Brexpiprazole, a Serotonin-Dopamine Activity Modulator, Can Sensitize Glioma Stem Cells to Osimertinib, a Third-Generation EGFR-TKI, via Survivin Reduction

 ,
,
Abstract
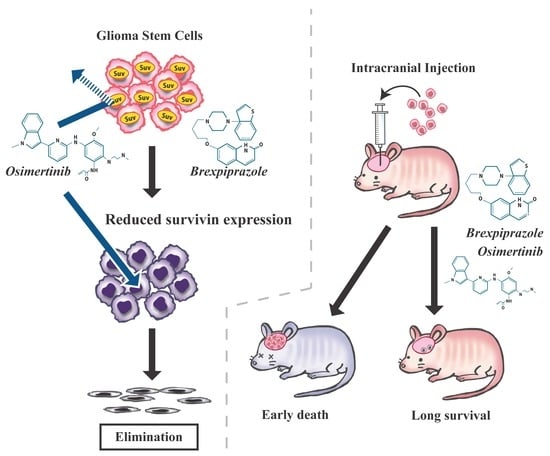
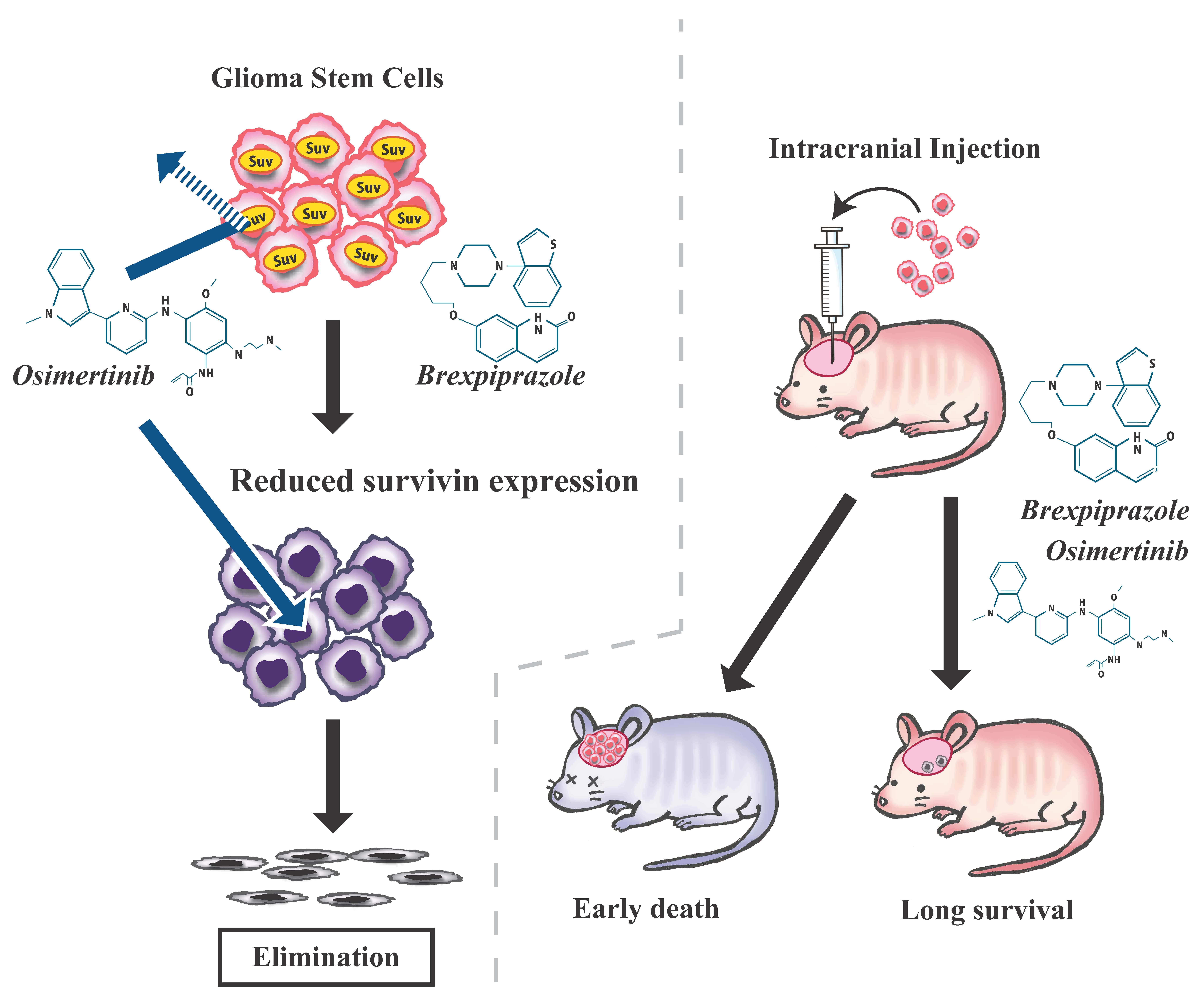
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
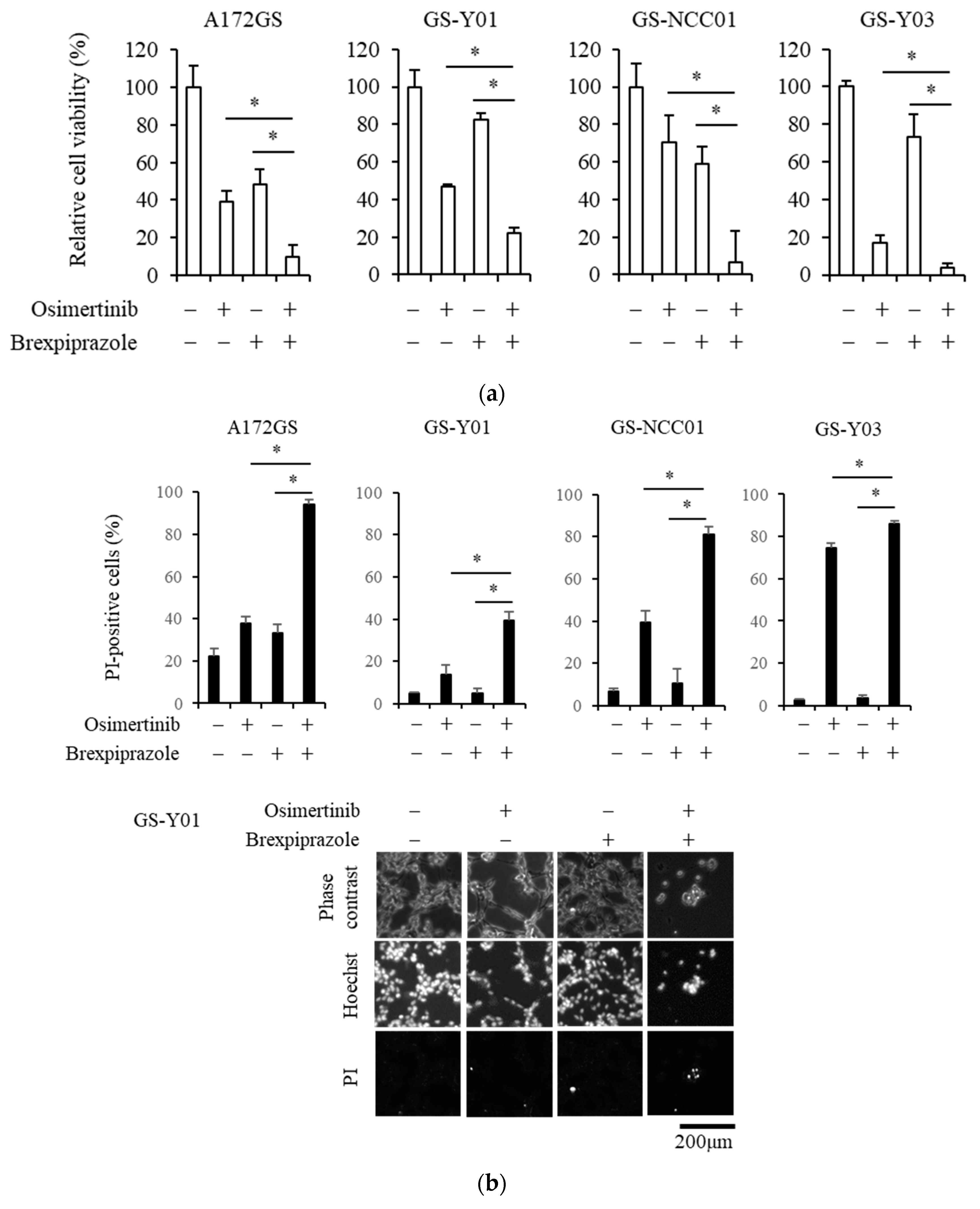
2.1. Brexpiprazole Sensitizes GSCs to Osimertinib
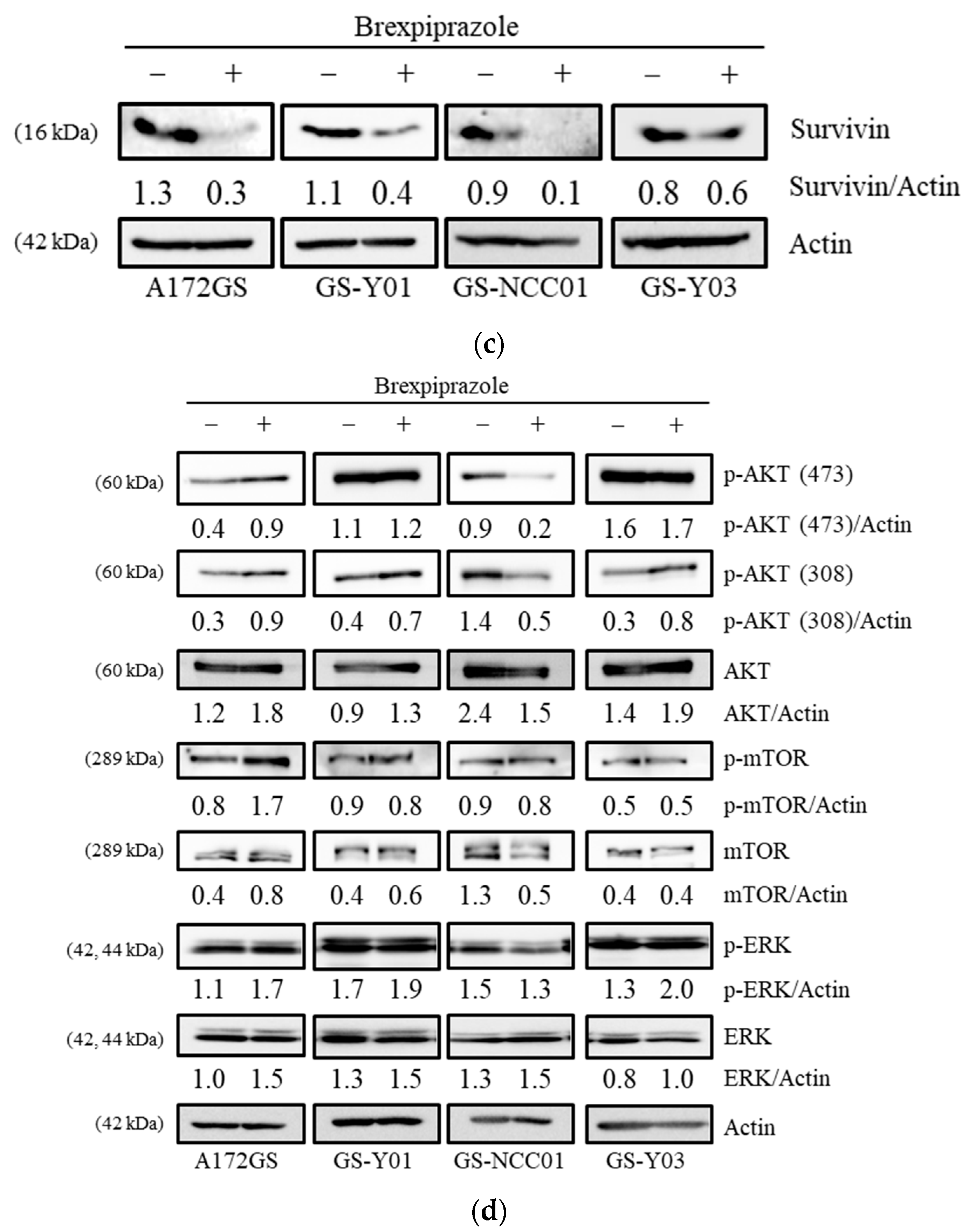
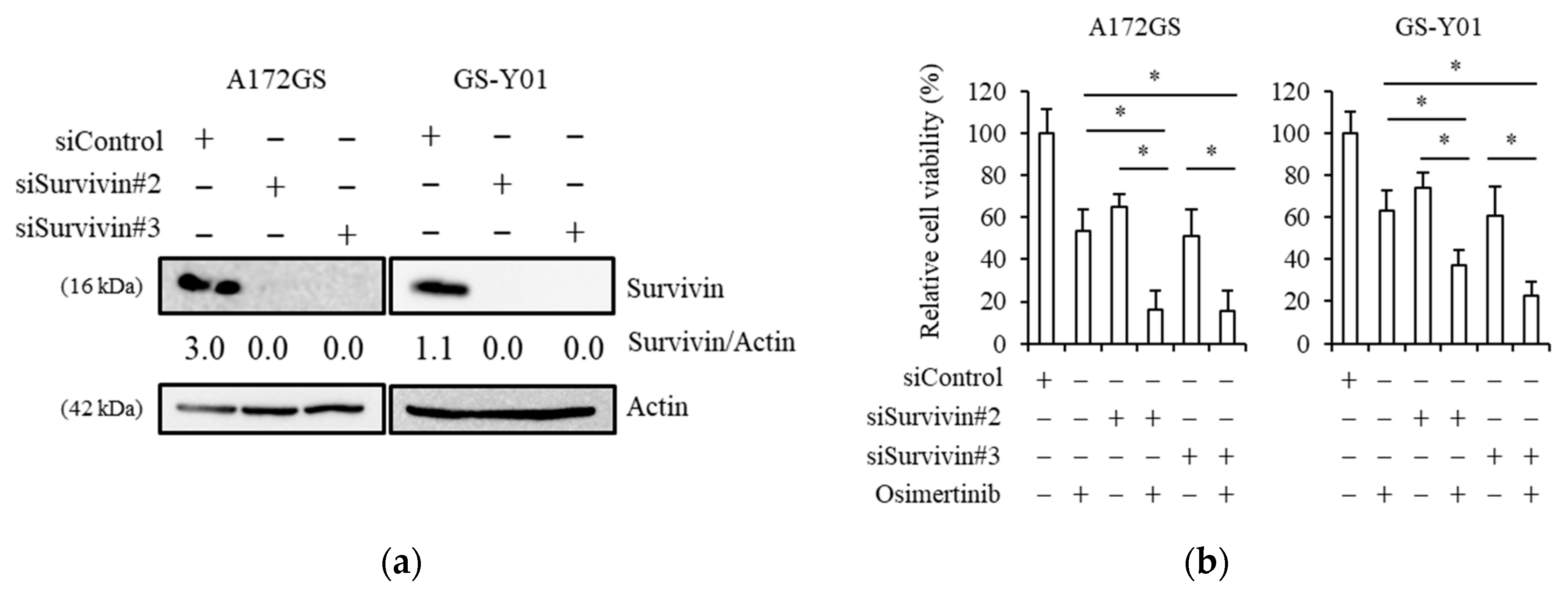
2.2. Pharmacological Inhibition of Survivin Sensitizes GSCs to Osimertinib
2.3. Genetic Inhibition of Survivin Mimics Brexpiprazole Treatment
2.4. Brexpiprazole Sensitizes GSCs to Osimertinib in Vivo.
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Cell Viability and Cell Death Assays
4.4. Immunoblot Analysis
4.5. Gene Silencing with siRNA.
4.6. Mouse Study
4.7. Transfection of Plasmid
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Brennan, C.W.; DeAngelis, L.M.; Omuro, A.M. Emerging therapies for glioblastoma. JAMA Neurol. 2014, 71, 1437–1444. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Temozolomide: Therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev. Neurother. 2010, 10, 1537–1544. [Google Scholar] [CrossRef]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Sacca, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells—Much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Lathia, J.; Williams, B.J.; Wu, Q.; Gallagher, J.; Androutsellis-Theotokis, A.; Giles, A.J.; Yang, C.; Zhuang, Z.; Gilbert, M.R.; et al. The p38 signaling pathway mediates quiescence of glioma stem cells by regulating epidermal growth factor receptor trafficking. Oncotarget 2017, 8, 33316–33328. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.T.; Wikstrand, C.J.; Bigner, D.D. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr. Relat. Cancer 2001, 8, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ballard, P.; Yates, J.W.T.; Yang, Z.; Kim, D.W.; Yang, J.C.H.; Cantarini, M.; Pickup, K.; Jordan, A.; Hickey, M.; Grist, M.; et al. Preclinical Comparison of Osimertinib with Other EGFR-TKIs in EGFR-Mutant NSCLC Brain Metastases Models, and Early Evidence of Clinical Brain Metastases Activity. Clin. Cancer Res. 2016, 22, 5130–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, G.; Tsai, C.M.; Shepherd, F.A.; Ahn, M.J.; Bazhenova, L.; Crino, L.; de Marinis, F.; Felip, E.; Morabito, A.; Hodge, R.; et al. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: Pooled data from two phase II trials. Ann. Oncol. 2018, 29, 687–693. [Google Scholar] [CrossRef]
- Aladeen, T.; Westphal, E.; Lee, Y.; Rong, C.; Rainka, M.; Capote, H.; McIntyre, R.S. The use of brexpiprazole amongst individuals with insufficient outcomes with aripiprazole or bupropion: A case series. Perspect. Psychiatr. Care 2018, 54, 507–513. [Google Scholar] [CrossRef]
- Frankel, J.S.; Schwartz, T.L. Brexpiprazole and cariprazine: Distinguishing two new atypical antipsychotics from the original dopamine stabilizer aripiprazole. Ther. Adv. Psychopharmacol. 2017, 7, 29–41. [Google Scholar] [CrossRef]
- Correll, C.U.; Skuban, A.; Ouyang, J.; Hobart, M.; Pfister, S.; McQuade, R.D.; Nyilas, M.; Carson, W.H.; Sanchez, R.; Eriksson, H. Efficacy and Safety of Brexpiprazole for the Treatment of Acute Schizophrenia: A 6-Week Randomized, Double-Blind, Placebo-Controlled Trial. Am. J. Psychiatry 2015, 172, 870–880. [Google Scholar] [CrossRef]
- Suzuki, S.; Okada, M.; Kuramoto, K.; Takeda, H.; Sakaki, H.; Watarai, H.; Sanomachi, T.; Seino, S.; Yoshioka, T.; Kitanaka, C. Aripiprazole, an Antipsychotic and Partial Dopamine Agonist, Inhibits Cancer Stem Cells and Reverses Chemoresistance. Anticancer Res. 2016, 36, 5153–5161. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamamoto, M.; Togashi, K.; Sanomachi, T.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. In vitro and in vivo anti-tumor effects of brexpiprazole, a newly-developed serotonin-dopamine activity modulator with an improved safety profile. Oncotarget 2019, 10, 3547–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Okamoto, I.; Hatashita, E.; Kuwata, K.; Yamaguchi, H.; Kita, A.; Yamanaka, K.; Ono, M.; Nakagawa, K. Overcoming Erlotinib Resistance in EGFR Mutation-Positive Non-Small Cell Lung Cancer Cells by Targeting Survivin. Mol. Cancer Ther. 2012, 11, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Okamoto, I.; Okamoto, W.; Tanaka, K.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res. 2010, 70, 10402–10410. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006, 66, 10100–10111. [Google Scholar] [CrossRef] [PubMed]
- Bajetto, A.; Porcile, C.; Pattarozzi, A.; Scotti, L.; Aceto, A.; Daga, A.; Barbieri, F.; Florio, T. Differential role of EGF and BFGF in human GBM-TIC proliferation: Relationship to EGFR-tyrosine kinase inhibitor sensibility. J. Biol. Regul. Homeost. Agents 2013, 27, 143–154. [Google Scholar] [PubMed]
- Yamamoto, M.; Suzuki, S.; Togashi, K.; Sanomachi, T.; Seino, S.; Kitanaka, C.; Okada, M. AS602801 Sensitizes Ovarian Cancer Stem Cells to Paclitaxel by Down-regulating MDR1. Anticancer Res. 2019, 39, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.H.; Shu, Y.; Chen, P.; Wu, J.N.; Zhu, L.H.; Yuan, R.X.; Long, W.G.; Zhu, Y.M.; Li, J. YM155 sensitizes non-small cell lung cancer cells to EGFR-tyrosine kinase inhibitors through the mechanism of autophagy induction. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3786–3798. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef]
- Pang, L.Y.; Saunders, L.; Argyle, D.J. Epidermal growth factor receptor activity is elevated in glioma cancer stem cells and is required to maintain chemotherapy and radiation resistance. Oncotarget 2017, 8, 72494–72512. [Google Scholar] [CrossRef] [Green Version]
- Marie, Y.; Carpentier, A.F.; Omuro, A.M.; Sanson, M.; Thillet, J.; Hoang-Xuan, K.; Delattre, J.Y. EGFR tyrosine kinase domain mutations in human gliomas. Neurology 2005, 64, 1444–1445. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Bianco, R.; Shin, I.; Ritter, C.A.; Yakes, F.M.; Basso, A.; Rosen, N.; Tsurutani, J.; Dennis, P.A.; Mills, G.B.; Arteaga, C.L. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene 2003, 22, 2812–2822. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Lu, K.V.; Zhu, S.; Dia, E.Q.; Vivanco, I.; Shackleford, G.M.; Cavenee, W.K.; Mellinghoff, I.K.; Cloughesy, T.F.; Sawyers, C.L.; et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006, 66, 7864–7869. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Cheng, C.K.; Nicolaides, T.P.; Hackett, C.S.; Knight, Z.A.; Shokat, K.M.; Weiss, W.A. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007, 67, 7960–7965. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O.; Cuatrecasas, M.; Benavides, M.; Segura, P.P.; Berrocal, A.; Erill, N.; Colomer, A.; Quintana, M.J.; Balaña, C.; Gil, M.; et al. Efficacy of erlotinib in patients with relapsed gliobastoma multiforme who expressed EGFRVIII and PTEN determined by immunohistochemistry. J. Neuro-Oncol. 2013, 116, 413–419. [Google Scholar] [CrossRef] [Green Version]
- De Groot, J.F.; Gilbert, M.R.; Aldape, K.; Hess, K.R.; Hanna, T.A.; Ictech, S.; Groves, M.D.; Conrad, C.; Colman, H.; Puduvalli, V.K.; et al. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J. Neuro-Oncol. 2008, 90, 89–97. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E., 2nd; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neuro-Oncol. 2010, 96, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Pourzia, A.L.; Nourian, A.A.; Williams, K.J.; Nathanson, D.; Babic, I.; Villa, G.R.; Tanaka, K.; Nael, A.; Yang, H.; et al. De-repression of PDGFRbeta transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov. 2013, 3, 534–547. [Google Scholar] [CrossRef]
- Iwanami, A.; Gini, B.; Zanca, C.; Matsutani, T.; Assuncao, A.; Nael, A.; Dang, J.; Yang, H.; Zhu, S.; Kohyama, J.; et al. PML mediates glioblastoma resistance to mammalian target of rapamycin (mTOR)-targeted therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 4339–4344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhu, S.; Kozono, D.; Ng, K.; Futalan, D.; Shen, Y.; Akers, J.C.; Steed, T.; Kushwaha, D.; Schlabach, M.; et al. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget 2014, 5, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.R.; Gladkov, O.A.; Gartner, E.; Vladimirov, V.; Crown, J.; Steinberg, J.; Jie, F.; Keating, A. Phase II, multicenter, open-label, randomized study of YM155 plus docetaxel as first-line treatment in patients with HER2-negative metastatic breast cancer. Breast Cancer Res. Treat. 2015, 149, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Thomas, A.; Rajan, A.; Chun, G.; Lopez-Chavez, A.; Szabo, E.; Spencer, S.; Carter, C.A.; Guha, U.; Khozin, S.; et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 2601–2606. [Google Scholar] [CrossRef]
- Kudchadkar, R.; Ernst, S.; Chmielowski, B.; Redman, B.G.; Steinberg, J.; Keating, A.; Jie, F.; Chen, C.; Gonzalez, R.; Weber, J. A phase 2, multicenter, open-label study of sepantronium bromide (YM155) plus docetaxel in patients with stage III (unresectable) or stage IV melanoma. Cancer Med. 2015, 4, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Citrome, L.; Ota, A.; Nagamizu, K.; Perry, P.; Weiller, E.; Baker, R.A. The effect of brexpiprazole (OPC-34712) and aripiprazole in adult patients with acute schizophrenia: Results from a randomized, exploratory study. Int. Clin. Psychopharmacol. 2016, 31, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.B.; Robinson, D.M.; Clayton, A.H. Clinical role of brexpiprazole in depression and schizophrenia. Ther. Clin. Risk Manag. 2017, 13, 299–306. [Google Scholar] [CrossRef]
- Stahl, S.M. Mechanism of action of brexpiprazole: Comparison with aripiprazole. CNS Spectr. 2016, 21, 1–6. [Google Scholar] [CrossRef]
- Citrome, L. The ABC’s of dopamine receptor partial agonists—Aripiprazole, brexpiprazole and cariprazine: The 15-min challenge to sort these agents out. Int. J. Clin. Pract. 2015, 69, 1211–1220. [Google Scholar] [CrossRef]
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Sunayama, J.; Matsuda, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Tachibana, K.; Tomiyama, A.; Kayama, T.; Kitanaka, C. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011, 29, 1942–1951. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Matsuda, K.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Okada, M.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Seino, S.; Yamashita, H.; Kitanaka, C. A Small-molecule Kinase Inhibitor, CEP-1347, Inhibits Survivin Expression and Sensitizes Ovarian Cancer Stem Cells to Paclitaxel. Anticancer Res. 2018, 38, 4535–4542. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Okada, M.; Shibuya, K.; Seino, M.; Sato, A.; Takeda, H.; Seino, S.; Yoshioka, T.; Kitanaka, C. JNK suppression of chemotherapeutic agents-induced ROS confers chemoresistance on pancreatic cancer stem cells. Oncotarget 2015, 6, 458–470. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. Brexpiprazole, a Serotonin-Dopamine Activity Modulator, Can Sensitize Glioma Stem Cells to Osimertinib, a Third-Generation EGFR-TKI, via Survivin Reduction. Cancers 2019, 11, 947. https://doi.org/10.3390/cancers11070947
Suzuki S, Yamamoto M, Sanomachi T, Togashi K, Sugai A, Seino S, Yoshioka T, Kitanaka C, Okada M. Brexpiprazole, a Serotonin-Dopamine Activity Modulator, Can Sensitize Glioma Stem Cells to Osimertinib, a Third-Generation EGFR-TKI, via Survivin Reduction. Cancers. 2019; 11(7):947. https://doi.org/10.3390/cancers11070947
Chicago/Turabian StyleSuzuki, Shuhei, Masahiro Yamamoto, Tomomi Sanomachi, Keita Togashi, Asuka Sugai, Shizuka Seino, Takashi Yoshioka, Chifumi Kitanaka, and Masashi Okada. 2019. "Brexpiprazole, a Serotonin-Dopamine Activity Modulator, Can Sensitize Glioma Stem Cells to Osimertinib, a Third-Generation EGFR-TKI, via Survivin Reduction" Cancers 11, no. 7: 947. https://doi.org/10.3390/cancers11070947