Exp. Pathol. 2014, 7, e2527. [Google Scholar]
- Tang, J.; Cui, J.; Chen, R.; Guo, K.; Kang, X.; Li, Y.; Gao, D.; Sun, L.; Xu, C.; Chen, J. A three-dimensional cell biology model of human hepatocellular carcinoma in vitro. Tumor Biol. 2011, 32, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. Ecm microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.H.; Ryeom, S.; Kim, C.F. Lung stem cell differentiation in mice directed by endothelial cells via a bmp4-nfatc1-thrombospondin-1 axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef]
- Petersen, O.W.; Rønnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064–9068. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J. Single lgr5 stem cells build crypt–villus structures in vitro without a mesenchymal niche. Nature 2009, 459, e262. [Google Scholar] [CrossRef]
- Huch, M.; Dorrell, C.; Boj, S.F.; Van Es, J.H.; Li, V.S.W.; Van De Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J. In vitro expansion of single lgr5+ liver stem cells induced by wnt-driven regeneration. Nature 2013, 494, e247. [Google Scholar] [CrossRef]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.J.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, e951. [Google Scholar] [CrossRef]
- Jung, P.; Sato, T.; Merlos-Suárez, A.; Barriga, F.M.; Iglesias, M.; Rossell, D.; Auer, H.; Gallardo, M.; Blasco, M.A.; Sancho, E. Isolation and in vitro expansion of human colonic stem cells. Nat. Med. 2011, 17, e1225. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P. Human primary liver cancer–derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, e1424. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jiao, Y.; Qin, S.; Zhao, W.; Chu, Q.; Wu, K. Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp. Hematol. Oncol. 2018, 7, e30. [Google Scholar] [CrossRef]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef]
- Li, L.; Knutsdottir, H.; Hui, K.; Weiss, M.J.; He, J.; Philosophe, B.; Cameron, A.M.; Wolfgang, C.L.; Pawlik, T.M.; Ghiaur, G. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 2019, 4, e121490. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 2018, 172, 373–386. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Ochiai, M.; Yoshihara, Y.; Maru, Y.; Tetsuya, M.; Izumiya, M.; Imai, T.; Hippo, Y. Kras-driven heterotopic tumor development from hepatobiliary organoids. Carcinogenesis 2019, 40, 1142–1152. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S. Generation of tumor-reactive t cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 2018, 174, 1586–1598. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3d tumor microenvironment models. BMC Cancer 2018, 18, e335. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, e560. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Acuña, R.; Quirós, M.; Farkas, A.E.; Dedhia, P.H.; Huang, S.; Siuda, D.; García-Hernández, V.; Miller, A.J.; Spence, J.R.; Nusrat, A. Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nat. Cell Biol. 2017, 19, e1326. [Google Scholar] [CrossRef] [PubMed]
- Broguiere, N.; Isenmann, L.; Hirt, C.; Ringel, T.; Placzek, S.; Cavalli, E.; Ringnalda, F.; Villiger, L.; Züllig, R.; Lehmann, R. Growth of epithelial organoids in a defined hydrogel. Adv. Mater. 2018, 30, e1801621. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, E.; Sahai, E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026781. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Van Huizen, N.A.; van den Braak, R.R.J.C.; Doukas, M.; Dekker, L.J.M.; Ijzermans, J.N.M.; Luider, T.M. Up-regulation of collagen proteins in colorectal liver metastasis compared with normal liver tissue. J. Biol. Chem. 2019, 294, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Schwabe, R.F. Nf-κb in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, e108. [Google Scholar] [CrossRef]
- Sirica, A.E.; Gores, G.J. Desmoplastic stroma and cholangiocarcinoma: Clinical implications and therapeutic targeting. Hepatology 2014, 59, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.T.P.; Fan, S.T.; Wong, J. Risk factors, prevention, and management of postoperative recurrence after resection of hepatocellular carcinoma. Ann. Surg. 2000, 232, e10. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Morton, S.D.; Strazzabosco, M.; Fabris, L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: An opportunity for new therapeutic strategies. Transl. Gastrointest. Cancer 2013, 2, e130. [Google Scholar]
- Naim, A.; Pan, Q.; Baig, M.S. Matrix metalloproteinases (mmps) in liver diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef]
- Rombouts, K.; Carloni, V. The fibrotic microenvironment as a heterogeneity facet of hepatocellular carcinoma. Fibrogenesis Tissue Repair 2013, 6, e17. [Google Scholar] [CrossRef]
- Govaere, O.; Komuta, M.; Berkers, J.; Spee, B.; Janssen, C.; de Luca, F.; Katoonizadeh, A.; Wouters, J.; van Kempen, L.C.; Durnez, A. Keratin 19: A key role player in the invasion of human hepatocellular carcinomas. Gut 2014, 63, 674–685. [Google Scholar] [CrossRef]
- Giannelli, G.; Fransvea, E.; Bergamini, C.; Marinosci, F.; Antonaci, S. Laminin-5 chains are expressed differentially in metastatic and nonmetastatic hepatocellular carcinoma. Clin. Cancer Res. 2003, 9, 3684–3691. [Google Scholar]
- Govaere, O.; Wouters, J.; Petz, M.; Vandewynckel, Y.P.; Van den Eynde, K.; Verhulst, S.; Dollé, L.; Gremeaux, L.; Ceulemans, A.; Nevens, F. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J. Hepatol. 2016, 64, 609–617. [Google Scholar] [CrossRef]
- Théret, N.; Musso, O.; Turlin, B.; Lotrian, D.; Bioulac-Sage, P.; Campion, J.P.; Boudjéma, K.; Clément, B. Increased extracellular matrix remodeling is associated with tumor progression in human hepatocellular carcinomas. Hepatology 2001, 34, 82–88. [Google Scholar] [CrossRef]
- Donato, M.F.; Colombo, M.; Matarazzo, M.; Paronetto, F. Distribution of basement membrane components in human hepatocellular carcinoma. Cancer 1989, 63, 272–279. [Google Scholar] [CrossRef]
- Fang, M.; Yuan, J.P.; Peng, C.W.; Pang, D.W.; Li, Y. Quantum dots-based in situ molecular imaging of dynamic changes of collagen iv during cancer invasion. Biomaterials 2013, 34, 8708–8717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ye, J.; Weng, X.; Liu, F.; He, L.; Zhou, D.; Liu, Y. Comparative transcriptome analysis reveals that the extracellular matrix receptor interaction contributes to the venous metastases of hepatocellular carcinoma. Cancer Genet. 2015, 208, 482–491. [Google Scholar] [CrossRef]
- Zhao, X.L.; Sun, T.; Che, N.; Sun, D.; Zhao, N.; Dong, X.Y.; Gu, Q.; Yao, Z.; Sun, B.C. Promotion of hepatocellular carcinoma metastasis through matrix metalloproteinase activation by epithelial-mesenchymal transition regulator twist1. J. Cell. Mol. Med. 2011, 15, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Riener, M.O.; Fritzsche, F.R.; Soll, C.; Pestalozzi, B.C.; Probst-Hensch, N.; Clavien, P.A.; Jochum, W.; Soltermann, A.; Moch, H.; Kristiansen, G. Expression of the extracellular matrix protein periostin in liver tumours and bile duct carcinomas. Histopathology 2010, 56, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C. Allosteric inhibition of lysyl oxidase–like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, e1009. [Google Scholar] [CrossRef]
- Zhao, G.; Cui, J.; Qin, Q.; Zhang, J.; Liu, L.; Deng, S.; Wu, C.; Yang, M.; Li, S.; Wang, C. Mechanical stiffness of liver tissues in relation to integrin β1 expression may influence the development of hepatic cirrhosis and hepatocellular carcinoma. J. Surg. Oncol. 2010, 102, 482–489. [Google Scholar] [CrossRef]
- Høgdall, D.; Lewinska, M.; Andersen, J.B. Desmoplastic tumor microenvironment and immunotherapy in cholangiocarcinoma. Trends Cancer 2018, 4, 239–255. [Google Scholar] [CrossRef]
- Aishima, S.I.; Taguchi, K.I.; Terashi, T.; Matsuura, S.; Shimada, M.; Tsuneyoshi, M. Tenascin expression at the invasive front is associated with poor prognosis in intrahepatic cholangiocarcinoma. Mod. Pathol. 2003, 16, e1019. [Google Scholar] [CrossRef]
- Shirabe, K.; Shimada, M.; Kajiyama, K.; Hasegawa, H.; Gion, T.; Ikeda, Y.; Takenaka, K.; Sugimachi, K. Expression of matrix metalloproteinase-9 in surgically resected intrahepatic cholangiocarcinoma. Surgery 1999, 126, 842–846. [Google Scholar] [CrossRef]
- Hirashita, T.; Iwashita, Y.; Ohta, M.; Komori, Y.; Eguchi, H.; Yada, K.; Kitano, S. Expression of matrix metalloproteinase-7 is an unfavorable prognostic factor in intrahepatic cholangiocarcinoma. J. Gastrointest. Surg. 2012, 16, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Thelen, A.; Scholz, A.; Weichert, W.; Wiedenmann, B.; Neuhaus, P.; Geßner, R.; Benckert, C.; Jonas, S. Tumor-associated angiogenesis and lymphangiogenesis correlate with progression of intrahepatic cholangiocarcinoma. Am. J. Gastroenterol. 2010, 105, e1123. [Google Scholar] [CrossRef] [PubMed]
- Bergeat, D.; Fautrel, A.; Turlin, B.; Merdrignac, A.; Rayar, M.; Boudjema, K.; Coulouarn, C.; Sulpice, L. Impact of stroma loxl2 overexpression on the prognosis of intrahepatic cholangiocarcinoma. J. Surg. Res. 2016, 203, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, T.; Isomoto, H.; Gores, G.; Smoot, R. Yap and the hippo pathway in cholangiocarcinoma. J. Gastroenterol. 2019, 54, 489–491. [Google Scholar] [CrossRef]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by yap and taz. Nat. Rev. Mol. Cell Biol. 2012, 13, e591. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. Yap/taz at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, e320. [Google Scholar] [CrossRef]
- Dobrokhotov, O.; Samsonov, M.; Sokabe, M.; Hirata, H. Mechanoregulation and pathology of yap/taz via hippo and non-hippo mechanisms. Clin. Transl. Med. 2018, 7, 1–14. [Google Scholar] [CrossRef]
- Van Haele, M.; Moya, I.M.; Karaman, R.; Rens, G.; Snoeck, J.; Govaere, O.; Nevens, F.; Verslype, C.; Topal, B.; Monbaliu, D. Yap and taz heterogeneity in primary liver cancer: An analysis of its prognostic and diagnostic role. Int. J. Mol. Sci. 2019, 20, 638. [Google Scholar] [CrossRef]
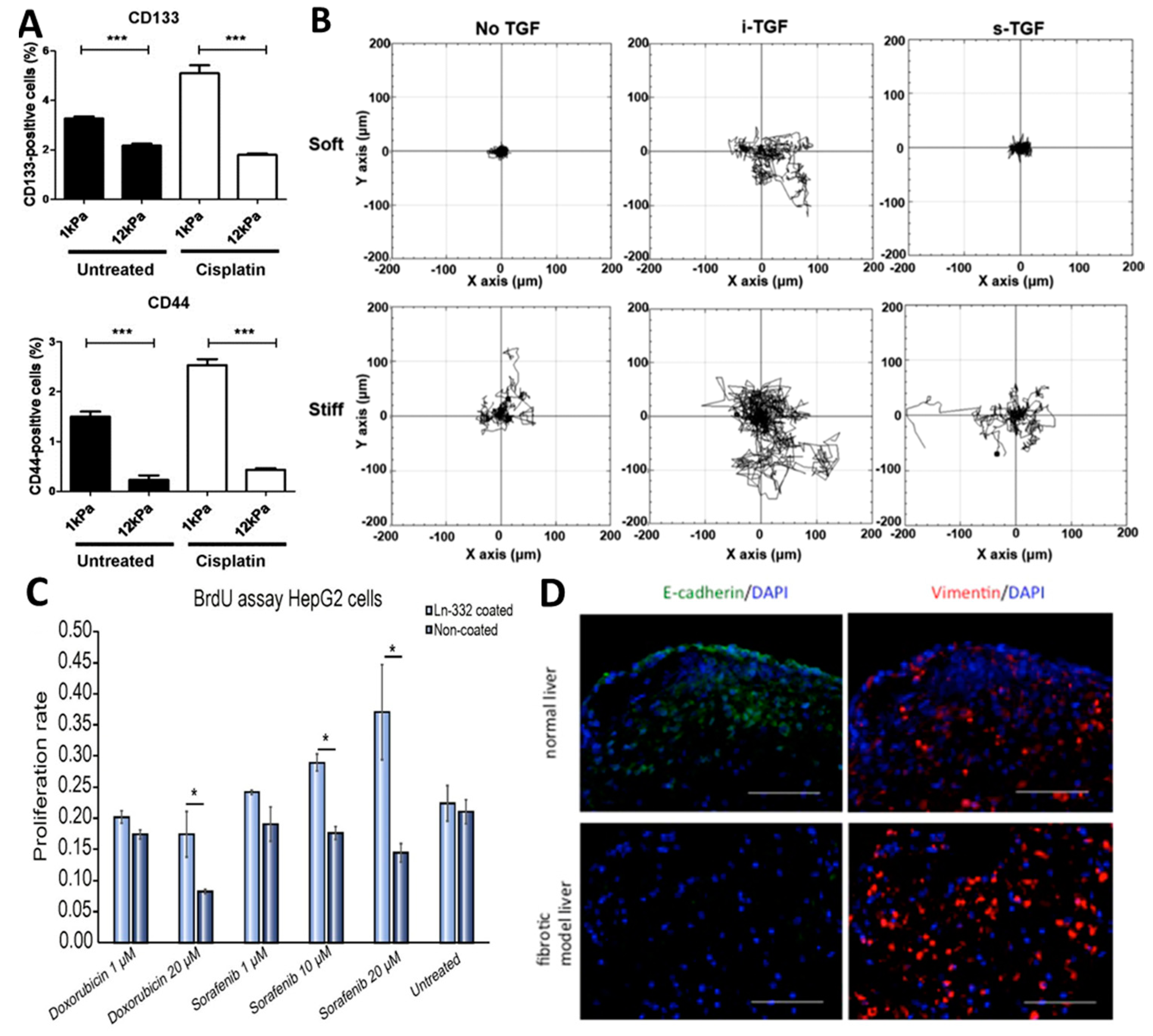
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef]
- Kornek, M.; Raskopf, E.; Tolba, R.; Becker, U.; Klöckner, M.; Sauerbruch, T.; Schmitz, V. Accelerated orthotopic hepatocellular carcinomas growth is linked to increased expression of pro-angiogenic and prometastatic factors in murine liver fibrosis. Liver Int. 2008, 28, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.Z.; Gu, S.S.; Chen, X.T.; He, L.J.; Wang, K.P.; Liu, X.Q. Immobilized transforming growth factor-beta 1 in a stiffness-tunable artificial extracellular matrix enhances mechanotransduction in the epithelial mesenchymal transition of hepatocellular carcinoma. ACS Appl. Mater. Interfaces 2019, 11, 14660–14671. [Google Scholar] [CrossRef] [PubMed]
- Chaijan, S.; Roytrakul, S.; Mutirangura, A.; Leelawat, K. Matrigel induces l-plastin expression and promotes l-plastin-dependent invasion in human cholangiocarcinoma cells. Oncol. Lett. 2014, 8, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Ott, H.C.; Matthiesen, T.S.; Goh, S.K.; Black, L.D.; Kren, S.M.; Netoff, T.I.; Taylor, D.A. Perfusion-decellularized matrix: Using nature’s platform to engineer a bioartificial heart. Nat. Med. 2008, 14, e213. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.H.; Batchelder, C.A.; Lee, C.I.; Tarantal, A.F. Decellularized rhesus monkey kidney as a three-dimensional scaffold for renal tissue engineering. Tissue Eng. Part A 2010, 16, 2207–2216. [Google Scholar] [CrossRef]
- Schwarz, S.; Koerber, L.; Elsaesser, A.F.; Goldberg-Bockhorn, E.; Seitz, A.M.; Dürselen, L.; Ignatius, A.; Walther, P.; Breiter, R.; Rotter, N. Decellularized cartilage matrix as a novel biomatrix for cartilage tissue-engineering applications. Tissue Eng. Part A 2012, 18, 2195–2209. [Google Scholar] [CrossRef]
- Verstegen, M.M.A.; Willemse, J.; Van Den Hoek, S.; Kremers, G.J.; Luider, T.M.; van Huizen, N.A.; Willemssen, F.E.J.A.; Metselaar, H.J.; Ijzermans, J.N.M.; van der Laan, L.J.W. Decellularization of whole human liver grafts using controlled perfusion for transplantable organ bioscaffolds. Stem Cells Dev. 2017, 26, 1304–1315. [Google Scholar] [CrossRef]
- Miyauchi, Y.; Yasuchika, K.; Fukumitsu, K.; Ishii, T.; Ogiso, S.; Minami, T.; Kojima, H.; Yamaoka, R.; Katayama, H.; Kawai, T. A novel three-dimensional culture system maintaining the physiological extracellular matrix of fibrotic model livers accelerates progression of hepatocellular carcinoma cells. Sci. Rep. 2017, 7, e9827. [Google Scholar] [CrossRef]
- Salim, M.S.; Issa, A.M.; Farrag, A.R.H.; Gabr, H. Decellularized liver bioscaffold: A histological and immunohistochemical comparison between normal, fibrotic and hepatocellular carcinoma. Clin. Exp. Hepatol. 2019, 5, e35. [Google Scholar] [CrossRef]
- Steele, N.G.; Chakrabarti, J.; Wang, J.; Biesiada, J.; Holokai, L.; Chang, J.; Nowacki, L.M.; Hawkins, J.; Mahe, M.; Sundaram, N. An organoid-based preclinical model of human gastric cancer. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 161–184. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Comparison | 2D Cell Lines | Spheroids | Organoids |
|---|---|---|---|
| Origin of cells | Immortalized cell lines | Often immortalized cell lines or tumour biopsies | Tissue-specific stem cells |
| Morphology | Sheet-like flat monolayer | Cell-clusters within 3D environment | Self-organizing, mimicking organ structure |
| Drug sensitivity | Very effective due to 2D morphology | More resistant compared to 2D, better in vivo drug response predictor | Patient-specific responses and matched controls for personalized therapy and best in vivo drug response predictor |
| Throughput drug screening | Suitable for high-throughput screening | Less suitable for high-throughput drug screening | Less suitable for high-throughput drug screening |
| Resource costs | Low | Medium | Medium-high |
| Heterogeneity | Cell-line derived from a single cell | Related to initial cell population | Related to patient used for cell isolation |
| In vivo features | Cellular properties | Cell-cell interactions, hypoxia, drug penetration, production of ECM | Cell-cell interactions, mutational landscape of original tumour, cellular heterogeneity |
| Long-term expansion | Immortalized for easy expansion | In few cases long-term reported, loss of heterogeneity | Robust long-term expansion with maintenance of heterogeneity |
| (Bio)materials for culture | Plastic or biomaterial coatings | Wide variety of (bio) materials | Primarily Matrigel/BME |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Tienderen, G.S.; Groot Koerkamp, B.; IJzermans, J.N.M.; van der Laan, L.J.W.; Verstegen, M.M.A. Recreating Tumour Complexity in a Dish: Organoid Models to Study Liver Cancer Cells and their Extracellular Environment. Cancers 2019, 11, 1706. https://doi.org/10.3390/cancers11111706
van Tienderen GS, Groot Koerkamp B, IJzermans JNM, van der Laan LJW, Verstegen MMA. Recreating Tumour Complexity in a Dish: Organoid Models to Study Liver Cancer Cells and their Extracellular Environment. Cancers. 2019; 11(11):1706. https://doi.org/10.3390/cancers11111706
Chicago/Turabian Stylevan Tienderen, Gilles S., Bas Groot Koerkamp, Jan N. M. IJzermans, Luc J. W. van der Laan, and Monique M. A. Verstegen. 2019. "Recreating Tumour Complexity in a Dish: Organoid Models to Study Liver Cancer Cells and their Extracellular Environment" Cancers 11, no. 11: 1706. https://doi.org/10.3390/cancers11111706