Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis

Abstract
:1. Introduction
1.1. Tight Regulation Over the Central Dogma of Molecular Biology is Essential for Cell Survival
1.2. Proteotoxic Stress: A Secondary Hallmark of Cancer
1.3. Endoplasmic Reticulum (ER) Stress is Closely Linked to Oxidative Stress in Cancer
1.4. Aneuploidy Contributes to Proteotoxic Stress
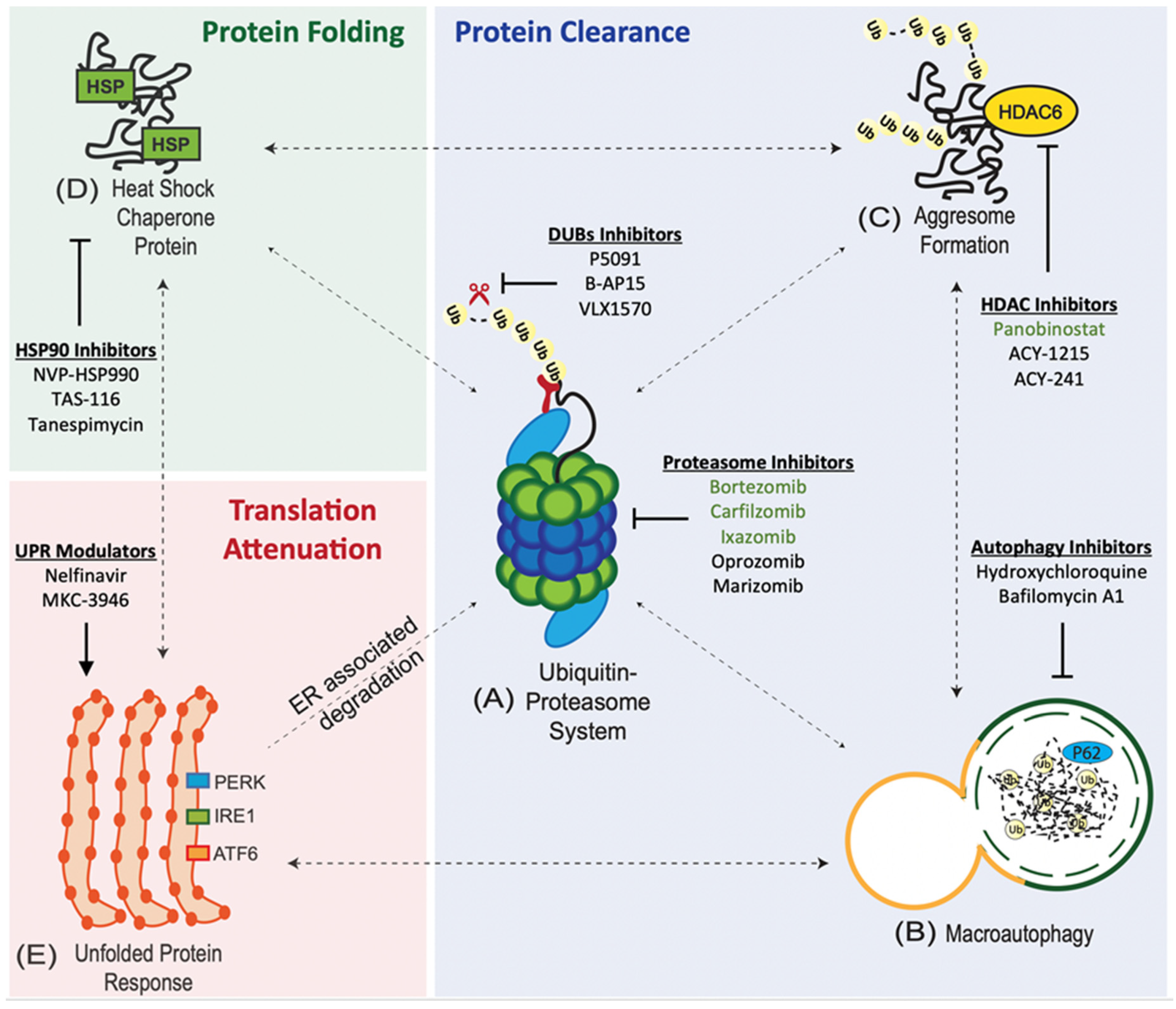
2. Overview of the Protein Quality Control System
2.1. Ubiquitin Proteasome System (UPS)
2.2. Macroautophagy (Autophagy-Lysosome System)
2.3. Aggresome Pathway
2.4. Heat-Shock Protein (HSP) Chaperone System
2.5. The Integrated Stress Response (ISR)
2.6. Endoplasmic Stress and the Unfolded Protein Response (UPR)
3. Exploiting Proteotoxic Stress in Hematologic Malignancies: Multiple Myeloma
3.1. Proteasome Inhibitors (PIs)
3.2. Autophagy Inhibitors
3.3. Blocking the Aggresomal Pathway through HDAC6 Inhibition
3.4. Heat Shock Protein 90 (HSP90) Inhibitors
3.5. Unfolded Protein Response (UPR) Modulators
4. Exploiting Proteotoxic Stress in Solid Tumors: Triple Negative Breast Cancer (TNBC)
4.1. Proteasome Inhibition in TNBC
4.2. Cellular Senescence and Proteotoxic Stress: A Double-Edged Sword?
5. A Look at What’s on the Horizon
5.1. Disrupting the Protein Secretory Pathway to Increase Proteotoxic Stress
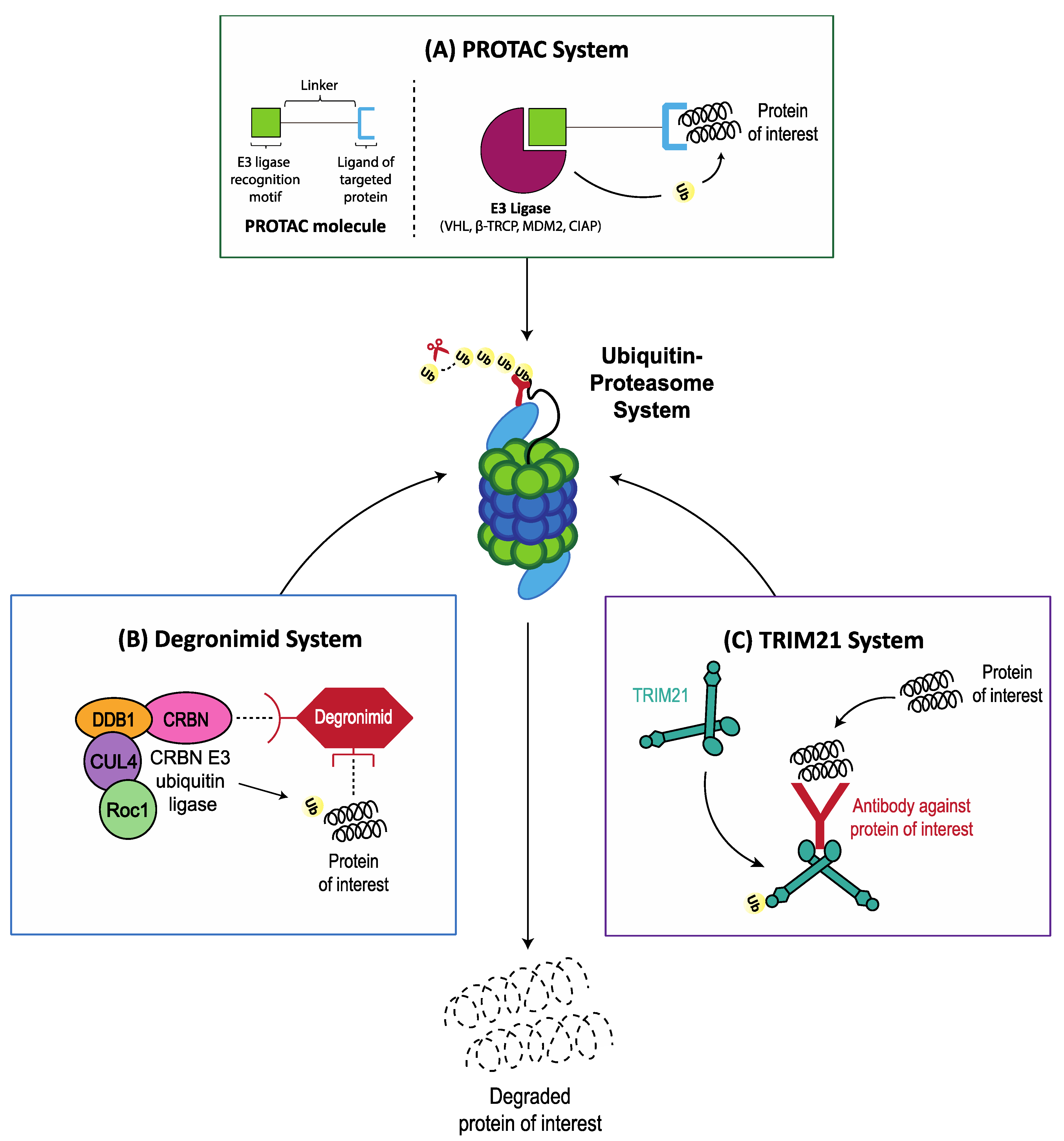
5.2. Hijacking E3 Ligases for Specific Target Protein Degradation via the Ubiquitin Proteasome System
5.2.1. PROTAC System
5.2.2. Degronimids
5.2.3. TRIM21 System
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokhale, S.; Nyayanit, D.; Gadgil, C. A systems view of the protein expression process. Syst. Synth. Biol. 2011, 5, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Cenci, S.; Sitia, R. Managing and exploiting stress in the antibody factory. FEBS Lett. 2007, 581, 3652–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggero, D. Translational control in cancer etiology. Cold Spring Harbor Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L.; et al. mTORC1 Coordinates Protein Synthesis and Immunoproteasome Formation via PRAS40 to Prevent Accumulation of Protein Stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- McConkey, D.J. The integrated stress response and proteotoxicity in cancer therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Varadharaj, S.; Khanderao, G.D.; Davidson, C.J.; Kannan, S.; Firpo, M.A.; Zweier, J.L.; Benjamin, I.J. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid. Redox Signal. 2011, 14, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Muthusamy, V.R.; Whitehead, K.J.; Wang, L.; Gomes, A.V.; Litwin, S.E.; Kensler, T.W.; Abel, E.D.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 100, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Dejeans, N.; Tajeddine, N.; Beck, R.; Verrax, J.; Taper, H.; Gailly, P.; Calderon, P.B. Endoplasmic reticulum calcium release potentiates the ER stress and cell death caused by an oxidative stress in MCF-7 cells. Biochem. Pharmacol. 2010, 79, 1221–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Suzuki-Karasaki, Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells. Free Radic. Biol. Med. 2013, 61, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Wang, J.; Luo, B.; Li, X.; Lu, W.; Yang, J.; Hu, Y.; Huang, P.; Wen, S. Inhibition of cancer growth in vitro and in vivo by a novel ROS-modulating agent with ability to eliminate stem-like cancer cells. Cell Death Dis. 2017, 8, e2887. [Google Scholar] [CrossRef]
- Guang, M.H.Z.; Bianchi, G. Targeting Protein Synthesis and Degradation in Multiple Myeloma: A Look at What’s on the Horizon. Am. J. Hematol. Oncol. 2017, 13, 11. [Google Scholar]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Storchova, Z. Aneuploidy and proteotoxic stress in cancer. Mol. Cell. Oncol. 2015, 2, e976491. [Google Scholar] [CrossRef] [Green Version]
- Torres, E.M.; Dephoure, N.; Panneerselvam, A.; Tucker, C.M.; Whittaker, C.A.; Gygi, S.P.; Dunham, M.J.; Amon, A. Identification of aneuploidy-tolerating mutations. Cell 2010, 143, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Oromendia, A.B.; Dodgson, S.E.; Amon, A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012, 26, 2696–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, A.; Ohori, M.; Iwai, K.; Nakayama, Y.; Nambu, T.; Morishita, D.; Kawamoto, T.; Miyamoto, M.; Hirayama, T.; Okaniwa, M.; et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat. Commun. 2015, 6, 7668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, P.; Bahlis, N.J. Genomic instability in multiple myeloma: Mechanisms and therapeutic implications. Expert Opin. Biol. Ther. 2013, 13, S69–S82. [Google Scholar] [CrossRef]
- Meister, S.; Schubert, U.; Neubert, K.; Herrmann, K.; Burger, R.; Gramatzki, M.; Hahn, S.; Schreiber, S.; Wilhelm, S.; Herrmann, M.; et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007, 67, 1783–1792. [Google Scholar] [CrossRef]
- Selvaraju, K.; Mazurkiewicz, M.; Wang, X.; Gullbo, J.; Linder, S.; D’Arcy, P. Inhibition of proteasome deubiquitinase activity: A strategy to overcome resistance to conventional proteasome inhibitors? Drug Resist. Updat. 2015, 21, 20–29. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Beere, H.M. “The stress of dying”: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117, 2641–2651. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.; Enenkel, C. Intracellular Dynamics of the Ubiquitin-Proteasome-System. F1000Res 2015, 4, 367. [Google Scholar] [CrossRef] [PubMed]
- Coux, O.; Tanaka, K.; Goldberg, A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef] [PubMed]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Akopian, T.N.; Castillo, V.; Goldberg, A.L. Proteasome active sites allosterically regulate each other, suggesting a cyclical bite-chew mechanism for protein breakdown. Mol. Cell 1999, 4, 395–402. [Google Scholar] [CrossRef]
- Mirabella, A.C.; Pletnev, A.A.; Downey, S.L.; Florea, B.I.; Shabaneh, T.B.; Britton, M.; Verdoes, M.; Filippov, D.V.; Overkleeft, H.S.; Kisselev, A.F. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem. Biol. 2011, 18, 608–618. [Google Scholar] [CrossRef]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 26979. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, G.; Richardson, P.G.; Anderson, K.C. Promising therapies in multiple myeloma. Blood 2015, 126, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Barrio, S.; Stühmer, T.; Teufel, E.; Barrio-Garcia, C.; Chatterjee, M.; Schreder, M.; Das Gupta, M.; Rosenwald, A.; Tibes, R.; Braggio, E.; et al. Parallel Evolution of Multiple PSMB5 mutations in a Myeloma Patient Treated with Bortezomib. Blood 2016, 128, 3282. [Google Scholar]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016, 5. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Metcalf, D.J.; Garcia-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Simms-Waldrip, T.; Fu, C.; Sakamoto, K.M. Role of the aggresome pathway in cancer: Targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008, 68, 2557–2560. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Couillault, C.; Fourquet, P.; Pophillat, M.; Ewbank, J.J. A UPR-independent infection-specific role for a BiP/GRP78 protein in the control of antimicrobial peptide expression in C. elegans epidermis. Virulence 2012, 3, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Fok, J.H.; Davies, F.E. Heat shock proteins in multiple myeloma. Oncotarget 2014, 5, 1132–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Dever, T.E.; Feng, L.; Wek, R.C.; Cigan, A.M.; Donahue, T.F.; Hinnebusch, A.G. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 1992, 68, 585–596. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef]
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.R.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A.L. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burwick, N.; Aktas, B.H. The eIF2-alpha kinase HRI: A potential target beyond the red blood cell. Expert Opin. Ther. Targets 2017, 21, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Auner, H.W.; Moody, A.M.; Ward, T.H.; Kraus, M.; Milan, E.; May, P.; Chaidos, A.; Driessen, C.; Cenci, S.; Dazzi, F.; et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS ONE 2013, 8, e74415. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R.; Ron, D. Conformational disease. Nat. Cell Biol. 2000, 2, E207–E209. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar] [CrossRef]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Effect of autophagy on multiple myeloma cell viability. Mol. Cancer Ther. 2009, 8, 1974–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, T.; Miyazawa, K.; Moriya, S.; Ohtomo, T.; Che, X.F.; Naito, M.; Itoh, M.; Tomoda, A. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: Crosstalk among proteasome, autophagy-lysosome and ER stress. Int. J. Oncol. 2011, 38, 643–654. [Google Scholar] [CrossRef]
- Qiao, L.; Zhang, J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci. Lett. 2009, 456, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467. [Google Scholar] [CrossRef] [PubMed]
- Cenci, S.; Mezghrani, A.; Cascio, P.; Bianchi, G.; Cerruti, F.; Fra, A.; Lelouard, H.; Masciarelli, S.; Mattioli, L.; Oliva, L.; et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. EMBO J. 2006, 25, 1104–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014, 12. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.-L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Scott, K.; Hayden, P.J.; Will, A.; Wheatley, K.; Coyne, I. The role of bortezomib treatment for patients with multiple myeloma. Cochrane Database Syst. Rev. 2016, 4. [Google Scholar] [CrossRef]
- Siegel, D.S.; Martin, T.; Wang, M.; Vij, R.; Jakubowiak, A.J.; Lonial, S.; Trudel, S.; Kukreti, V.; Bahlis, N.; Alsina, M.; et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurchla, M.A.; Garcia-Gomez, A.; Hornick, M.C.; Ocio, E.M.; Li, A.; Blanco, J.F.; Collins, L.; Kirk, C.J.; Piwnica-Worms, D.; Vij, R.; et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia 2013, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, K.; Misund, K.; Starheim, K.K.; Holien, T.; Johansson, I.; Darvekar, S.; Buene, G.; Waage, A.; Bjorkoy, G.; Sundan, A. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016, 7, 70845–70856. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [Green Version]
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S.; et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Khong, T.; Spencer, A. Targeting HSP 90 induces apoptosis and inhibits critical survival and proliferation pathways in multiple myeloma. Mol. Cancer Ther. 2011, 10, 1909–1917. [Google Scholar] [CrossRef]
- Suzuki, R.; Hideshima, T.; Mimura, N.; Minami, J.; Ohguchi, H.; Kikuchi, S.; Yoshida, Y.; Gorgun, G.; Cirstea, D.; Cottini, F.; et al. Anti-tumor activities of selective HSP90alpha/beta inhibitor, TAS-116, in combination with bortezomib in multiple myeloma. Leukemia 2015, 29, 510–514. [Google Scholar] [CrossRef]
- Richardson, P.G.; Chanan-Khan, A.A.; Lonial, S.; Krishnan, A.Y.; Carroll, M.P.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: Results of a phase 1/2 study. Br. J. Haematol. 2011, 153, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Staff, T.M.B. Bristol-Myers Squibb Halts Development of Tanespimycin. Available online: https://myelomabeacon.org/news/2010/07/22/tanespimycin-development-halted/ (accessed on 22 July 2010).
- Park, H.R.; Tomida, A.; Sato, S.; Tsukumo, Y.; Yun, J.; Yamori, T.; Hayakawa, Y.; Tsuruo, T.; Shin-ya, K. Effect on tumor cells of blocking survival response to glucose deprivation. J. Natl. Cancer Inst. 2004, 96, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Manevich, Y.; He, L.; Xiong, Y.; Bowers, R.R., Jr.; Hutchens, S.; Tew, K.D. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009, 69, 7626–7634. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.; Bader, J.; Overkleeft, H.; Driessen, C. Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood Cancer J. 2013, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Driessen, C.; Kraus, M.; Joerger, M.; Rosing, H.; Bader, J.; Hitz, F.; Berset, C.; Xyrafas, A.; Hawle, H.; Berthod, G.; et al. Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: A phase I trial (SAKK 65/08). Haematologica 2016, 101, 346–355. [Google Scholar] [CrossRef]
- Raedler, L. Velcade (Bortezomib) Receives 2 New FDA Indications: For Retreatment of Patients with Multiple Myeloma and for First-Line Treatment of Patients with Mantle-Cell Lymphoma. Am. Health Drug Benefits 2015, 8, 135–140. [Google Scholar]
- Chao, A.; Wang, T.-H. Molecular mechanisms for synergistic effect of proteasome inhibitors with platinum-based therapy in solid tumors. Taiwan. J. Obstetr. Gynecol. 2016, 55, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Weyburne, E.S.; Wilkins, O.M.; Sha, Z.; Williams, D.A.; Pletnev, A.A.; de Bruin, G.; Overkleeft, H.S.; Goldberg, A.L.; Cole, M.D.; Kisselev, A.F. Inhibition of the Proteasome beta2 Site Sensitizes Triple-Negative Breast Cancer Cells to beta5 Inhibitors and Suppresses Nrf1 Activation. Cell Chem. Biol. 2017, 24, 218–230. [Google Scholar] [CrossRef]
- Shen, S.; Du, X.J.; Liu, J.; Sun, R.; Zhu, Y.H.; Wang, J. Delivery of bortezomib with nanoparticles for basal-like triple-negative breast cancer therapy. J. Control. Release 2015, 208, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, H.; von Kobbe, C.; Sanchez-Estevez, C.; Julian-Tendero, M.; Palacios, J.; Moreno-Bueno, G. Inhibition of paclitaxel-induced proteasome activation influences paclitaxel cytotoxicity in breast cancer cells in a sequence-dependent manner. Cell Cycle 2007, 6, 2662–2668. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Kuhnhardt, D.; Kiewe, P.; Lehenbauer-Dehm, S.; Schippinger, W.; Greil, R.; Lange, W.; Preiss, J.; Niederle, N.; Brossart, P.; et al. A phase I/II study of bortezomib and capecitabine in patients with metastatic breast cancer previously treated with taxanes and/or anthracyclines. Ann. Oncol. 2008, 19, 871–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Yeh, M.H.; Yu, M.C.; Wei, Y.L.; Chen, W.S.; Chen, J.Y.; Shih, C.Y.; Tu, C.Y.; Chen, C.H.; Hsia, T.C.; et al. Lapatinib-induced NF-kappaB activation sensitizes triple-negative breast cancer cells to proteasome inhibitors. Breast Cancer Res. 2013, 15, R108. [Google Scholar] [CrossRef] [PubMed]
- Meissner, T.; Mark, A.; Williams, C.; Berdel, W.E.; Wiebe, S.; Kerkhoff, A.; Wardelmann, E.; Gaiser, T.; Muller-Tidow, C.; Rosenstiel, P.; et al. Metastatic triple-negative breast cancer patient with TP53 tumor mutation experienced 11 months progression-free survival on bortezomib monotherapy without adverse events after ending standard treatments with grade 3 adverse events. Cold Spring Harbor Mol. Case Stud. 2017, 3. [Google Scholar] [CrossRef]
- Tseng, L.M.; Liu, C.Y.; Chang, K.C.; Chu, P.Y.; Shiau, C.W.; Chen, K.F. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012, 14, R68. [Google Scholar] [CrossRef]
- Deshmukh, R.R.; Kim, S.; Elghoul, Y.; Dou, Q.P. P-Glycoprotein Inhibition Sensitizes Human Breast Cancer Cells to Proteasome Inhibitors. J. Cell. Biochem. 2017, 118, 1239–1248. [Google Scholar] [CrossRef]
- Clemens, J.; Seckinger, A.; Hose, D.; Theile, D.; Longo, M.; Haefeli, W.E.; Burhenne, J.; Weiss, J. Cellular uptake kinetics of bortezomib in relation to efficacy in myeloma cells and the influence of drug transporters. Cancer Chemother Pharmacol. 2015, 75, 281–291. [Google Scholar] [CrossRef]
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013, 71, 1357–1368. [Google Scholar] [CrossRef]
- Petrocca, F.; Altschuler, G.; Tan, S.M.; Mendillo, M.L.; Yan, H.; Jerry, D.J.; Kung, A.L.; Hide, W.; Ince, T.A.; Lieberman, J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 2013, 24, 182–196. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Kavanagh, E.L.; Lindsay, S.; Halasz, M.; Gubbins, L.C.; Weiner-Gorzel, K.; Guang, M.H.Z.; McGoldrick, A.; Collins, E.; Henry, M.; Blanco-Fernández, A.; et al. Protein and chemotherapy profiling of extracellular vesicles harvested from therapeutic induced senescent triple negative breast cancer cells. Oncogenesis 2017, 6, e388. [Google Scholar] [CrossRef] [PubMed]
- Gey, C.; Seeger, K. Metabolic changes during cellular senescence investigated by proton NMR-spectroscopy. Mech. Ageing Dev. 2013, 134, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Alimirah, F.; Desprez, P.-Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Lessard, F.; Gaumont-Leclerc, M.F.; Bardeesy, N.; Ferbeyre, G. Cellular senescence and protein degradation: Breaking down cancer. Cell Cycle 2014, 13, 1840–1858. [Google Scholar] [CrossRef]
- Matos, L.; Gouveia, A.M.; Almeida, H. ER Stress Response in Human Cellular Models of Senescence. J. Gerontol. Ser. A 2015, 70, 924–935. [Google Scholar] [CrossRef]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–425. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Stratford, F.L.; Trougakos, I.P.; Friguet, B.; Rivett, A.J.; Gonos, E.S. Central role of the proteasome in senescence and survival of human fibroblasts: Induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 2003, 278, 28026–28037. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; Jensen, S.S.; Lim, J.W. Proteomic profiling of exosomes: Current perspectives. Proteomics 2008, 8, 4083–4099. [Google Scholar] [CrossRef] [PubMed]
- Gercel-Taylor, C.; Atay, S.; Tullis, R.H.; Kesimer, M.; Taylor, D.D. Nanoparticle analysis of circulating cell-derived vesicles in ovarian cancer patients. Anal. Biochem. 2012, 428, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biological function. Immunol. Lett. 2006, 107, 102–108. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef]
- Desdín-Micó, G.; Mittelbrunn, M. Role of exosomes in the protection of cellular homeostasis. Cell Adhes. Migr. 2017, 11, 127–134. [Google Scholar] [CrossRef]
- Pink, D.; Donnelier, J.; Lewis, J.; Braun, J.E.A. Cargo-Loading of Misfolded Proteins into Extracellular Vesicles: The Role of J Proteins. bioRxiv 2018. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Overview of the Secretory Pathway. In Molecular Cell Biology, 4th ed.; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Salaun, C.; James, D.J.; Greaves, J.; Chamberlain, L.H. Plasma membrane targeting of exocytic SNARE proteins. Biochim. Biophys. Acta 2004, 1693, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Maglathlin, R.; Lerner, A.; Taunton, J.; Kirk, C.J.; Arastu-Kapur, S. Blocking Protein Secretion and Degradation Is a Novel Treatment Strategy For Malignant Cells With High Protein Load. Blood 2013, 122, 4439. [Google Scholar]
- Meister, S.; Frey, B.; Lang, V.R.; Gaipl, U.S.; Schett, G.; Schlotzer-Schrehardt, U.; Voll, R.E. Calcium channel blocker verapamil enhances endoplasmic reticulum stress and cell death induced by proteasome inhibition in myeloma cells. Neoplasia 2010, 12, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Born, E.J.; Hartman, S.V.; Holstein, S.A. Targeting HSP90 and monoclonal protein trafficking modulates the unfolded protein response, chaperone regulation and apoptosis in myeloma cells. Blood Cancer J. 2013, 3, e167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Ma, X.; Iyer, L.; Chaulagain, C.; Comenzo, R.L. One siRNA pool targeting the λ constant region stops λ light-chain production and causes terminal endoplasmic reticulum stress. Blood 2014, 123, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Crews, C.M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Kitaguchi, R.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg. Med. Chem. 2011, 19, 6768–6778. [Google Scholar] [CrossRef]
- Hines, J.; Gough, J.D.; Corson, T.W.; Crews, C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA 2013, 110, 8942–8947. [Google Scholar] [CrossRef] [Green Version]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- James, L.C.; Keeble, A.H.; Khan, Z.; Rhodes, D.A.; Trowsdale, J. Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc. Natl. Acad. Sci. USA 2007, 104, 6200–6205. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clift, D.; McEwan, W.A.; Labzin, L.I.; Konieczny, V.; Mogessie, B.; James, L.C.; Schuh, M. A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 2017, 171, 1692–1706 e1618. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Class | Drug Name | Mechanism of Action | Study Design | Status |
|---|---|---|---|---|
| PI | Bortezomib | Proteasome inhibition; Apoptosis via caspase 8/9; UPR apoptotic response | Single use approval | FDA approved |
| Carfilzomib | Irreversible proteasome inhibition | Single use approval; Combination treatment DEX and/or LEN | FDA approved | |
| Ixazomib | Oral Proteasome inhibitor | Combination treatment with LEN and DEX | FDA approved | |
| Oprozomib | Proteasome inhibition; Apoptosis via caspase 8/9 | Single agent | Phase IB/II | |
| Marizomib | Pan-Proteasome inhibition; Apoptosis via caspase 8/9; UPR apoptotic response | Single agent | Phase I | |
| UPR Modulators | MKC-3946 | Inhibition of XBP1 splicing by IRE1α endoribonuclease domain inhibition | Combination treatment with bortezomib | Preclinical |
| Nelfinavir | Activation of PERK apoptotic pathway; Upregulation of CHOP; Inhibition of AKT phosphorylation | Combination treatment with bortezomib | Preclinical | |
| Combination treatment with bortezomib in R/R and progressive MM | Phase I | |||
| HDACi | Panobinostat | Broad spectrum inhibitor of HDAC leading to aggresome disruption; Apoptosis via caspase 8/9; UPR apoptotic response | Combination treatment with bortezomib and DEX (where 2 or more treatment options have been used prior) | FDA approved |
| ACY-1215 (Ricolinstat) | Selective inhibition of HDAC6 leading to aggresome disruption; Apoptosis via caspase 8/9; UPR apoptotic response | Combination treatment with bortezomib | Preclinical | |
| Combination treatment with carfilzomib | Preclinical | |||
| Combination treatment with LEN and DEX | Phase IB | |||
| Autophagy Inhibitors | Hydroxy-chloroquine | Inhibition of autophagy by increased lysosomal pH | Combination treatment with bortezomib R/R MM | Phase 1 |
| Combination treatment with carfilzomib | Preclinical | |||
| Bafilomycin A1 | Inhibition of autophagy by prevention of autophagosome/lysosome fusion | Combination treatment with bortezomib | Preclinical | |
| HSP Inhibitors | NVP-HSP990 | HSP90 inhibitor; Disruption of AKT, JAK/STAT pathways | Single agent | Preclinical |
| TAS-116 | HSP90 inhibitor Induction of apoptosis Disruption of AKT & ERK | Single agent; Combination treatment with bortezomib | Preclinical | |
| Tanespimycin | HSP90 inhibitor Induction UPR | Combination treatment with bortezomib | Phase I/II |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho Zhi Guang, M.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. https://doi.org/10.3390/cancers11010066
Ho Zhi Guang M, Kavanagh EL, Dunne LP, Dowling P, Zhang L, Lindsay S, Bazou D, Goh CY, Hanley C, Bianchi G, et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers. 2019; 11(1):66. https://doi.org/10.3390/cancers11010066
Chicago/Turabian StyleHo Zhi Guang, Matthew, Emma L. Kavanagh, Luke Paul Dunne, Paul Dowling, Li Zhang, Sinéad Lindsay, Despina Bazou, Chia Yin Goh, Cathal Hanley, Giada Bianchi, and et al. 2019. "Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis" Cancers 11, no. 1: 66. https://doi.org/10.3390/cancers11010066