A Therapeutic Strategy for Chemotherapy-Resistant Gastric Cancer via Destabilization of Both β-Catenin and RAS

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
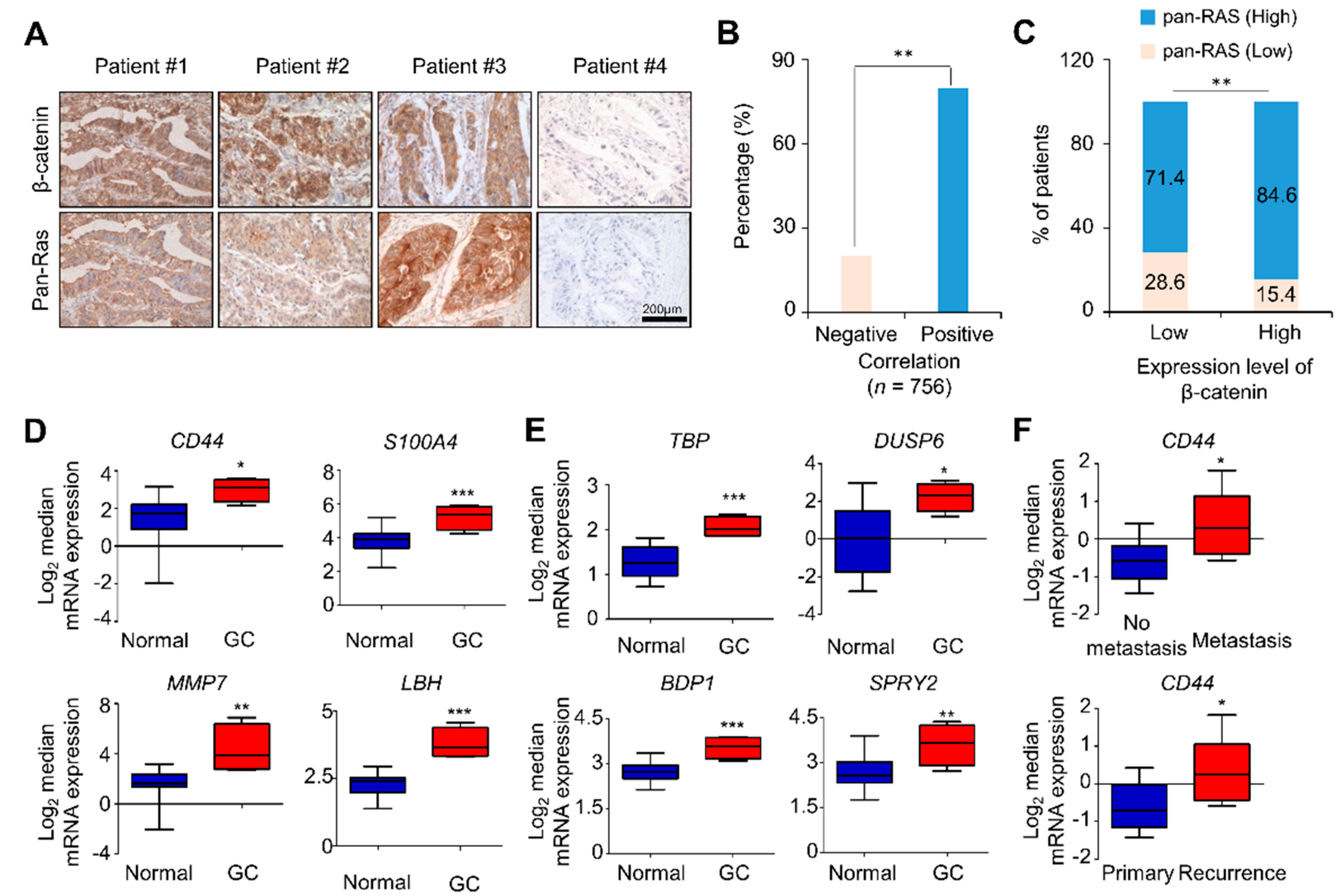
2.1. Both β-Catenin and RAS Levels as well as CSC Markers were Increased in Tissues of AGC Patients
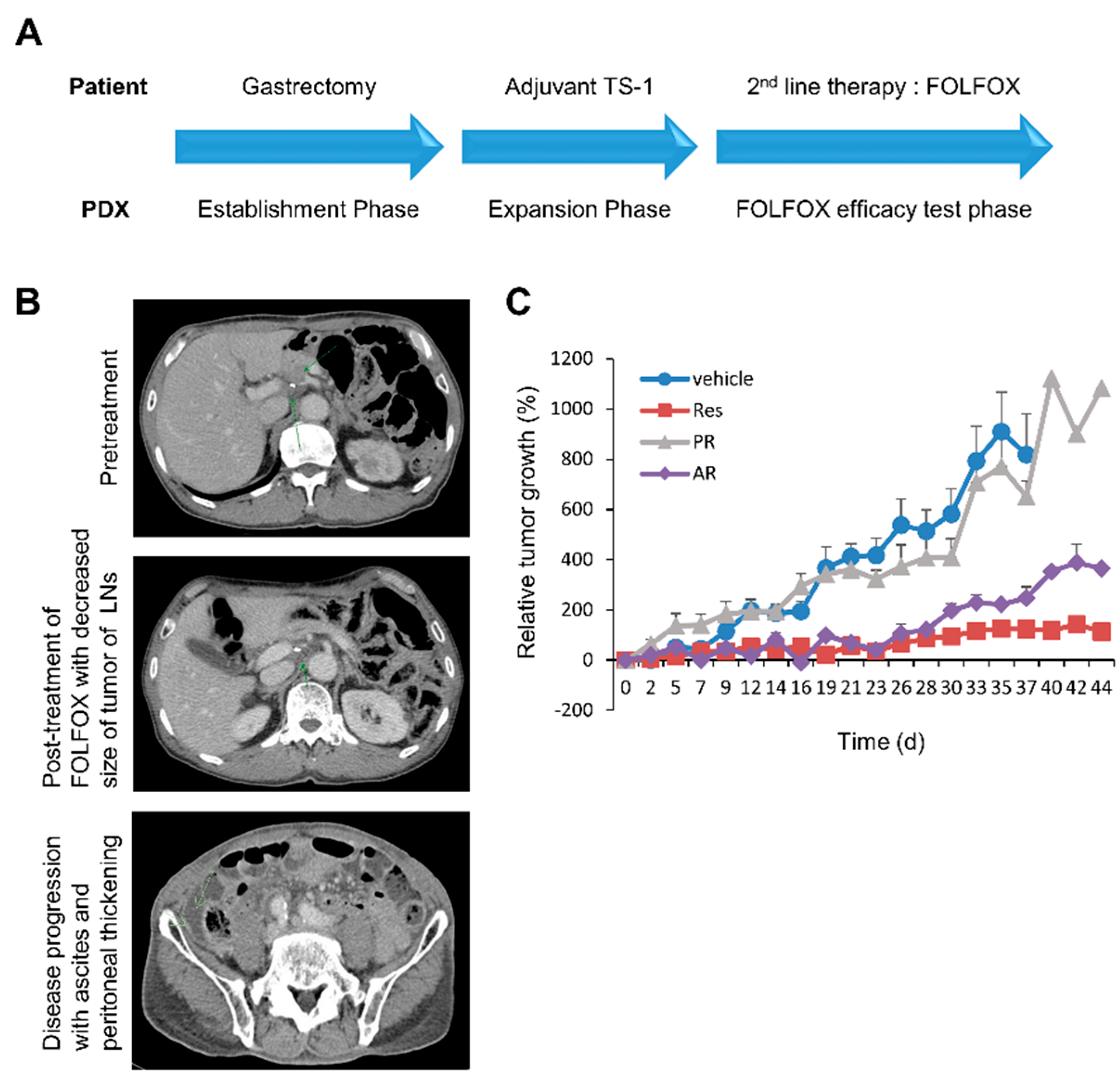
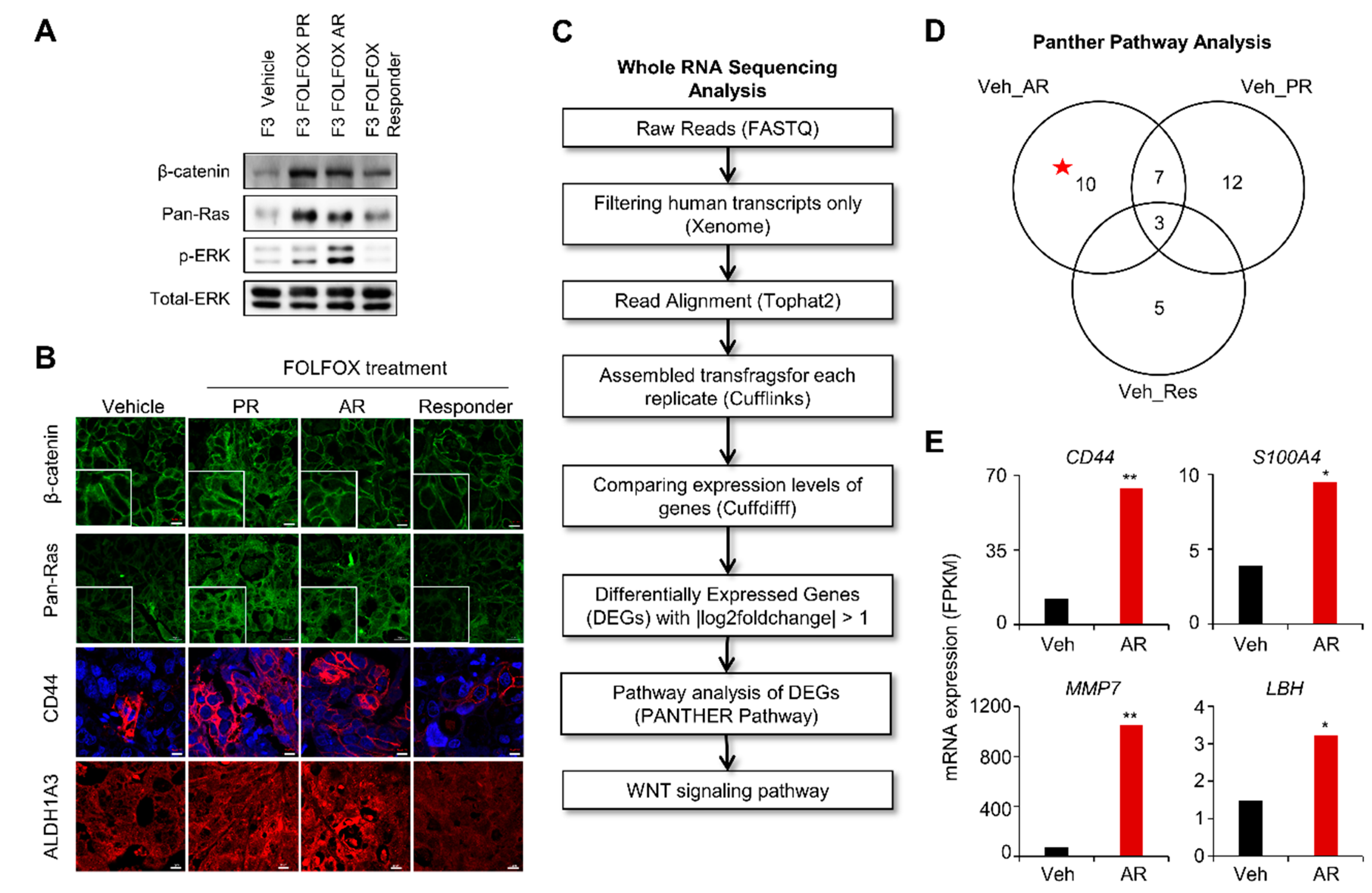
2.2. The Wnt/β-catenin and RAS-ERK Pathways were Activated with Increases of both β-catenin and pan-RAS Levels in Tissues of Chemotherapy-Resistant PDX Tumor
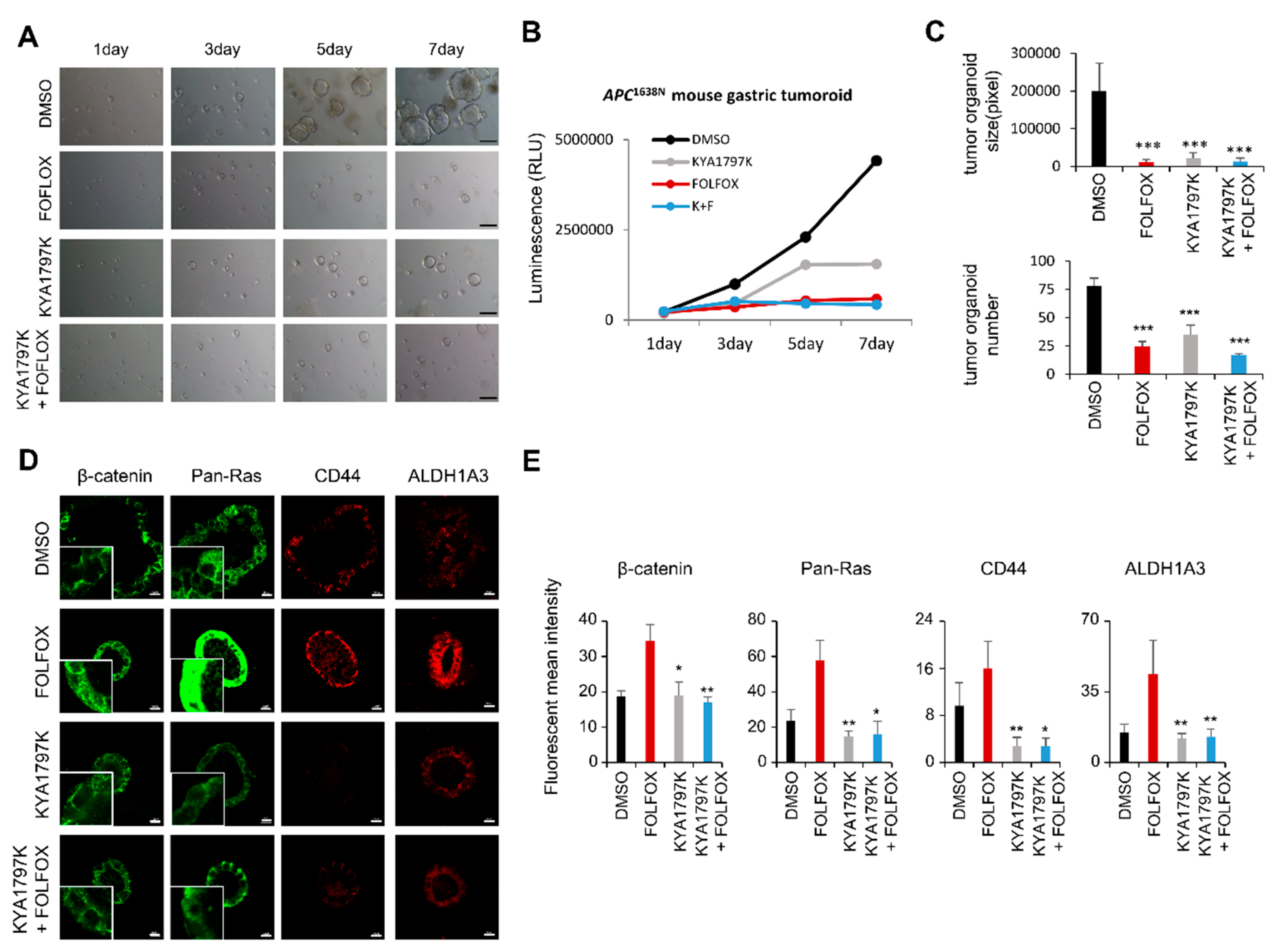
2.3. Destabilization of both β-catenin and RAS Inhibited Transformation of GC Cells, and Suppressed Tumoroids Derived from Gastric Adenoma Tissues of Apc1638N Mice
2.4. KYA1797K Overcame Paclitaxel Resistance of PDX Tumors of FOLFOX-Resistant GC Tissues
3. Discussion
4. Materials and Methods
4.1. Patients and TMA of AGC Patients
4.2. PDX Experiments
4.3. Whole RNA Sequencing Analysis of PDX Model
4.4. Cell Culture and Drug Treatment
4.5. Tumor Organoid
4.6. Mice and Maintenance
4.7. Immunoblotting Analysis
4.8. Immunohistochemistry
4.9. Cell Proliferation and Colony Formation Assays
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yuasa, Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat. Rev. Cancer 2003, 3, 592–600. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Lunet, N.; Barros, H. Helicobacter pylori infection and gastric cancer: Facing the enigmas. Int. J. Cancer 2003, 106, 953–960. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Choi, Y.Y.; An, J.Y.; Shin, H.B.; Jo, A.; Choi, H.; Seo, S.H.; Bang, H.J.; Cheong, J.H.; Hyung, W.J.; et al. The benefit of microsatellite instability is attenuated by chemotherapy in stage II and stage III gastric cancer: Results from a large cohort with subgroup analyses. Int. J. Cancer 2015, 137, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Song, S.; Lee, J.S.; Yao, Y.; Wei, Q.; Ajani, J.A. Gastric cancer-molecular and clinical dimensions. Nat. Rev. Clin. Oncol. 2013, 10, 643–655. [Google Scholar] [CrossRef]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef]
- Sokolova, O.; Bozko, P.M.; Naumann, M. Helicobacter pylori suppresses glycogen synthase kinase 3beta to promote beta-catenin activity. J. Biol. Chem. 2008, 283, 29367–29374. [Google Scholar] [CrossRef]
- Lee, J.H.; Abraham, S.C.; Kim, H.S.; Nam, J.H.; Choi, C.; Lee, M.C.; Park, C.S.; Juhng, S.W.; Rashid, A.; Hamilton, S.R.; et al. Inverse relationship between APC gene mutation in gastric adenomas and development of adenocarcinoma. Am. J. Pathol. 2002, 161, 611–618. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [CrossRef]
- Kim, M.A.; Lee, H.S.; Lee, H.E.; Jeon, Y.K.; Yang, H.K.; Kim, W.H. EGFR in gastric carcinomas: Prognostic significance of protein overexpression and high gene copy number. Histopathology 2008, 52, 738–746. [Google Scholar] [CrossRef]
- Oliveira, C.; Pinto, M.; Duval, A.; Brennetot, C.; Domingo, E.; Espin, E.; Armengol, M.; Yamamoto, H.; Hamelin, R.; Seruca, R.; et al. BRAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogene 2003, 22, 9192–9196. [Google Scholar] [CrossRef]
- Mita, H.; Toyota, M.; Aoki, F.; Akashi, H.; Maruyama, R.; Sasaki, Y.; Suzuki, H.; Idogawa, M.; Kashima, L.; Yanagihara, K.; et al. A novel method, digital genome scanning detects KRAS gene amplification in gastric cancers: Involvement of overexpressed wild-type KRAS in downstream signaling and cancer cell growth. BMC Cancer 2009, 9, 198. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Liu, J.B.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018. [Google Scholar]
- D’Abaco, G.M.; Whitehead, R.H.; Burgess, A.W. Synergy between Apc min and an activated ras mutation is sufficient to induce colon carcinomas. Mol. Cell Biol. 1996, 16, 884–891. [Google Scholar] [CrossRef]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl. Acad. Sci. 2006, 103, 14122–14127. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.S.; Jeong, W.J.; Park, J.; Kim, T.I.; Min do, S.; Choi, K.Y. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/beta-catenin signaling. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Jeong, W.J.; Yoon, J.; Park, J.C.; Lee, S.H.; Lee, S.H.; Kaduwal, S.; Kim, H.; Yoon, J.B.; Choi, K.Y. Ras stabilization through aberrant activation of Wnt/beta-catenin signaling promotes intestinal tumorigenesis. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Cha, P.H.; Cho, Y.H.; Lee, S.K.; Lee, J.; Jeong, W.J.; Moon, B.S.; Yun, J.H.; Yang, J.S.; Choi, S.; Yoon, J.; et al. Small-molecule binding of the axin RGS domain promotes beta-catenin and Ras degradation. Nat. Chem. Biol. 2016, 12, 593–600. [Google Scholar] [CrossRef]
- Cho, Y.H.; Cha, P.H.; Kaduwal, S.; Park, J.C.; Lee, S.K.; Yoon, J.S.; Shin, W.; Kim, H.; Ro, E.J.; Koo, K.H.; et al. KY1022, a small molecule destabilizing Ras via targeting the Wnt/beta-catenin pathway, inhibits development of metastatic colorectal cancer. Oncotarget 2016, 7, 81727–81740. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, J.G.; Jung, H.K.; Kim, J.H.; Jeong, W.K.; Jeon, T.J.; Kim, J.M.; Kim, Y.I.; Ryu, K.W.; Kong, S.H.; et al. Clinical practice guidelines for gastric cancer in Korea: An evidence-based approach. J. Gastric Cancer 2014, 14, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van den Born, M. Detection of beta-catenin localization by immunohistochemistry. Methods Mol. Biol. 2008, 468, 91–98. [Google Scholar] [PubMed]
- Prior, I.A.; Hancock, J.F. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol 2012, 23, 145–153. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Thomas, P. PANTHER pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Wilke, H.J.; Van Cutsem, E. Current treatments and future perspectives in colorectal and gastric cancer. Ann. Oncol. 2003, 14 (Suppl. 2), ii49–ii55. [Google Scholar] [CrossRef]
- Wielenga, V.J.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am. J. Pathol. 1999, 154, 515–523. [Google Scholar] [CrossRef]
- Wu, D.; Mou, Y.P.; Chen, K.; Cai, J.Q.; Zhou, Y.C.; Pan, Y.; Xu, X.W.; Zhou, W.; Gao, J.Q.; Chen, D.W.; et al. Aldehyde dehydrogenase 3A1 is robustly upregulated in gastric cancer stem-like cells and associated with tumorigenesis. Int. J. Oncol. 2016, 49, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Wu, W.; Cheng, T.; Schlitter, A.M.; Qian, C.; Bruns, P.; Jian, Z.; Jager, C.; Regel, I.; Raulefs, S.; et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut. 2016, 65, 647–657. [Google Scholar] [CrossRef] [PubMed]
- du Bois, A.; Lück, H.-J.; Meier, W.; Adams, H.-P.; Möbus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schröder, W.; et al. A Randomized Clinical Trial of Cisplatin/Paclitaxel Versus Carboplatin/Paclitaxel as First-Line Treatment of Ovarian Cancer. J. Nat. Cancer Inst. 2003, 95, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, A.; Boku, N.; Tamura, F.; Muro, K.; Shimada, Y.; Saigenji, K.; Akazawa, S.; Kitajima, M.; Kanamaru, R.; Taguchi, T. An early phase II study of a 3-hour infusion of paclitaxel for advanced gastric cancer. Am. J. Clin. Oncol. 1998, 21, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.Y.; Lee, J.E.; Kim, H.; Sim, M.H.; Kim, K.K.; Lee, G.; Kim, H.I.; An, J.Y.; Hyung, W.J.; Kim, C.B.; et al. Establishment and characterisation of patient-derived xenografts as paraclinical models for gastric cancer. Sci. Rep. 2016, 6, 22172. [Google Scholar] [CrossRef]
- Conway, T.; Wazny, J.; Bromage, A.; Tymms, M.; Sooraj, D.; Williams, E.D.; Beresford-Smith, B. Xenome--a tool for classifying reads from xenograft samples. Bioinformatics 2012, 28, i172–i178. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Mi, H.; Poudel, S.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016, 44, D336–D342. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guo, H.; Song, Y.; Zhao, X.; Shi, Y.; Lu, Y.; Hu, S.; Nie, Y.; Fan, D.; Wu, K. Loss of vinculin and membrane-bound beta-catenin promotes metastasis and predicts poor prognosis in colorectal cancer. Mol. Cancer 2014, 13, 263. [Google Scholar] [CrossRef] [PubMed]
- Azzarello, D.; Giuffre, C.; Panuccio, V.; Giannicola, R.; Del Medico, P.; Zavettieri, M.; Raffaele, M.; Maisano, R.; Falzea, A.; Nardi, M. First line chemotherapy with FOLFOX 4 in elderly patients (>65 years) with advanced or metastatic gastric cancer (A/MGC): A pilot study. J. Clin. Oncol. 2006, 24, 14095. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, W.-J.; Lee, J.E.; Cho, Y.-H.; Lee, G.; Seo, M.-k.; Lee, S.-K.; Hwang, J.-H.; Min, D.S.; Noh, S.H.; Paik, S.; et al. A Therapeutic Strategy for Chemotherapy-Resistant Gastric Cancer via Destabilization of Both β-Catenin and RAS. Cancers 2019, 11, 496. https://doi.org/10.3390/cancers11040496
Ryu W-J, Lee JE, Cho Y-H, Lee G, Seo M-k, Lee S-K, Hwang J-H, Min DS, Noh SH, Paik S, et al. A Therapeutic Strategy for Chemotherapy-Resistant Gastric Cancer via Destabilization of Both β-Catenin and RAS. Cancers. 2019; 11(4):496. https://doi.org/10.3390/cancers11040496
Chicago/Turabian StyleRyu, Won-Ji, Jae Eun Lee, Yong-Hee Cho, Gunho Lee, Mi-kyoung Seo, Sang-Kyu Lee, Jeong-Ha Hwang, Do Sik Min, Sung Hoon Noh, Soonmyung Paik, and et al. 2019. "A Therapeutic Strategy for Chemotherapy-Resistant Gastric Cancer via Destabilization of Both β-Catenin and RAS" Cancers 11, no. 4: 496. https://doi.org/10.3390/cancers11040496