Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies


1
Section on Medical Neuroendocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA
2
Neuro-Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to the work.
Cancers 2019, 11(4), 436; https://doi.org/10.3390/cancers11040436
Submission received: 14 March 2019
/
Revised: 25 March 2019
/
Accepted: 26 March 2019
/
Published: 28 March 2019
(This article belongs to the Special Issue Pheochromocytoma (PHEO) and Paraganglioma (PGL))
{kind=link}
Abstract
:Pheochromocytoma and paraganglioma (PCPGs) are rare neuroendocrine tumors that arise from the chromaffin tissue of adrenal medulla and sympathetic ganglia. Although metastatic PCPGs account for only 10% of clinical cases, morbidity and mortality are high because of the uncontrollable mass effect and catecholamine level generated by these tumors. Despite our expanding knowledge of PCPG genetics, the clinical options to effectively suppress PCPG progression remain limited. Several recent translational studies revealed that PCPGs with different molecular subtypes exhibit distinctive oncogenic pathways and spectrum of therapy resistance. This suggests that therapeutics can be adjusted based on the signature molecular and metabolic pathways of PCPGs. In this review, we summarized the latest findings on PCPG genetics, novel therapeutic targets, and perspectives for future personalized medicine.
1. Introduction
Pheochromocytomas and paragangliomas (PGPCs) are catecholamine-producing tumors that arise from adrenal medulla, or from extra-adrenal ganglial sympathetic/parasympathetic chains (of chromaffin or non-chromaffin origin), respectively. Tumor-associated secretion of catecholamine causes symptoms of hyperactivity in the sympathetic nervous system including paroxysmal hypertension, headache and diaphoresis. PCPGs result from genetic abnormalities, mostly disruption/mutation in single disease-related genes [1]. Approximately 30–35% of patients with PCPG carry germline mutations in over 20 susceptible genes [2]. In pediatric patients, or in patients who developed the origin tumor in their childhood, approximately 69–87.5% of cases carry germline mutations [3]. Germline mutations may lead to clinical syndromes with symptoms that affect multiple organs, such as von Hippel-Lindau disease, multiple endocrine neoplasia type 2 syndrome, and neurofibromatosis type 1 [4]. On the other hand, somatic mutations in key oncogenic pathways, such as SDHx, VHL, HIF2A, H-RAS, NF1, RET, or MAX, predispose PCPG formation [5].
Despite our expanding understanding of PCPG genetics and transcriptomics, therapies against this malignancy, especially those against PCPG metastatic lesions, are limited. In addition to surgical resection and radiation therapy, combination chemotherapy that includes cyclophosphamide–vincristine–dacarbazine (CVD) is recommended for advanced PCPG. However, retrospective studies showed that CVD-based treatment provides limited benefit to patient quality of life and overall survival [6]. There is an urgent need to decipher the molecular signature of PCPG for optimized therapeutic regimens, which may result in improved selectivity and efficacy of treatment. In this review, we summarized the latest reports on PCPG genetics, clinical findings and management, and emerging targeted therapies against PCPG subtypes.
2. Genetics of PCPGs
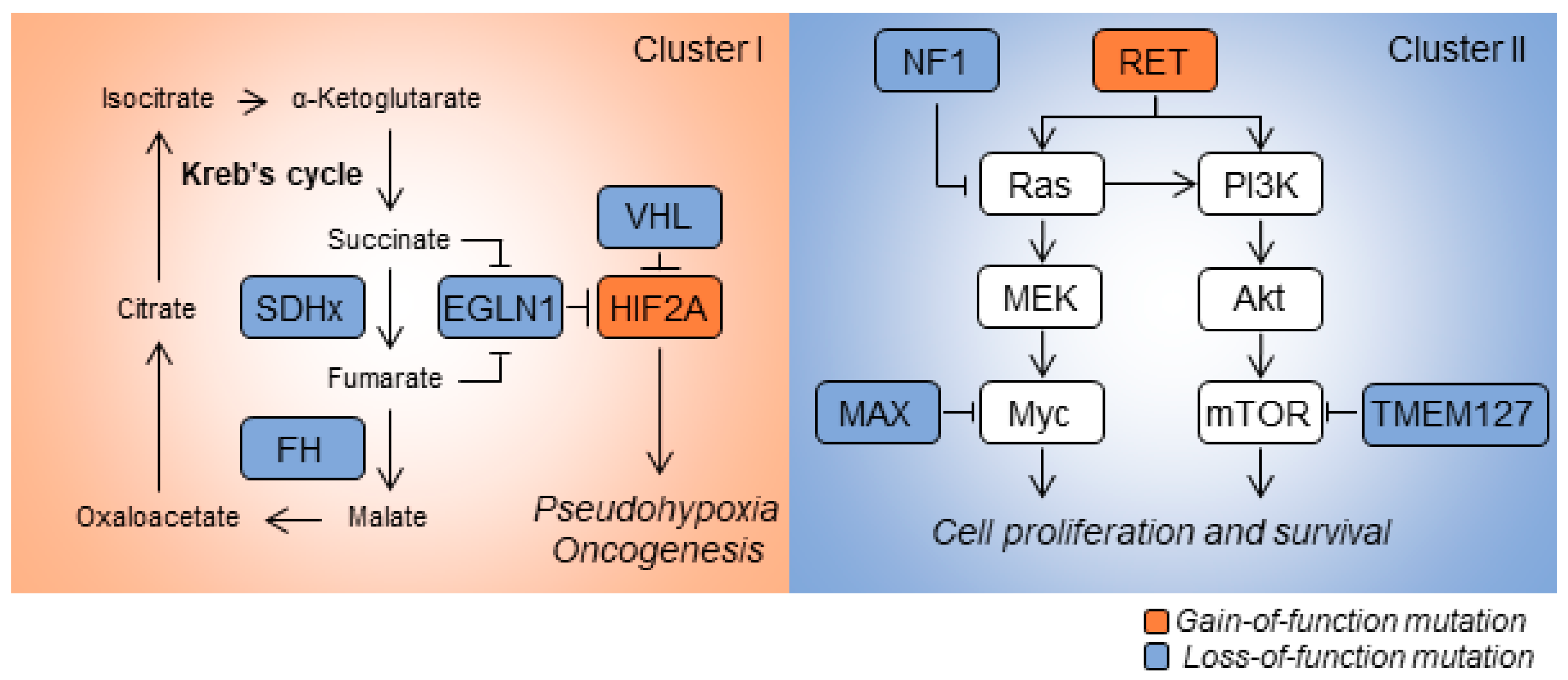
Transcriptomic analysis of patient-derived specimens revealed distinctive gene-expression signatures among histologically similar PCPGs. Based on mRNA-expression signatures, PCPGs can be divided into two main categories: Cluster I and Cluster II diseases (Figure 1). Cluster I disease exhibits metabolic reprogramming and pseudo hypoxic signaling commonly linked to mutations in oxygen-sensing genes or those encoding key enzymes in the Krebs cycle such as VHL, SDHx, HIF2A, EGLN1/2 and FH. Cluster I disease is further stratified into respective subgroups based on differentially-expressed genes. PCPGs showing mutation of SDHx and VHL are sub-characterized into Cluster IA and Cluster IB, respectively [5]. In contrast, Cluster II PCPGs are commonly related to genetic mutations affecting kinase signaling, gene translation, protein synthesis and neural differentiation; the genes showing mutations include NF1, RET, KIF1Bβ, TMEM127 and MAX. Cluster II disease is further categorized into Cluster 2A (in which patient show mutations in RET, NF1, and TMEM127 ), Cluster 2B (sporadic tumors) and Cluster 2C (patients with mutations in 3.7% VHL and 11.1% RET, and sporadic tumors) [5]. Recent findings show that mutations in the Wnt/Hedgehog pathway are involved in a new molecular subtype of PCPGs [7]. Fishbein et al. discovered that the in-frame RNA fusion transcripts of the UBTF-MAML3 gene and somatic CSDE1 mutation may drive activation of the Wnt and Hedgehog pathways, and trigger PCPG oncogenesis [8]. In addition to assessing mutations in coding sequences, analysis of somatic copy-number alterations and miRNA profiling are increasingly used to determine sub-clusters in PCPGs [9].
2.1. SDHx
Germline mutations in SDHx are attributed to approximately half of hereditary PCPGs and are detected in 15% of total patients [10]. Germline mutations in SDHx are commonly accompanied by the loss of heterozygosity on the other healthy allele, which leads to substantial loss of SDH catalytic activity [11]. Familial PCPGs, caused by SDHx germline mutations, usually show earlier onsets and more severe clinical presentations (including bilateral or multiple tumors) compared with those observed in sporadic cases [12]. In 2000, SDHC and SDHD were first identified as susceptibility genes for hereditary PCPGs [13,14]. SDHC mutations account for 6% of PCPGs, and patients usually present head and neck paragangliomas (HNPGL), while PHEO and PGL occur far less frequently [15]. SDHD-mutant PCPGs typically show multiple HNPGL, but PGL and PHEO in other locales have also been described; less than 5% of patients with SDHD mutations develop metastatic lesions [15]. Overall, the penetrance of SDHD-mutant PCPGs is approximately 71% at age 60 and increases to 90% in the following 10 years [16]. Germline mutations of SDHD exhibit ‘parent-of-origin’ expression phenotype, with tumor onset only when mutations are inherited from the paternal DNA [17,18]. This phenomenon has also been described in other PCPG predisposition genes such as SDHAF2 and MAX [19,20,21]. In 2001, mutations in SDHB were also discovered in patients with familial PCPG [22]. SDHB-mutant tumors can occur at adrenal, extra-adrenal and pelvic locations, but mainly develop in the abdomen. Several studies demonstrated that compared with other molecular subtypes, SDHB-mutant PCPGs are associated with increased incidence of early onset (25–30 years old), increased metastatic risk and poor prognosis [23]. In 2009, SDHAF2, also known as SDH5, was identified as the driver gene for HNPGL without PHEO, which occurs via compromised flavination of the SDH complex [19]. In patients with familial PCPGs who carry SDHAF2 germline mutations, 91% present with more than one HNPGL, and no metastatic tumors have been reported [24]. Mutations in SDHA have not been identified as a cancer susceptibility gene in PCPG until recently [20]. Approximately 3% of patients with sporadic PCPG carry SDHA germline mutations [25]. Somatic mutations in SDHx are rare and occur in approximately 1% of patients with PCPG [5].
SDHx genes encode succinate dehydrogenase (SDH), also known as mitochondrial complex II. SDH consists of four subunits: SDHA, SDHB, SDHC and SDHD. SDHA is a flavoprotein that contains a flavin adenine dinucleotide (FAD) cofactor. SDHB contains three iron-sulfur clusters, which assist electron transfer via the SDH complex. SDHC and SDHD subunits anchor the entire SDH complex to the inner mitochondrial membrane. Mechanistically, SDHA converts succinate into fumarate, which converts FAD to FADH2. The electrons from FADH2 are then transferred via iron-sulfur clusters in SDHB, eventually forming the ubiquinone pool via SDHC/D subunits. SDH complex plays key roles in energy metabolism by participating in both the Krebs cycle and electron transport chain. Deleterious mutations in SDHx lead to deficiencies in energy metabolism and accumulation of succinate, which promotes susceptibility to PCPGs, renal cell carcinoma and mitochondrial encephalopathy. Studies using in vivo and in vitro models have shown that loss of succinate dehydrogenase activity results in: (i) abnormal activation of hypoxia-signaling pathway in the presence of oxygen (pseudohypoxia) and angiogenesis [26]; (ii) increased production of reactive oxygen species (ROS) [27]; and (iii) impeded repair and hypermethylation of DNA [28]. The distinctive signatures in tumor biology have supplied valuable clues for developing future molecular-targeted therapeutics against SDHx-mutant PCPGs.
2.2. Von Hippel-Lindau (VHL)
Germline mutations in the VHL gene cause the von Hippel-Lindau syndrome (VHL disease). VHL disease is an autosomal dominant disorder associated with retinal, cerebellar, brainstem and spinal hemangioblastoma, as well as with neuroendocrine tumors, renal cell carcinoma (RCC) and multiple pancreatic cysts [29]. PHEO is present in approximately 7–20% of patients with VHL, who are then diagnosed with VHL syndrome type 2; patients diagnosed with type 1 VHL do not present with PHEO [30]. PHEO usually occurs as bilateral or multifocal tumors in the second decade of life in patients with VHL. Although VHL mutations lead to early onset of symptoms, they rarely develop into metastatic disease. In addition to the VHL syndrome, Chuvash polycythemia is a type of inherited hematopoetic disease caused by a specific germline VHL mutation (p.R200W). The mutation leads to activation of the hypoxia inducible factors (HIF) signaling pathway under normal oxygen level and increased concentration of erythropoietin, causing overproduction of red blood cells [31]. Germline VHL pathogenic mutations are also reported in patients with PHEO and polycythemia, causing by stabilized HIF-2α and elevated production of erythropoietin [32].
Approximately 14% of sporadic PCPGs are found in patients carrying somatic VHL mutations, and this is consistently accompanied by the loss of the 3p chromosome [5]. Our previous study has shown that somatic VHL gene mutations are also involved in tumorigenesis in hereditary MEN 2A-associatd PHEO [33]. Somatic VHL mutations play roles in HNPGL by stimulating the HIF-1α/miR-210 pathway [34]. Although the relationship between somatic VHL mutations and prognosis is unclear, different VHL variants may contribute to the differential clinical phenotype and prognosis. In our recent study, we established a VHL knockout mouse model and found that retinal hemangioblastomas are derived from the hemangioblast cell lineage [35].
VHL is a tumor-suppressor gene that is located on chromosome 3p25.3 and encodes the pVHL protein. The pVHL protein functions as an E3 ligase that ubiquitinates its client proteins. For example, pVHL recognizes the hydroxylated HIF-α oxygen-sensing domains (ODD) domain and recruits other components of the E3 ligase complex such as Elongin B, Elongin C, RBx 1 and Cul2. The VHL-Elongin B/C (VBC) complex processes HIF-α for ubiquitination and subsequent proteasomal degradation. Under hypoxic conditions, VHL recognition of HIF-α is compromised due to reduced ODD hydroxylation. HIF-α is then stabilized and initiates transcription of hypoxia-related genes. Pathogenic VHL mutations lead to compromised VBC activity and abnormal oxygen sensing. Consequent transcription of hypoxia-related genes, such as EPO and VEGFA, serves as oncogenic factors for VHL-related symptoms such as hemangioblastomas and PHEO. Moreover, mutations in VHL may disrupt the binding of Elongin C and p53, leading to deregulation of cellular apoptosis and consequent tumorigenesis [36].
2.3. HIF2A
Hypoxia inducible factors (HIFs), transcriptional factors that govern cellular responses to low oxygen, were first described by Semenza in 1995 [37]. HIFs are composed of α and β subunits. The α subunits are nuclear factors that are sensitive to the oxygen level in the microenvironment, whereas β subunits are constitutively expressed and serve as cofactors for HIF-α. Under normoxia, the ODD in HIF-α are rapidly hydroxylated by prolyl hydroxylase, which alters the conformation of HIF-α. Hydroxylated HIF-α is recognized by the VBC complex and is rapidly degraded via the ubiquitin proteasome pathway [38]. Under hypoxic or pseudohypoxic conditions, the function of prolyl hydroxylase is compromised, leading to stabilization and accumulation of HIF-α. HIF-α is then translocated into the nucleus as a heterodimer HIF-β, initiating transcriptional activation of hypoxia-related genes involved in biological reactions such as angiogenesis, glycolysis and erythropoiesis.
Overexpression of HIF-1/2α is frequently identified in most human cancers, and activation of tumorigenesis and angiogenesis [39]. Low oxygen concentration activates the hypoxia-signaling pathway in tumors, especially in regions with minimal oxygen penetration. On the other hand, the hypoxia pathway can also be activated under normoxia due to genetic abnormalities in key regulatory genes of the oxygen-sensing pathway. Elevated expression of HIF-1α is associated with poor outcomes in multiple human cancers such as those of head and neck, breast and colorectal cancers [40]. HIF-2α overexpression is associated with higher metastatic potential and with metastases-presenting tumors such as melanoma and glioma [41]. HIF-2α overexpression may be preferentially linked with metastatic progression and poor prognosis in patients [41].
Mutations in HIF2A have been identified in human diseases such as polycythemia, PCPG and somatostatinoma [42,43]. HIF2A mutations present as somatic mutations or somatic mosaicism, affecting multiple lineages of somatic cells [44]. HIF2A mutations are mainly located on exon 12, resulting in amino-acid substitutions in the ODD domain of HIF-2α. Alterations in peptide sequences lead to compromised prolyl hydroxylation, VBC recognition and transcription of hypoxia-related genes. Accordingly, HIF2A-mutated PHEO/PGLs show increased expression of hypoxia-related genes such as EPO, EDN1 and VEGFA, which may be linked to polycythemia and oncogenesis [43].
2.4. Neurofibromin 1 (NF1)
The NF1 syndrome, also known as von Recklinghausen disease, is caused by germline mutations in NF1. Mutations in NF1 are involved in numerous types of tumors such as desmoplastic melanoma, glioblastomas, neuroblastomas, PCPGs, gastrointestinal tumors, ovarian tumors and urinary tract transitional cell carcinoma [45]. Approximately 0.1–6% of patients with NF1 present with PHEO [46]. Patients with NF1 usually develop PHEO after their third decade of life. Approximately 80% of these patients present solitary adrenal tumors, and 10% present with bilateral adrenal tumors. Additionally, over 10% of patients with NF1 and PHEO develop metastatic tumors, and most of these metastatic tumors are distant from the primary location [47]. A common feature observed in patients with NF1-related PCPGs is a significantly up-regulated level of catecholamine in plasma and urine [48].
Somatic mutations in NF1 occur in 20–25% of patients with PCPGs. NF1 is the most frequently occurring susceptibility gene in all sporadic PCPGs. An integrative genomic study has shown that 26% of sporadically occurring tumors show loss of one allele in NF1. Additionally, 91% of tumors in patients with NF1-related PCPGs show somatic truncating mutations on the other wild-type allele [48]. However, a genetic-mapping study has shown that only 20% of patients with NF1-related PCPGs show deletion of the other allele [5], indicating that other molecular pathways may be involved in NF1-mediated oncogenesis.
NF1 is a tumor-suppressor gene located on chromosome 17q11.2. The NF1 gene spans approximately 300 kb in genomic DNA, contains 58 coding exons and encodes 2818 amino acids. Currently, genetic detection and characterization of NF1 mutations in patients is challenging because of the large size of the NF1 gene, presence of multiple pseudogenes, and a wide spectrum of mutations without obvious hotspots. NF1 encodes neurofibromin, a GTPase-activating protein (GAP) that negatively regulates the Ras/MAPK pathway. The 20 to 27 exons of NF1 encode a GAP-associated domain, which hydrolyses Ras-GTP to its inactive GDP-bound form, thereby deactivating the Ras signaling pathway. Loss-of-function mutations in NF1 lead to uncontrollable activation of kinase and tumorigenesis. Several genetically-engineered NF1 mouse models have shown pigmentary lesions, skeletal abnormalities, and tumors.
2.5. RET
Germline mutations in RET are linked with multiple endocrine neoplasia type 2 (MEN2). MEN2 is a rare autosomal dominant syndrome that is classified into MEN2A (Sipple syndrome), MEN2B (Gorlin syndrome) and familial medullary thyroid carcinoma (FMTC). Patients with MEN2 have a nearly 100% risk for developing medullary thyroid carcinoma (MTC) and 57% risk for developing PHEO [49]. Additionally, patients with MEN2A can also develop primary hyperparathyroidism, while those with MEN2B can develop Marfanoid habitus, mucosal neuromas and ganglioneuromatosis. Although mutations in RET have been detected on all exons, 95% of patients with MEN2A carry RET mutations on exon 10 (codons 609, 611, 618 and 620) or exon 11 (codon 634). Similarly, most mutations in patients with MEN2B occur on exon 16 (codon 918) [50]. The most common RET mutations in PHEO-related syndrome usually occur on exon 10, 11, 13 and 16. However, penetrance and age of onset are not necessarily associated with types of RET mutations [51]. Carriers of codon 634 germline mutations present with much younger mean age of onset, and have a higher risk of developing PHEO, than do carriers of other mutations. In patients with MEN2, most PCPG-related PHEOs occur on the adrenal glands, and more than half of these are bilateral; parasympathetic head and neck PGLs have been found, but are very rare [52]. These patients rarely develop metastatic PCPGs, and mean age of onset is approximately 36 years old [53].
The RET proto-oncogene is located on chromosome 10q11.2 and contains 21 exons. RET encodes transmembrane receptor tyrosine kinase (RTK), which binds to growth factors such as glial derived neurotrophic factor (GDNF). The RET protein contains an extracellular portion, a single transmembrane domain and an intracellular portion. There are 12 autophosphorylation sites on the intracellular portion, and phosphorylated tyrosine may be the docking site for multiple intracellular-signaling pathway proteins, including those involved in cell growth and differentiation [54]. Genetic alterations in RET include gain-of-function mutations, which lead to constitutive RTK activation and tumorigenesis such as those observed in patients with MEN2A.
2.6. MAX
Germline mutations in MAX were first implicated in susceptibility to hereditary PHEO in a whole-exome sequencing study. Loss-of-function mutations in MAX are also a risk for metastatic PHEO [21]. Most of the MAX mutations occur on the highly-conserved basic helix-loop-helix leucine-zipper (bHLHZ) domain. Loss of heterozygosity on the wild-type allele is also detected in the tumors of patients with germline missense mutations in MAX. Although metastatic PHEOs are rare, except in patients carrying SDHB mutations, Mendez found that approximately 37% of patients with MAX mutations present with metastases at diagnosis [21]; this suggests MAX mutations may be risk factors for metastatic disease. Somatic MAX mutations are detected in patients with sporadic PCPGs at an incidence of 1.65% [55]. Tumors with MAX mutations show substantial upregulation of normetanephrine expression, with almost normal or slighted increased levels of metanephrine.
The MAX gene is located on chromosome 14q23, which encodes the transcriptional regulator MAX. MAX belongs to the family of bHLHZ transcriptional factors. It can form heterodimers with MYC or MAX dimerization protein 1 (MXD1), which controls the transcription of numerous downstream genes that regulate cellular proliferation, differentiation and apoptosis [56]. The highly-conserved bHLHZ domain of MAX is vital for the protein-DNA and protein-protein interactions. Furthermore, casein kinase II phosphorylation sites on MAX modulate DNA-binding kinetics of MAX-MAX or Myc-MAX dimerization [57]. Therefore, alteration in MAX, especially mutations on the bHLHZ domain and casein kinase II phosphorylation sites, can induce the dysfunction of the MYC/MAX/MXD1 axis and consequent tumorigenesis.
2.7. Harvey Rat Sarcoma Viral Oncogene Homologue (HRAS)
The first somatic mutation in HRAS in a patient with pheochromocytoma was reported by Yoshimoto et al. in 1992 [58]. Missense gain-of-function mutations in HRAS have been detected in various types of human tumors; the hotspots for HRAS mutations are G13R and Q61K [1]. Until now, HRAS somatic mutations were found in approximately 5% of sporadic patients with PCPGs and present as mostly benign tumors [1]. No germline HRAS mutation has been discovered in patients with PCPGs thus far. The other two proteins in the RAS family, NRAS and KRAS, have never been described as susceptibility factors for PCPGs.
HRAS is located on the chromosome 11p15.5. HRAS encodes GTPase HRas, also known as transforming protein p21. HRas is activated via binding to GTP. The activity of HRas can be inactivated by GTP hydrolysis to GDP [9]. Activation of the HRas signaling pathway stimulates downstream pathways such as Ras/Raf/Erk and PI3K/Akt/mTOR, which are vital for cellular proliferation and oncogenic transformation.
3. Current Therapies and Limitations
The goal of anti-PCPG therapies is to effectively control tumor growth and other disease-related symptoms. Alpha-blockers, calcium channel blockers, or β-blockers are the first line treatment to control hypertension and prevent hypertensive crisis. When β blockers are used without prior alpha blockade, there is a theoretical risk of hypertensive crisis due to alpha adrenergic receptor mediated vasoconstriction without the opposition of the β2-adrenergic receptor mediated vasodilation. For benign and locally invasive PCPGs, surgical intervention, including minimal invasion endoscopic surgery, is considered standard therapy. Laparoscopic surgery can be used for patients with bilateral and extra-adrenal PCPGs, with laparotomy showing similar outcomes. For multifocal and metastatic cases, and for tumors larger than 7 to 8 cm, surgical procedures are usually preferable for ensuring complete removal of all suspected tumors. When surgery is not applicable, radio-and/or chemotherapies are considered alternative approaches. For the metaiodobenzylguanidine (MIBG) scintigraphy-positive patients, 131iodine-meta-iodobenzylguanidine (131I-MIBG) therapy is considered a priority. MIBG positive patients with metastatic PCPG have been demonstrated to benefit from 131I-MIBG-based treatment, showing symptomatic and hormonal responses [59]. However, dose-dependent side effects of this therapy, such as severe thrombocytopenia, hypothyroidism and neutropenia, are also observed [60]. Most importantly, 131I-MIBG-based treatment is less likely to achieve complete response. In a study that included 243 patients, 3% of patients showed complete response, while 27% and 52% of patients showed partial response and stable disease, respectively [61].
Overexpression of somatostatin receptors in PCPGs promotes application of radiolabeled somatostatin agonists, for imaging and treatment of the PCPGs patients. 123I-Tyr-octreotide and 111In-pentetreotide were first introduced as the radiolabeled somatostatin agonists. However, the 90Y and 177Lu peptide-labelled somatostatin radionuclides were recommended by European centers to replace the old ones, due to higher uptake ratio and less side effects [62,63]. Besides, the 90Y is more effective on larger tumors due to higher energy β emission, while the 177Lu is favorable for smaller tumors. Less side effects were also found in 177Lu compared to 90Y, especially in the aspect of renal toxicity [64]. A successful phase III clinical trial NETTER-1 regarding the 177Lu-DOTATATE showed to prolong the median progression-free survival to 40 months in mid-gut neuroendocrine tumors, compared to a long-acting somatostatin analogue, octreotide-LAR (median progression-free survival: 8.4 months) [65]. For inoperable PCPGs patients, the 177Lu-DOTATATE is under a phase II clinical trial to evaluate the safety, tolerability and overall survival (NCT03206060). However, radiolabeled somatostatin agonists are only applied for somatostatin receptor positive patients and side effects still need further evaluation.
Chemotherapy is another valuable treatment modality for controlling tumor growth in patients with metastatic PCPGs. Most traditional chemotherapy regimens, such as those using cyclophosphamide, vincristine and dacarbazine (CVD), have been used to treat patients with PCPGs over the past 30 years. Although clinical studies have shown that 33–57% of patients with PCPGs respond to CVD or similar regimens, a 22 year-long follow-up study found there were no significant differences in patient survival between CVD responders and CVD non-responders. Overall, the present options of chemotherapy do not provide survival benefits for advanced PCPG, and their value remains limited [6].
4. Targeted Molecular Therapies
Current knowledge of signatures involved in the molecular signaling, metabolism and resistance mechanisms of PCPGs suggests that therapeutic regimens can be optimized to each molecular subtype. Profiling of gene expression and methylation can serve as a powerful tool for characterizing disease clusters and for guiding targeted therapy for improved selectivity and efficacy. In the following sections, we introduce the latest advances in targeted therapeutics against PHEO/PGL.
4.1. Antiangiogenic Therapies
Antiangiogenic therapies have been proposed for targeting pseudohypoxic and angiogenic phenotypes in Cluster I PCPGs, which are commonly accompanied by mutations in SDH or VHL [66]. Humanized VEGF-A monoclonal antibodies (such as bevacizumab) and tyrosine kinase inhibitors (such as sunitinib and sorafenib) are used in current antiangiogenic therapies. These regimens are approved by the FDA for the treatment of patients with advanced renal cell carcinoma, which includes patients with mutations in SDHB [67,68]. Interestingly, several case studies on sunitinib have shown partial response or stable disease in patients with Cluster I PCPGs. This indicates that patients with Cluster I PCPGs may show improved responses to antiangiogenic therapies [69,70,71,72,73,74]. Several ongoing clinical trials are aiming to further validate the efficacy of sunitinib-based therapy in patients with progressive PCPGs. For example, a randomized double-blind phase II clinical trial, called the FIRSTMAPPP (First Randomized STudy in MAlignant Progressive Pheochromocytomas and Paragangliomas) study (NCT01371201), is currently conducting recruitment to evaluate the efficacy of sunitinib vs placebo in patients with progressive malignant PCPGs. A single arm, nonrandomized phase II study (NCT00843037) aims to evaluate the response and toxicity profile of sunitinib in a cohort of 25 patients with malignant PCPGs. Another tyrosine kinase inhibitor, Axitinib (AG-013736), is currently under evaluation in a phase II nonrandomized clinical trial including 14 patients with PCPGs (NCT01967576). Moreover, a phase II clinical trial is ongoing to determine the efficacy of Lenvatinib, a multiple kinase inhibitor against VEGFR1, VEGFR2 and VEGFR3 in patients with metastatic or advanced PCPGs (NCT03008369).
4.2. Hypoxia-Inducible Factor (HIF) Inhibitors
The abnormal activation of hypoxia signaling is a hallmark of Cluster I PCPGs. HIF inhibitors may potentially be used in therapy against Cluster I PCPGs. HIF inhibitors, such as PX-12 and PX-478, have been studied in various tumor xenograft models [75,76]. Recently, PT2339 and PT2385, two selective HIF-2α antagonists, were developed and evaluated for their anti-tumor effects. PT2399 showed a stronger suppression effect than that of sunitinib in cell lines derived from VHL-mutated clear cell renal cell carcinomas (ccRCCs) [77]. An ongoing phase I clinical trial (NCT02293980), designed to evaluate the efficacy of PT2385, indicated that complete response, partial response and stable disease were achieved in 2%, 12% and 52% of patients with ccRCCs [78]. A phase II clinical trial (NCT03108066) is currently ongoing to evaluate the use of PT2385 in patients with VHL-associated ccRCCs. These compounds have not been evaluated in patients with PCPGs; however, the tumor-suppressing effects of these compounds on HIF-driven solid tumors are promising, suggesting that HIF-2α inhibitors can be used to treat patients with Cluster I PCPGs in the future. Recently, anthracyclines (daunorubicin, doxorubicin, epirubicin and idarubicin) have been reported to suppress cell growth of metastatic PCPGs by inhibiting both HIF-1 and 2α, indicating a new therapeutic option for patients with metastatic PCPGs, especially those with alterations in HIF pathways [79].
4.3. mTOR Inhibitors
Hyperactivation of kinase activity is commonly detected in the Ras/Raf/Erk or PI3K/Akt/mTOR pathways of patients with Cluster II PCPGs and mutations in RET, NF1, TMEM127 and MAX, [46,55,80,81,82]. Inhibitors of pro-survival kinase signaling have been proposed for targeted therapeutics. For example, treatment with mTORC1 inhibitor everolimus (RAD001) has been evaluated in patients with progressive PHEO. However, this therapy showed unfavorable results, with disease progression in all four recruited patients [83]. In another phase II study (NCT01152827), five out of seven patients with PCPGs achieved stable disease [84]. In 2013, a selective ATP-competitive dual mTORC1/2 small molecule inhibitor was evaluated in a mouse model of sporadic PHEO, and PHEO associated with VHL or SDHB mutations. The results showed promising therapeutic effects of AZD8055, indicated by decreased tumor size and metastatic burden in athymic nude mice [85]. Moreover, combining AZD8055 with an Erk inhibitor AEZS-131 may prevent the compensatory feedback loop and overcome resistance [86].
4.4. DNA Demethylation
Mutations in SDHx result in accumulation of succinate, an oncometabolite that inhibits 2-oxoglutarate (2-OG)-dependent dioxygenases, resulting in a global DNA and histone hypermethylation phenotype [87,88]. Demethylating agents may rectify the hypermethylation phenotype in SDH- or FH-mutated PCPGs. For example, DNA-demethylating agent decitabine suppresses cellular proliferation and metastasis in SDHB-knockout chromaffin cells [87]. SGI-110, a DNA methyltransferase inhibitor, is currently under investigation in a phase II non-randomized trial (NCT03165721) for treatment of patients with PCPGs associated with SDH deficiency. Further preclinical studies are needed to assess the safety profile and therapeutic efficacy of these compounds before proceeding to clinical trials.
4.5. DNA-Alkylating Agents
Temozolomide (TMZ) is an FDA-approved DNA-alkylating agent used for treatment of glioblastoma in combination with radiotherapy. TMZ generates DNA alkylation at O6-guanine, N7-guanine and N3-adenine, which causes base-pair mismatch and leads to the death of tumor cells. In some tumor cells, the expression of O6-methylguanine-DNA methyltransferase (MGMT) can directly remove the alkyl group from O6-guanine, resulting in resistance to TMZ. However, tumors with mutations in genes encoding Krebs-cycle enzymes, such as IDH1/2 and SDHx, often show CpG island methylator phenotype (CIMP), which results in hypermethylation of the MGMT promoter and reduced expression of MGMT [87,89]. Loss of MGMT expression predisposes patients to a better therapeutic response to TMZ because of reduced methyltransferase activity. Several studies have shown the remarkable sensitivity of IDH1/2-mutant glioblastoma to TMZ [90,91]. Similarly, TMZ exerts strong therapeutic effect on metastatic neuroendocrine carcinoma, especially that with mutations in SDHB [92]. A phase II clinical trial (NCT00165230) is currently evaluating the efficacy of TMZ combined with thalidomide in therapy against neuroendocrine tumors. Additionally, one out of three patients with PCPGs shows response to radiotherapy [93]. Clinical trials with larger patient cohorts are needed to further evaluate the efficacy of TMZ in patients with PCPGs.
4.6. PARP Inhibitors
Mutations in enzymes encoding Krebs-cycle enzymes, such as SDHx, are associated with hereditary PCPGs that are characterized by increased level of succinate. High level of succinate serves as an intrinsic inhibitor of homologous recombination (HR)-based DNA repair; this occurs via inhibition of the lysine demethylases KDM4A and KDM4B [94]. Moreover, SDH deficiency in Cluster I PCPGs is associated with alterations in NAD+/NADH metabolism and potentiation of the PARP-mediated DNA repair pathways [95]. These findings indicate that SDH-deficient tumor cells are highly sensitive to treatment with PARP inhibitors. Combinations of PARP inhibitors with other genotoxic agents may be a promising approach for treating patients with Cluster I PCPGs. Olaparib, an FDA-proved PARP inhibitor, markedly potentiates the therapeutic effect of TMZ in SDHB-mutant preclinical models; this occurs via induction of DNA lesions and inhibition of tumor growth in vitro and in vivo [94,95].
4.7. Histone Deacetylase Inhibitors
Histone deacetylase (HDAC) inhibitors were also reported to have anti-tumor effect in PCPGs. HDAC inhibitors have been shown to induce cell cycle arrest and apoptosis in PCPGs through activation of Notch1 signaling or inhibition of nuclear factor erythroid 2-related factor 2/heme oxygenase 1(Nrf2/HO-1) pathway [96,97,98,99]. Additionally, our previous study demonstrated that HDAC inhibitors improved the stability of SDHB protein, and therefore supported the function of mitochondrial complex II, which might limit disease progression of PCPGs with SDHB deficiency [100].
4.8. Immunotherapy
The pseudo-hypoxia phenotype may alter the immune system through inactivation of cytotoxic T-cell lymphocytes, activation of immune-suppressive monocytes and increased expression of the immune checkpoint protein programmed death-ligand 1 (PD-L1) and its receptor [101,102,103]. Thus, immunotherapy has been considered as a candidate therapeutic approach for Cluster I PCPGs. A study of 14 patients with progressive metastatic PCPGs treated with interferon alpha-2b resulted in 12 patients with disease stabilization and two with partial responses [104]. Two phase II clinical trials of checkpoint inhibitors (Nivolumab, ipilimumab and pembrolizumab) are currently ongoing in patients with rare tumors, including metastasis PCPGs (NCT02834013, NCT02721732).
4.9. Other Potential Therapies
Our previously study indicated that cells with high baseline level of reactive oxygen species (ROS), such as IDH-mutated glioma, dependency on antioxidative pathways are crucial to maintain ROS homeostasis. Blockade of antioxidative pathways showed promising therapeutic effects in IDH-mutated cancers [105]. Similarly, evidence has shown that the deficiency in SDH and accumulation of succinate may lead to elevated generation of ROS [27,106]. Our recent data indicated that SDHB deficient PCPG cells developed addiction to the Nrf2 antioxidative pathway and Nrf2 blockade might be a novel therapeutic approach to this type of PCPGs.
5. Future Directions
Despite our increased understanding of PCPG biology and advancements in translational medicine, the underlying pathogenetic mechanisms and molecular pathways of PCPG require further investigation. Cell-based and preclinical mouse models do not fully recapitulate the molecular subtypes of human cancers, posing a challenge in studies on PCPG. PCPG cell lines, derived from heterozygous NF1-knockout mouse (MTT and MPC cells) and representative rat pheochromocytoma (PC12 cells), are widely accepted and used in molecular biology studies; however, generating cell lines from patient-derived PCPGs remains challenging. Currently, only one progenitor cell line (hPheo1 cells) derived from a human pheochromocytoma tumor has been established successfully [107]. This illustrates an urgent need to develop patient-derived cell lines, especially those for modeling Cluster I PCPG in vitro.
With the rapid development of genomic sequencing platforms, large-scale sequencing projects have illustrated the genomics, methylomics and epigenenomic changes of PCPG. The concept of personalized medicine has been brought into vision to treating individuals based on their specific genetic and micro-environmental background. By altering specific signaling pathways, enzymes and receptors, targeted therapies can be optimized for each individual, with reduced side effects with respect to normal tissues. To this end, tumoral genetic and molecular profiles should be investigated in future clinical studies and trials. On the other hand, continuous investigation of molecular mechanisms involved in PCPG oncogenesis is highly important. Detailed understanding of PCPG genetics and key oncogenic pathways will lead to novel therapeutic targets. Overall, understanding the genetic background, developing effective molecular-targeted agents and optimizing the design of clinical trials will improve prognosis and survival in patients with PCPG.
6. Conclusions
In this review, we briefly summarized the latest knowledge of PCPG molecular subtypes and their implications to clinical management. PCPGs generate tumors with genetic alterations; therefore, detailed genetic analysis should be recommended for all patients with PCPGs to better characterize the potential therapeutic vulnerabilities in each case. Therapeutic regimens with long-term efficacy are needed to improve patient survival and quality of life. Development of patient-derived cell lines and disease-relevant preclinical animal models will generate novel therapeutic targets for future management of PCPGs.
Funding
This study was supported by the Neuro-Oncology Branch, Center for Cancer Research, National Cancer Institute and Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda MD USA 20892.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crona, J.; Delgado Verdugo, A.; Maharjan, R.; Stalberg, P.; Granberg, D.; Hellman, P.; Bjorklund, P. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J. Clin. Endocrinol. Metab. 2013, 98, E1266–E1271. [Google Scholar] [PubMed]
- Luchetti, A.; Walsh, D.; Rodger, F.; Clark, G.; Martin, T.; Irving, R.; Sanna, M.; Yao, M.; Robledo, M.; Neumann, H.P.; et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int. J. Endocrinol. 2015, 2015, 138573. [Google Scholar] [CrossRef] [PubMed]
- King, K.S.; Prodanov, T.; Kantorovich, V.; Fojo, T.; Hewitt, J.K.; Zacharin, M.; Wesley, R.; Lodish, M.; Raygada, M.; Gimenez-Roqueplo, A.P.; et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: Significant link to SDHB mutations. J. Clin. Oncol. 2011, 29, 4137–4142. [Google Scholar] [CrossRef] [PubMed]
- Erlic, Z.; Neumann, H.P. Familial pheochromocytoma. Hormones (Athens) 2009, 8, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libe, R.; de Reynies, A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Abraham, J.; Hung, E.; Averbuch, S.; Merino, M.; Steinberg, S.M.; Pacak, K.; Fojo, T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: Recommendation from a 22-year follow-up of 18 patients. Cancer 2008, 113, 2020–2028. [Google Scholar] [CrossRef]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Castro-Vega, L.J.; Letouze, E.; Burnichon, N.; Buffet, A.; Disderot, P.H.; Khalifa, E.; Loriot, C.; Elarouci, N.; Morin, A.; Menara, M.; et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6044. [Google Scholar] [CrossRef]
- Gill, A.J.; Benn, D.E.; Chou, A.; Clarkson, A.; Muljono, A.; Meyer-Rochow, G.Y.; Richardson, A.L.; Sidhu, S.B.; Robinson, B.G.; Clifton-Bligh, R.J. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2010, 41, 805–814. [Google Scholar] [CrossRef]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Vicha, A.; Taieb, D.; Pacak, K. Current views on cell metabolism in SDHx-related pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2014, 21, R261–277. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef]
- Jafri, M.; Maher, E.R. The genetics of phaeochromocytoma: Using clinical features to guide genetic testing. Eur. J. Endocrinol. 2012, 166, 151–158. [Google Scholar] [CrossRef]
- Pasini, B.; Stratakis, C.A. SDH mutations in tumorigenesis and inherited endocrine tumours: Lesson from the phaeochromocytoma-paraganglioma syndromes. J. Intern. Med. 2009, 266, 19–42. [Google Scholar] [CrossRef]
- Bayley, J.P.; Oldenburg, R.A.; Nuk, J.; Hoekstra, A.S.; van der Meer, C.A.; Korpershoek, E.; McGillivray, B.; Corssmit, E.P.; Dinjens, W.N.; de Krijger, R.R.; et al. Paraganglioma and pheochromocytoma upon maternal transmission of SDHD mutations. BMC Med. Genet. 2014, 15, 111. [Google Scholar] [CrossRef]
- Van der Mey, A.G.; Maaswinkel-Mooy, P.D.; Cornelisse, C.J.; Schmidt, P.H.; van de Kamp, J.J. Genomic imprinting in hereditary glomus tumours: Evidence for new genetic theory. Lancet 1989, 2, 1291–1294. [Google Scholar] [CrossRef]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef]
- Burnichon, N.; Briere, J.J.; Libe, R.; Vescovo, L.; Riviere, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Benit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [Green Version]
- Comino-Mendez, I.; Gracia-Aznarez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.J.; Leton, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef]
- Timmers, H.J.; Kozupa, A.; Eisenhofer, G.; Raygada, M.; Adams, K.T.; Solis, D.; Lenders, J.W.; Pacak, K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 779–786. [Google Scholar] [CrossRef]
- Kunst, H.P.; Rutten, M.H.; de Monnink, J.P.; Hoefsloot, L.H.; Timmers, H.J.; Marres, H.A.; Jansen, J.C.; Kremer, H.; Bayley, J.P.; Cremers, C.W. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin. Cancer Res. 2011, 17, 247–254. [Google Scholar] [CrossRef]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.P.; et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–1476. [Google Scholar] [CrossRef]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.H.; Fan, T.W.; Lane, A.N.; Sourbier, C.; Singh, A.; et al. SDHB-Deficient Cancers: The Role of Mutations That Impair Iron Sulfur Cluster Delivery. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef]
- de Cubas, A.A.; Korpershoek, E.; Inglada-Perez, L.; Letouze, E.; Curras-Freixes, M.; Fernandez, A.F.; Comino-Mendez, I.; Schiavi, F.; Mancikova, V.; Eisenhofer, G.; et al. DNA Methylation Profiling in Pheochromocytoma and Paraganglioma Reveals Diagnostic and Prognostic Markers. Clin. Cancer Res. 2015, 21, 3020–3030. [Google Scholar] [CrossRef] [Green Version]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Friedrich, C.A. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum. Mol. Genet. 2001, 10, 763–767. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capodimonti, S.; Teofili, L.; Martini, M.; Cenci, T.; Iachininoto, M.G.; Nuzzolo, E.R.; Bianchi, M.; Murdolo, M.; Leone, G.; Larocca, L.M. Von hippel-lindau disease and erythrocytosis. J. Clin. Oncol. 2012, 30, e137–e139. [Google Scholar] [CrossRef]
- Koch, C.A.; Huang, S.C.; Zhuang, Z.; Stolle, C.; Stolle, C.; Azumi, N.; Chrousos, G.P.; Vortmeyer, A.O.; Pacak, K. Somatic VHL gene deletion and point mutation in MEN 2A-associated pheochromocytoma. Oncogene 2002, 17, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Merlo, A.; de Quiros, S.B.; de Santa-Maria, I.S.; Pitiot, A.S.; Balbin, M.; Astudillo, A.; Scola, B.; Aristegui, M.; Quer, M.; Suarez, C.; et al. Identification of somatic VHL gene mutations in sporadic head and neck paragangliomas in association with activation of the HIF-1alpha/miR-210 signaling pathway. J. Clin. Endocrinol. Metab. 2013, 98, E1661–E1666. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shepard, M.J.; Zhang, C.; Dong, L.; Walker, D.; Guedez, L.; Park, S.; Wang, Y.; Chen, S.; Pang, Y.; et al. Deletion of the von Hippel-Lindau Gene in Hemangioblasts Causes Hemangioblastoma-like Lesions in Murine Retina. Cancer Res. 2018, 78, 1266–1274. [Google Scholar] [CrossRef]
- Roe, J.S.; Kim, H.; Lee, S.M.; Kim, S.T.; Cho, E.J.; Youn, H.D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 2006, 22, 395–405. [Google Scholar] [CrossRef]
- Semenza, G.L.; Rue, E.A.; Iyer, N.V.; Pang, M.G.; Kearns, W.G. Assignment of the hypoxia-inducible factor 1alpha gene to a region of conserved synteny on mouse chromosome 12 and human chromosome 14q. Genomics 1996, 34, 437–439. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Amar, L.; Baudin, E.; Burnichon, N.; Peyrard, S.; Silvera, S.; Bertherat, J.; Bertagna, X.; Schlumberger, M.; Jeunemaitre, X.; Gimenez-Roqueplo, A.P.; et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 3822–3828. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Percy, M.J.; Furlow, P.W.; Lucas, G.S.; Li, X.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N. Engl. J. Med. 2008, 358, 162–168. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef]
- Yang, C.; Hong, C.S.; Prchal, J.T.; Balint, M.T.; Pacak, K.; Zhuang, Z. Somatic mosaicism of EPAS1 mutations in the syndrome of paraganglioma and somatostatinoma associated with polycythemia. Hum. Genome. Var. 2015, 2, 15053. [Google Scholar] [CrossRef] [PubMed]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Bausch, B.; Borozdin, W.; Mautner, V.F.; Hoffmann, M.M.; Boehm, D.; Robledo, M.; Cascon, A.; Harenberg, T.; Schiavi, F.; Pawlu, C.; et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 2007, 92, 2784–2792. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Welander, J.; Larsson, C.; Backdahl, M.; Hareni, N.; Sivler, T.; Sivler, T.; Brauckhoff, M.; Soderkvist, P.; Gimm, O. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum. Mol. Genet. 2012, 21, 5406–5416. [Google Scholar] [CrossRef] [Green Version]
- Correia, M.J.; Lopes, L.O.; Bugalho, M.J.; Cristina, L.; Santos, A.I.; Bordalo, A.D.; Pinho, B.; da Silva, H.L.; Goncalves, M.D.; Ribeiro, C.; et al. Multiple endocrine neoplasia type 2A. Study of a family. Rev. Port. Cardiol. 2000, 19, 11–31. [Google Scholar]
- Bergsland, E.K. The evolving landscape of neuroendocrine tumors. Semin. Oncol. 2013, 40, 4–22. [Google Scholar] [CrossRef]
- Machens, A.; Brauckhoff, M.; Holzhausen, H.J.; Thanh, P.N.; Lehnert, H.; Dralle, H. Codon-specific development of pheochromocytoma in multiple endocrine neoplasia type 2. J. Clin. Endocrinol. Metab. 2005, 90, 3999–4003. [Google Scholar] [CrossRef]
- Boedeker, C.C.; Erlic, Z.; Richard, S.; Kontny, U.; Gimenez-Roqueplo, A.P.; Cascon, A.; Robledo, M.; de Campos, J.M.; van Nederveen, F.H.; de Krijger, R.R.; et al. Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J. Clin. Endocrinol. Metab. 2009, 94, 1938–1944. [Google Scholar] [CrossRef]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef]
- Plaza-Menacho, I.; Barnouin, K.; Goodman, K.; Martinez-Torres, R.J.; Borg, A.; Murray-Rust, J.; Mouilleron, S.; Knowles, P.; McDonald, N.Q. Oncogenic RET kinase domain mutations perturb the autophosphorylation trajectory by enhancing substrate presentation in trans. Mol. Cell 2014, 53, 738–751. [Google Scholar] [CrossRef]
- Burnichon, N.; Cascon, A.; Schiavi, F.; Morales, N.P.; Comino-Mendez, I.; Abermil, N.; Inglada-Perez, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Bousset, K.; Henriksson, M.; Luscher-Firzlaff, J.M.; Litchfield, D.W.; Luscher, B. Identification of casein kinase II phosphorylation sites in Max: Effects on DNA-binding kinetics of Max homo- and Myc/Max heterodimers. Oncogene 1993, 8, 3211–3220. [Google Scholar]
- Yoshimoto, K.; Iwahana, H.; Fukuda, A.; Sano, T.; Katsuragi, K.; Kinoshita, M.; Saito, S.; Itakura, M. ras mutations in endocrine tumors: Mutation detection by polymerase chain reaction-single strand conformation polymorphism. Jpn. J. Cancer Res. 1992, 83, 1057–1062. [Google Scholar] [CrossRef]
- Safford, S.D.; Coleman, R.E.; Gockerman, J.P.; Moore, J.; Feldman, J.M.; Leight, G.S., Jr.; Tyler, D.S.; Olson, J.A., Jr. Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery 2003, 134, 956–962. [Google Scholar] [CrossRef]
- Fitzgerald, P.A.; Goldsby, R.E.; Huberty, J.P.; Price, D.C.; Hawkins, R.A.; Veatch, J.J.; Dela Cruz, F.; Jahan, T.M.; Linker, C.A.; Damon, L.; et al. Malignant pheochromocytomas and paragangliomas: A phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG). Ann. NY Acad. Sci. 2006, 1073, 465–490. [Google Scholar] [CrossRef]
- van Hulsteijn, L.T.; Niemeijer, N.D.; Dekkers, O.M.; Corssmit, E.P. (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. (Oxf) 2014, 80, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.; Jermann, E.; Behe, M.; Goetze, M.; Bucher, H.C.; Roser, H.W.; Heppeler, A.; Mueller-Brand, J.; Maecke, H.R. DOTATOC: A powerful new tool for receptor-mediated radionuclide therapy. Eur. J. Nucl. Med. 1997, 24, 792–795. [Google Scholar] [PubMed]
- Kwekkeboom, D.J.; Bakker, W.H.; Kam, B.L.; Teunissen, J.J.; Kooij, P.P.; de Herder, W.W.; Feelders, R.A.; van Eijck, C.H.; de Jong, M.; Srinivasan, A.; et al. Treatment of patients with gastro-entero-pancreatic (GEP) tumours with the novel radiolabelled somatostatin analogue [177Lu-DOTA(0),Tyr3]octreotate. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Mueller-Brand, J.; Baum, R.P.; Pavel, M.E.; Horsch, D.; O–Dorisio, M.S.; O–Dorisio, T.M.; Howe, J.R.; Cremonesi, M.; Kwekkeboom, D.J.; et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 800–816. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Igaz, P.; Burnichon, N.; Amar, L.; Libe, R.; Badoual, C.; Tissier, F.; Bertherat, J.; Plouin, P.F.; Jeunemaitre, X.; et al. Rationale for anti-angiogenic therapy in pheochromocytoma and paraganglioma. Endocr. Pathol. 2012, 23, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nose, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef]
- Tuthill, M.; Barod, R.; Pyle, L.; Cook, T.; Chew, S.; Gore, M.; Maxwell, P.; Eisen, T. A report of succinate dehydrogenase B deficiency associated with metastatic papillary renal cell carcinoma: Successful treatment with the multi-targeted tyrosine kinase inhibitor sunitinib. BMJ Case Rep. 2009, 2009. [Google Scholar] [CrossRef]
- Joshua, A.M.; Ezzat, S.; Asa, S.L.; Evans, A.; Broom, R.; Freeman, M.; Knox, J.J. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J. Clin. Endocrinol. Metab. 2009, 94, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Hahn, N.M.; Reckova, M.; Cheng, L.; Baldridge, L.A.; Cummings, O.W.; Sweeney, C.J. Patient with malignant paraganglioma responding to the multikinase inhibitor sunitinib malate. J. Clin. Oncol. 2009, 27, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Cabanillas, M.E.; Santarpia, L.; Jonasch, E.; Kyle, K.L.; Lano, E.A.; Matin, S.F.; Nunez, R.F.; Perrier, N.D.; Phan, A.; et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: Targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J. Clin. Endocrinol. Metab. 2009, 94, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, K.; Miura, T.; Shioji, G.; Tsuboi, N. Sunitinib treatment for refractory malignant pheochromocytoma. Neuro. Endocrinol. Lett. 2012, 33, 260–264. [Google Scholar] [PubMed]
- Ayala-Ramirez, M.; Chougnet, C.N.; Habra, M.A.; Palmer, J.L.; Leboulleux, S.; Cabanillas, M.E.; Caramella, C.; Anderson, P.; Al Ghuzlan, A.; Waguespack, S.G.; et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 2012, 97, 4040–4050. [Google Scholar] [CrossRef]
- Welsh, S.J.; Williams, R.R.; Birmingham, A.; Newman, D.J.; Kirkpatrick, D.L.; Powis, G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol. Cancer Ther. 2003, 2, 235–243. [Google Scholar] [PubMed]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z.; et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313–22324. [Google Scholar] [CrossRef] [Green Version]
- Burnichon, N.; Lepoutre-Lussey, C.; Laffaire, J.; Gadessaud, N.; Molinie, V.; Hernigou, A.; Plouin, P.F.; Jeunemaitre, X.; Favier, J.; Gimenez-Roqueplo, A.P. A novel TMEM127 mutation in a patient with familial bilateral pheochromocytoma. Eur. J. Endocrinol. 2011, 164, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Attie, T.; Pelet, A.; Edery, P.; Eng, C.; Mulligan, L.M.; Amiel, J.; Boutrand, L.; Beldjord, C.; Nihoul-Fekete, C.; Munnich, A.; et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum. Mol. Genet. 1995, 4, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell. Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Druce, M.R.; Kaltsas, G.A.; Fraenkel, M.; Gross, D.J.; Grossman, A.B. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: Experience with the mTOR inhibitor everolimus (RAD001). Horm. Metab. Res. 2009, 41, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Kim, T.W.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.K.; Lee, H.J.; Lee, K.W.; Bang, Y.J. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giubellino, A.; Bullova, P.; Nolting, S.; Turkova, H.; Powers, J.F.; Liu, Q.; Guichard, S.; Tischler, A.S.; Grossman, A.B.; Pacak, K. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: In vitro and in vivo studies in female athymic nude mice. Endocrinology 2013, 154, 646–655. [Google Scholar] [CrossRef]
- Matro, J.; Giubellino, A.; Pacak, K. Current and future therapeutic approaches for metastatic pheochromocytoma and paraganglioma: Focus on SDHB tumors. Horm. Metab. Res. 2013, 45, 147–153. [Google Scholar] [CrossRef]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I., Jr.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- SongTao, Q.; Lei, Y.; Si, G.; YanQing, D.; HuiXia, H.; XueLin, Z.; LanXiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulke, M.H.; Stuart, K.; Enzinger, P.C.; Ryan, D.P.; Clark, J.W.; Muzikansky, A.; Vincitore, M.; Michelini, A.; Fuchs, C.S. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J. Clin. Oncol. 2006, 24, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NAD(+)/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, Z.; Zhan, Y.; Li, H.; Wu, S. Role of histone acetylation in activation of nuclear factor erythroid 2-related factor 2/heme oxygenase 1 pathway by manganese chloride. Toxicol. Appl. Pharmacol. 2017, 336, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Li, Q.Z.; Chen, L.; Chen, B.D.; Zhang, C.; Wang, X.; Li, W.P. HPOB, an HDAC6 inhibitor, attenuates corticosterone-induced injury in rat adrenal pheochromocytoma PC12 cells by inhibiting mitochondrial GR translocation and the intrinsic apoptosis pathway. Neurochem. Int. 2016, 99, 239–251. [Google Scholar] [CrossRef]
- Cayo, M.A.; Cayo, A.K.; Jarjour, S.M.; Chen, H. Sodium butyrate activates Notch1 signaling, reduces tumor markers, and induces cell cycle arrest and apoptosis in pheochromocytoma. Am. J. Transl. Res. 2009, 1, 178–183. [Google Scholar]
- Adler, J.T.; Hottinger, D.G.; Kunnimalaiyaan, M.; Chen, H. Histone deacetylase inhibitors upregulate Notch-1 and inhibit growth in pheochromocytoma cells. Surgery 2008, 144, 956–961. [Google Scholar] [CrossRef]
- Yang, C.; Matro, J.C.; Huntoon, K.M.; Ye, D.Y.; Huynh, T.T.; Fliedner, S.M.; Breza, J.; Zhuang, Z.; Pacak, K. Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J. 2012, 26, 4506–4516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1alpha driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef]
- Labiano, S.; Palazon, A.; Bolanos, E.; Azpilikueta, A.; Sanchez-Paulete, A.R.; Morales-Kastresana, A.; Quetglas, J.I.; Perez-Gracia, J.L.; Gurpide, A.; Rodriguez-Ruiz, M.; et al. Hypoxia-induced soluble CD137 in malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology 2016, 5, e1062967. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Terroir, M.; Leboulleux, S.; Deschamps, F.; Al Ghuzlan, A.; Hescot, S.; Tselikas, L.; Borget, I.; Caramella, C.; Deandreis, D.; et al. Interferon-alpha Treatment for Disease Control in Metastatic Pheochromocytoma/Paraganglioma Patients. Horm. Cancer 2017, 8, 330–337. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Ghayee, H.K.; Bhagwandin, V.J.; Stastny, V.; Click, A.; Ding, L.H.; Mizrachi, D.; Zou, Y.S.; Chari, R.; Lam, W.L.; Bachoo, R.M.; et al. Progenitor cell line (hPheo1) derived from a human pheochromocytoma tumor. PLoS ONE 2013, 8, e65624. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic illustrations of cancer-associated mutations in pheochromocytomas and paragangliomas (PCPGs). Cluster I PCPGs exhibit dysfunction in the Krebs cycle and hypoxia sensing pathways. Loss-of-function mutations in SDHx, FH, EGLN1 or VHL are commonly identified in this disease cluster. HIF2A mutations that activate hypoxia signaling are also found in Cluster I disease. Cluster II PCPGs exhibit abnormal kinase activity. This is caused by mutations of major regulators in the feedback loop, such as NF1, MAX and TMEM127. Gain-of-function mutations in RET prompt cellular proliferation and survival by initiating kinase pathways such as Ras/MEK and PI3K/Akt.
Figure 1.
Schematic illustrations of cancer-associated mutations in pheochromocytomas and paragangliomas (PCPGs). Cluster I PCPGs exhibit dysfunction in the Krebs cycle and hypoxia sensing pathways. Loss-of-function mutations in SDHx, FH, EGLN1 or VHL are commonly identified in this disease cluster. HIF2A mutations that activate hypoxia signaling are also found in Cluster I disease. Cluster II PCPGs exhibit abnormal kinase activity. This is caused by mutations of major regulators in the feedback loop, such as NF1, MAX and TMEM127. Gain-of-function mutations in RET prompt cellular proliferation and survival by initiating kinase pathways such as Ras/MEK and PI3K/Akt.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pang, Y.; Liu, Y.; Pacak, K.; Yang, C. Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers 2019, 11, 436. https://doi.org/10.3390/cancers11040436
AMA Style
Pang Y, Liu Y, Pacak K, Yang C. Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers. 2019; 11(4):436. https://doi.org/10.3390/cancers11040436
Chicago/Turabian StylePang, Ying, Yang Liu, Karel Pacak, and Chunzhang Yang. 2019. "Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies" Cancers 11, no. 4: 436. https://doi.org/10.3390/cancers11040436
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.