Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer

 , and
, and
Abstract
:1. Introduction
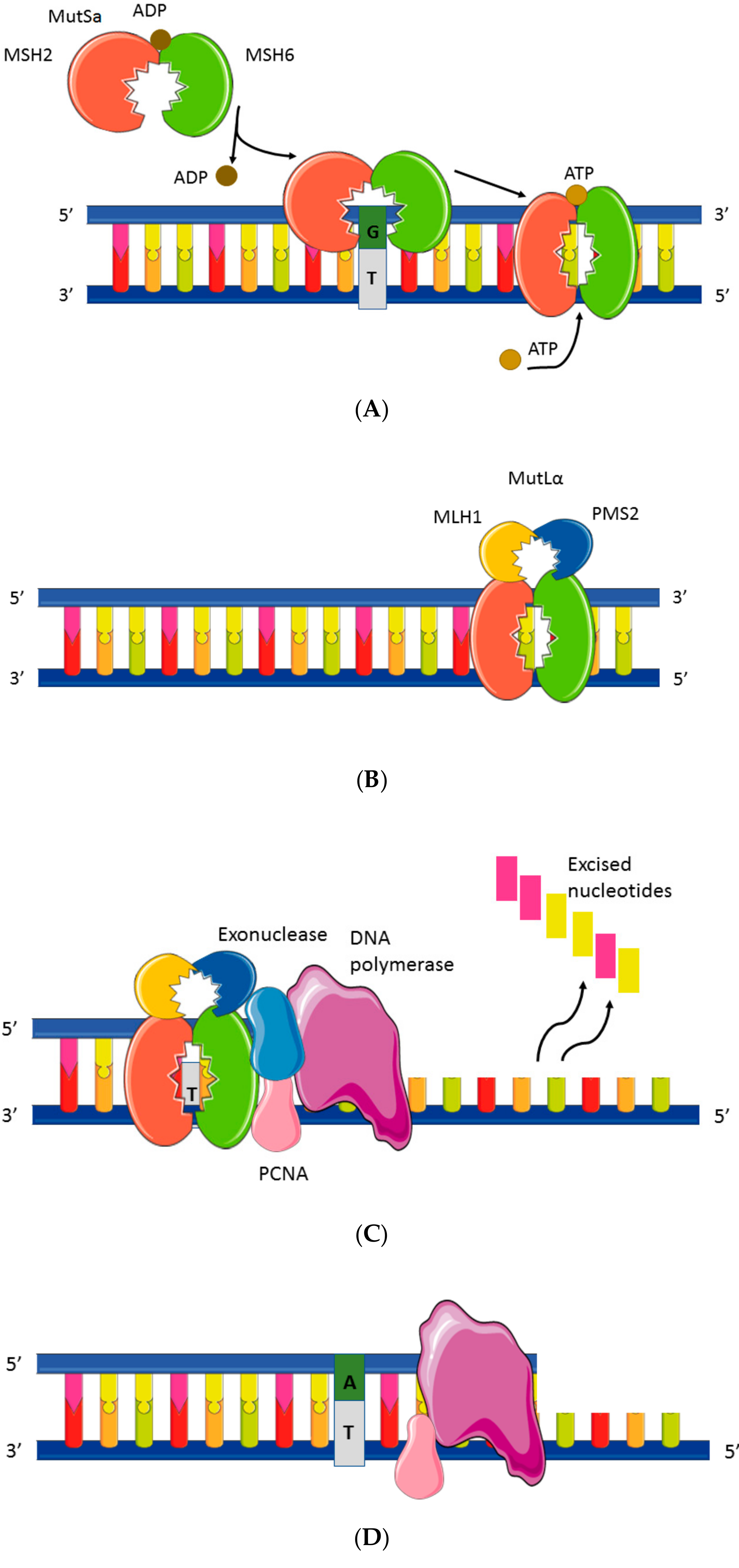
2. Mismatch Repair System and Microsatellite Instability Testing
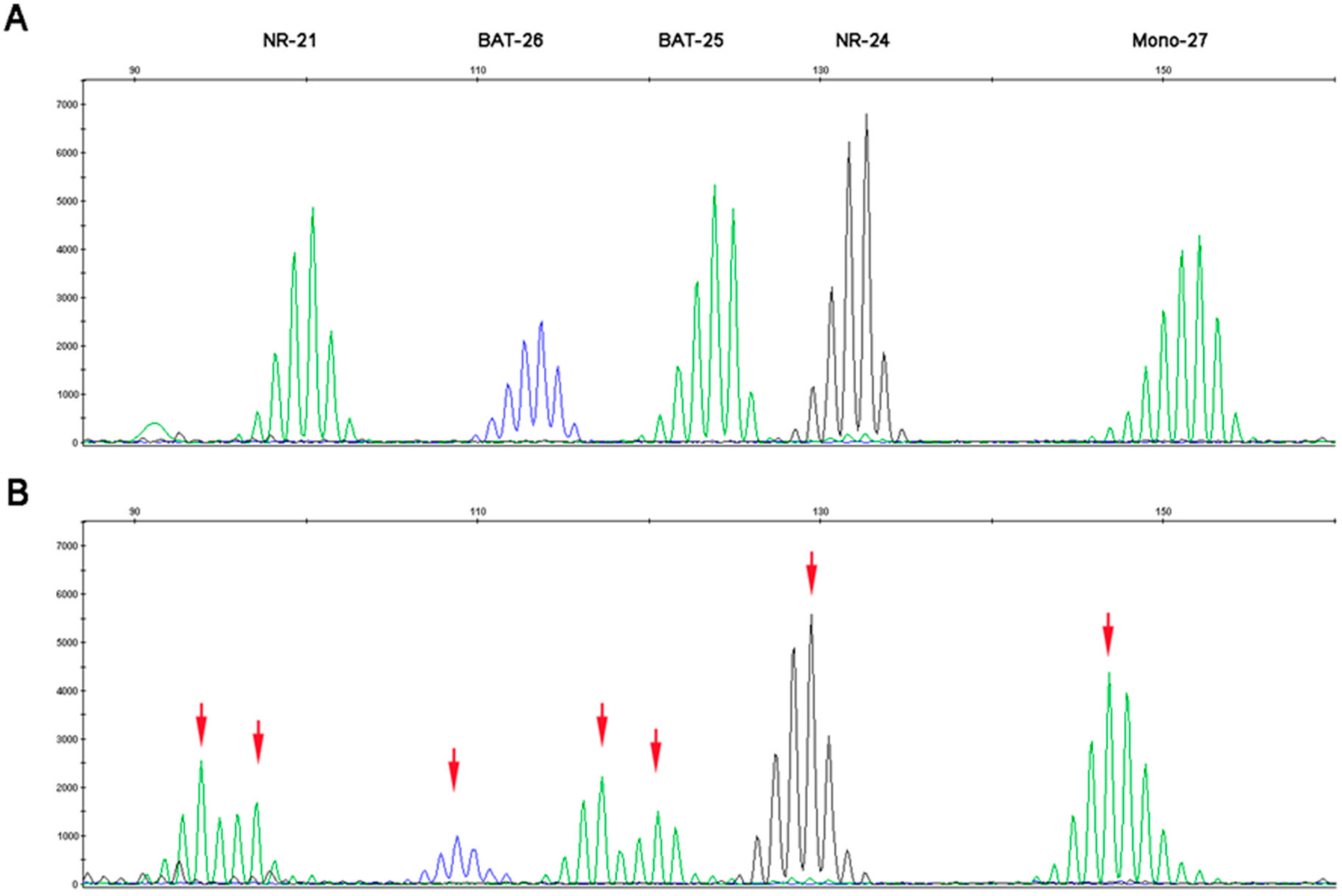
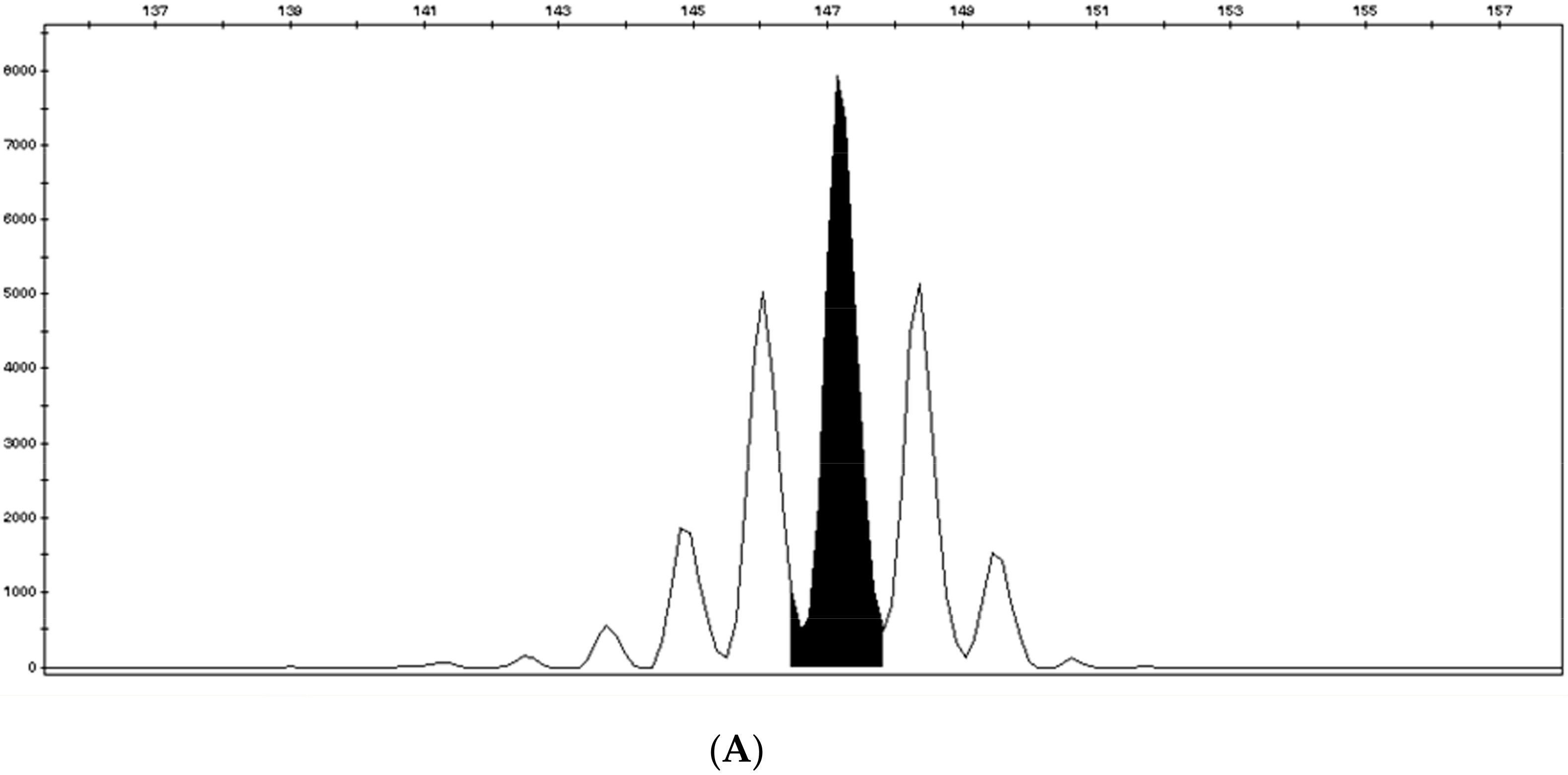
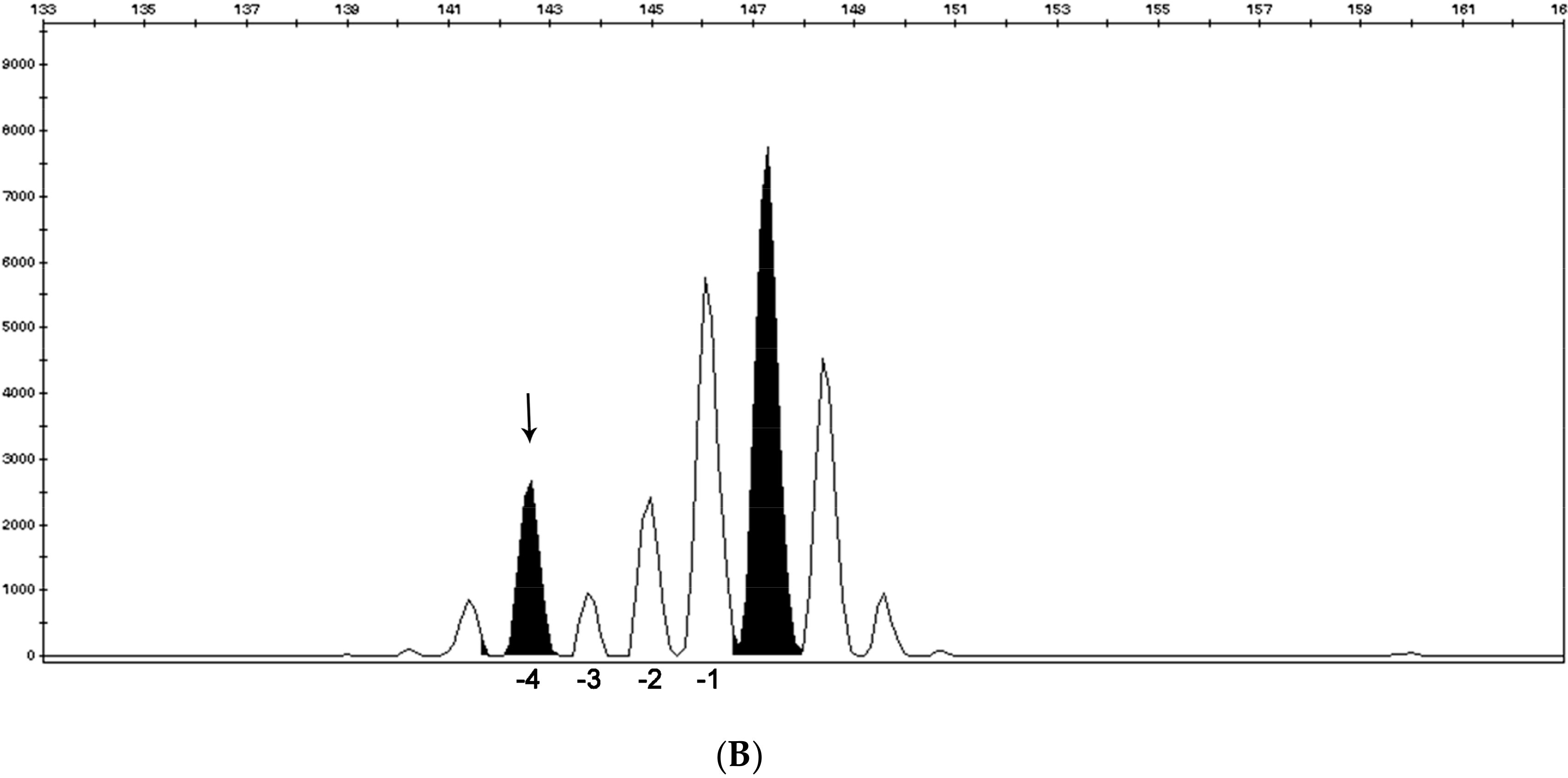
2.1. Microsatellite Instability Testing
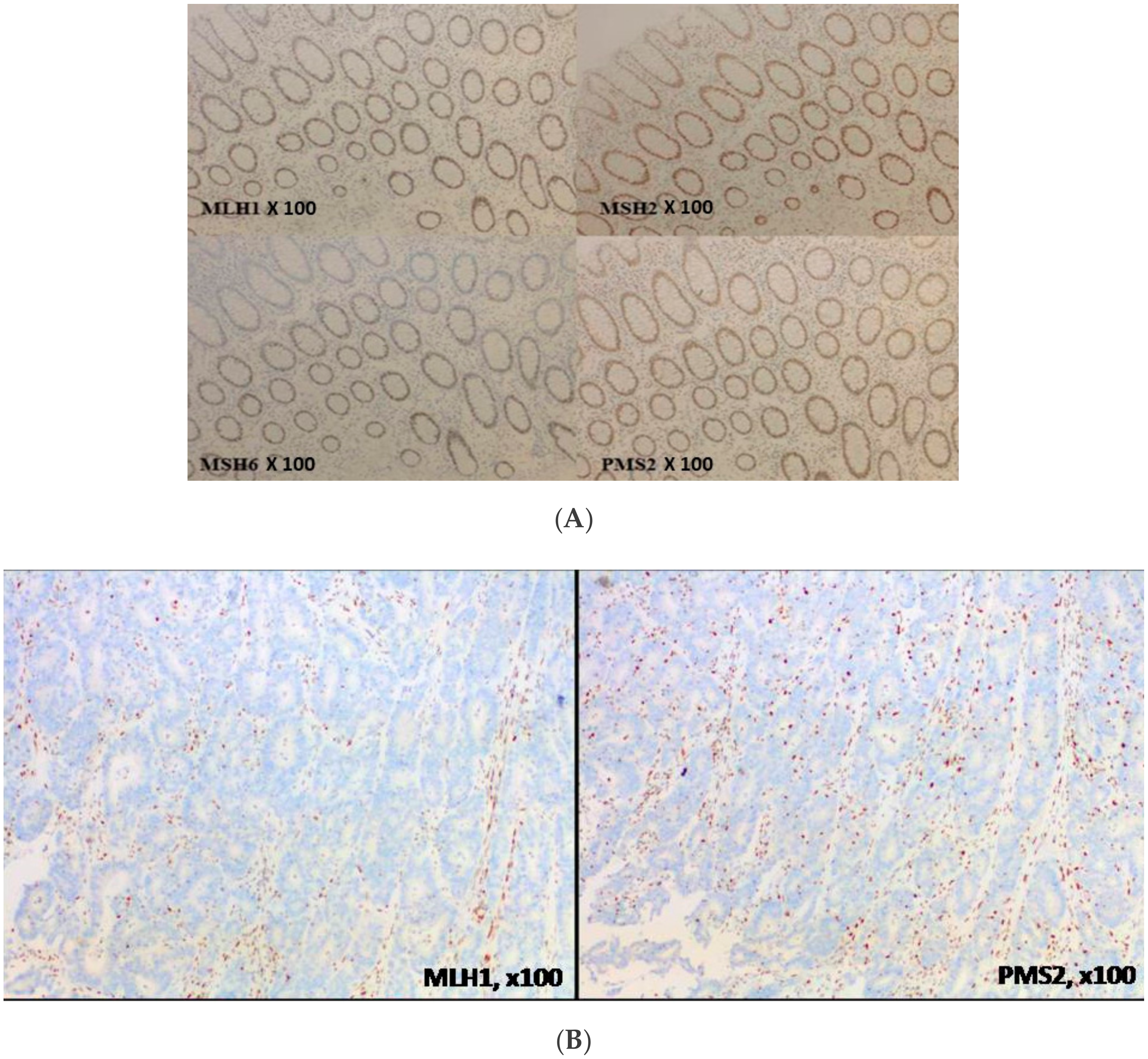
2.2. MMR Protein Testing
2.3. Comparison of Molecular MSI Testing and MMR Proteins Immunohistochemistry
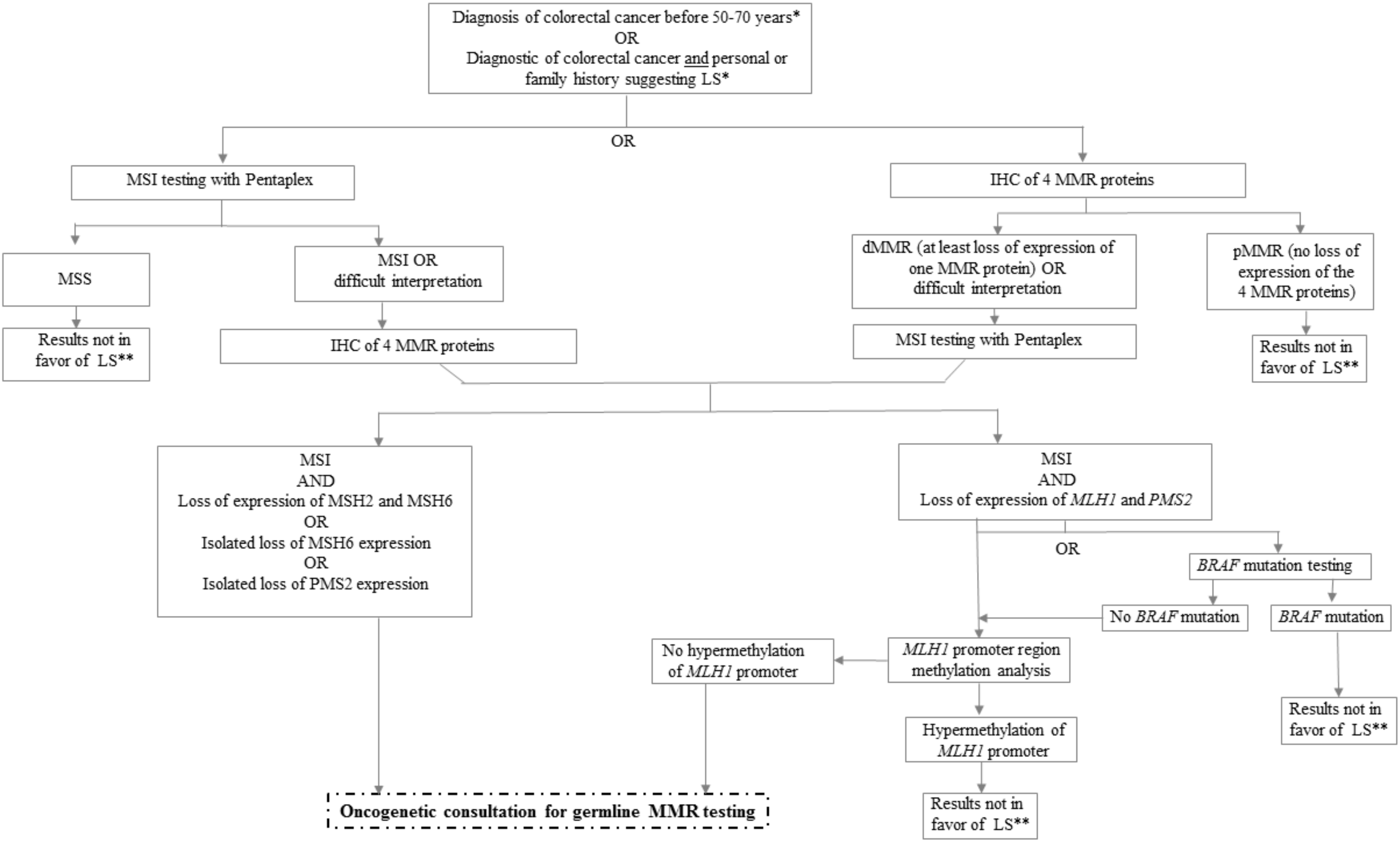
3. Challenges for Determination of the dMMR/MSI Mechanism
3.1. How to Classify Sporadic versus Germline dMMR/MSI Colorectal Cancer?
3.2. MLH1 Promoter Hypermethylation
3.3. Challenge in Determination of Lynch Syndrome
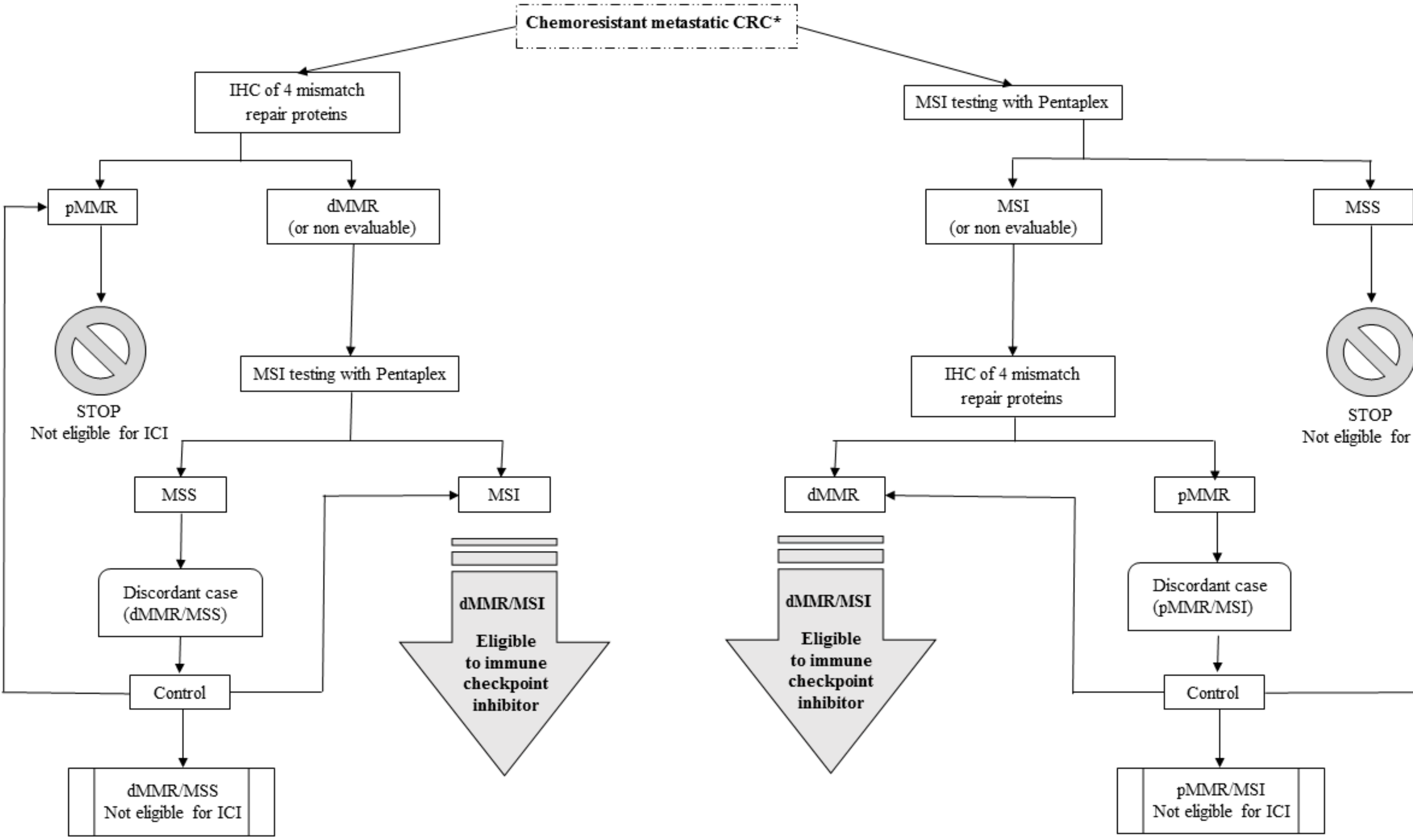
4. Discordance Between MMR Immunohistochemistry and DNA Microsatellites Testing
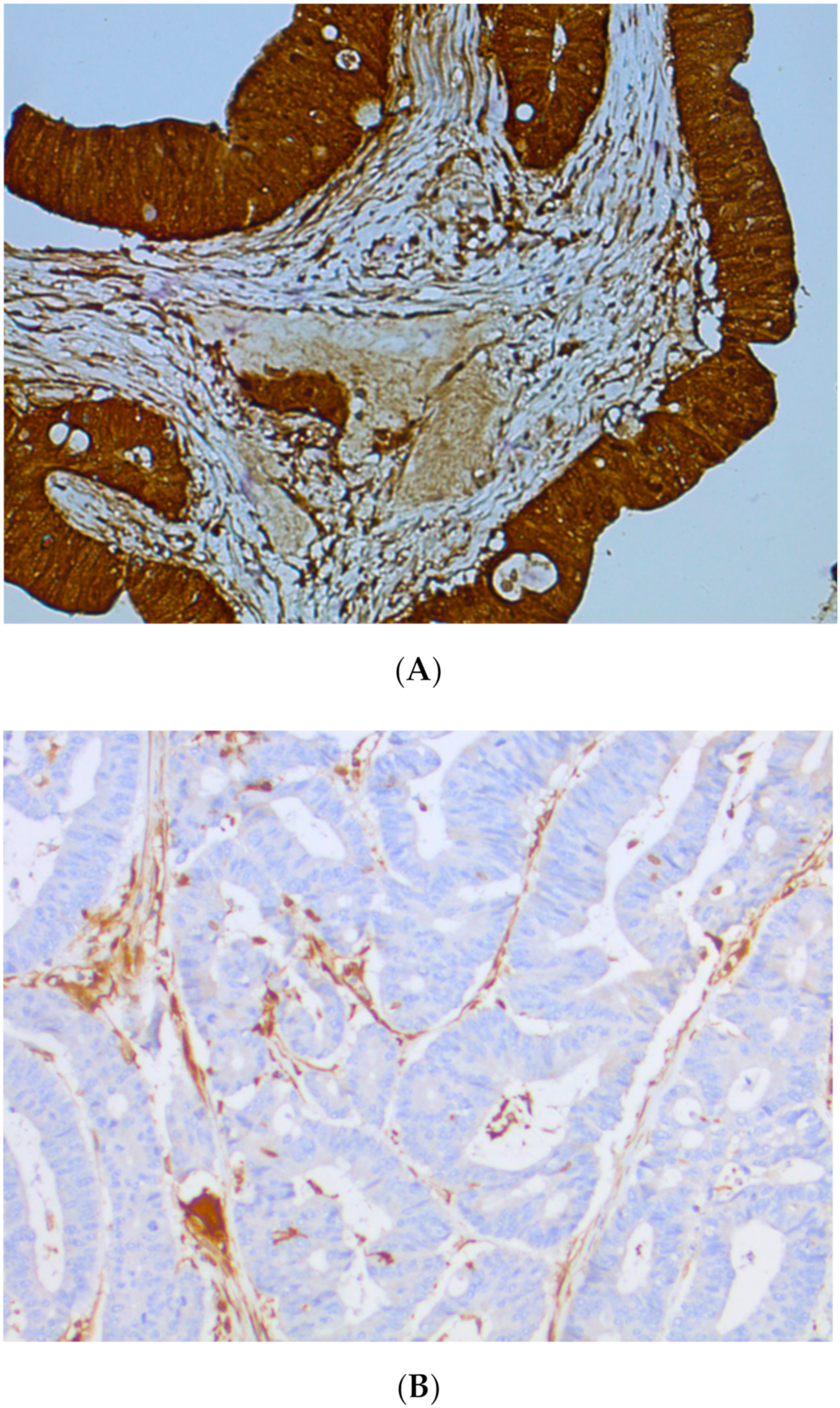
5. Focus on Tumor Heterogeneity
6. Other Markers of Microsatellite Instability and Response to Immune Checkpoint Inhibitors
7. Perspectives of MMR Immunohistochemistry and DNA Microsatellites Testing
7.1. The Role of HSP110 Protein in the Diagnosis of Microsatellite Status
7.2. A Larger Panel of Microsatellites for Better Detection of Instability
7.3. Tumor Circulating DNA to Overcome Tumor Heterogeneity
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| APC | adenomatosis polyposis coli |
| APCs | antigen-presenting cells |
| ATP | adenosine triphosphate |
| CRC | colorectal caner |
| ctDNA | circulating tumor DNA |
| dMMR | deficient mismatch repair |
| ICI | immune checkpoint inhibitor |
| IHC | immunohistochemistry |
| LS | Lynch syndrome |
| mCRC | metastatic colorectal cancer |
| MHC | major histocompatibility complex |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| MSS | microsatellite stability |
| NGS | next-generation sequencing |
| NCI | national cancer institute |
| PCNA | proliferating cell nuclear antigen |
| PCR | polymerase chain reaction |
| PD-L1 | programmed death ligand |
| pMMR | proficient mismatch repair |
| TMB | tumor mutational burden |
| LAG-3 | lymphocyte activation gene 3 |
| TIM-3 | mucin domain-containing protein-3 |
References
- Tariq, K.; Ghias, K. Colorectal cancer carcinogenesis: A review of mechanisms. Cancer Biol. Med. 2016, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Snover, D.C. Update on the serrated pathway to colorectal carcinoma. Hum. Pathol. 2011, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R. Mismatch Repair. J. Biol. Chem. 2015, 290, 26395. [Google Scholar] [CrossRef] [PubMed]
- Kawasoe, Y.; Tsurimoto, T.; Nakagawa, T.; Masukata, H.; Takahashi, T.S. MutSα maintains the mismatch repair capability by inhibiting PCNA unloading. Elife 2016, 5, e15155. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Sargent, D.J. Molecular Pathways: Microsatellite Instability in Colorectal Cancer: Prognostic, Predictive, and Therapeutic Implications. Clin. Cancer Res. 2012, 18, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Clendenning, M.; Sotamaa, K.; Prior, T.; Westman, J.A.; et al. Feasibility of Screening for Lynch Syndrome Among Patients with Colorectal Cancer. J. Clin. Oncol. 2008, 26, 5783. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N. Engl. J. Med. 2003, 349, 247. [Google Scholar] [CrossRef] [PubMed]
- Tougeron, D.; Mouillet, G.; Trouilloud, I.; Lecomte, T.; Coriat, R.; Aparicio, T.; Des Guetz, G.; Lécaille, C.; Artru, P.; Sickersen, G.; et al. Efficacy of Adjuvant Chemotherapy in Colon Cancer with Microsatellite Instability: A Large Multicenter AGEO Study. J. Natl. Cancer Inst. 2016, 108, djv438. [Google Scholar] [CrossRef]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN and FOCUS studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Tougeron, D.; Sueur, B.; Sefrioui, D.; Gentilhomme, L.; Lecomte, T.; Aparicio, T.; DES Guetz, G.; Artru, P.; De La Fouchardiere, C.; Moulin, V.; et al. A large multicenter study evaluating prognosis and chemosensitivity of metastatic colorectal cancers with microsatellite instability. J. Clin. Oncol. 2017, 35, 3536. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.A.; Möslein, G.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Blanco, I.; Bulow, S.; Burn, J.; Capella, G.; et al. Recommendations to improve identification of hereditary and familial colorectal cancer in Europe. Fam. Cancer 2010, 9, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: Development of International Criteria for the Determination of Microsatellite Instability in Colorectal Cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Scarisbrick, J.J.; Mitchell, T.J.; Calonje, E.; Orchard, G.; Russell-Jones, R.; Whittaker, S.J. Microsatellite Instability Is Associated with Hypermethylation of the hMLH1 Gene and Reduced Gene Expression in Mycosis Fungoides. J. Investig. Dermatol. 2003, 121, 894–901. [Google Scholar] [CrossRef]
- Murphy, K.M.; Zhang, S.; Geiger, T.; Hafez, M.J.; Bacher, J.; Berg, K.D.; Eshleman, J.R. Comparison of the Microsatellite Instability Analysis System and the Bethesda Panel for the Determination of Microsatellite Instability in Colorectal Cancers. J. Mol. Diagn. 2006, 8, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, N.; Duval, A.; Reperant, M.; Vaury, C.; Furlan, D.; Leroy, K.; Seruca, R.; Iacopetta, B.; Hamelin, R. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology 2002, 123, 1804–1811. [Google Scholar] [CrossRef]
- Wong, Y.F.; Cheung, T.H.; Lo, K.W.K.; Yim, S.F.; Chan, L.K.Y.; Buhard, O.; Duval, A.; Chung, T.K.H.; Hamelin, R. Detection of microsatellite instability in endometrial cancer: Advantages of a panel of five mononucleotide repeats over the National Cancer Institute panel of markers. Carcinogenesis 2006, 27, 951–955. [Google Scholar] [CrossRef]
- Acharya, S.; Wilson, T.; Gradia, S.; Kane, M.F.; Guerrette, S.; Marsischky, G.T.; Kolodner, R.; Fishel, R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 1996, 93, 13629–13634. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, D.; Tan, S.A.; Liu, X.; Lai, J. Sigmoid Colon Adenocarcinoma with Isolated Loss of PMS2 Presenting in a Patient with Synchronous Prostate Cancer with Intact MMR: Diagnosis and Analysis of the Family Pedigree. Anticancer Res. 2018, 38, 4847–4852. [Google Scholar] [CrossRef] [PubMed]
- Verma, L.; Kane, M.F.; Brassett, C.; Schmeits, J.; Evans, D.G.R.; Kolodner, R.D.; Maher, E.R. Mononucleotide microsatellite instability and germline MSH6 mutation analysis in early onset colorectal cancer. J. Med. Genet. 1999, 36, 678–682. [Google Scholar] [PubMed]
- INCA. Tests Somatiques Recherchant une Déficience du Système MMR au Sein des Tumeurs du Spectre du Syndrome de Lynch; Institut National du Cancer: Boulogne-Billancourt, France, 2016. [Google Scholar]
- Pyatt, R.; Chadwick, R.B.; Johnson, C.K.; Adebamowo, C.; de la Chapelle, A.; Prior, T.W. Polymorphic Variation at the BAT-25 and BAT-26 Loci in Individuals of African Origin: Implications for Microsatellite Instability Testing. Am. J. Pathol. 1999, 155, 349–353. [Google Scholar] [CrossRef]
- Zhang, L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J. Mol. Diagn. 2008, 10, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; Panarelli, N.C.; Rennert, H.; Sherr, D.L.; Yantiss, R.K. Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am. J. Surg. Pathol. 2010, 34, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Shia, J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J. Mol. Diagn. 2008, 10, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, L.I.H.; Ligtenberg, M.J.L.; Willems, R.W.; Hermens, R.P.M.G.; Blokx, W.A.M.; Dubois, S.V.; van der Linden, H.; Meijer, J.W.R.; Mlynek-Kersjes, M.L.; Hoogerbrugge, N.; et al. Interpretation of Immunohistochemistry for Mismatch Repair Proteins is Only Reliable in a Specialized Setting. Am. J. Surg. Pathol. 2008, 32, 1246. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Capo-Chichi, J.M.; Spence, T.; Grenier, S.; Stockley, T.; Kamel-Reid, S.; Serra, S.; Sabatini, P.; Chetty, R. Heterogenous loss of mismatch repair (MMR) protein expression: A challenge for immunohistochemical interpretation and microsatellite instability (MSI) evaluation. J. Pathol. Clin. Res. 2019, 5, 115–129. [Google Scholar] [CrossRef]
- Snowsill, T.; Coelho, H.; Huxley, N.; Jones-Hughes, T.; Briscoe, S.; Frayling, I.M.; Hyde, C. Molecular testing for Lynch syndrome in people with colorectal cancer: Systematic reviews and economic evaluation. Health Technol. Assess. Winch. Engl. 2017, 21, 1–238. [Google Scholar] [CrossRef]
- Goel, A.; Nagasaka, T.; Hamelin, R.; Boland, C.R. An Optimized Pentaplex PCR for Detecting DNA Mismatch Repair-Deficient Colorectal Cancers. PLoS ONE 2010, 5, e9393. [Google Scholar] [CrossRef]
- Xicola, R.M.; Llor, X.; Pons, E.; Castells, A.; Alenda, C.; Piñol, V.; Andreu, M.; Castellví-Bel, S.; Payá, A.; Jover, R.; et al. Performance of Different Microsatellite Marker Panels for Detection of Mismatch Repair–Deficient Colorectal Tumors. J. Natl. Cancer Inst. 2007, 99, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Kambara, T.; Simms, L.A.; Whitehall, V.L.J.; Spring, K.J.; Wynter, C.V.A.; Walsh, M.D.; Barker, M.A.; Arnold, S.; McGivern, A.; Matsubara, N.; et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004, 53, 1137–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutlewska, K.; Lubinski, J.; Kurzawski, G. Germline deletions in the EPCAM gene as a cause of Lynch syndrome—Literature review. Hered. Cancer Clin. Pract. 2013, 11, 9. [Google Scholar] [CrossRef]
- Ward, R.L.; Dobbins, T.; Lindor, N.M.; Rapkins, R.W.; Hitchins, M.P. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet. Med. 2013, 15, 25–35. [Google Scholar] [CrossRef]
- Crépin, M.; Dieu, M.C.; Lejeune, S.; Escande, F.; Boidin, D.; Porchet, N.; Morin, G.; Manouvrier, S.; Mathieu, M.; Buisine, M.P. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum. Mutat. 2012, 33, 180–188. [Google Scholar] [CrossRef]
- Chadwick, R.B.; Meek, J.E.; Prior, T.W.; Peltomaki, P.; de la Chapelle, A. Polymorphisms in a pseudogene highly homologous to PMS2. Hum. Mutat. 2000, 16, 530. [Google Scholar] [CrossRef]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef]
- Sinicrope, F.A. Lynch Syndrome–Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.T.; Buchanan, D.D.; Thompson, B.; Young, J.P.; Spurdle, A.B. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: A literature review assessing utility of tumour features for MMR variant classification. J. Med. Genet. 2012, 49, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Dámaso, E.; Castillejo, A.; Arias, M.D.M.; Canet-Hermida, J.; Navarro, M.; Del Valle, J.; Campos, O.; Fernández, A.; Marín, F.; Turchetti, D.; et al. Primary constitutional MLH1 epimutations: A focal epigenetic event. Br. J. Cancer 2018, 119, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Nguyen, T.P.; Leung, H.C.E.; Nagasaka, T.; Rhees, J.; Hotchkiss, E.; Arnold, M.; Banerji, P.; Koi, M.; Kwok, C.T.; et al. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int. J. Cancer 2011, 128, 869–878. [Google Scholar] [CrossRef]
- Hitchins, M.P.; Ward, R.L. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J. Med. Genet. 2009, 46, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Pineda, M.; Mur, P.; Iniesta, M.D.; Borràs, E.; Campos, O.; Vargas, G.; Iglesias, S.; Fernández, A.; Gruber, S.B.; Lázaro, C.; et al. MLH1 methylation screening is effective in identifying epimutation carriers. Eur. J. Hum. Genet. 2012, 20, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Carethers, J.M. Differentiating Lynch-like from Lynch Syndrome. Gastroenterology 2014, 146, 602–604. [Google Scholar] [CrossRef]
- Antelo, M.; Golubicki, M.; Roca, E.; Mendez, G.; Carballido, M.; Iseas, S.; Cuatrecasas, M.; Moreira, L.; Sanchez, A.; Carballal, S.; et al. Lynch-like syndrome is as frequent as Lynch syndrome in early-onset nonfamilial nonpolyposis colorectal cancer. Int. J. Cancer 2019, 145, 705–713. [Google Scholar] [CrossRef]
- Rodríguez–Soler, M.; Pérez–Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barberá, V.M.; Juárez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of Cancer in Cases of Suspected Lynch Syndrome without Germline Mutation. Gastroenterology 2013, 144, 926–932. [Google Scholar] [CrossRef]
- Geurts-Giele, W.R.R.; Leenen, C.H.M.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.B.M.; Kuipers, E.J.; Goverde, A.; van den Ouweland, A.M.W.; van Lier, M.G.F.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Vissers, L.E.L.M.; Venkatachalam, R.; Bodmer, D.; Hoenselaar, E.; Goossens, M.; Haufe, A.; Kamping, E.; Niessen, R.C.; Hogervorst, F.B.L.; et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum. Mutat. 2011, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Haraldsdottir, S.; de la Chapelle, A.; Jonasson, J.G.; Liyanarachchi, S.; Frankel, W.L.; Rafnar, T.; Stefansson, K.; Pritchard, C.C.; Hampel, H. Clinical Characteristics of Colorectal Cancer Patients with Double Somatic Mismatch Repair Mutations Compared to Lynch Syndrome. J. Med. Genet. 2019, 56, 462. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Burgart, L.J.; Leontovich, O.; Goldberg, R.M.; Cunningham, J.M.; Sargent, D.J.; Walsh-Vockley, C.; Petersen, G.M.; Walsh, M.D.; Leggett, B.A.; et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J. Clin. Oncol. 2002, 20, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Hatch, S.B. Microsatellite Instability Testing in Colorectal Carcinoma: Choice of Markers Affects Sensitivity of Detection of Mismatch Repair-Deficient Tumors. Clin. Cancer Res. 2005, 11, 2180–2187. [Google Scholar] [CrossRef]
- Piñol, V.; Castells, A.; Andreu, M.; Castellví-Bel, S.; Alenda, C.; Llor, X.; Xicola, R.M.; Rodríguez-Moranta, F.; Payá, A.; Jover, R.; et al. Accuracy of Revised Bethesda Guidelines, Microsatellite Instability, and Immunohistochemistry for the Identification of Patients with Hereditary Nonpolyposis Colorectal Cancer. JAMA 2005, 293, 1986–1994. [Google Scholar] [CrossRef]
- Watson, N.; Grieu, F.; Morris, M.; Harvey, J.; Stewart, C.; Schofield, L.; Goldblatt, J.; Iacopetta, B. Heterogeneous Staining for Mismatch Repair Proteins during Population-Based Prescreening for Hereditary Nonpolyposis Colorectal Cancer. J. Mol. Diagn. 2007, 9, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Chi, Y.; Chen, W.; Chen, X.; Wei, P.; Sheng, W.; Zhou, X.; Shi, D. Immunohistochemistry and microsatellite instability analysis in molecular subtyping of colorectal carcinoma based on mismatch repair competency. Int. J. Clin. Exp. Med. 2015, 8, 20988. [Google Scholar]
- Chen, M.; Chen, J.; Hu, J.; Chen, Q.; Yu, L.; Liu, B.; Qian, X.; Yang, M. Comparison of microsatellite status detection methods in colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 1431–1438. [Google Scholar]
- Cohen, R.; Hain, E.; Buhard, O.; Guilloux, A.; Bardier, A.; Kaci, R.; Bertheau, P.; Renaud, F.; Bibeau, F.; Fléjou, J.F. 537P Assessment of local clinical practice for testing of mismatch repair deficiency in metastatic colorectal cancer: The need for new diagnostic guidelines prior to immunotherapy. Ann. Oncol. 2018, 29, mdy281-083. [Google Scholar] [CrossRef]
- Jaffrelot, M.; Laurenty, A.P.; Fares, N.; Staub, A.; Bonnet, D.; Danjoux, M.; Vande Perre, P.; Meilleroux, J.; Chipoulet, E.; Toulas, C.; et al. Fiabilité de l’étude du phénotype MMR tumoral: Étude à partir d’une cohorte de 4 948 cas de tests MSI et analyse des phénotypes atypiques. In Proceedings of the JFHOD, Paris, France, 21–24 March 2019. [Google Scholar]
- Wang, Y. Differences in Microsatellite Instability Profiles between Endometrioid and Colorectal Cancers. J. Mol. Diagn. 2017, 19, 57–64. Available online: https://www-ncbi-nlm-nih-gov.gate2.inist.fr/pmc/articles/PMC5225298/ (accessed on 30 June 2019). [CrossRef]
- Goldstein, J.B.; Wu, W.; Borras, E.; Masand, G.; Cuddy, A.; Mork, M.E.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.; Taggart, M.W.; et al. Can Microsatellite Status of Colorectal Cancer Be Reliably Assessed after Neoadjuvant Therapy? Clin. Cancer Res. 2017, 23, 5246–5254. [Google Scholar] [CrossRef] [PubMed]
- Tachon, G.; Frouin, E.; Karayan-Tapon, L.; Auriault, M.L.; Godet, J.; Moulin, V.; Wang, Q.; Tougeron, D. Heterogeneity of mismatch repair defect in colorectal cancer and its implications in clinical practice. Eur. J. Cancer 2018, 95, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Lonardi, S.; Wong, K.Y.M.; Morse, M.; McDermott, R.S.; Hill, A.G.; Hendlisz, A.; Lenz, H.J.; Leach, J.W.; Moss, R.A.; et al. Combination of nivolumab (nivo) + ipilimumab (ipi) in the treatment of patients (pts) with deficient DNA mismatch repair (dMMR)/high microsatellite instability (MSI-H) metastatic colorectal cancer (mCRC): CheckMate 142 study. J. Clin. Oncol. 2017, 35, 3531. [Google Scholar] [CrossRef]
- Diaz, L.A.; Le, D.T.; Yoshino, T.; Andre, T.; Bendell, J.C.; Koshiji, M.; Zhang, Y.; Kang, S.P.; Lam, B.; Jäger, D. KEYNOTE-177: Randomized phase III study of pembrolizumab versus investigator-choice chemotherapy for mismatch repair-deficient or microsatellite instability-high metastatic colorectal carcinoma. J. Clin. Oncol. 2017, 35, TPS815. [Google Scholar] [CrossRef]
- Standard Chemotherapy vs. Immunotherapie in 2nd Line Treatment of MSI Colorectal Mestastatic Cancer. Full Text View. Available online: https://clinicaltrials.gov/ct2/show/NCT03186326 (accessed on 18 August 2019).
- Interest of iRECIST Evaluation for DCR for Evaluation of Patients with Deficient MMR and /or MSI Metastatic Colorectal Cancer Treated with Nivolumab and Ipilimumab. Full Text View. Available online: https://clinicaltrials.gov/ct2/show/NCT03350126 (accessed on 18 August 2019).
- Buhard, O.; Lagrange, A.; Guilloux, A.; Colas, C.; Chouchène, M.; Wanherdrick, K.; Coulet, F.; Guillerm, E.; Dorard, C.; Marisa, L.; et al. HSP110 T17 simplifies and improves the microsatellite instability testing in patients with colorectal cancer. J. Med. Genet. 2016, 53, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Huang, Y.; Fang, X.; Liu, C.; Deng, W.; Zhong, C.; Xu, J.; Xu, D.; Yuan, Y. A Novel and Reliable Method to Detect Microsatellite Instability in Colorectal Cancer by Next-Generation Sequencing. J. Mol. Diagn. 2018, 20, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Joost, P.; Veurink, N.; Holck, S.; Klarskov, L.; Bojesen, A.; Harbo, M.; Baldetorp, B.; Rambech, E.; Nilbert, M. Heterogenous mismatch-repair status in colorectal cancer. Diagn. Pathol. 2014, 9, 126. [Google Scholar] [CrossRef]
- Kim, K.; Kim, J.E.; Hong, Y.S.; Ahn, S.M.; Chun, S.M.; Hong, S.M.; Jang, S.J.; Yu, C.S.; Kim, J.C.; Kim, T.W. Paired Primary and Metastatic Tumor Analysis of Somatic Mutations in Synchronous and Metachronous Colorectal Cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2017, 49, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Jesinghaus, M.; Wolf, T.; Pfarr, N.; Muckenhuber, A.; Ahadova, A.; Warth, A.; Goeppert, B.; Sers, C.; Kloor, M.; Endris, V.; et al. Distinctive Spatiotemporal Stability of Somatic Mutations in Metastasized Microsatellite-stable Colorectal Cancer. Am. J. Surg. Pathol. 2015, 39, 1140–1147. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal cancer: Genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef]
- Jeantet, M.; Tougeron, D.; Tachon, G.; Cortes, U.; Archambaut, C.; Fromont, G.; Karayan-Tapon, L. High Intra- and Inter-Tumoral Heterogeneity of RAS Mutations in Colorectal Cancer. Int. J. Mol. Sci. 2016, 17, 2015. [Google Scholar] [CrossRef] [PubMed]
- Chapusot, C.; Martin, L.; Bouvier, A.M.; Bonithon-Kopp, C.; Ecarnot-Laubriet, A.; Rageot, D.; Ponnelle, T.; Laurent Puig, P.; Faivre, J.; Piard, F. Microsatellite instability and intratumoural heterogeneity in 100 right-sided sporadic colon carcinomas. Br. J. Cancer 2002, 87, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, W.; Ma, J.; Liu, Y.; Liang, J.; Wu, Y.; Yang, X.; Xu, E.; Li, Y.; Xi, Y. Screening of MSI detection loci and their heterogeneity in East Asian colorectal cancer patients. Cancer Med. 2019, 8, 2157–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, D.A.; George, T.J.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Tougeron, D.; Fauquembergue, E.; Rouquette, A.; Le Pessot, F.; Sesboüé, R.; Laurent, M.; Berthet, P.; Mauillon, J.; Di Fiore, F.; Sabourin, J.C.; et al. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod. Pathol. 2009, 22, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Sæterdal, I.; Gjertsen, M.K.; Straten, P.; Eriksen, J.A.; Gaudernack, G. A TGFβRII frameshift-mutation-derived CTL epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol. Immunother. 2001, 50, 469–476. [Google Scholar]
- Colle, R.; Cohen, R.; Cochereau, D.; Duval, A.; Lascols, O.; Lopez-Trabada, D.; Afchain, P.; Trouilloud, I.; Parc, Y.; Lefevre, J.H.; et al. Immunotherapy and patients treated for cancer with microsatellite instability. Bull. Cancer (Paris) 2017, 104, 42–51. [Google Scholar] [CrossRef] [Green Version]
- De Guillebon, E.; Roussille, P.; Frouin, E.; Tougeron, D. Anti program death-1/anti program death-ligand 1 in digestive cancers. World J. Gastrointest. Oncol. 2015, 7, 95–101. [Google Scholar] [CrossRef]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Silberman, R.; Steiner, D.F.; Lo, A.A.; Gomez, A.; Zehnder, J.L.; Chu, G.; Suarez, C.J. Complete and Prolonged Response to Immune Checkpoint Blockade in POLE-Mutated Colorectal Cancer. JCO Precis. Oncol. 2019, 3, 1–5. [Google Scholar] [CrossRef]
- Vanderwalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marginean, E.C.; Melosky, B. Is There a Role for Programmed Death Ligand-1 Testing and Immunotherapy in Colorectal Cancer with Microsatellite Instability? Part II-The Challenge of Programmed Death Ligand-1 Testing and Its Role in Microsatellite Instability-High Colorectal Cancer. Arch. Pathol. Lab. Med. 2018, 142, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Borel, V.; Adelman, C.A.; Schindler, D.; Boulton, S.J. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015, 29, 2532–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.W.; Ayoub, N. Overexpression of KDM4 lysine demethylases disrupts the integrity of the DNA mismatch repair pathway. Biol. Open 2015, 4, 498. [Google Scholar] [CrossRef] [PubMed]
- Puccini, A.; Lenz, H.J.; Marshall, J.L.; Arguello, D.; Raghavan, D.; Korn, W.M.; Weinberg, B.A.; Poorman, K.; Heeke, A.L.; Philip, P.A.; et al. Impact of Patient Age on Molecular Alterations of Left-Sided Colorectal Tumors. Oncologist 2019, 24, 319–326. [Google Scholar] [CrossRef]
- Dorard, C.; de Thonel, A.; Collura, A.; Marisa, L.; Svrcek, M.; Lagrange, A.; Jego, G.; Wanherdrick, K.; Joly, A.L.; Buhard, O.; et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat. Med. 2011, 17, 1283–1289. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.J.; Rhee, Y.Y.; Oh, S.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Expression status of wild-type HSP110 correlates with HSP110 T17 deletion size and patient prognosis in microsatellite-unstable colorectal cancer. Mod. Pathol. 2014, 27, 443–453. [Google Scholar] [CrossRef]
- Salipante, S.J.; Scroggins, S.M.; Hampel, H.L.; Turner, E.H.; Pritchard, C.C. Microsatellite Instability Detection by Next Generation Sequencing. Clin. Chem. 2014, 60, 1192–1199. [Google Scholar] [CrossRef]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. MSIsensor: Microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014, 30, 1015. [Google Scholar] [CrossRef] [PubMed]
- Kautto, E.A.; Bonneville, R.; Miya, J.; Yu, L.; Krook, M.A.; Reeser, J.W.; Roychowdhury, S. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017, 8, 7452. [Google Scholar] [CrossRef] [PubMed]
- Baudrin, L.G.; Deleuze, J.F.; How-Kit, A. Molecular and Computational Methods for the Detection of Microsatellite Instability in Cancer. Front Oncol. 2018, 8, 621. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.; Yang, J.; Lang, J.; Jiang, Z.; Wang, W.; Yuan, D.; Wang, X.; Tian, G. Monitoring microsatellite instability (MSI) in circulating tumor DNA by next-generation DNA-seq. J. Clin. Oncol. 2018, 36, 12025. [Google Scholar] [CrossRef]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.T.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Cabel, L.; Proudhon, C.; Romano, E.; Girard, N.; Lantz, O.; Stern, M.H.; Pierga, J.Y.; Bidard, F.C. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2018, 15, 639–650. [Google Scholar] [CrossRef]
- Mattos-Arruda, L.D.; Weigelt, B.; Cortes, J.; Won, H.H.; Ng, C.K.Y.; Nuciforo, P.; Bidard, F.C.; Aura, C.; Saura, C.; Peg, V.; et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: A proof-of-principle. Ann. Oncol. 2014, 25, 1729. [Google Scholar] [CrossRef]
- Day, D.; Frentzas, S.; Naidu, C.A.; Segelov, E.; Green, M. Current Utility and Future Applications of ctDNA in Colorectal Cancer. In Advances in the Molecular Understanding of Colorectal Cancer; Segelov, E., Ed.; IntechOpen: London, UK, 2019; Chapter 4; ISBN 978-1-78985-060-4. [Google Scholar] [Green Version]
- Fader, A.N.; Diaz, L.A.; Armstrong, D.K.; Tanner, E.J.; Uram, J.; Eyring, A.; Wang, H.; Fisher, G.; Greten, T.; Le, D. Preliminary results of a phase II study: PD-1 blockade in mismatch repair–deficient, recurrent or persistent endometrial cancer. Gynecol. Oncol. 2016, 141, 206–207. [Google Scholar] [CrossRef]
- Cohen, R.; Hain, E.; Buhard, O.; Guilloux, A.; Bardier, A.; Kaci, R.; Bertheau, P.; Renaud, F.; Bibeau, F.; Fléjou, J.F.; et al. Association of Primary Resistance to Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer with Misdiagnosis of Microsatellite Instability or Mismatch Repair Deficiency Status. JAMA Oncol. 2019, 5, 551–555. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
| Mismatch Repair Gene | Mutation Frequency |
|---|---|
| MSH2 | 50% |
| MLH1 | 30–40% |
| MSH6 | 7–10% |
| PMS2 | <5% |
| EPCAM | 1–3% |
| Constitutional MLH1 epimutation | 1–3% |
| Series * | Number of Patients | Population | MMR IHC | Molecular MSI Testing | Discordance Rates |
|---|---|---|---|---|---|
| Lindor NM et al., 2002 [54] | 1144 | From multiple centers from the Cooperative Family Registry for Colon Cancer Studies: USA, Australia, and Canada | 2 proteins (MLH1 and MSH2) | 10 markers: BAT25, BAT26, BAT40, BAT34C4, D5S346, D17S250, ACTC, D18S55, D10S197, and MYCL. or 6 markers: D5S346, TP53, D18S34, D18S49, D18S61, ACTC and BAT 26 | 2.4% |
| Hatch et al., 2005 [55] | 262 | CRC with complete resection | 4 proteins (MLH1, MSH2, MSH6 and PMS2) | NCI panel (D5S346, BAT25, BAT26, D2S123, and D17S250) | 5.4% |
| Pinol et al., 2005 [56] | 1222 | CRC in Spain | 2 proteins (MSH2 and MLH1) | BAT26 ± BAT-25, D5S346, D2S123, and D17S250 | 2.8% |
| Watson et al., 2007 [57] | Cohort 1: 68 Cohort 2: 208 | CRC patients younger than 60 years (BRAF mutated CRC are excluded in cohort 1) | 4 proteins (MLH1, MSH2, MSH6 and PMS2) | Single microsatellite: BAT26 | Cohort 1: 1.4% Cohort 2: 1% |
| Yuan L et al., 2015 [58] | 296 | CRC patients fulfilled revised Bethesda criteria | 4 proteins (MLH1, MSH2, MSH6 and PMS2) | Bethesda panel | 1% |
| Chen et al., 2018 [59] | 569 | Chinese monocentric study with only CRC | 4 proteins (MLH1, MSH2, MSH6 and PMS2) | Bethesda panel | 8.1% |
| Cohen et al., abstract ESMO 2018 [60] | 92 | CRC only | 4 proteins (MLH1, MSH2, MSH6 and PMS2) | Pentaplex panel | 9.1% |
| Jaffrelot M et al., abstract JFHOD 2019 [61] | 2528 | Patients with dMMR tumors (CRC, endometrium, non-colorectal digestive cancers and others) | 4 proteins (MLH1, MSH2, MSH6 and PMS2) | Pentaplex panel | 1.1% |
| Causes of Discordance | Quality Criteria to Prevent Discordance | |
|---|---|---|
| MMR IHC | Molecular DNA testing | |
| Low tumor cells [62] | Selection of a specific area with the highest rate of tumor cells | Macrodissection or selection of tumor sections enriched in tumor cells (≥20%) |
| Pre-analytical difficulties [28] | Use formol 4% (not Bouin’s fixative), protocol standardization efforts, participation in national and international quality assessment | Protocol standardization efforts, participation in national and international quality assessment |
| Non-expert physician [29] | Participation in training sessions and request for rereading by expert if necessary | |
| Neoadjuvant treatment [63] | Testing on pretherapeutic samples | |
| Polymorphisms in non-Caucasian ethnic groups | - | Testing of paired tumor and non-tumor tissues |
| Discordance of tumor biopsy | Testing of the complete surgical resection | |
| Heterogeneous IHC pattern (Tumor heterogeneity suspected) [64] | Multiple sampling | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evrard, C.; Tachon, G.; Randrian, V.; Karayan-Tapon, L.; Tougeron, D. Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers 2019, 11, 1567. https://doi.org/10.3390/cancers11101567
Evrard C, Tachon G, Randrian V, Karayan-Tapon L, Tougeron D. Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers. 2019; 11(10):1567. https://doi.org/10.3390/cancers11101567
Chicago/Turabian StyleEvrard, Camille, Gaëlle Tachon, Violaine Randrian, Lucie Karayan-Tapon, and David Tougeron. 2019. "Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer" Cancers 11, no. 10: 1567. https://doi.org/10.3390/cancers11101567