Anti-Invasion and Antiangiogenic Effects of Stellettin B through Inhibition of the Akt/Girdin Signaling Pathway and VEGF in Glioblastoma Cells

 , , , and
, , , and 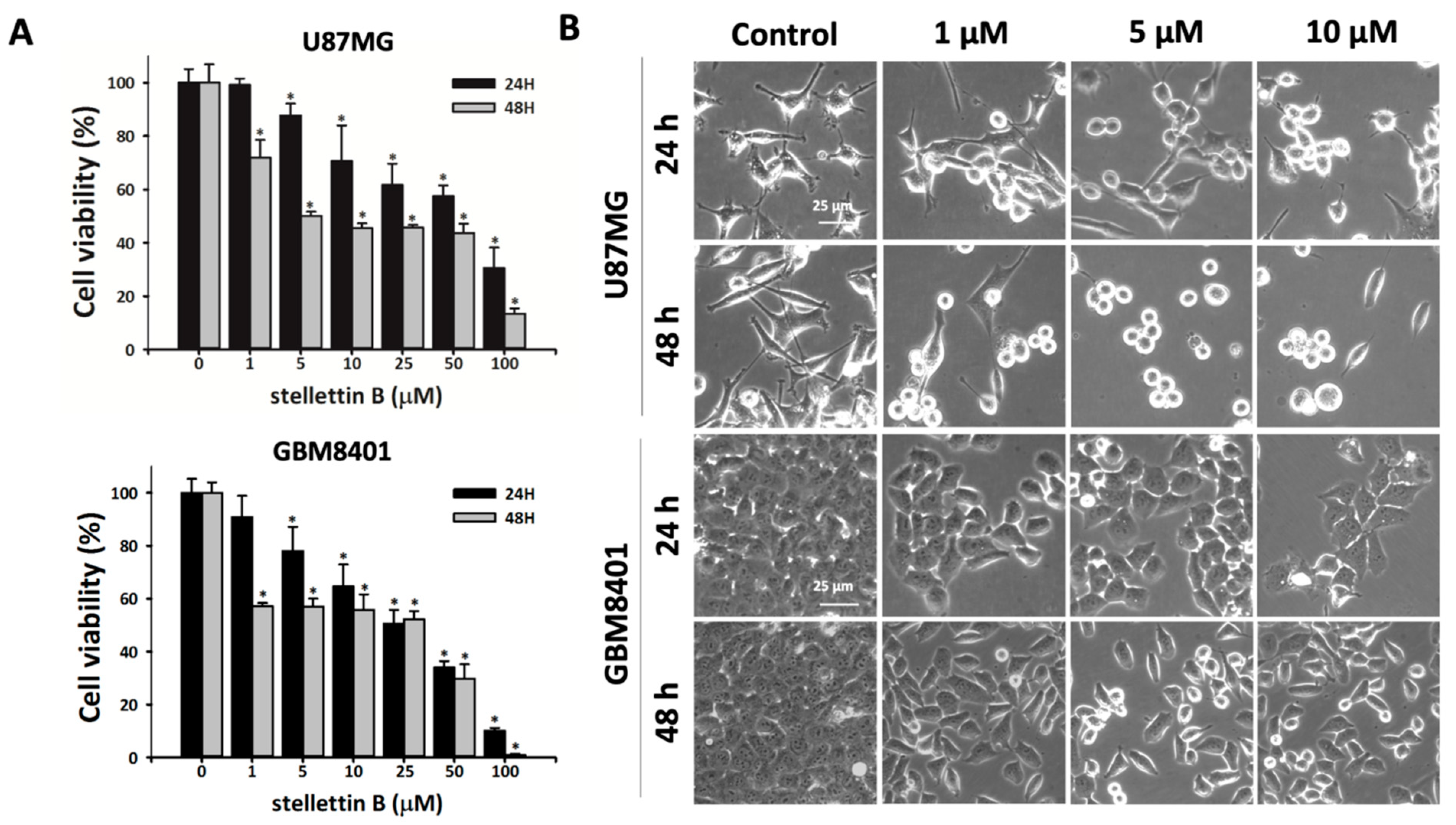{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cell Death Rate and Morphological Changes in Stellettin B–Treated Human Glioblastoma Cells
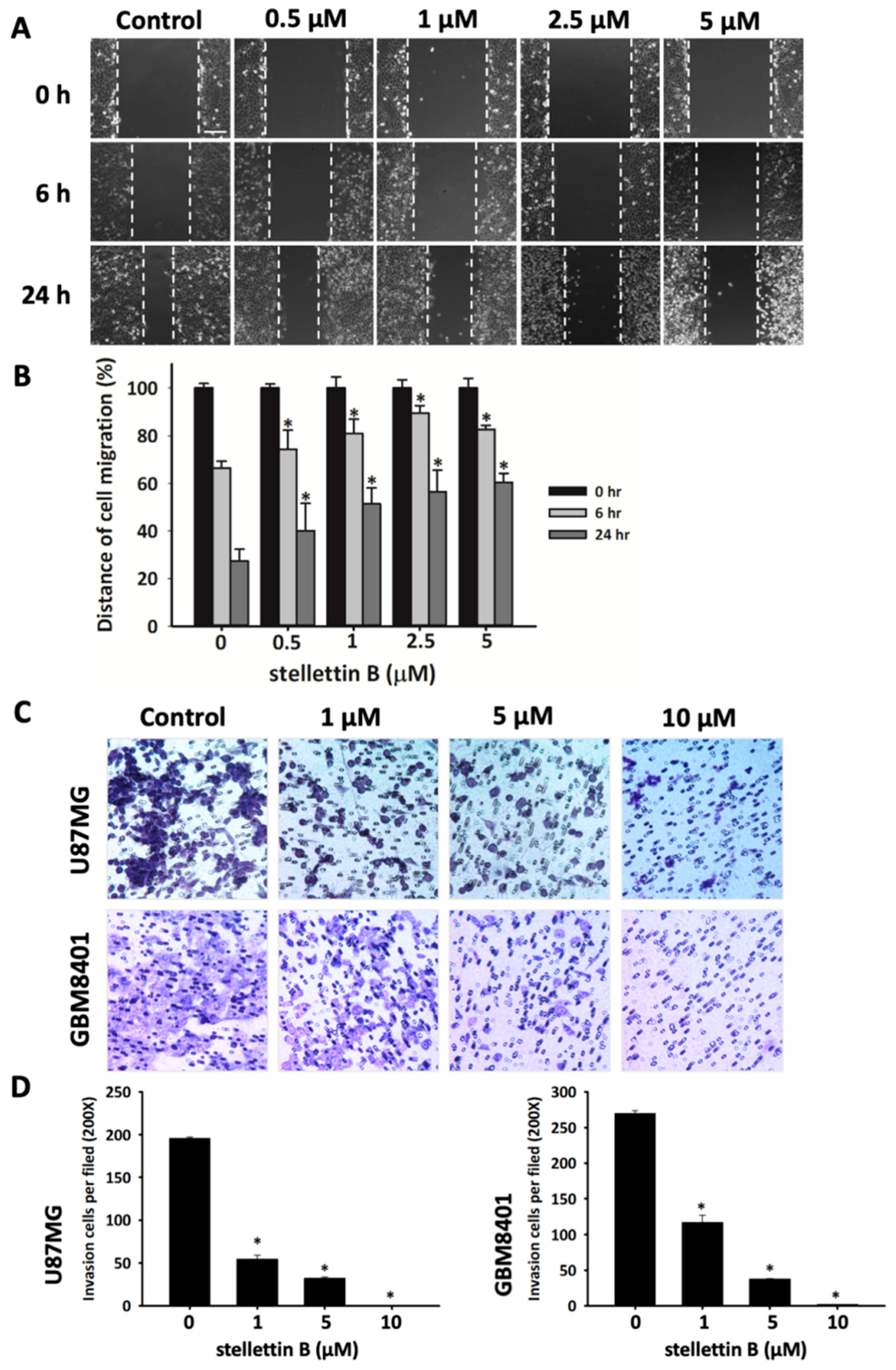
2.2. Stellettin B Suppresses Migration in Glioblastoma Cells
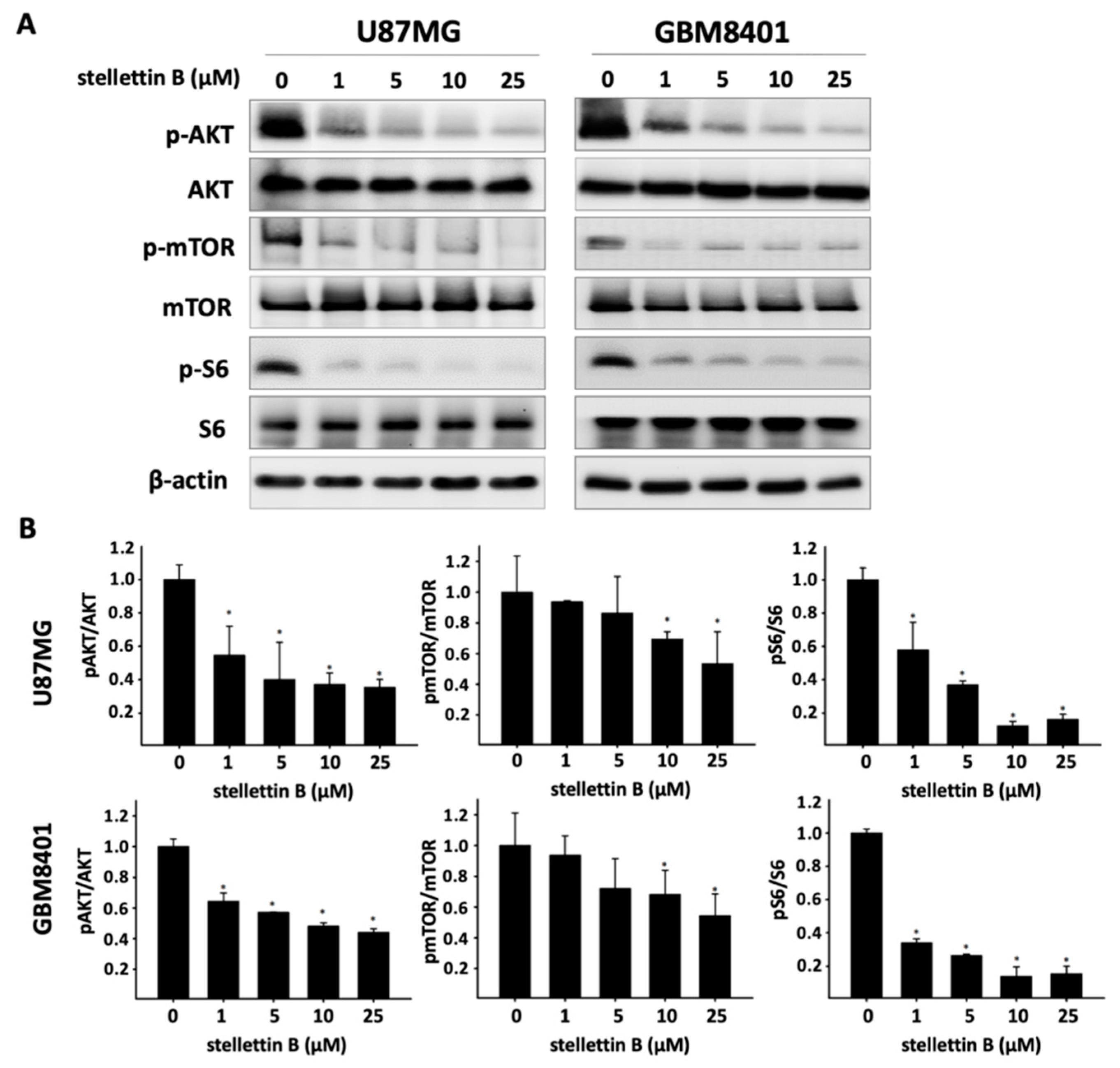
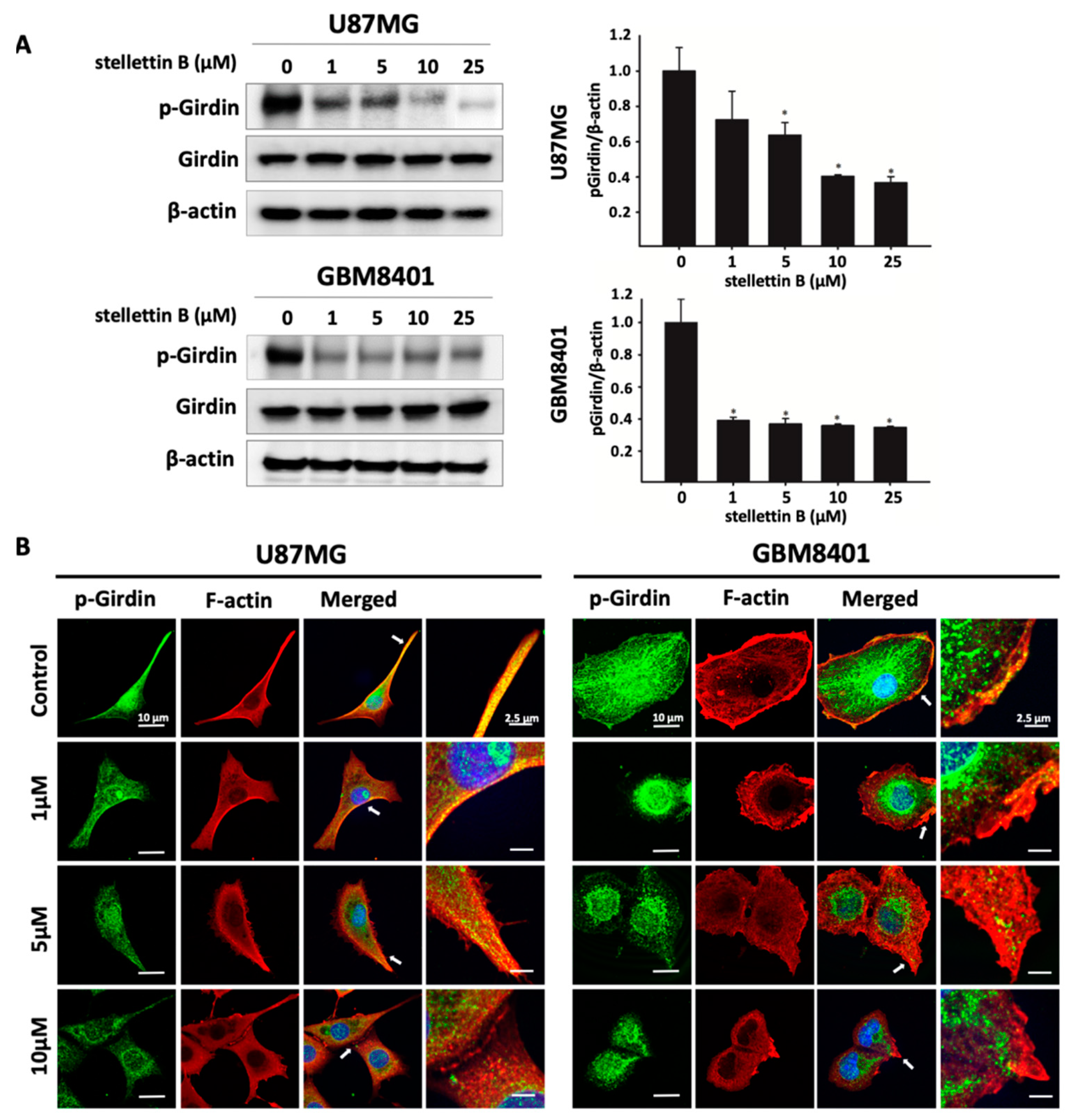
2.3. Stellettin B Suppresses Akt/mTOR/Girdin Signaling and Affects Cell Movement in p-Girdin/F-Actin Interaction in Glioblastoma Cell Lines
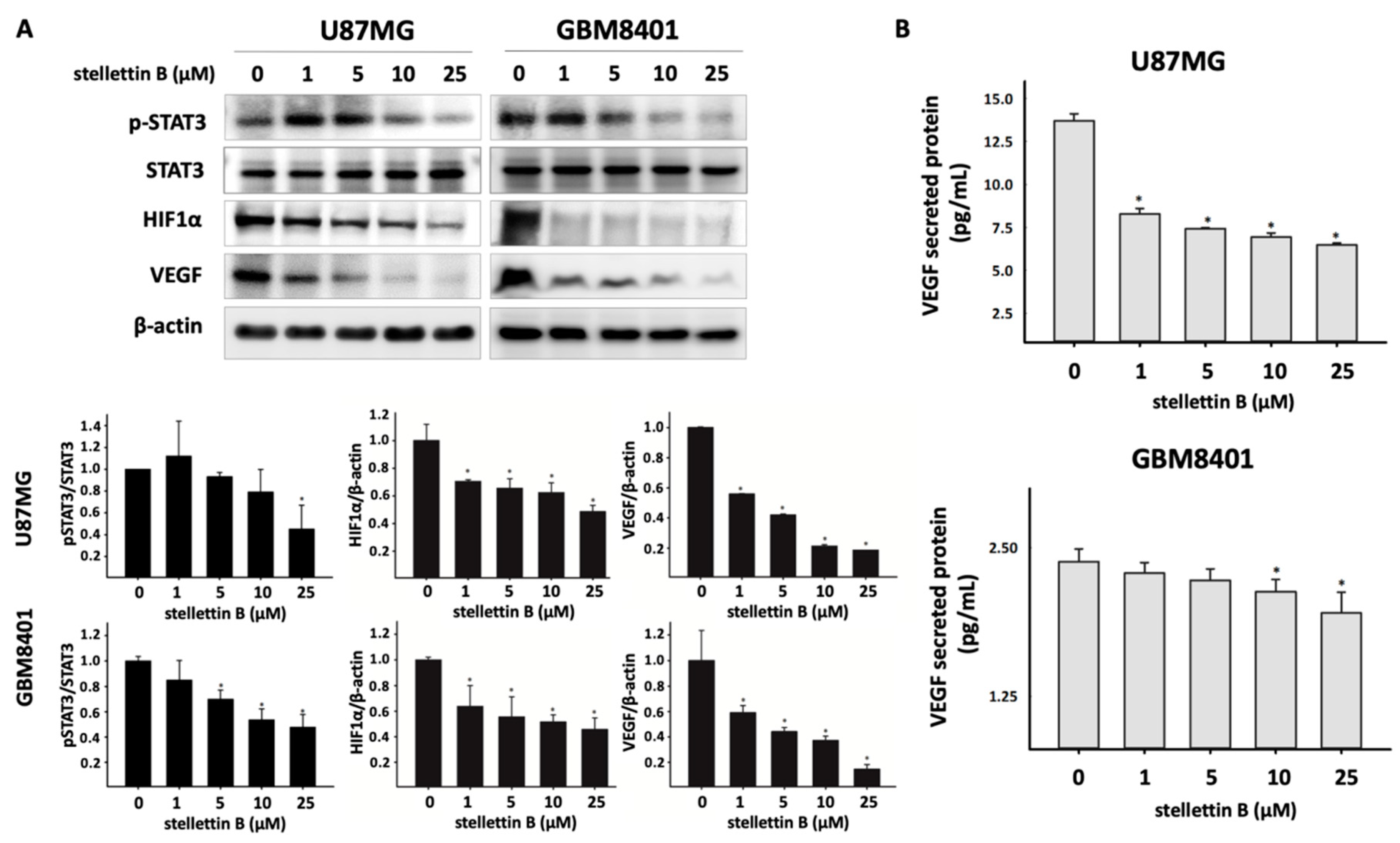
2.4. Stellettin B Suppresses VEGF Expression and Secretion through Downregulated Stat3 and HIF-1α in Glioblastoma
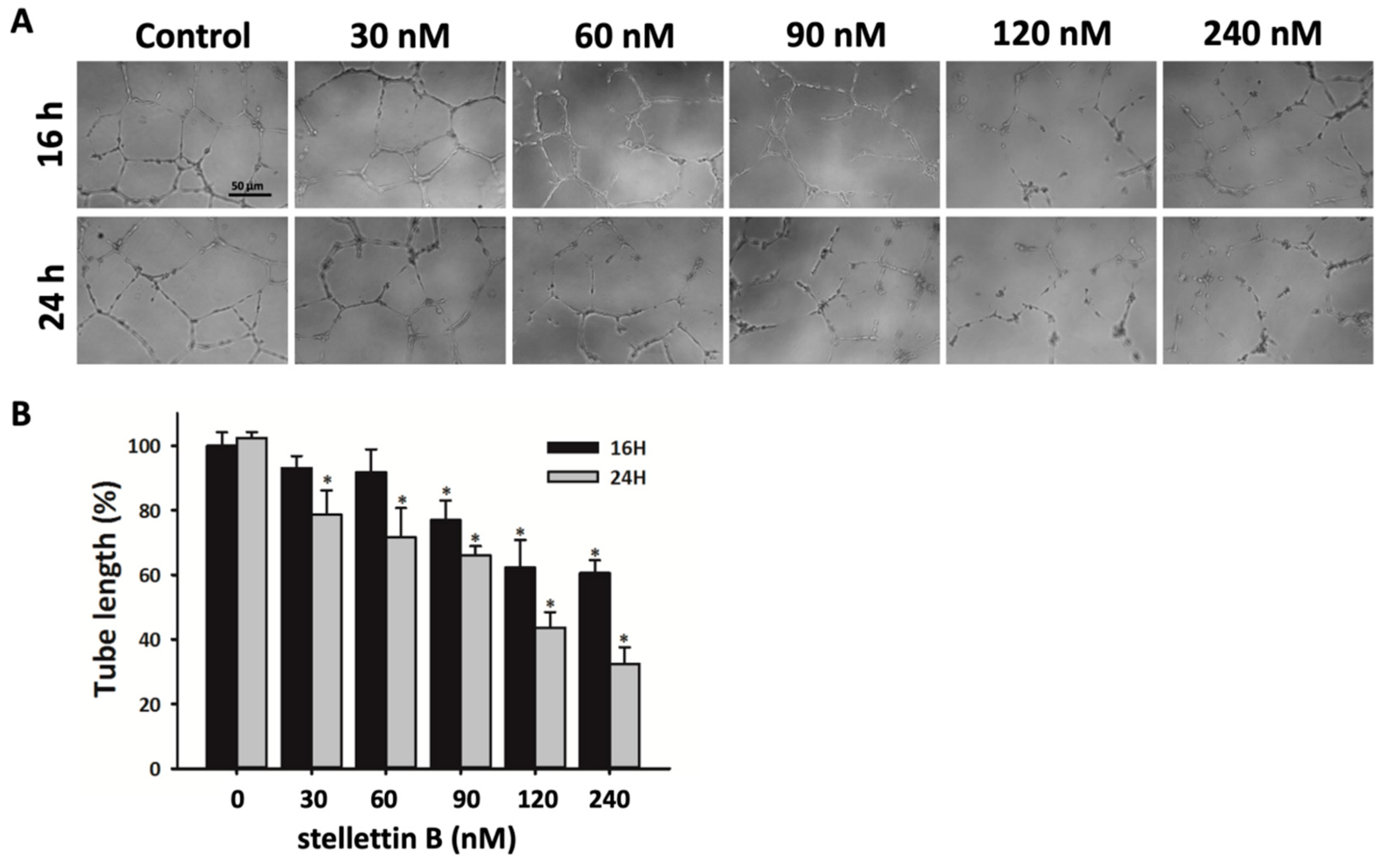
2.5. Stellettin B Inhibits Capillary Structure Formation of HUVECs
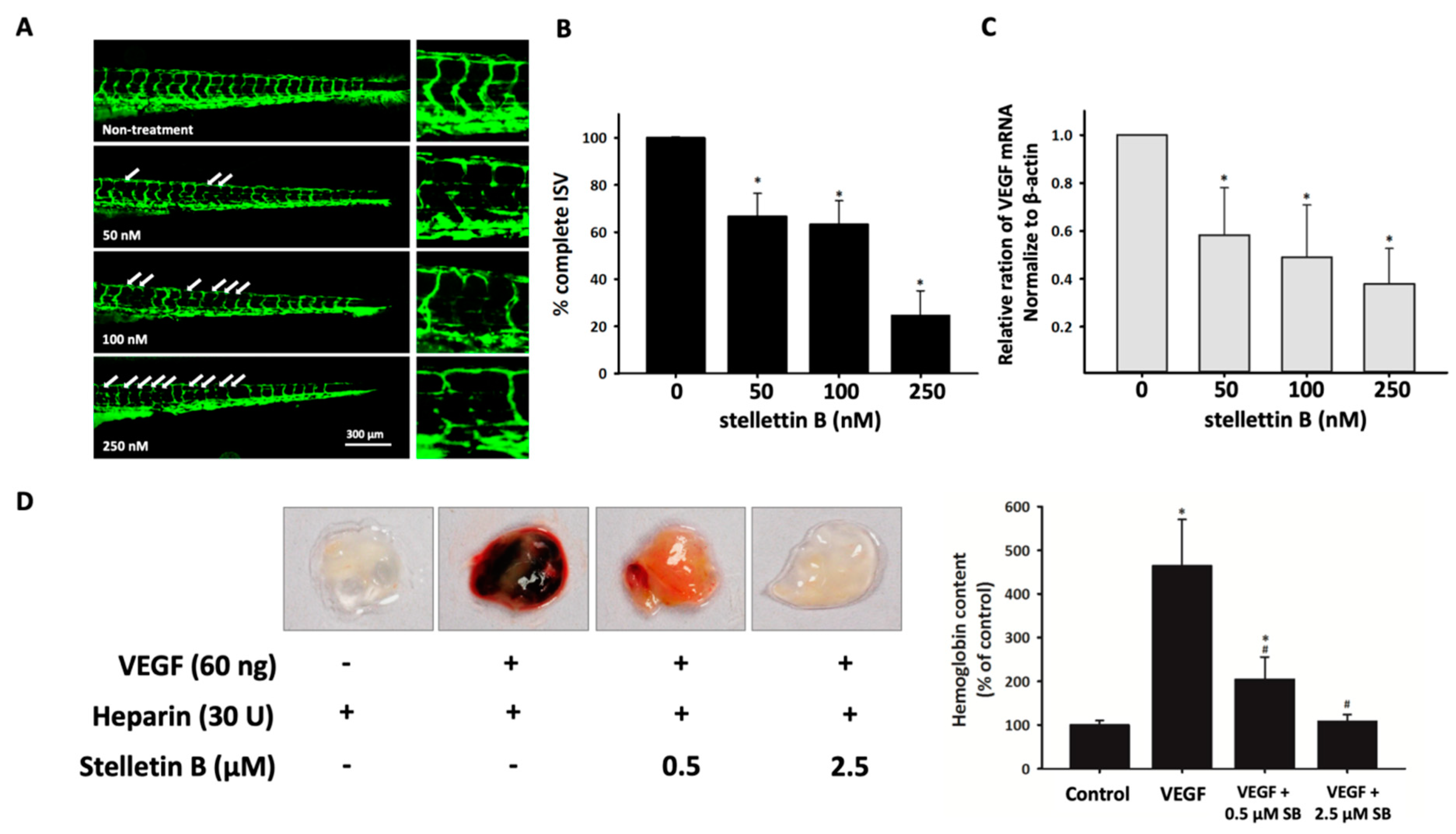
2.6. Stellettin B Inhibits Angiogenesis in In Vivo Zebrafish Embryogenesis and In Vivo Murine Matrigel Plug Models
3. Discussion
4. Materials and Methods
4.1. Compound
4.2. Cell Culture
4.3. MTT Assay for Cell Viability Analysis
4.4. Endothelial Cell Tube Formation Assay
4.5. Wound Healing Migration Assay
4.6. Transwell Chamber Invasion Assay
4.7. Signaling Pathway Detection through Western Blotting
4.8. Antiangiogenesis-Related Gene Detection Using Quantitative Real-Time Polymerase Chain Reaction (PCR)
4.9. Observation of p-Girdin Localized with F-Actin Using Confocal Microscopy
4.10. ELISA of VEGF
4.11. In Vivo Zebrafish ISV Angiogenesis Assay
4.12. In Vivo Mouse Matrigel Plug Angiogenesis Assay
4.13. Hemoglobin Measurement Using Drabkin’s Reagent
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Roa, W.; Brasher, P.; Bauman, G.; Anthes, M.; Bruera, E.; Chan, A.; Fisher, B.; Fulton, D.; Gulavita, S.; Hao, C. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: A prospective randomized clinical trial. J. Clin. Oncol. 2004, 22, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro-Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Lee, H.Y. Brain angiogenesis in developmental and pathological processes: Mechanism and therapeutic intervention in brain tumors. FEBS J. 2009, 276, 4653–4664. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.L.; McKee, T.C.; Cardellina, J.H.; Leid, M.; Boyd, M.R. Cytotoxic triterpenes from a marine sponge, Stelletta sp. J. Nat. Prod. 1996, 59, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-A.; Zhou, Q.; Guo, W.-Z.; Qiu, Y.; Wang, R.; Jin, M.; Zhang, W.; Li, K.; Yamori, T.; Dan, S. In vitro antitumor activity of stellettin B, a triterpene from marine sponge Jaspis stellifera, on human glioblastoma cancer SF295 cells. Mar. Drugs 2014, 12, 4200–4213. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin B induces G1 arrest, apoptosis and autophagy in human non-small cell lung cancer A549 cells via blocking PI3K/Akt/mTOR pathway. Sci. Rep. 2016, 6, 27071. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Q.; Zhang, L.; Zhong, Y.; Fan, G.; Zhang, Z.; Wang, R.; Jin, M.; Qiu, Y.; Kong, D. Stellettin B induces apoptosis in human chronic myeloid leukemia cells via targeting PI3K and Stat5. Oncotarget 2017, 8, 28906. [Google Scholar] [CrossRef]
- Chin, Y.R.; Toker, A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.-H.; Liu, L.-Z. AKT signaling in regulating angiogenesis. Curr. Cancer Drug Targets 2008, 8, 19–26. [Google Scholar] [CrossRef]
- Xie, Q.; Mittal, S.; Berens, M.E. Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro-Oncol. 2014, 16, 1575–1584. [Google Scholar] [CrossRef]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Betensky, R.A.; Batchelor, T.T.; Bernay, D.C.; Louis, D.N.; Nutt, C.L. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006, 65, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Doss, B.; Lim, C.T.; Voituriez, R.; Ladoux, B. Single cell rigidity sensing: A complex relationship between focal adhesion dynamics and large-scale actin cytoskeleton remodeling. Cell Adhes. Migr. 2016, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Murakami, H.; Asai, N.; Morone, N.; Watanabe, T.; Kawai, K.; Murakumo, Y.; Usukura, J.; Kaibuchi, K.; Takahashi, M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 2005, 9, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, Y.; Ong, A.; Notani, D.; Mittal, Y.; Lam, M.; Mi, X.; Ghosh, P. STAT3 upregulates GIV/Girdin expression, and GIV enhances STAT3 activation in a positive feedback loop during wound healing and tumor invasion/metastasis. J. Biol. Chem. 2012. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, F.; Zhang, Y.; Wang, X.; Shi, J.; Wei, H.; Sun, F.; Yu, Y. Girdin protein: A potential metastasis predictor associated with prognosis in lung cancer. Exp. Ther. Med. 2018, 15, 2837–2843. [Google Scholar] [CrossRef]
- Choi, J.-S.; Kim, K.H.; Oh, E.; Shin, Y.K.; Seo, J.; Kim, S.-H.; Park, S.; Choi, Y.-L. Girdin protein expression is associated with poor prognosis in patients with invasive breast cancer. Pathology 2017, 49, 618–626. [Google Scholar] [CrossRef]
- Gu, F.; Wang, L.; He, J.; Liu, X.; Zhang, H.; Li, W.; Fu, L.; Ma, Y. Girdin, an actin-binding protein, is critical for migration, adhesion, and invasion of human glioblastoma cells. J. Neurochem. 2014, 131, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The role of hypoxia in glioblastoma invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Schmidt, N.O.; Westphal, M.; Hagel, C.; Ergün, S.; Stavrou, D.; Rosen, E.M.; Lamszus, K. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int. J. Cancer 1999, 84, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000. [Google Scholar] [CrossRef] [PubMed]
- Luwor, R.B.; Stylli, S.S.; Kaye, A.H. The role of Stat3 in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Yu, M.O.; Park, K.-J.; Chi, S.-G.; Park, D.-H.; Chung, Y.-G. Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery 2010, 67, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Armento, A.; Ehlers, J.; Schötterl, S.; Naumann, U. Molecular Mechanisms of Glioma Cell Motility. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Weathers, S.-P.; de Groot, J. VEGF manipulation in glioblastoma. Oncology 2015, 29, 720–727. [Google Scholar]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552. [Google Scholar] [CrossRef]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.-H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.-W.; Ye, S.-K. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. Faseb J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef]
- Qutub, A.A.; Popel, A.S. Elongation, proliferation & migration differentiate endothelial cell phenotypes and determine capillary sprouting. BMC Syst. Biol. 2009, 3, 13. [Google Scholar]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213. [Google Scholar] [CrossRef]
- Ma, A.C.; Lin, R.; Chan, P.-K.; Leung, J.C.; Chan, L.Y.; Meng, A.; Verfaillie, C.M.; Liang, R.; Leung, A.Y. The role of survivin in angiogenesis during zebrafish embryonic development. BMC Dev. Biol. 2007, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Coltrini, D.; Di Salle, E.; Ronca, R.; Belleri, M.; Testini, C.; Presta, M. Matrigel plug assay: Evaluation of the angiogenic response by reverse transcription-quantitative PCR. Angiogenesis 2013, 16, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Ichikawa, T.; Kurozumi, K.; Date, I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011, 28, 13–24. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef]
- Wen, S.; Stolarov, J.; Myers, M.P.; Su, J.D.; Wigler, M.H.; Tonks, N.K.; Durden, D.L. PTEN controls tumor-induced angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 4622–4627. [Google Scholar] [CrossRef] [Green Version]
- Talasila, K.M.; Soentgerath, A.; Euskirchen, P.; Rosland, G.V.; Wang, J.; Huszthy, P.C.; Prestegarden, L.; Skaftnesmo, K.O.; Sakariassen, P.Ø.; Eskilsson, E. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 2013, 125, 683–698. [Google Scholar] [CrossRef] [Green Version]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shao, N.; Luo, G.; Li, L.; Zheng, L.; Nilsson-Ehle, P.; Xu, N. Mutations of PTEN gene in gliomas correlate to tumor differentiation and short-term survival rate. Anticancer Res. 2010, 30, 981–985. [Google Scholar]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the AKT pathway in glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Carico, C.; Nuño, M.; Mukherjee, D.; Elramsisy, A.; Dantis, J.; Hu, J.; Rudnick, J.; John, S.Y.; Black, K.L.; Bannykh, S.I. Loss of PTEN is not associated with poor survival in newly diagnosed glioblastoma patients of the temozolomide era. PLoS ONE 2012, 7, e33684. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Asai, N.; Enomoto, A.; Maeda, K.; Kato, T.; Ishida, M.; Jiang, P.; Watanabe, T.; Usukura, J.; Kondo, T. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat. Cell Biol. 2008, 10, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Enomoto, A.; Jijiwa, M.; Kato, T.; Hasegawa, T.; Ishida, M.; Sato, T.; Asai, N.; Murakumo, Y.; Takahashi, M. An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 2008, 68, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kaneko, N.; Asai, N.; Enomoto, A.; Isotani-Sakakibara, M.; Kato, T.; Asai, M.; Murakumo, Y.; Ota, H.; Hikita, T. Girdin is an intrinsic regulator of neuroblast chain migration in the rostral migratory stream of the postnatal brain. J. Neurosci. 2011, 31, 8109–8122. [Google Scholar] [CrossRef]
- Natsume, A.; Kato, T.; Kinjo, S.; Enomoto, A.; Toda, H.; Shimato, S.; Ohka, F.; Motomura, K.; Kondo, Y.; Miyata, T. Girdin maintains the stemness of glioblastoma stem cells. Oncogene 2012, 31, 2715–2724. [Google Scholar] [CrossRef]
- Garcia-Marcos, M.; Jung, B.H.; Ear, J.; Cabrera, B.; Carethers, J.M.; Ghosh, P. Expression of GIV/Girdin, a metastasis-related protein, predicts patient survival in colon cancer. FASEB J. 2011, 25, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Lorente, G.; Syriani, E.; Morales, M. Actin filaments at the leading edge of cancer cells are characterized by a high mobile fraction and turnover regulation by profilin I. PLoS ONE 2014, 9, e85817. [Google Scholar] [CrossRef]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-Y.; Huang, H.; Nagane, M.; Ji, X.-D.; Wang, D.; Shih, C.; Arap, W.; Huang, C.-M.; Cavenee, W.K. Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 8502–8507. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Zhang, J.; Ellis, L.M.; Semenza, G.L.; Evans, D.B.; Watowich, S.S.; Gallick, G.E. HIF-1α, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 2005, 24, 3110. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Ahn, S.H.; Kong, D.-S.; Lee, H.W.; Nam, D.-H. The role of STAT3 in glioblastoma progression through dual influences on tumor cells and the immune microenvironment. Mol. Cell. Endocrinol. 2017, 451, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Knizetova, P.; Ehrmann, J.; Hlobilkova, A.; Vancova, I.; Kalita, O.; Kolar, Z.; Bartek, J. Autocrine regulation of glioblastoma cell-cycle progression, viability and radioresistance through the VEGF-VEGFR2 (KDR) interplay. Cell Cycle 2008, 7, 2553–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, H.A.; Figg, W.D.; Jaeckle, K.; Wen, P.Y.; Kyritsis, A.P.; Loeffler, J.S.; Levin, V.A.; Black, P.M.; Kaplan, R.; Pluda, J.M. Phase II trial of the antiangiogenic agent thalidomide in patients with recurrent high-grade gliomas. J. Clin. Oncol. 2000, 18, 708. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Wenger, K.J.; Wagner, M.; You, S.J.; Franz, K.; Harter, P.N.; Burger, M.C.; Voss, M.; Ronellenfitsch, M.W.; Fokas, E.; Steinbach, J.P. Bevacizumab as a last-line treatment for glioblastoma following failure of radiotherapy, temozolomide and lomustine. Oncol. Lett. 2017, 14, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.R.; Omuro, A.M.; Ravelo, A.; Sommer, N.; Guerin, A.; Ionescu-Ittu, R.; Shi, S.; Macalalad, A.; Uhm, J.H. Overall survival in patients with glioblastoma before and after bevacizumab approval. Curr. Med. Res. Opin. 2018, 34, 813–820. [Google Scholar] [CrossRef]
- Gramatzki, D.; Roth, P.; Rushing, E.; Weller, J.; Andratschke, N.; Hofer, S.; Korol, D.; Regli, L.; Pangalu, A.; Pless, M. Bevacizumab may improve quality of life, but not overall survival in glioblastoma: An epidemiological study. Ann. Oncol. 2018, 29, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; Hall, D.E.; Porter, S.; Barbu, K.; Cannon, C.; Horner, H.C.; Janatpour, M.; Liaw, C.W.; Manning, K.; Morales, J.; et al. A cell culture model of the blood-brain barrier. J. Cell Biol. 1991, 115, 1725–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, H.; Clark, K.; Bressler, J.P. In vitro models to study the blood brain barrier. Methods Mol. Biol. 2011, 758, 153–168. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.-Y.; Chen, N.-F.; Lin, P.-Y.; Su, J.-H.; Chen, B.-H.; Kuo, H.-M.; Sung, C.-S.; Sung, P.-J.; Wen, Z.-H.; Chen, W.-F. Anti-Invasion and Antiangiogenic Effects of Stellettin B through Inhibition of the Akt/Girdin Signaling Pathway and VEGF in Glioblastoma Cells. Cancers 2019, 11, 220. https://doi.org/10.3390/cancers11020220
Cheng S-Y, Chen N-F, Lin P-Y, Su J-H, Chen B-H, Kuo H-M, Sung C-S, Sung P-J, Wen Z-H, Chen W-F. Anti-Invasion and Antiangiogenic Effects of Stellettin B through Inhibition of the Akt/Girdin Signaling Pathway and VEGF in Glioblastoma Cells. Cancers. 2019; 11(2):220. https://doi.org/10.3390/cancers11020220
Chicago/Turabian StyleCheng, Shu-Yu, Nan-Fu Chen, Pi-Yu Lin, Jui-Hsin Su, Bing-Hung Chen, Hsiao-Mei Kuo, Chun-Sung Sung, Ping-Jyun Sung, Zhi-Hong Wen, and Wu-Fu Chen. 2019. "Anti-Invasion and Antiangiogenic Effects of Stellettin B through Inhibition of the Akt/Girdin Signaling Pathway and VEGF in Glioblastoma Cells" Cancers 11, no. 2: 220. https://doi.org/10.3390/cancers11020220