Novel STAT3 Inhibitor LDOC1 Targets Phospho-JAK2 for Degradation by Interacting with LNX1 and Regulates the Aggressiveness of Lung Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
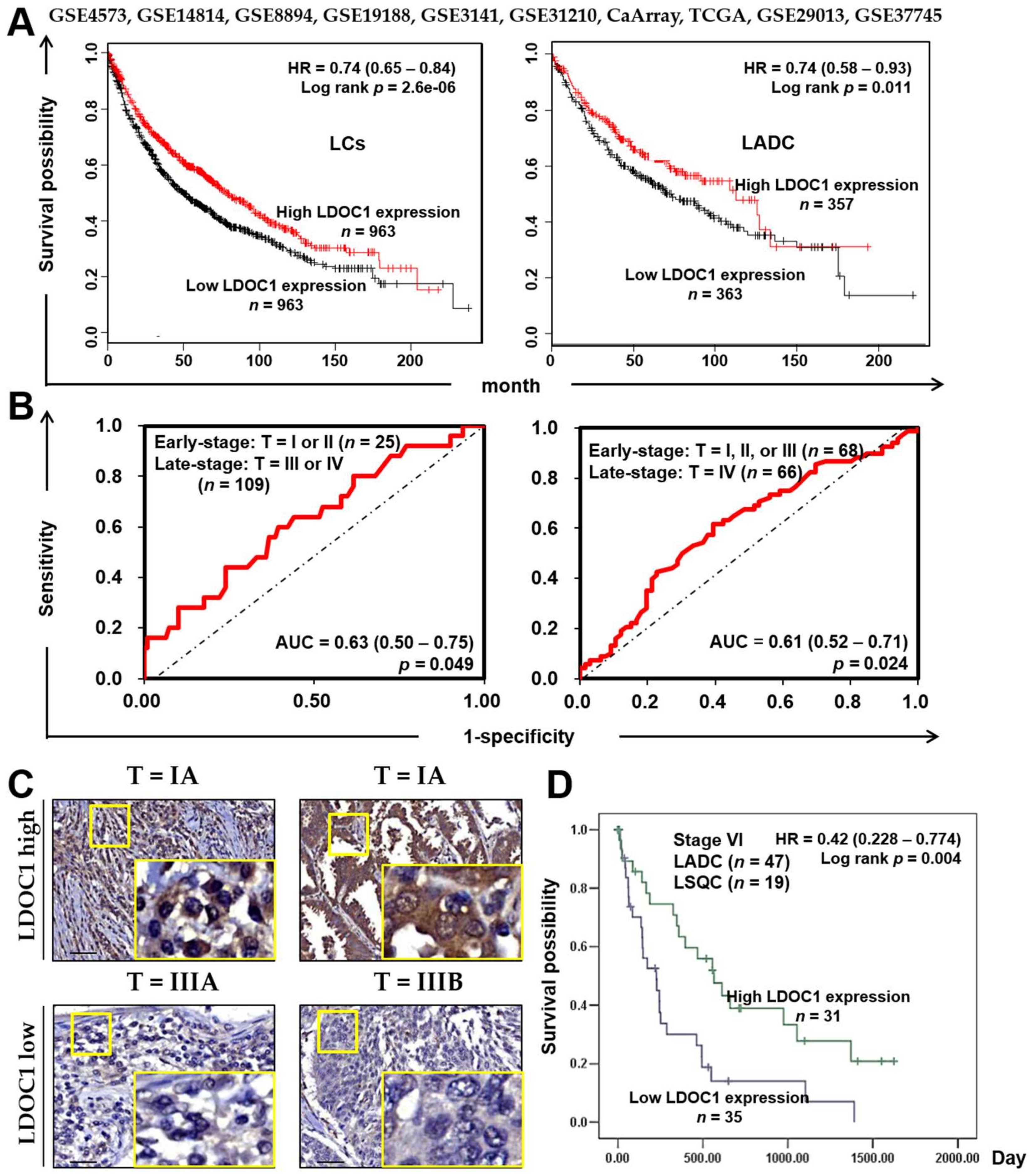
2.1. LDOC1 Was Silenced by Promoter Hypermethylation in a Cigarette Smoke Condensate (CSC)-Exposed BEAS-2B Cell Line and Was Associated with the Clinical Outcome of Patients with Lung Cancer
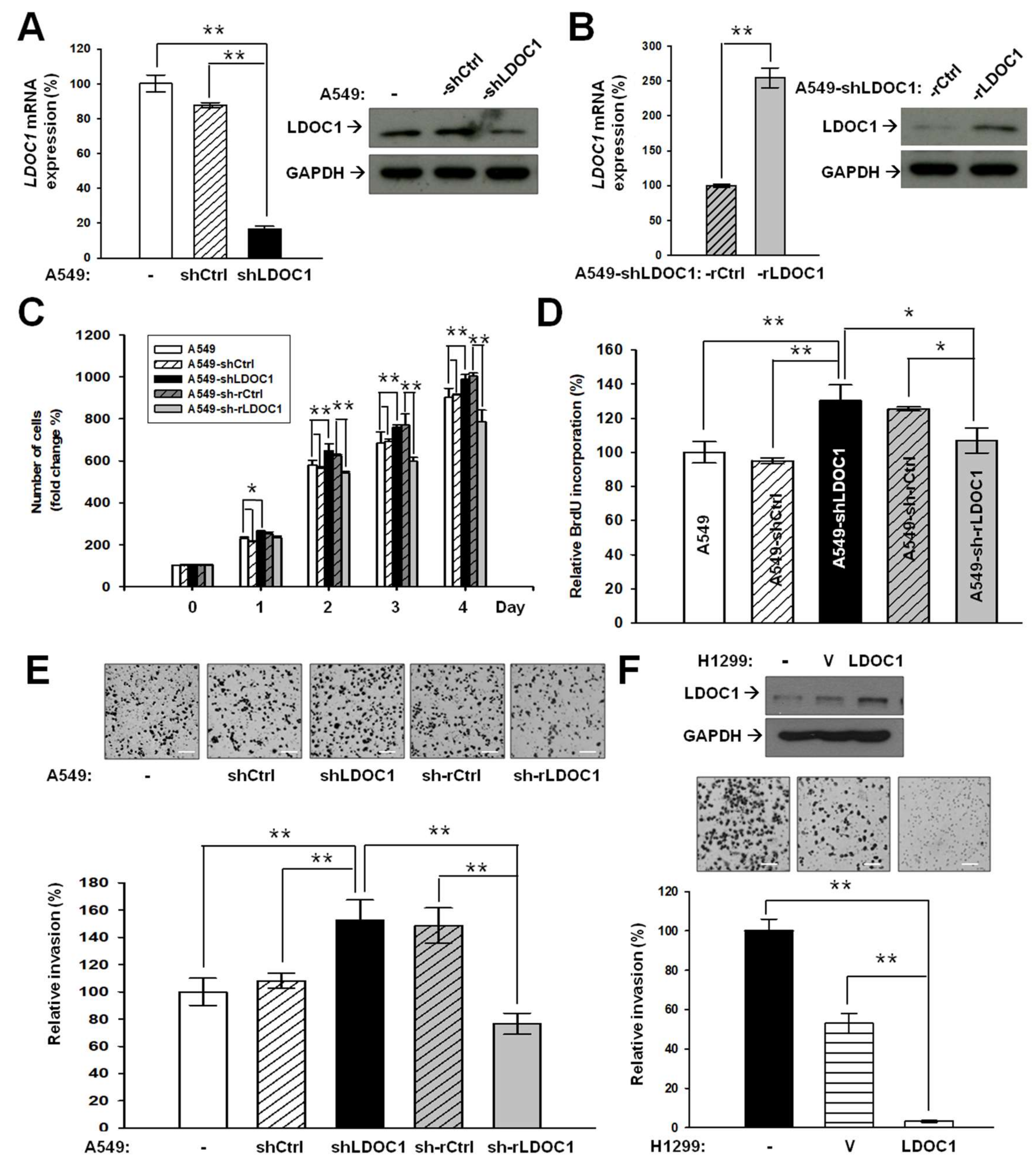
2.2. LDOC1 Knockdown Mediates Malignancy Progression of Lung Cancer In Vitro
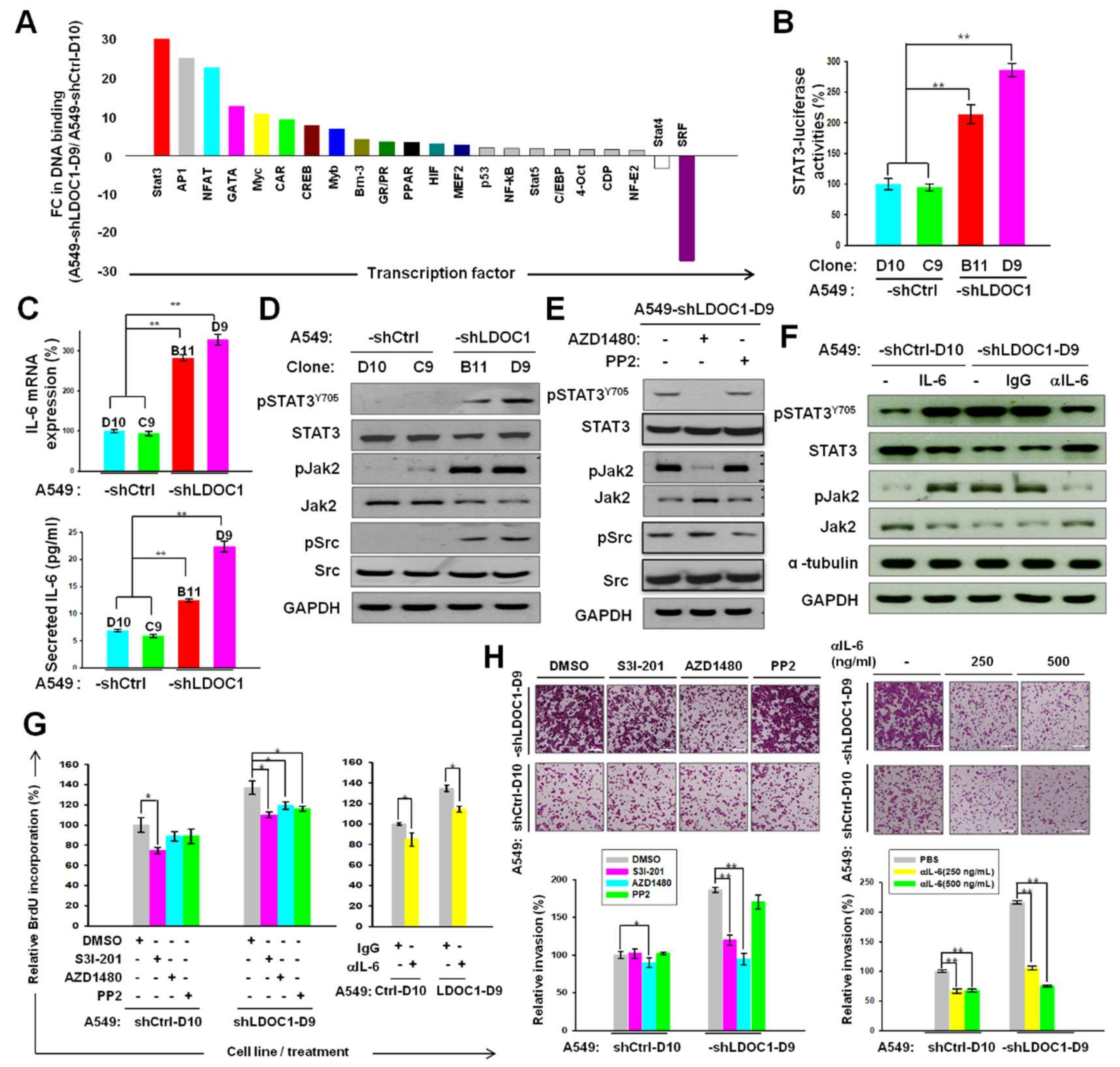
2.3. LDOC1 Knockdown Activated a Reciprocal Loop of IL-6/JAK2/STAT3, through Which LDOC1 Mediated the Aggressiveness of Lung Cancer Cells
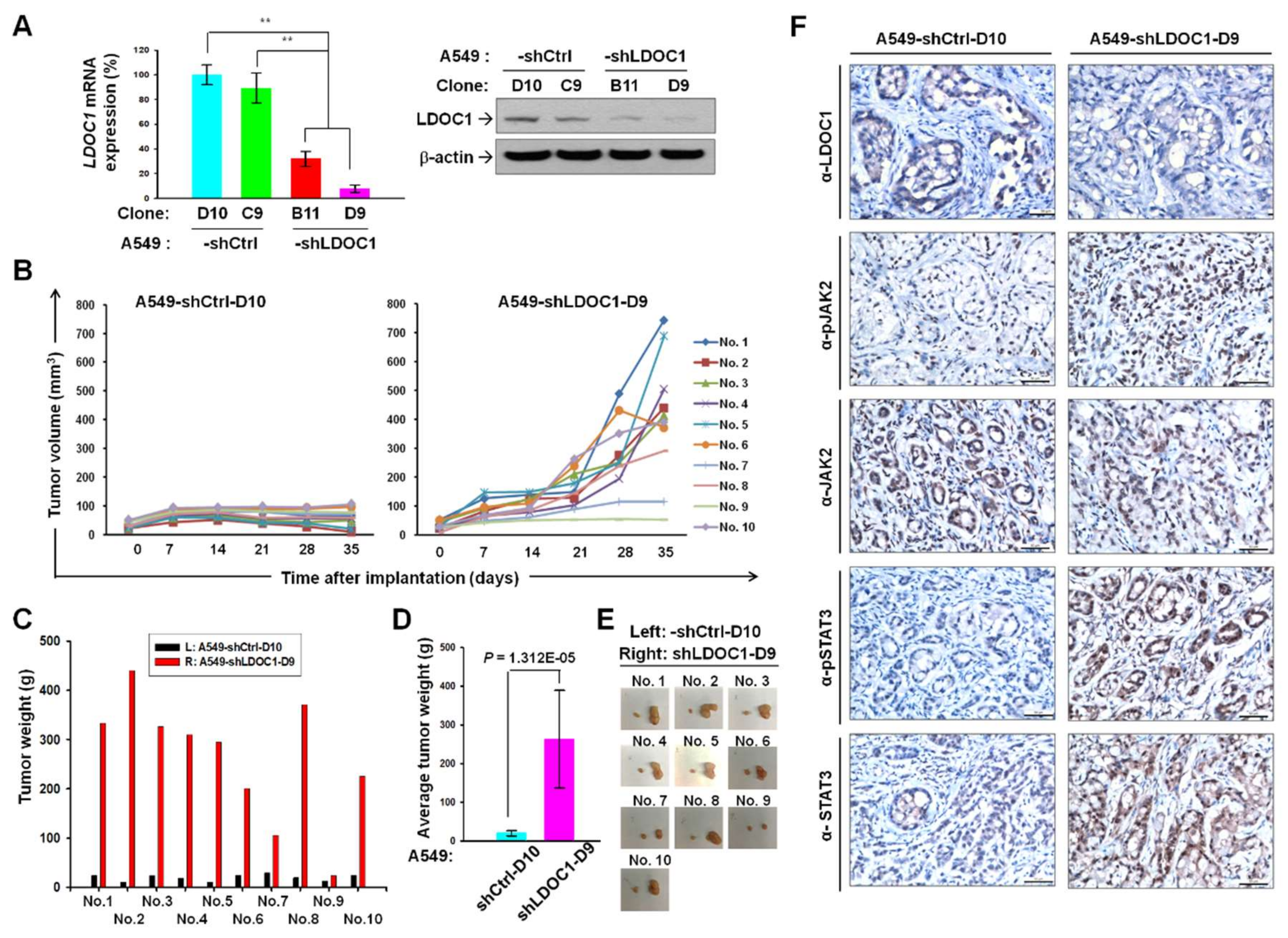
2.4. LDOC1 Knockdown Augmented Tumorigenesis and Phosphorylated JAK2 and STAT3 in Xenograft Tumor Model of Lung Cancer
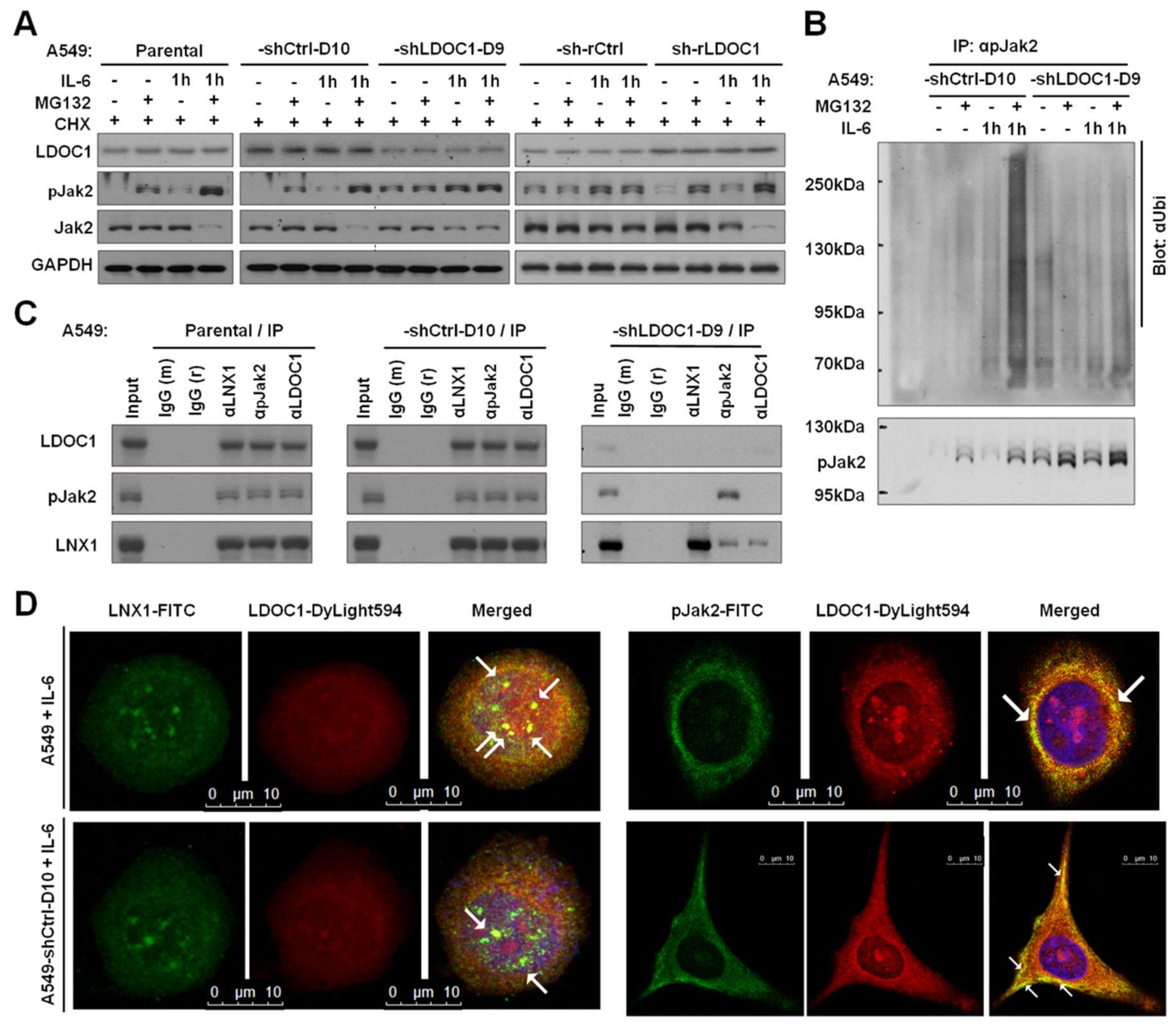
2.5. LDOC1 Associated with LNX1 and pJAK2 to Facilitate Ubiquitination–Proteasomal Degradation of pJAK2
3. Discussion
4. Methods
4.1. Chemicals and Reagents
4.2. Preparation and Treatment of Cigarette Smoke Condensate (CSC)
4.3. Cell Lines and Transfection
4.4. Bisulfite Conversion of Genomic DNA and Quantitative Methylation-Specific PCR (qMSP)
4.5. Quantitative Real-Time PCR (qPCR)
4.6. Study Subjects and Human Tissue Microarray (TMA) Constructs
4.7. Immunohistochemistry (IHC) and Quantitative Staining Measurement of IHC
4.8. Cell Viability and Proliferation Assay
4.9. BrdU Incorporation Assay
4.10. Trans-Well Invasion Assay
4.11. Soft Agar Assay
4.12. Transcription Factor (TF) Profiling Array
4.13. Luciferase Reporter Assay
4.14. IL-6 ELISA Assay
4.15. Immunoprecipitation and Western Blot Analysis
4.16. Mouse Xenogaft Tumor Model
4.17. Immunofluorescence Assay (IFA) and Confocal Microscopy
4.18. Statistical Analysis
4.19. Availability of Data and Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–146. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.; Daley, A.; Begh, R.; Aveyard, P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: Systematic review of observational studies with meta-analysis. BMJ 2010, 340, b5569. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wong, T.S.; Chan, J.Y.; Lu, S.C.; Lin, P.; Cheng, A.J.; Chen, Y.J.; Chang, J.S.; Hsiao, S.H.; Leu, Y.W.; et al. Epigenetic regulation of the X-linked tumour suppressors BEX1 and LDOC1 in oral squamous cell carcinoma. J. Pathol. 2013, 230, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Pan, K.L.; Tang, Y.C.; Tsai, M.H.; Cheng, A.J.; Shen, M.Y.; Cheng, Y.M.; Huang, T.T.; Lin, P. LDOC1 silenced by cigarette exposure and involved in oral neoplastic transformation. Oncotarget 2015, 6, 25188–25201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaki, K.; Manabe, T.; Hanzawa, H.; Maass, N.; Tsukada, T. Yamaguchi, K. Identification of a novel gene, LDOC1, down-regulated in cancer cell lines. Cancer Lett. 1999, 140, 227–234. [Google Scholar] [CrossRef]
- Thoompumkal, I.J.; Rehna, K.; Anbarasu, K.; Mahalingam, S. Leucine Zipper Down-regulated in Cancer-1 (LDOC1) interacts with Guanine nucleotide binding protein-like 3-like (GNL3L) to modulate Nuclear Factor-kappa B (NF-κB) signaling during cell proliferation. Cell Cycle 2016, 15, 3251–3267. [Google Scholar] [CrossRef]
- Thiebaut, R.; Esmiol, S.; Lecine, P.; Mahfouz, B.; Hermant, A.; Nicoletti, C.; Parnis, S.; Perroy, J.; Borg, J.P.; Pascoe, L.; et al. Characterization and Genetic Analyses of New Genes Coding for NOD2 Interacting Proteins. PLoS ONE 2016, 11, e0165420. [Google Scholar] [CrossRef]
- Buchholtz, M.L.; Juckstock, J.; Weber, E.; Mylonas, I.; Dian, D.; Bruning, A. Loss of LDOC1 expression by promoter methylation in cervical cancer cells. Cancer Investig. 2013, 31, 571–577. [Google Scholar] [CrossRef]
- Buchholtz, M.L.; Bruning, A.; Mylonas, I.; Juckstock, J. Epigenetic silencing of the LDOC1 tumor suppressor gene in ovarian cancer cells. Arch. Gynecol. Obstet. 2014, 290, 149–154. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Q.; Li, Z.; Ma, X.; Wu, L.; Ji, H.; Qin, G. LDOC1 inhibits proliferation and promotes apoptosis by repressing NF-κB activation in papillary thyroid carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 146. [Google Scholar] [CrossRef]
- Yong, B.C.; Lu, J.C.; Xie, X.B.; Su, Q.; Tan, P.X.; Tang, Q.L.; Wang, J.; Huang, G.; Han, J.; Xu, H.W.; et al. LDOC1 regulates Wnt5a expression and osteosarcoma cell metastasis and is correlated with the survival of osteosarcoma patients. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duzkale, H.; Schweighofer, C.D.; Coombes, K.R.; Barron, L.L.; Ferrajoli, A.; O’Brien, S.; Wierda, W.G.; Pfeifer, J.; Majewski, T.; Czerniak, B.A.; et al. LDOC1 mRNA is differentially expressed in chronic lymphocytic leukemia and predicts overall survival in untreated patients. Blood 2011, 117, 4076–4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko-Ishino, T.; Ishino, F. Evolution of brain functions in mammals and LTR retrotransposon-derived genes. Uirusu 2016, 66, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Henke, C.; Strissel, P.L.; Schubert, M.T.; Mitchell, M.; Stolt, C.C.; Faschingbauer, F.; Beckmann, M.W.; Strick, R. Selective expression of sense and antisense transcripts of the sushi-ichi-related retrotransposon—Derived family during mouse placentogenesis. Retrovirology 2015, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Imakawa, K.; Nakagawa, S. The phylogeny of placental evolution through dynamic integrations of retrotransposons. Prog. Mol. Biol. Transl. Sci. 2017, 145, 89–109. [Google Scholar] [PubMed]
- Naruse, M.; Ono, R.; Irie, M.; Nakamura, K.; Furuse, T.; Hino, T.; Oda, K.; Kashimura, M.; Yamada, I.; Wakana, S.; et al. Sirh7/Ldoc1 knockout mice exhibit placental P4 overproduction and delayed parturition. Development 2014, 141, 4763–4771. [Google Scholar] [CrossRef] [Green Version]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef]
- Chang, C.H.; Hsiao, C.F.; Yeh, Y.M.; Chang, G.C.; Tsai, Y.H.; Chen, Y.M.; Huang, M.S.; Chen, H.L.; Li, Y.J.; Yang, P.C.; et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int. J. Cancer 2013, 132, 1977–1985. [Google Scholar] [CrossRef]
- Songur, N.; Kuru, B.; Kalkan, F.; Ozdilekcan, C.; Cakmak, H.; Hizel, N. Serum interleukin-6 levels correlate with malnutrition and survival in patients with advanced non-small cell lung cancer. Tumori 2004, 90, 196–200. [Google Scholar] [CrossRef]
- Jiang, R.; Jin, Z.; Liu, Z.; Sun, L.; Wang, L.; Li, K. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol. Diagn. Ther. 2011, 15, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Harada, D.; Takigawa, N.; Kiura, K. The role of STAT3 in non-small cell lung cancer. Cancers 2014, 6, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhang, Y.; Li, Y.; Lv, T.; Liu, J.; Wang, X. Prognostic significance of STAT3 expression and its correlation with chemoresistance of non-small cell lung cancer cells. Acta Histochem. 2012, 114, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008, 18, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagstrom, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, K.S.; Alberobello, A.T.; Kallakury, B.; Weng, M.T.; Wang, Y.; Giaccone, G. The Janus kinases inhibitor AZD1480 attenuates growth of small cell lung cancers in vitro and in vivo. Clin. Cancer Res. 2013, 19, 6777–6786. [Google Scholar] [CrossRef]
- Xin, H.; Herrmann, A.; Reckamp, K.; Zhang, W.; Pal, S.; Hedvat, M.; Zhang, C.; Liang, W.; Scuto, A.; Weng, S.; et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011, 71, 6601–6610. [Google Scholar] [CrossRef] [PubMed]
- Looyenga, B.D.; Hutchings, D.; Cherni, I.; Kingsley, C.; Weiss, G.J.; Mackeigan, J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS ONE 2012, 7, e30820. [Google Scholar] [CrossRef] [PubMed]
- Baffoe-Bonnie, A.B.; Smith, J.R.; Stephan, D.A.; Schleutker, J.; Carpten, J.D.; Kainu, T.; Gillanders, E.M.; Matikainen, M.; Teslovich, T.M.; Tammela, T.; et al. A major locus for hereditary prostate cancer in Finland: Localization by linkage disequilibrium of a haplotype in the HPCX region. Hum. Genet. 2005, 117, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Lin, J.H.; Hsu, T.W.; Su, K.; Li, A.F.; Hsu, H.S.; Hung, S.C. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int. J. Cancer 2015, 136, 547–559. [Google Scholar] [PubMed]
- Liu, X.; Du, L.; Feng, R. c-Src regulates cell cycle proteins expression through protein kinase B/glycogen synthase kinase 3 beta and extracellular signal-regulated kinases 1/2 pathways in MCF-7 cells. Acta Biochim. Biophys. Sin. 2013, 45, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M.; et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef] [Green Version]
- Chaib, I.; Karachaliou, N.; Pilotto, S.; Codony Servat, J.; Cai, X.; Li, X.; Drozdowskyj, A.; Servat, C.C.; Yang, J.; Hu, C.; et al. Co-activation of STAT3 and YES-Associated Protein 1 (YAP1) Pathway in EGFR-Mutant NSCLC. J. Natl. Cancer Inst. 2017, 109, djx014. [Google Scholar] [CrossRef]
- Codony-Servat, C.; Codony-Servat, J.; Karachaliou, N.; Molina, M.A.; Chaib, I.; Ramirez, J.L.; de Los Llanos Gil, M.; Solca, F.; Bivona, T.G.; Rosell, R. Activation of signal transducer and activator of transcription 3 (STAT3) signaling in EGFR mutant non-small-cell lung cancer (NSCLC). Oncotarget 2017, 8, 47305–47316. [Google Scholar] [CrossRef] [Green Version]
- Koh, J.; Jang, J.Y.; Keam, B.; Kim, S.; Kim, M.Y.; Go, H.; Kim, T.M.; Kim, D.W.; Kim, C.W.; Jeon, Y.K.; et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1alpha and STAT3. Oncoimmunology 2016, 5, e1108514. [Google Scholar] [CrossRef]
- Cuyas, E.; Perez-Sanchez, A.; Micol, V.; Menendez, J.A.; Bosch-Barrera, J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle 2016, 15, 3413–3418. [Google Scholar] [CrossRef]
- Qu, Z.; Sun, F.; Zhou, J.; Li, L.; Shapiro, S.D.; Xiao, G. Interleukin-6 prevents the initiation but enhances the progression of lung cancer. Cancer Res. 2015, 75, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Syu, S.H.; Liu, K.J.; Chu, P.Y.; Yang, W.C.; Lin, P.; Shieh, W.Y. Interleukin-1 beta transactivates epidermal growth factor receptor via the CXCL1-CXCR2 axis in oral cancer. Oncotarget 2015, 6, 38866–38880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Chang, J.S.; Syu, S.H.; Wong, T.S.; Chan, J.Y.; Tang, Y.C.; Yang, Z.P.; Yang, W.C.; Chen, C.T.; Lu, S.C.; et al. IL-1beta promotes malignant transformation and tumor aggressiveness in oral cancer. J. Cell Physiol. 2015, 230, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, D.; Saharinen, P.; Junttila, I.; Hilton, D.J.; Silvennoinen, O. Regulation of JAK2 through the ubiquitin-proteasome pathway involves phosphorylation of JAK2 on Y1007 and interaction with SOCS-1. Mol. Cell Biol. 2002, 22, 3316–3326. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-H.; Yang, J.-R.; Chen, C.-Y.; Tsai, M.-H.; Hung, P.-F.; Chen, S.-J.; Chiang, S.-L.; Chang, H.; Lin, P. Novel STAT3 Inhibitor LDOC1 Targets Phospho-JAK2 for Degradation by Interacting with LNX1 and Regulates the Aggressiveness of Lung Cancer. Cancers 2019, 11, 63. https://doi.org/10.3390/cancers11010063
Lee C-H, Yang J-R, Chen C-Y, Tsai M-H, Hung P-F, Chen S-J, Chiang S-L, Chang H, Lin P. Novel STAT3 Inhibitor LDOC1 Targets Phospho-JAK2 for Degradation by Interacting with LNX1 and Regulates the Aggressiveness of Lung Cancer. Cancers. 2019; 11(1):63. https://doi.org/10.3390/cancers11010063
Chicago/Turabian StyleLee, Chia-Huei, Ji-Rui Yang, Chih-Yu Chen, Ming-Hsien Tsai, Pin-Feng Hung, Shin-Jih Chen, Shang-Lun Chiang, Han Chang, and Pinpin Lin. 2019. "Novel STAT3 Inhibitor LDOC1 Targets Phospho-JAK2 for Degradation by Interacting with LNX1 and Regulates the Aggressiveness of Lung Cancer" Cancers 11, no. 1: 63. https://doi.org/10.3390/cancers11010063