
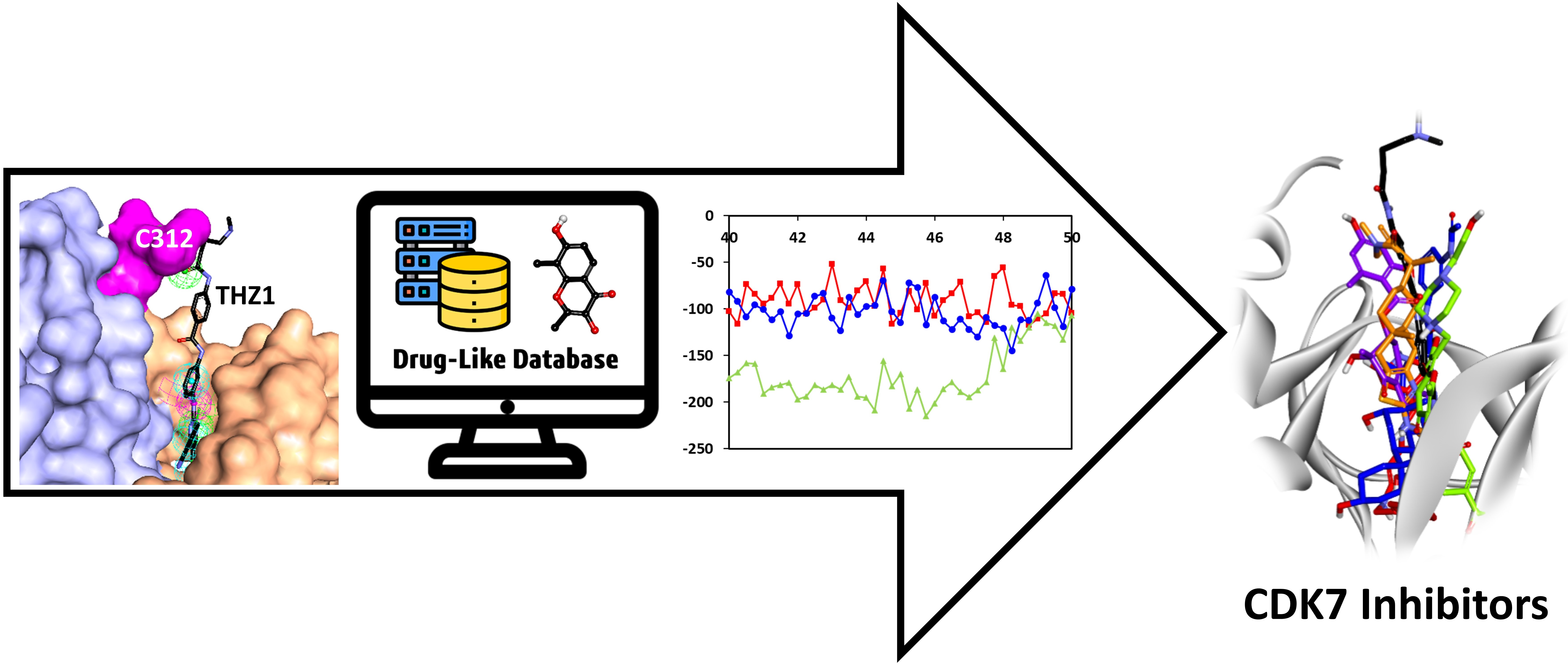
Identification of CDK7 Inhibitors from Natural Sources Using Pharmacoinformatics and Molecular Dynamics Simulations

 ,
,  ,
,  , ,
, ,
Abstract
:
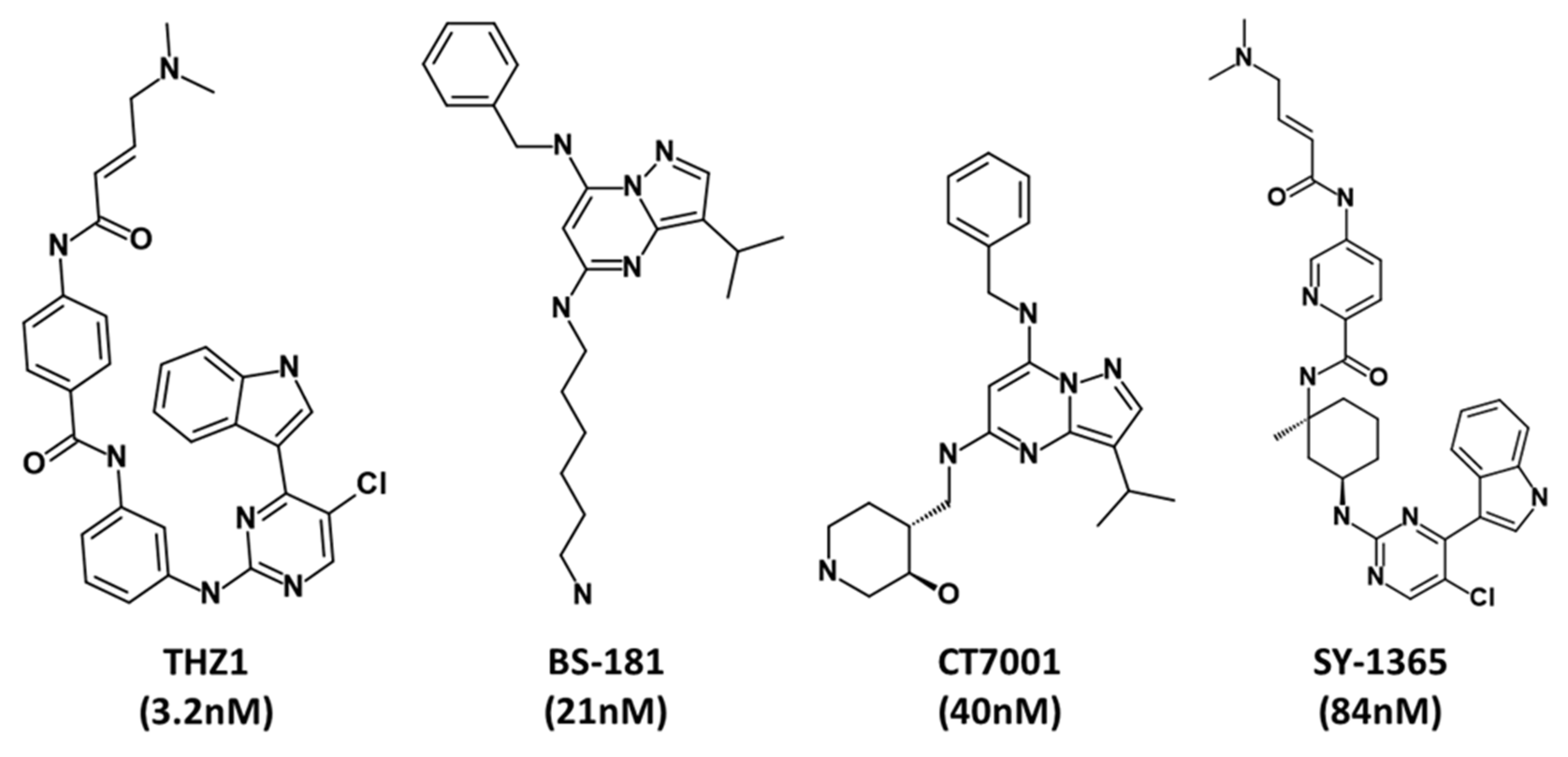
1. Introduction
2. Materials and Methods
2.1. Ligand-Based Pharmacophore Generation
2.2. Structure-Based Pharmacophore Generation
2.3. Validation of the Pharmacophore
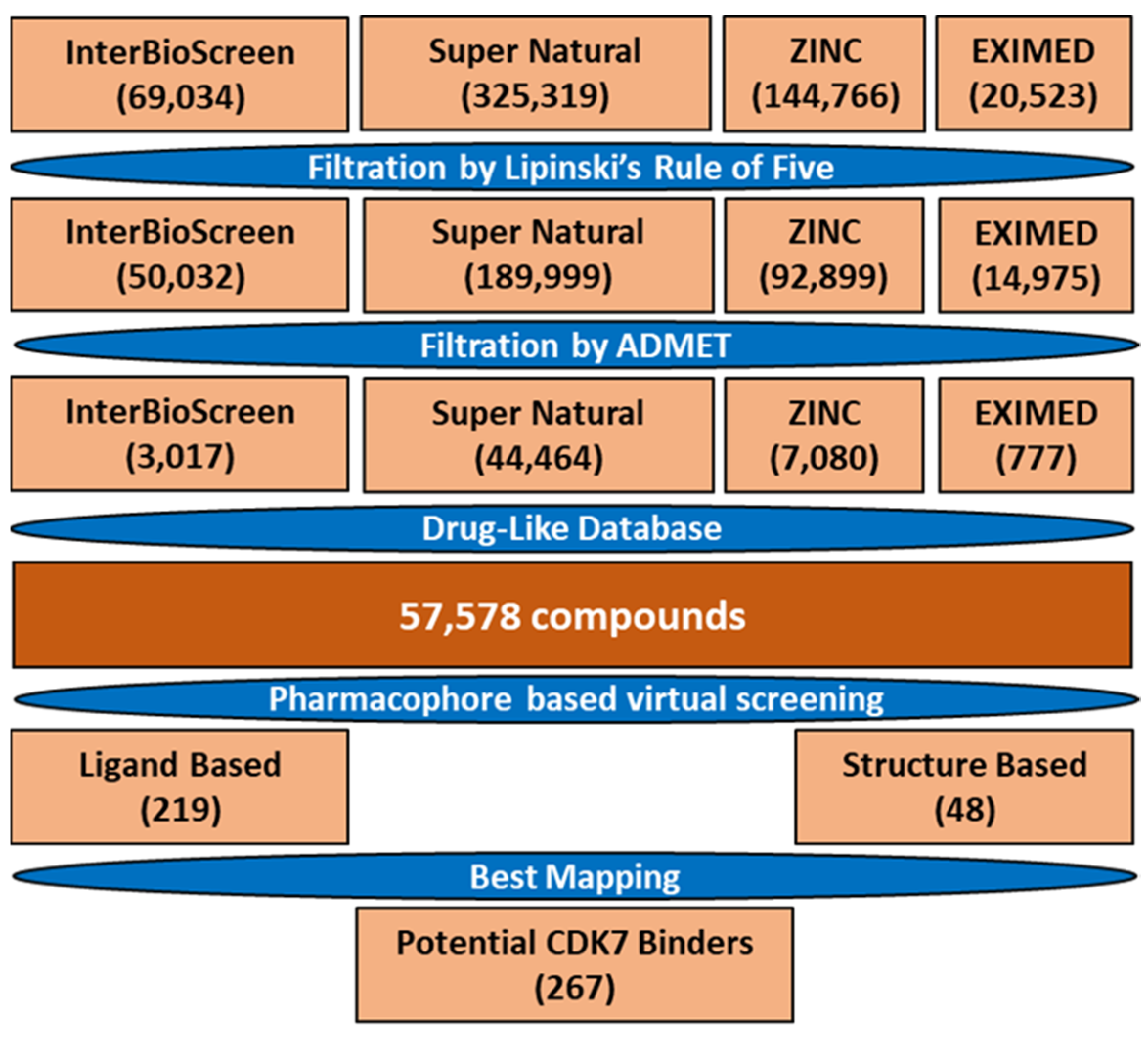
2.4. Drug-like Database Generation and Virtual Screening
2.5. Molecular Docking
2.6. Molecular Dynamics Simulation
2.7. Binding Free Energy Calculations
2.8. In Silico Specificity over CDK2
2.9. In Silico Prediction of Pharmacokinetic Properties
3. Results
3.1. Ligand-Based Pharmacophore Generation
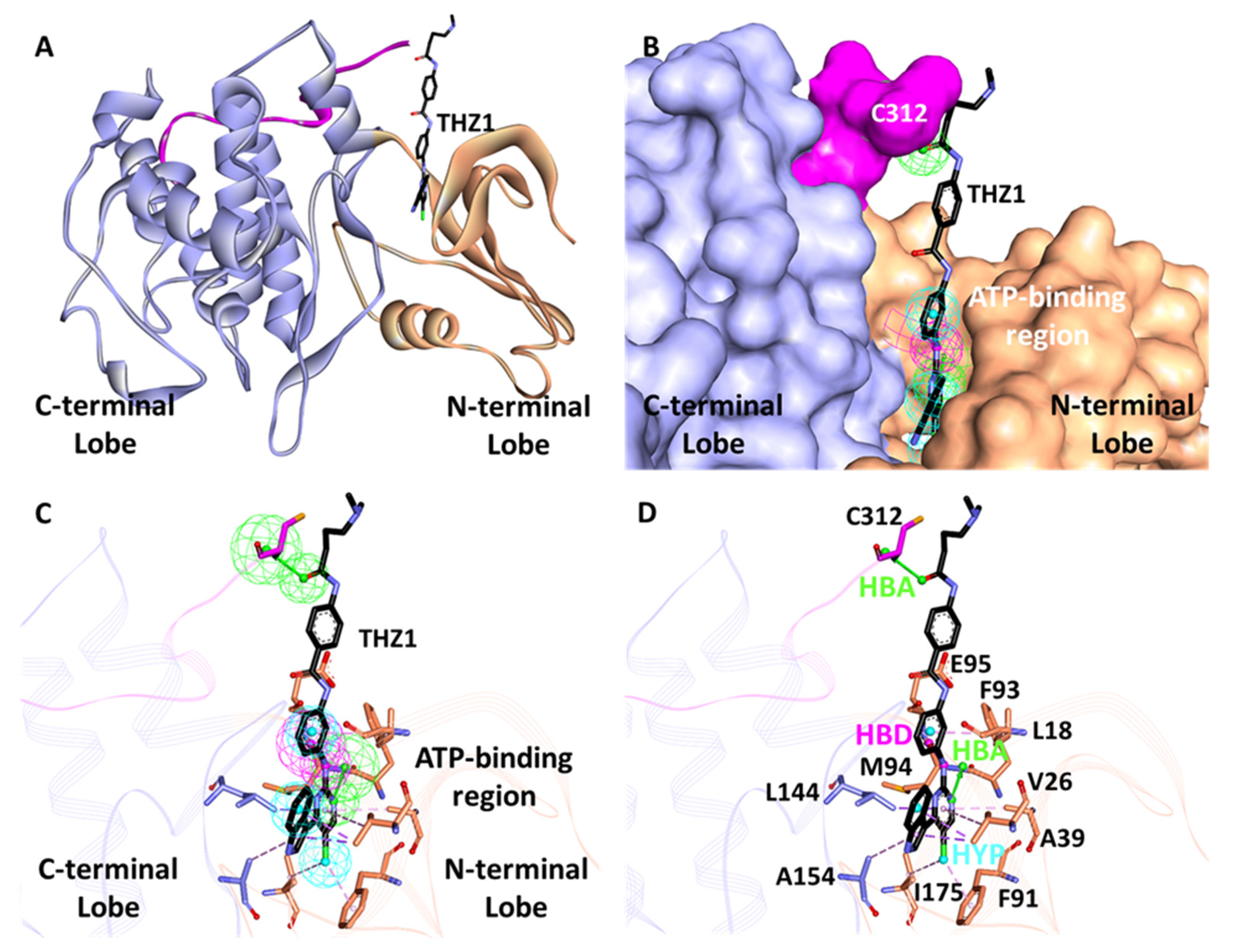
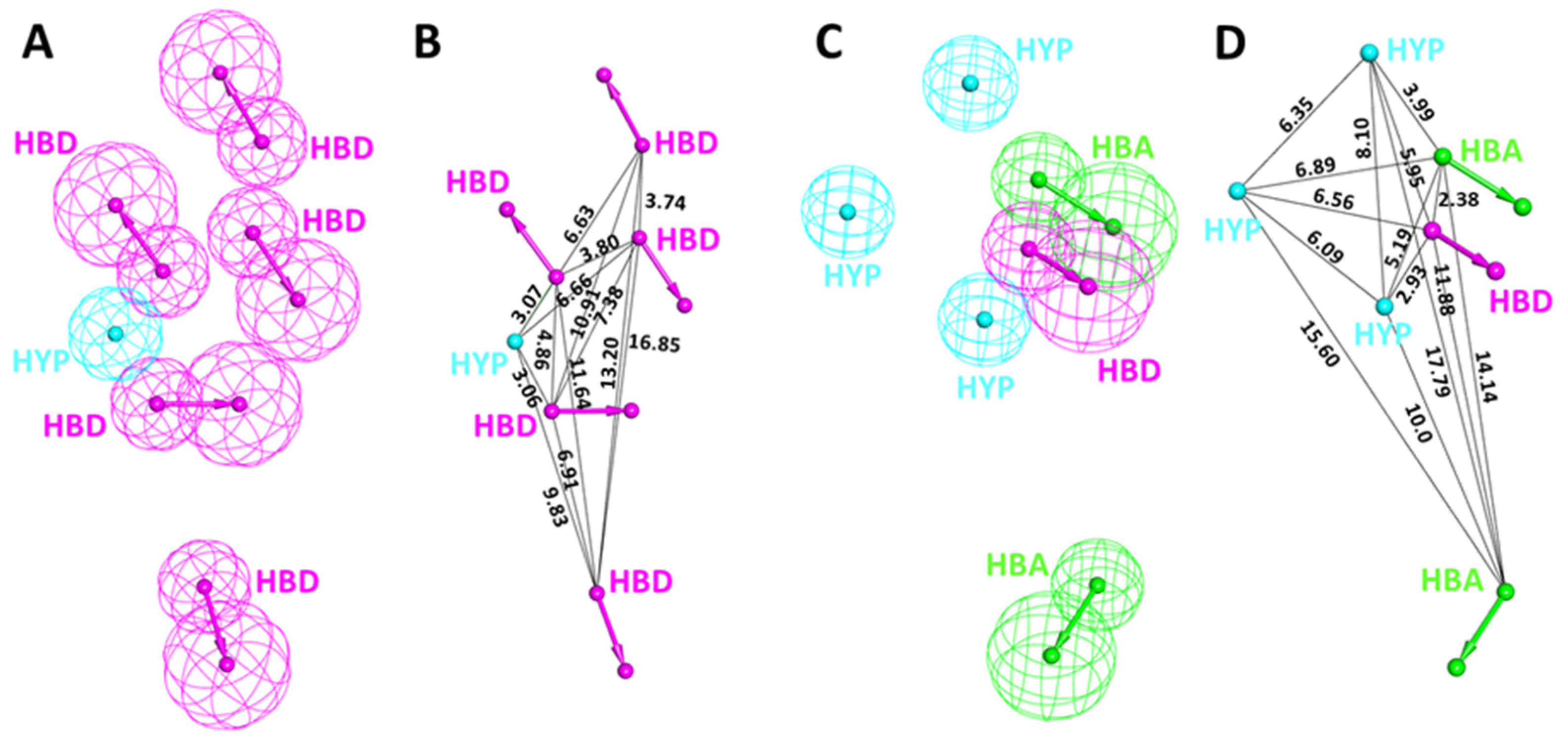
3.2. Structure-Based Pharmacophore Generation
3.3. Pharmacophore Validation
Hypothesis Comparison
3.4. Drug-like Database and Virtual Screening
3.5. Molecular Docking
3.6. Molecular Dynamics Simulations
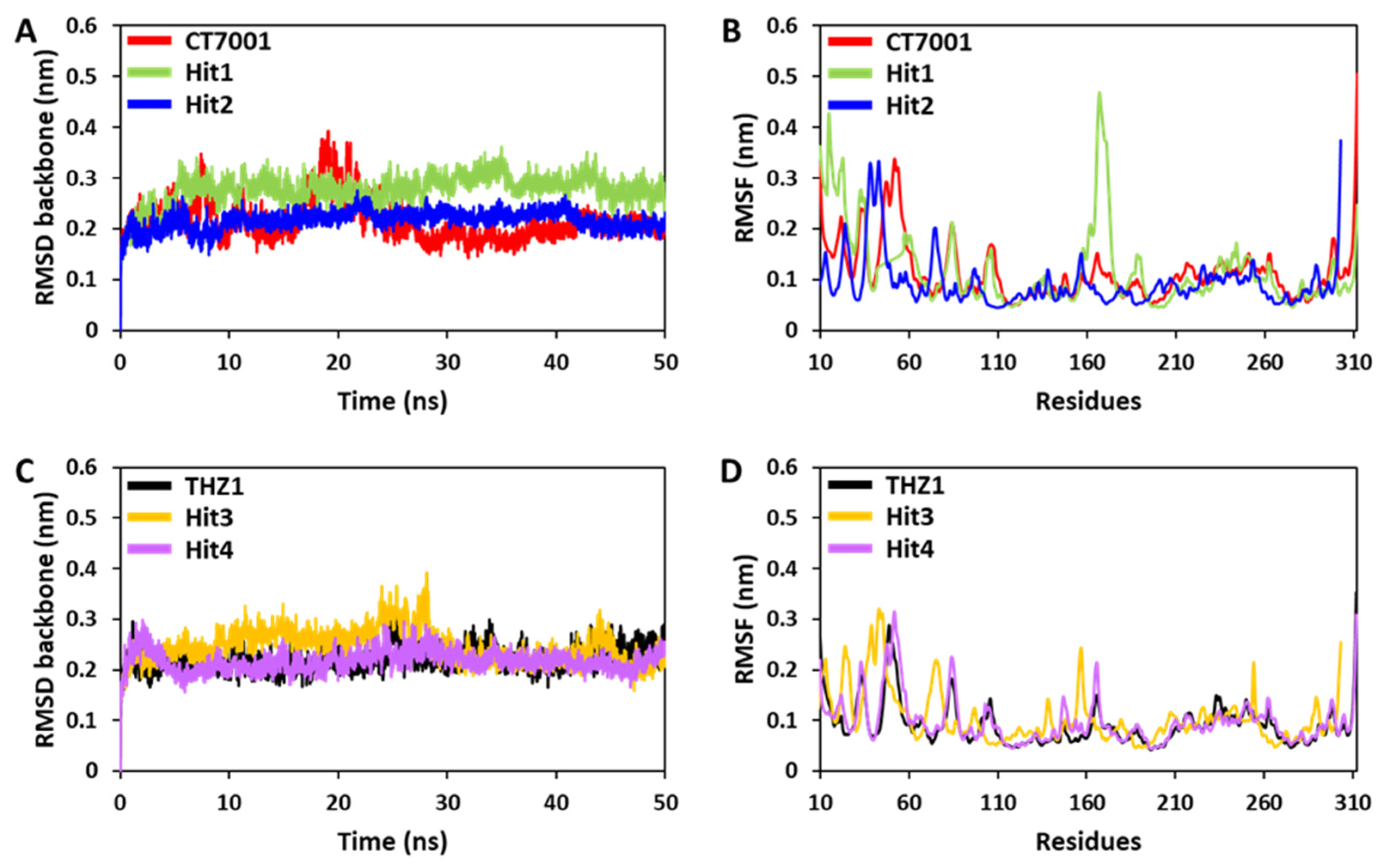
3.6.1. Root Mean Square Deviation and Fluctuations
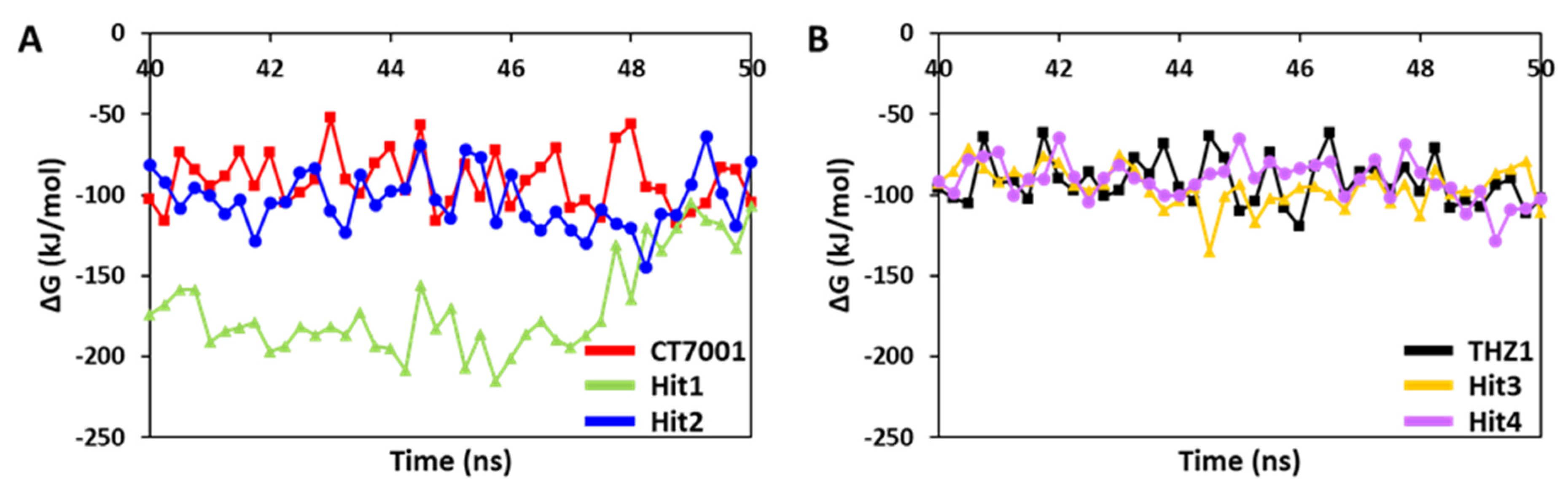
3.6.2. Binding Free Energy Analysis
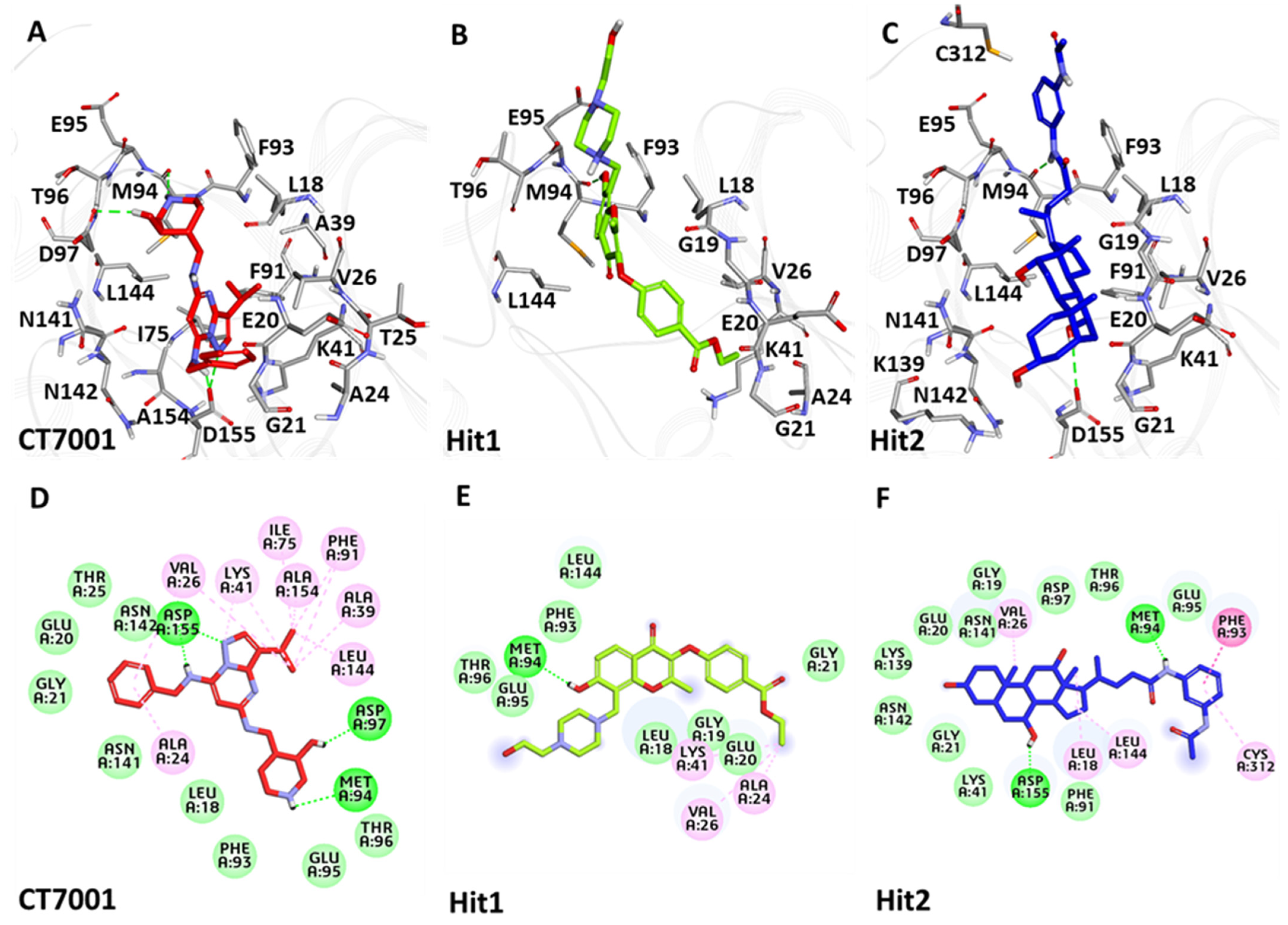
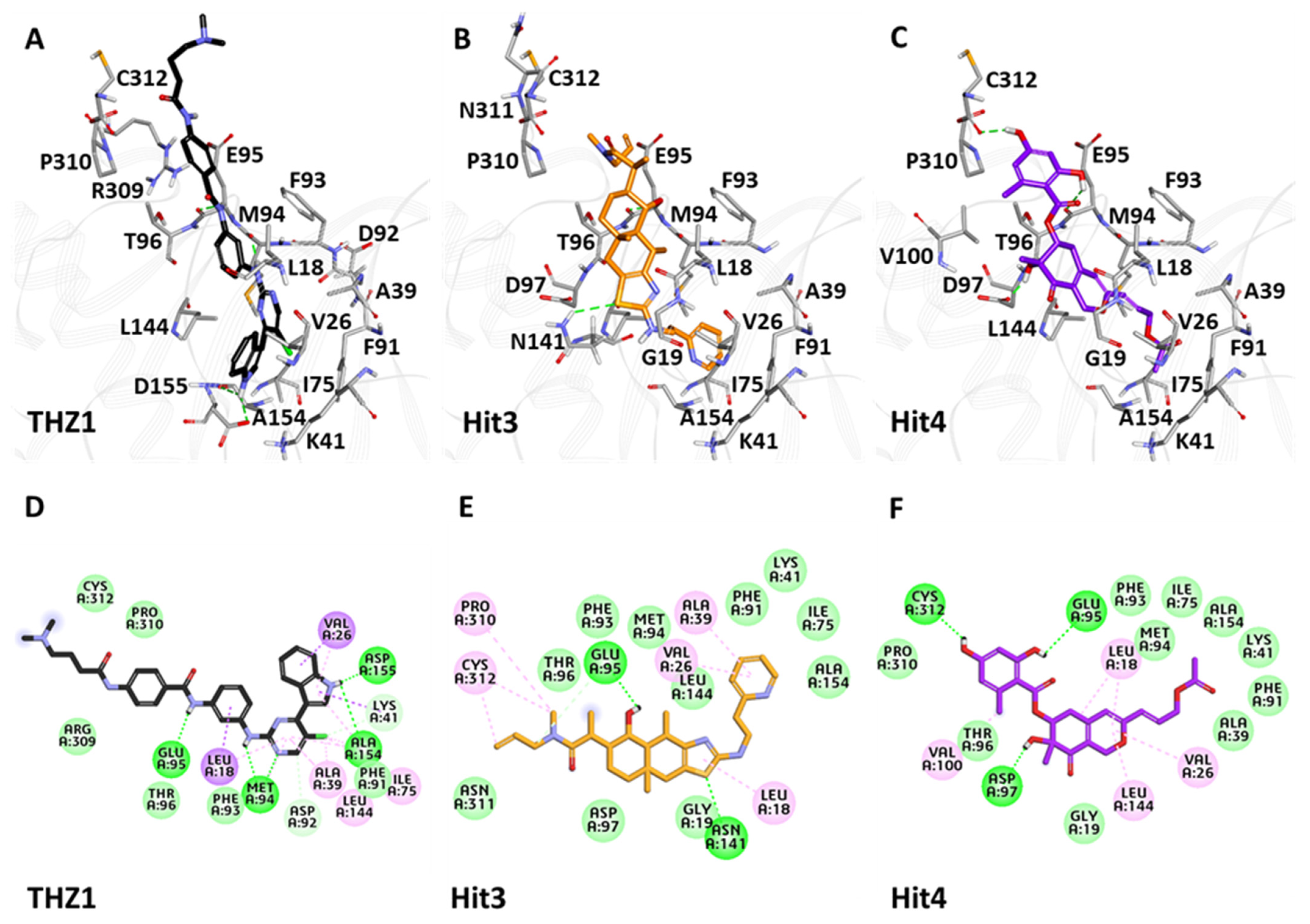
3.6.3. Binding Mode Analysis
3.7. Specificity of Inhibitors and Hits with CDK7 over CDK2
3.8. In Silico Prediction of Pharmacokinetic Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cy-clin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2019, 10, 118–136. [Google Scholar] [CrossRef]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassan, J.P.; Jaquenoud, M.; Fry, A.M.; Frutiger, S.; Hughes, G.J.; Nigg, E. In vitro assembly of a functional human CDK7-cyclin H complex requires MAT1, a novel 36 kDa RING finger protein. EMBO J. 1995, 14, 5608–5617. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.-M.; Afshar, M.; Martin, F.; Cavadore, J.; Labbé, J.; Dorée, M. Dual phosphorylation of the T-loop in cdk7: Its role in controlling cyclin H binding and CAK activity. EMBO J. 1997, 16, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Shilatifard, A. Inhibit Globally, Act Locally: CDK7 Inhibitors in Cancer Therapy. Cancer Cell 2014, 26, 158–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, Y.; Rajpara, S.M.; Reza, S.M.; Lees, E.; Shuman, S.; Mathews, M.B.; Pe’Ery, T. Three RNA Polymerase II Carboxyl-terminal Domain Kinases Display Distinct Substrate Preferences. J. Biol. Chem. 2001, 276, 10913–10920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover-Cutter, K.; Larochelle, S.; Erickson, B.; Zhang, C.; Shokat, K.; Fisher, R.P.; Bentley, D.L. TFIIH-Associated Cdk7 Kinase Functions in Phosphorylation of C-Terminal Domain Ser7 Residues, Promoter-Proximal Pausing, and Termination by RNA Polymerase II. Mol. Cell. Biol. 2009, 29, 5455–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkova, J.; Zemanova, M.; Bartek, J. Expression of CDK7/CAK in normal and tumour cells of diverse histogenesis, cell-cycle position and differentiation. Int. J. Cancer 1996, 66, 732–737. [Google Scholar] [CrossRef]
- Kim, J.; Cho, Y.-J.; Ryu, J.-Y.; Hwang, I.; Han, H.D.; Ahn, H.J.; Kim, W.Y.; Cho, H.; Chung, J.-Y.; Hewitt, S.M.; et al. CDK7 is a reliable prognostic factor and novel therapeutic target in epithelial ovarian cancer. Gynecol. Oncol. 2020, 156, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.-C.; Liu, J.; Zhang, M.; Zhang, S.; et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef] [Green Version]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef]
- Lu, P.; Geng, J.; Zhang, L.; Wang, Y.; Niu, N.; Fang, Y.; Liu, F.; Shi, J.; Zhang, Z.-G.; Sun, Y.-W.; et al. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene 2019, 38, 3932–3945. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Abduljabbar, R.; Lai, C.-F.; Periyasamy, M.; Harrod, A.; Gemma, C.; Steel, J.H.; Patel, N.; Busonero, C.; Jerjees, D.; et al. Expression of CDK7, Cyclin H, and MAT1 Is Elevated in Breast Cancer and Is Prognostic in Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2016, 22, 5929–5938. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, M.; Zhang, X.; Huang, H.; Huang, J.; Ke, J.; Ding, H.; Xiao, J.; Shan, X.; Liu, Q.; et al. Upregulation of CDK7 in gastric cancer cell promotes tumor cell proliferation and predicts poor prognosis. Exp. Mol. Pathol. 2016, 100, 514–521. [Google Scholar] [CrossRef]
- Naseh, G.; Mohammadifard, M.; Mohammadifard, M. Upregulation of cyclin-dependent kinase 7 and matrix metalloproteinase-14 expression contribute to metastatic properties of gastric cancer. IUBMB Life 2016, 68, 799–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, F.H.; Law, C.; Tang, T.C.; Cheng, C.L.; Chin, D.W.; Tam, W.V.; Wei, L.; Wong, C.C.L.; Ng, I.O.; Wong, C. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology 2019, 69, 2502–2517. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, R.; Wu, Y.; Diao, P.; Zhang, W.; Li, J.; Li, Z.; Wang, Y.; Cheng, J.; Yang, J. Overexpression of CDK7 is associated with unfavourable prognosis in oral squamous cell carcinoma. Pathology 2019, 51, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Ding, X.; Xu, G.; Chen, G.; Cao, Y.; Peng, C.; Shen, S.; Lv, Y.; Wang, L.; Zou, X. CDK7 inhibitor THZ1 inhibits MCL1 synthesis and drives cholangiocarcinoma apoptosis in combination with BCL2/BCL-XL inhibitor ABT-263. Cell Death Dis. 2019, 10, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lu, L.; Jiang, G.; Chen, Z.; Li, J.; An, P.; Chen, L.; Du, J.; Wang, H.-S. Targeting CDK7 increases the stability of Snail to promote the dissemination of colorectal cancer. Cell Death Differ. 2019, 26, 1442–1452. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.P. Cdk7: A kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2018, 10, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 inhibitors as anti-cancer drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Lolli, G.; Lowe, E.D.; Brown, N.R.; Johnson, L.N. The Crystal Structure of Human CDK7 and Its Protein Recognition Properties. Structure 2004, 12, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, T.; Zhang, X.; Wu, X.; Jiang, S. Cyclin-dependent kinase 7 inhibitors in cancer therapy. Futur. Med. Chem. 2020, 12, 813–833. [Google Scholar] [CrossRef]
- Diab, S.; Yu, M.; Wang, S. CDK7 Inhibitors in Cancer Therapy: The Sweet Smell of Success? J. Med. Chem. 2020, 63, 7458–7474. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorganic Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Ali, S.; Heathcote, D.A.; Kroll, S.H.B.; Jogalekar, A.S.; Scheiper, B.; Patel, H.; Brackow, J.; Siwicka, A.; Fuchter, M.; Periyasamy, M.; et al. The Development of a Selective Cyclin-Dependent Kinase Inhibitor That Shows Antitumor Activity. Cancer Res. 2009, 69, 6208–6215. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Ainscow, E.; Peall, A.; Thomson, S.; Leishman, A.; Elaine, S.; Ali, S.; Coombes, R.; Barrett, A.; Bahl, A.K. CT7001, a Novel Orally Bio-Available CDK7 Inhibitor, Is Highly Active in in-Vitro and in-Vivo Models of AML. Blood 2017, 130, 2645. [Google Scholar] [CrossRef]
- Patel, H.; Periyasamy, M.; Sava, G.; Bondke, A.; Slafer, B.W.; Kroll, S.H.B.; Barbazanges, M.; Starkey, R.; Ottaviani, S.; Harrod, A.; et al. ICEC0942, an Orally Bioavailable Selective Inhibitor of CDK7 for Cancer Treatment. Mol. Cancer Ther. 2018, 17, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, B.; Agrawal, N.; Kaushik, S. Pharmacophore Development. In Encyclopedia of Bioinformatics and Computational Biology: ABC of Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2019; Volume 1–3, pp. 677–687. [Google Scholar]
- Kumar, V.; Kumar, R.; Parate, S.; Yoon, S.; Lee, G.; Kim, D.; Lee, K.W. Identification of ACK1 Inhibitors as Anticancer Agents by using Computer-Aided Drug Designing. J. Mol. Struct. 2021, 1235, 130200. [Google Scholar] [CrossRef]
- Greber, B.J.; Perez-Bertoldi, J.M.; Lim, K.; Iavarone, A.T.; Toso, D.B.; Nogales, E. The cryoelectron microscopy structure of the human CDK-activating kinase. Proc. Natl. Acad. Sci. USA 2020, 117, 22849–22857. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Hong, J.; Lee, K. Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics. Molecules 2021, 26, 2114. [Google Scholar] [CrossRef]
- Zou, K.H.; O’Malley, A.J.; Mauri, L. Receiver-Operating Characteristic Analysis for Evaluating Diagnostic Tests and Predictive Models. Circulation 2007, 115, 654–657. [Google Scholar] [CrossRef] [Green Version]
- Guner, O.F. History and Evolution of the Pharmacophore Concept in Computer-Aided Drug Design. Curr. Top. Med. Chem. 2002, 2, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Computational Investigation Identified Potential Chemical Scaffolds for Hepa-ranase as Anticancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 5311. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Parate, S.; Yoon, S.; Lee, G.; Lee, K.W. Computational Simulations Identified Marine-Derived Natural Bioactive Compounds as Replication Inhibitors of SARS-CoV-2. Front. Microbiol. 2021, 12, 647295. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Jones, G.H.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapundzhi, F.; Prodanova, K.; Lazarova, M. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 2172, 70902. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Sapay, N.; Tieleman, D.P. Combination of the CHARMM27 force field with united-atom lipid force fields. J. Comput. Chem. 2010, 32, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Berry, M.; Fielding, B.; Gamieldien, J. Practical Considerations in Virtual Screening and Molecular Docking. In Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology: Algorithms and Software Tools; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 487–502. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A.; Open Source Drug Discovery Consortium. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Kumar, V.; Singh, S.; Goswami, B.C.; Camps, I.; Sekar, A.; Yoon, S.; Lee, K.W. Repurposing potential of Ayurvedic medicinal plants derived active principles against SARS-CoV-2 associated target proteins revealed by molecular docking, molecular dynamics and MM-PBSA studies. Biomed. Pharmacother. 2021, 137, 111356. [Google Scholar] [CrossRef] [PubMed]
- Maddox, S.; Hecht, D.; Gustafson, J.L. Enhancing the selectivity of kinase inhibitors in oncology: A chemical biology perspec-tive. Future Med. Chem. 2016, 8, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Norman, R.A.; Toader, D.; Ferguson, A.D. Structural approaches to obtain kinase selectivity. Trends Pharmacol. Sci. 2012, 33, 273–278. [Google Scholar] [CrossRef]
- Hazel, P.; Kroll, S.H.B.; Bondke, A.; Barbazanges, M.; Patel, H.; Fuchter, M.J.; Coombes, R.C.; Ali, S.; Barrett, A.G.M.; Freemont, P.S. Inhibitor Selectivity for Cyclin-Dependent Kinase 7: A Structural, Thermodynamic, and Modelling Study. ChemMedChem 2017, 12, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and Toxicity Prediction of Ceftazidime and Its Impurities. Front. Pharmacol. 2019, 10, 434. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Douguet, D.; Miteva, M.; Villoutreix, B.O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, srep46277. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef] [Green Version]
- Chohan, T.A.; Pan, Y.-L.; Qian, H.-Y.; Chen, J.-Z. Molecular simulation studies on the binding selectivity of 2-anilino-4-(thiazol-5-yl)-pyrimidines in complexes with CDK2 and CDK7. Mol. BioSyst. 2015, 12, 145–161. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef]
- Whittaker, S.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Arooj, M.; Sakkiah, S.D.; Kim, S.; Arulalapperumal, V.; Lee, K.W. A Combination of Receptor-Based Pharmacophore Modeling & QM Techniques for Identification of Human Chymase Inhibitors. PLoS ONE 2013, 8, e63030. [Google Scholar] [CrossRef] [Green Version]
- Maganti, L.; Grandhi, P.; Ghoshal, N. Integration of ligand and structure based approaches for identification of novel MbtI inhibitors in Mycobacterium tuberculosis and molecular dynamics simulation studies. J. Mol. Graph. Model. 2016, 70, 14–22. [Google Scholar] [CrossRef]
- Bhowmick, S.; AlFaris, N.A.; Altamimi, J.Z.; Alothman, Z.A.; Aldayel, T.S.; Wabaidur, S.M.; Islam, A. Screening and analysis of bioactive food compounds for modulating the CDK2 protein for cell cycle arrest: Multi-cheminformatics approaches for anticancer therapeutics. J. Mol. Struct. 2020, 1216, 128316. [Google Scholar] [CrossRef]
- Zeb, A.; Son, M.; Yoon, S.; Kim, J.H.; Park, S.J.; Lee, K.W. Computational Simulations Identified Two Candidate Inhibitors of Cdk5/p25 to Abrogate Tau-associated Neurological Disorders. Comput. Struct. Biotechnol. J. 2019, 17, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Kumar, V.; Lee, K.W. A computational drug repurposing approach in identifying the cephalosporin antibiotic and anti-hepatitis C drug derivatives for COVID-19 treatment. Comput. Biol. Med. 2021, 130, 104186. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.-Q.; Zhu, K.-K.; Zhang, J.; Song, J.-L.; Muehlmann, L.A.; Jiang, C.-S.; Liu, C.-L.; Zhang, H. Molecular-docking-guided design and synthesis of new IAA-tacrine hybrids as multifunctional AChE/BChE inhibitors. Bioorganic Chem. 2019, 83, 277–288. [Google Scholar] [CrossRef]
- Silva, L.R.; Guimarães, A.S.; Nascimento, J.D.; Nascimento, I.J.D.S.; da Silva, E.B.; McKerrow, J.H.; Cardoso, S.H.; da Silva-Júnior, E.F. Computer-aided design of 1,4-naphthoquinone-based inhibitors targeting cruzain and rhodesain cysteine proteases. Bioorganic Med. Chem. 2021, 41, 116213. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Features a | Rank b | Direct Hit c | Partial Hit d | Max Fit e |
|---|---|---|---|---|---|
| Hypo1 | HYA, HBD, HYP, HBD, HBD, HBD | 69.73 | 1111 | 0000 | 6 |
| Hypo2 | HYA, HYA, HBD, HYP, HBD, HBD, HBD | 69.51 | 1111 | 0000 | 7 |
| Hypo3 | HYA, HBD, HYP, HBD, HBD, HBD | 68.80 | 1111 | 0000 | 6 |
| Hypo4 | RA, HYA, HYP, HBD, HBD, HBD | 68.72 | 1111 | 0000 | 6 |
| Hypo5 | RA, HYA, HYP, HBD, HBD, HBD | 68.39 | 1111 | 0000 | 6 |
| Hypo6 | RA, HYA, HYP, HBD, HBD | 68.39 | 1111 | 0000 | 6 |
| Hypo7 | HYA, HBD, HBD, HBD, HBD, HBD | 68.18 | 1111 | 0000 | 6 |
| Hypo8 | HYA, HBD, HYP, HBD, HBD, HBD | 67.86 | 1111 | 0000 | 6 |
| Hypo9 | HYA, HBD, HYP, HBD, HBD, HBD | 67.79 | 1111 | 0000 | 6 |
| Hypo10 | HYA, HBD, HYP, HBD, HBD, HBD | 67.71 | 1111 | 0000 | 6 |
| Sr. No. | Number of Features | Features Set | Selectivity Score |
|---|---|---|---|
| Hypo1 | 6 | HBA, HBA, HBD, HYP, HYP, HYP | 10.31 |
| Hypo2 | 6 | HBA, HBD, HYP, HYP, HYP, HYP | 10.31 |
| Hypo3 | 6 | HBA, HBD, HYP, HYP, HYP, HYP | 10.31 |
| Hypo4 | 6 | HBA, HBA, HBD, HYP, HYP, HYP | 10.31 |
| Hypo5 | 6 | HBA, HBA, HBD, HYP, HYP, HYP | 10.31 |
| Hypo6 | 6 | HBA, HBA, HBD, HYP, HYP, HYP | 10.31 |
| Hypo7 | 6 | HBA, HBA, HYP, HYP, HYP, HYP | 9.39 |
| Hypo8 | 5 | HBA, HBD, HYP, HYP, HYP | 8.79 |
| Hypo9 | 5 | HBA, HBD, HYP, HYP, HYP | 8.79 |
| Hypo10 | 5 | HBA, HBD, HYP, HYP, HYP | 8.79 |
| Sr. No. | Parameters | Ligand-Based | Structure-Based | ||||
|---|---|---|---|---|---|---|---|
| Hypo1 | Hypo2 | Hypo7 | Hypo1 | Hypo3 | Hypo4 | ||
| 1 | Total number of compounds in the database (D) | 110 | 110 | 110 | 110 | 110 | 110 |
| 2 | Total number of active compounds in the database (A) | 6 | 6 | 6 | 6 | 6 | 6 |
| 3 | Total number of hits retrieved by pharmacophore model from the database (Ht) | 11 | 8 | 5 | 3 | 4 | 2 |
| 4 | Total number of active compounds in the hit list (Ha) | 5 | 5 | 4 | 2 | 3 | 2 |
| 5 | % Yield of active ((Ha/Ht) × 100) | 45.45 | 62.5 | 80 | 66.66 | 75 | 100 |
| 6 | % Ratio of actives ((Ha/A) × 100) | 83.33 | 83.33 | 66.66 | 33.33 | 50 | 33.33 |
| 7 | False negatives (A-Ha) | 1 | 1 | 1 | 4 | 3 | 4 |
| 8 | False positives (Ht-Ha) | 5 | 3 | 3 | 1 | 1 | 0 |
| 9 | Goodness of fit score (GF) | 0.51 | 0.65 | 0.75 | 0.57 | 0.68 | 0.83 |
| Inhibitors | van der Waals (kJ/mol) | Electrostatic (kJ/mol) | Polar Solvation (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy ΔGbind (kJ/mol) |
|---|---|---|---|---|---|
| Hit1 | −191.19 +/− 14.45 | −309.22 +/− 30.04 | 355.74 +/− 44.97 | −25.34 +/− 1.40 | −170.01 +/− 29.50 |
| Hit2 | −164.36 +/− 14.68 | −47.28 +/− 20.40 | 128.50 +/− 26.24 | −20.02 +/− 1.73 | −103.17 +/− 17.65 |
| Hit3 | −167.45 +/− 10.86 | −22.27 +/− 7.28 | 115.57 +/− 17.06 | −20.50 +/− 1.07 | −94.66 +/− 12.26 |
| Hit4 | −147.54 +/− 11.28 | −17.71 +/− 9.15 | 91.10 +/− 13.54 | −16.44 +/− 1.65 | −90.59 +/− 12.80 |
| THZ1 | −151.40 +/− 11.25 | −22.06 +/− 15.20 | 98.27 +/− 18.60 | −16.29 +/− 1.06 | −91.48 +/− 14.79 |
| CT7001 | −181.13 +/− 13.51 | −44.09 +/− 16.30 | 154.73 +/− 31.72 | −20.09 +/− 1.20 | −90.58 +/− 17.08 |
| ADMET Properties | Hit 1 | Hit 2 | Hit 3 | Hit 4 | CT7001 | THZ1 | Unit | |
|---|---|---|---|---|---|---|---|---|
| Absorption | Water solubility | −3.57 | −4.40 | −5.18 | −3.70 | −3.18 | −3.26 | log mol/L |
| Caco-2 permeability | 0.01 | 1.00 | 1.09 | 0.45 | 1.26 | 0.86 | log Papp in 10−6 cm/s | |
| IA (human) | 64.37 | 84.75 | 95.29 | 60.14 | 89.43 | 93.01 | % Absorbed | |
| Skin permeability | −2.74 | −2.73 | −3.14 | −2.79 | −2.73 | −2.73 | log Kp | |
| P-gp substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes/No | |
| P-gp I inhibitor | Yes | Yes | Yes | No | No | Yes | Yes/No | |
| P-gp II inhibitor | No | Yes | Yes | No | Yes | Yes | Yes/No | |
| Distribution | VDss (human) | 1.49 | 0.04 | 0.50 | −0.08 | 2.13 | −0.64 | log L/kg |
| BBBp | −1.33 | −0.67 | −0.83 | −1.40 | −0.84 | −1.26 | logBB | |
| CNSp | −3.69 | −2.12 | −2.92 | −3.50 | −2.66 | −2.2 | log PS | |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | No | Yes/No |
| CYP2D6 inhibitor | No | No | No | No | No | No | Yes/No | |
| CYP3A4 substrate | Yes | Yes | Yes | No | Yes | Yes | Yes/No | |
| CYP3A4 inhibitor | Yes | No | Yes | No | Yes | Yes | Yes/No | |
| Excretion | TC | 1.08 | 0.10 | 0.18 | 0.77 | 0.88 | 0.48 | log mL/min/kg |
| Toxicity | AMES toxicity | No | No | No | No | No | No | Yes/No |
| Max. tolerated dose (human) | 0.14 | −1.47 | −0.3 | 0.32 | 0.15 | 0.43 | log mg/kg/day | |
| hERG I inhibitor | No | No | No | No | No | No | Yes/No | |
| hERG II inhibitor | Yes | Yes | Yes | No | Yes | Yes | Yes/No | |
| Oral rat acute toxicity | 2.70 | 3.72 | 2.60 | 3.46 | 2.82 | 2.84 | LD50 mol/kg | |
| Hepatotoxicity | Yes | Yes | Yes | Yes | Yes | Yes | Yes/No | |
| Skin sensitization | No | No | No | No | No | No | Yes/No | |
| Inhibitor | Database ID | IUPAC Name | 2D Representation |
|---|---|---|---|
| Hit1 | ZINC20392430 | ethyl 4-[7-hydroxy-8-[[4-(2-hydroxyethyl)piperazin-1-ium-1-yl]methyl]-2-methyl-4-oxo-chromen-3-yl]oxybenzoate |  |
| Hit2 | SN00112175 | (4R)-N-(3-acetamidophenyl)-4-[(3R,5S,7R,8R,9S,10S,12S,13R,14S,17R)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]pentanamide |  |
| Hit3 | SN00004718 | (2S)-2-[(4S,4aS,5S,6S,8aS)-5-hydroxy-4,8a-dimethyl-2-[2-(2-pyridyl)ethylamino]-4a,5,6,7,8,9-hexahydro-4H-benzo[f][1,3]benzothiazol-6-yl]-N-allyl-N-methyl-propanamide |  |
| Hit4 | SN00262261 | [(6R,7R)-3-[(E)-3-acetoxyprop-1-enyl]-7-hydroxy-7-methyl-8-oxo-5,6-dihydro-1H-isochromen-6-yl] 2,4-dihydroxy-6-methyl-benzoate |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; Parate, S.; Thakur, G.; Lee, G.; Ro, H.-S.; Kim, Y.; Kim, H.J.; Kim, M.O.; Lee, K.W. Identification of CDK7 Inhibitors from Natural Sources Using Pharmacoinformatics and Molecular Dynamics Simulations. Biomedicines 2021, 9, 1197. https://doi.org/10.3390/biomedicines9091197
Kumar V, Parate S, Thakur G, Lee G, Ro H-S, Kim Y, Kim HJ, Kim MO, Lee KW. Identification of CDK7 Inhibitors from Natural Sources Using Pharmacoinformatics and Molecular Dynamics Simulations. Biomedicines. 2021; 9(9):1197. https://doi.org/10.3390/biomedicines9091197
Chicago/Turabian StyleKumar, Vikas, Shraddha Parate, Gunjan Thakur, Gihwan Lee, Hyeon-Su Ro, Yongseong Kim, Hong Ja Kim, Myeong Ok Kim, and Keun Woo Lee. 2021. "Identification of CDK7 Inhibitors from Natural Sources Using Pharmacoinformatics and Molecular Dynamics Simulations" Biomedicines 9, no. 9: 1197. https://doi.org/10.3390/biomedicines9091197