
Lipids and Lipoproteins in Health and Disease: Focus on Targeting Atherosclerosis
by
 , , , , and
, , , , and
Chih-Kuo Lee
1,2,3,† ,
,
Che-Wei Liao
1,4,†,
Shih-Wei Meng
1,5,†,
Wei-Kai Wu
1,6,
Jiun-Yang Chiang
1,3,7,* and
Ming-Shiang Wu
1,6,* 1
College of Medicine, National Taiwan University, Taipei 100, Taiwan
2
Department of Internal Medicine, National Taiwan University Hospital Hsin-Chu Branch, Hsin-Chu 300, Taiwan
3
Graduate Institute of Clinical Medicine, College of Medicine, National Taiwan University, Taipei 100, Taiwan
4
Department of Internal Medicine, National Taiwan University Cancer Center, Taipei 106, Taiwan
5
Division of Cardiology, Department of Internal Medicine, National Taiwan University Hospital Hsin-Chu Branch, Hsin-Chu 300, Taiwan
6
Division of Gastroenterology and Hepatology, Department of Internal Medicine, National Taiwan University Hospital, National Taiwan University College of Medicine, Taipei 100, Taiwan
7
Division of Cardiology, Department of Internal Medicine and Cardiovascular Center, National Taiwan University Hospital, Taipei 100, Taiwan
*
Authors to whom correspondence should be addressed.
†
Che-Wei Liao, Chih-Kuo Lee, and Shih-Wei Meng contributed equally to this work.
Biomedicines 2021, 9(8), 985; https://doi.org/10.3390/biomedicines9080985
Submission received: 7 July 2021
/
Revised: 1 August 2021
/
Accepted: 4 August 2021
/
Published: 9 August 2021
(This article belongs to the Special Issue Lipids and Lipoproteins in Health and Disease: Focus on Targeting Atherosclerosis)
Abstract
:Despite advances in pharmacotherapy, intervention devices and techniques, residual cardiovascular risks still cause a large burden on public health. Whilst most guidelines encourage achieving target levels of specific lipids and lipoproteins to reduce these risks, increasing evidence has shown that molecular modification of these lipoproteins also has a critical impact on their atherogenicity. Modification of low-density lipoprotein (LDL) by oxidation, glycation, peroxidation, apolipoprotein C-III adhesion, and the small dense subtype largely augment its atherogenicity. Post-translational modification by oxidation, carbamylation, glycation, and imbalance of molecular components can reduce the capacity of high-density lipoprotein (HDL) for reverse cholesterol transport. Elevated levels of triglycerides (TGs), apolipoprotein C-III and lipoprotein(a), and a decreased level of apolipoprotein A-I are closely associated with atherosclerotic cardiovascular disease. Pharmacotherapies aimed at reducing TGs, lipoprotein(a), and apolipoprotein C-III, and enhancing apolipoprotein A-1 are undergoing trials, and promising preliminary results have been reported. In this review, we aim to update the evidence on modifications of major lipid and lipoprotein components, including LDL, HDL, TG, apolipoprotein, and lipoprotein(a). We also discuss examples of translating findings from basic research to potential therapeutic targets for drug development.
1. Introduction
During the past decades, the risk of atherosclerotic cardiovascular disease (ASCVD) and mortality has been much reduced due to advances in pharmacotherapy, intervention devices, and techniques. [1,2] ASCVD risk is significantly reduced by controlling blood low-density lipoprotein cholesterol (LDL-C) level [3]. Statin is the drug of choice to treat hypercholesterolemia. Non-statin medication including PCSK9 inhibitors and ezetimibe would further reduce LDL-C level while added to a statin or act as statin alternatives. Bempedoic acid is a newly approved effective non-statin LDL-C lowering agent [4]. Bempedoic acid has been associated with increased incidence of hyperuricemia, gout, and elevated serum creatinine level. On-going trials will clarify its long-term effect on cardiovascular outcomes [5].
However, the risk of ASCVD has not been eliminated. In the CANTOS trial, patients with a history of myocardial infarction (MI) had a 20% 5-year rate of recurrent major cardiovascular events (MACEs) despite statin treatment [6]. These residual risks can be caused by many factors, and methods to modify these factors have been proposed in contemporary guidelines. For example, measuring the lipoprotein(a) (Lp(a)) level should be considered among high-risk patients for a more precise reclassification and identification [3,7]. Lipid and lipoprotein metabolism disorders remain an unsolved problem. Whilst most guidelines encourage achieving target levels of specific lipoproteins to reduce the risk of ASCVD, increasing evidence has shown that molecular modification of these lipoproteins also has a critical impact on their atherogenecity and may contribute to residual ASCVD risk (Figure 1). For example, native low-density lipoproteins (LDLs) are much less atherogenic than those that have been structurally modified, such as by oxidation [8]. Apolipoproteins also play important roles in modulating lipid homeostasis and may alter the functions of different lipoproteins. In this review, we aim to update the evidence on modifications of major lipid components, including LDL, high-density lipoprotein (HDL), triglycerides (TGs), apolipoprotein, and Lp(a). We also discuss examples of translating findings from basic research to potential therapeutic targets for drug development.
2. Low-Density Lipoprotein Cholesterol
An increased serum LDL level is a well-known major risk factor for atherosclerotic disease. LDL is the major cholesterol carrier in plasma, consisting mainly of cholesterol ester (29%), phospholipids (28%), protein (21%), free cholesterol (11%), and TGs (9%). The only static protein component of LDL is apolipoprotein B (apoB) [10]. Mediated by apoB, LDL binds to and is internalized by LDL receptors (LDLRs) expressed on endothelial cells, macrophages, monocytes, and smooth muscle cells [11]. However, since LDLRs are downregulated as the intracellular cholesterol level increases, the uptake of native LDL via LDLRs does not consequently cause a significant increase in total cholesterol. In contrast, macrophage uptake modifies LDL through scavenger receptors, which are not downregulated as the cholesterol level increases, leading to substantial cholesterol accumulation and subsequent foam cell formation [12]. Therefore, modified LDL is assumed to be more atherogenic than native LDL.
LDL can be modified in several ways and mainly by oxidation. Oxidation of LDL is generally thought to take place in the subendothelial space but not in the circulation, as the plasma is rich in antioxidants. LDL can be oxidized non-enzymatically by metal ions or enzymatically via various mechanisms in the arterial wall [8].
The oxidation of LDL results in a broad spectrum of oxidized LDL, from minimally oxidized LDL (mmLDL) to fully oxidized LDL (oxLDL) [13]. mmLDL represents the initial stage of LDL oxidation, with no or little change in apoB, and retaining the affinity to LDLRs [14]. mmLDL increases chemokines and cytokines and enhances interactions between monocytes and endothelial and inflammatory cell recruitment. Subsequently, LDL is further oxidized with more extensive modification of its proteins, losing recognition by LDLRs and shifting affinity to a variety of scavenger receptors, including class A (SR-AI and SR-AII), class B (CD36), class D (CD68), class E (LOX-1), and class G (SR-PSOX) [8,12,15,16,17,18].
In vitro, LDL can be oxidized by iron and copper [19], and this can be blocked by metal chelators [20]. Although elevated iron and copper levels have been detected in human carotid atherosclerotic lesions [21], the relationship between serum iron and copper levels with atherosclerosis is unclear. Not only do patients with hemochromatosis and Wilson disease not have a higher risk of atherosclerosis [22], epidemiological studies have also reported inconclusive results with regards to the relationship between plasma iron level and atherosclerosis [23]. Consequently, further studies are needed to elucidate the actual mechanism of transition ions in LDL oxidation in vivo.
Lipoxygenase is an intracellular enzyme that directly oxygenates poly-unsaturated fatty acids [24]. In vitro, 15-lipoxygenase has been shown to oxidizes LDL via direct and non-direct reactions [25]. In human atherosclerotic lesions and macrophage-rich areas of fatty streaks, elevated co-localization of oxidized LDL epitope, 15-lipoxygenase mRNA and protein, and abundant acetyl LDL receptor mRNA have been shown [26]. Further studies have reported compelling results, indicating that lipoxygenase may not only be involved in atherogenesis but also exert an anti-inflammatory effect [27].
Myeloperoxidase (MPO) is abundant in the azurophilic granules of leukocytes (neutrophils, monocytes), and it has been linked to inflammation and oxidative stress. Local release from resident macrophages and transcytosis of intra-luminally produced MPO by activated leukocytes has been shown to contribute to MPO production in the vascular wall [28,29]. Enzymatically active MPO has been shown in human vascular atherosclerotic lesions, indicating the involvement of MPO in atherosclerosis [30].
Glycation to LDL occurs non-enzymatically to the major amino acid of apoB, lysine, resulting in 2%–17% of LDL-lysine glycation [31]. In vivo, free radicals generated from glucose and Amadori products by glycation show the concurrence of glycation and oxidation, and glycated LDL is more susceptible to oxidation [32]. LDL glycation inevitably results in a certain level of LDL oxidation [33].
Nitric oxide (NO) plays an important role in vascular physiology. Despite the antioxidant property of NO, the concurrent formation of NO and superoxide anions potentiate peroxynitrite production and lipid peroxidation [34]. Part of the cell-mediated LDL oxidation in the arterial wall can be attributed to superoxide anions, peroxynitrite, or other reactive nitrogen intermediates produced by endothelial cells, smooth muscle cells, or macrophages [35].
LDL is a type of lipoprotein with heterogenous proteomic and lipidomic profiles and pathophysiological activity. Small dense LDL is a specific subclass of LDL considered to be more prominent in atherogenesis. In the placebo group of a large statin clinical trial, small dense LDL concentration but not large LDL was associated with higher cardiovascular risk [36]. Small dense LDL has several characteristics distinct from large LDL, including being enriched with apolipoprotein C-III (apoC-III) and glycated apoB [37], and having unsaturated cholesterol esters markedly susceptible to hydroperoxide formation under oxidative stress [38], which may be associated with their pathogenicity. In addition, compared with large LDL, small dense LDL has a longer residence time in the circulation, which is considered to increase the risk of atherosclerosis. The longer residence time may be due to more particle oxidation, modification, reduction in size, and increased arterial wall uptake [39].
Activated by oxLDL, endothelial cells can up-regulate adhesion molecules and chemokines, triggering the recruitment of monocytes, typically Ly6Chi monocytes into the arterial wall [40]. These recruited monocytes differentiate into macrophages and further oxidize LDL. The oxLDL is then recognized and internalized by various scavenger receptors, turning the macrophages into cholesterol-laden foam cells [41]. By engaging pattern recognition receptors, such as toll-like receptors (TLRs), several DAMPs (damage-associated molecular patterns, notably oxidation-specific epitopes, and cholesterol crystals) generated by modification of retained LDL trigger the expressions of pro-inflammatory and pro-thrombotic genes in macrophages. This also boosts the recruitment of inflammatory cells, including monocyte-macrophages, neutrophils, lymphocytes, and dendritic cells [42]. Specifically, the recognition of oxLDL by the combination of TLR4-TLR6 and CD36 can promote the NFκB-dependent expression of chemokines, leading to further recruitment of macrophages [43]. ApoB is covalently modified during oxidation and apoB derived LDL-associated antigens are the most frequently studied putative T cell antigens. oxLDL-specific IgG levels have been shown to be well correlated with atherosclerosis progression and regression in animal models, however, the findings have been inconsistent in human studies. Antibody-dependent complement activation has also been shown in human atherosclerotic lesions [44]. The persistent presence of lipid-derived DAMPs, inflammatory cytokines, and the recruitment of phagocytes has been shown to sustain inflammatory responses and facilitates cross-talk with other arterial cells such as mast cells, contributing to plaque formation [45].
3. High-Density Lipoprotein Cholesterol
The concept of high-density lipoprotein cholesterol (HDL-C) as a “good cholesterol” can be traced to the Framingham Heart Study [46]. Subsequently, numerous epidemiological studies have suggested an inverse relationship between HDL-C level and the risk of ASCVD [47]. However, later efforts, including interventional studies focusing on niacin, fibrates, and cholesteryl ester transfer protein (CETP) inhibitors to increase HDL-C level, as well as meta-analyses, have failed to demonstrate a protective effect of a high HDL-C level against ASCVD [48,49,50,51]. These findings, along with several Mendelian randomization analyses, have prompted research that is focused on the functional properties rather than the quantity of HDL-C.
HDL is composed of a central hydrophobic non-polar lipid core, consisting of primarily triacylglycerols and cholesterol esters. HDL-C particles are highly heterogeneous, ranging in size from 5 to 17 nm and density from 1.063–1.210 kg/L, within human plasma [52]. Through lipidomic and proteomic techniques, more than 200 additional lipids and 85 proteins have been identified in HDL particles. Moreover, multiple enzymes and even genetic material in the form of micro RNAs have also been identified. These variations result in different sizes, densities, and functions of HDL. Based on the size and density, HDL is commonly classified into five broad subclasses, ranging from the largest and least dense (termed HDL2b) to the smallest and most dense (termed HDL3b) [53].
HDL proteins are composed of apolipoproteins (e.g. apoE, apoCs, apoA-IV, apoA-V, apoJ, apoF, apoM, apoL1), enzymes, lipid transfer proteins, acute-phase response proteins (including serum amyloid A [SAA]), complement components, proteinase inhibitors, and other protein components. Apolipoprotein A-I (apoA-I) is the major structural protein component of HDL and is primarily synthesized in the liver (~80%) and intestine (~20%), and then secreted in a lipid-free state. The enzymes carried by HDL may include those for lipid metabolism (e.g. lecithin-cholesterol acyltransferase [LCAT] and phospholipid transfer protein [PLTP]), and for antioxidation (e.g. paraoxonase 1 [PON1], platelet-activating factor-acetyl hydrolase [PAF-AH] and glutathione selenoperoxidase [GSPx]) [53].
The most well-known pathway for the anti-atherogenic nature of HDL is the ability to promote cholesterol efflux from cells through reverse cholesterol transport (RCT), which transports excess cholesterol from peripheral tissues to the liver for excretion. In addition, due to the numerous proteins carried by HDL as mentioned above, it also exhibits anti-oxidative, anti-thrombotic, and anti-inflammatory properties in normal physiology [54].
4. Dysfunctional HDL
As mentioned earlier, HDL-C is a highly heterogeneous particle with various sizes and additional apoproteins and enzymes. Hence, many alterations and conditions may cause dysfunctional HDL. In brief, compositional changes, post-translational modifications of proteins, or alterations in lipids and other cargo molecules, may result in functional differences [55,56,57,58].
The most commonly reported post-translational modifications include oxidation, carbamylation, and glycation [56]. HDL oxidation takes place predominantly in inflammatory conditions and frequently involves MPO. Excessive MPO expression has been observed during the progression of atherosclerotic plaques, and with a maximum expression just before plaque rupture. Carbamylation, a non-enzymatic and irreversible post-translational modification, is caused by interactions between isocyanic acid and various amino groups of proteins. Carbamylation also occurs in an inflammatory environment as a result of MPO-dependent cyanate formation. Glycation occurs after lipids or proteins are exposed to sugar. In chronic hyperglycemia (i.e. poorly controlled diabetes), nonenzymatic glycation may occur and lead to alterations in HDL composition (lipids, apoproteins, and enzymes) and functionality [59].
Modification of HDL can affect the lipidome and proteome then finally alter the function of HDL. Several pathways have been identified such as SAA, LCAT, CETP, apolipoprotein M, sphingosine-1-phosphate (S1P), paraoxonase 1, and MPO [56]. Many inflammatory processes can cause alteration in these pathways and modify HDL, which eventually losses its atheroprotective effect.
SAA is a family of apolipoproteins that are primarily synthesized in the liver. It is one of the main proteins of the acute phase response and plays a pivotal role in innate immunity [60,61]. The level of SAA has been shown to be higher during acute and chronic inflammation, which eventually causes compositional changes of HDL and the loss of its anti-inflammatory and atheroprotective potential.
LCAT esterifies free cholesterol and phosphatidylcholine into cholesteryl esters, and it is an essential enzyme for lipoprotein metabolism. Various acute and chronic inflammatory conditions can decrease LCAT activity, and subsequently, change the composition and function of HDL. This dysfunctional HDL also loses its atheroprotective potential.
CETP mediates the transfer of cholesterol esters from HDL to apoB-containing lipoproteins in exchange for TGs. Studies have shown a decreased level and activity of CETP in patients with sepsis, cardiac surgery, and rheumatoid arthritis. This decrease in CETP may cause remodeling of HDL and may increase the risk of cardiovascular mortality.
Apolipoprotein M (apoM) is a plasma protein of the apolipoprotein family expressed in the liver and kidneys. As mentioned earlier, apoM is an additional apolipoprotein carried by HDL. Previous studies have shown a decrease in the apoM level in inflammatory conditions, including sepsis, diabetes, and autoimmune disease, which in turn decrease the anti-inflammatory effects of HDL.
S1P is a lysosphingolipid found in association with small and dense HDL particles. HDL-bound S1P is important for endothelial survival, angiogenesis, migration, NO production, and inhibition of inflammatory responses [62]. The level of S1P has been found to be lower in patients with coronary artery disease and MI. Similar results have also been shown in patients with diabetes mellitus, chronic kidney disease, and atherosclerosis.
PONs are a family of enzymes with antioxidative properties in mammals. They are composed of three different enzymes. HDL-associated PON1 possesses anti-atherogenic properties, protects LDL from oxidative modification, and promotes cholesterol efflux [63]. Many studies focusing on patients with obesity, diabetes mellitus, various autoimmune diseases, and infection have demonstrated decreased PON levels or activity, resulting in a possible decline in the anti-oxidative effect of HDL.
From a clinical perspective, many diseases have been correlated with dysfunctional HDL irrespective of its plasma level. For example, patients with acute coronary syndrome or stable coronary artery disease, have still be shown to exhibit reduced HDL-mediated cellular cholesterol efflux compared with healthy subjects even when matched by HDL-C level [64]. Alteration in the HDL function in such patients may be due to combinations of the abovementioned pathways. Other examples have also been reported, such as in patients with type 2 diabetes, in whom dysfunctional HDL may be caused by advanced glycation end products and increased oxidative stress with inflammation that reduces their ability to promote cholesterol efflux [59] or anti-inflammatory capacity [65]. In addition, HDL efflux capacity in dialytic patients has been shown to be dramatically reduced compared to matched subjects with normal kidney function, which could not be improved with statins [66]. In conclusion, current evidence supports that dysfunctional HDL, rather than HDL level, plays a more significant role in atherosclerosis. Further studies on the causality of the possible mechanisms of HDL modifications are needed.
5. Triglycerides
Hypertriglyceridemia is a prevalent condition observed in daily medical care. According to previous literature, its prevalence in the adult population is approximately 10% [67,68,69]. Moreover, the increasing trend of hypertriglyceridemia has been parallel to that of type 2 diabetes and obesity in the past decades [68]. Very low-density lipoprotein (VLDL) and chylomicrons, which are known as TG-rich lipoproteins (TRLs), are spherical particles with core lipids (TG and cholesterol esters), phospholipids, free cholesterol, and surface apolipoproteins. The origins of TGs are generally exogenous or endogenous. Exogenous TG is mostly obtained from daily diet and transported within chylomicrons, while endogenous TG circulates in VLDL and is mostly formed in the hepatobiliary system.
The levels of fasting and postprandial TGs depend on the balance between lipoprotein lipase-mediated lipolysis and uptake in the human liver. VLDL overproduction is the most common upstream cause of hypertriglyceridemia, and the inherited capacity of the lipoprotein lipase-mediated lipolysis pathway modulates the steady-state level. Comprehensive evaluations are strongly suggested when hypertriglyceridemia is suspected. Furthermore, insulin resistance, overweight, and type 2 diabetes mellitus may be detected simultaneously in this population. In clinical practice, these conditions are usually treated as metabolic syndrome, which comprises the aforementioned three conditions. Metabolic syndrome may increase VLDL levels, especially when free acids and insulin accumulate in the circulation [70]. An environment with an extremely high free acid concentration, hyperglycemia, and insulin resistance, would result in increased chylomicron secretion, while glucagon-like peptide 1 would play a counterbalancing role in the pathway [70]. Moreover, apoC-III has been shown to decrease the removal of remnants in individuals with high VLDL levels and a higher apoC-III concentration is an important factor leading to dyslipidemia [71].
TG-rich VLDL particles and metabolic remnants are the main transporters of TGs in human circulation. The plasma concentration of TGs has been shown to be parallel to the circulating apo B-containing TRL level, which is known to be associated with ASCVD formation [72]. A non-fasting TG level of 6.6 mmol/L was significantly associated with a 5-fold higher risk of acute coronary syndrome, a 3-fold increased risk of stroke, and a 2-fold increased adjusted risk of all-cause mortality compared to a level of 0.8 mmol/L in population-based cohort studies in Copenhagen [73,74]. These results show the importance of monitoring the TG level in primary ASCVD risk modification. In another study investigating secondary ASCVD risk after acute MI, TGs were found to be significantly associated with both short-term and long-term ASCVD outcomes. Furthermore, most patients in the study had been treated with statins, which further highlights the crucial role of TGs in secondary ASCVD prevention [75].
Several studies have used Mendelian randomization and shown that the association between TG concentrations and ASCVD may be causal. Nevertheless, the evidence needs to be interpreted with caution, because nearly all variants associated with TGs were also associated with the trends of HDL-C, LDL-C, and Lp(a) [76,77]. In another study, the authors used Mendelian randomization to show that TG-lowering lipoprotein lipase variants and LDL-C-lowering LDL receptor variants had similar effects on the ASCVD risk per unit change in apo-B [78]. Taken together, these studies demonstrated the causality of TRLs and their remnants on the ASCVD risk, partly due to the plasma level of apo B-containing particles. Another possible mechanism underlying the relationship between TGs and atherosclerosis is the deposition of cholesterol-ester-enriched smaller TRLs on the arterial walls and the subsequent initiation of pro-inflammatory/thrombotic pathways. Furthermore, high circulating TG levels have been associated with pathological HDL-C particles, which could lead to an increased risk of ASCVD [69]. In contrast, the correlation between circulating TG concentrations and ASCVD risk has varied among previous studies and was lost in several multivariate analyses [79]. Moreover, the correlation was reduced after adjusting for non-HDL-C or apoB in an epidemiological study [80].
Collectively, the aforementioned studies demonstrate that TRLs and their remnants play a crucial role in ASCVD risk assessment [81]. According to current clinical guidelines, lowering LDL-C remains the primary treatment goal in the management of dyslipidemia. In addition, clinicians should focus on modifications of TRLs, such as non-HDL-C and apoB, which are highly recommended in the updated guideline [7]. Previous studies on fibrates, niacin, and cholesteryl ester transfer protein inhibitors did not demonstrate a robust or convincing reduction in the risk of ASCVD in an optimal cholesterol-lowering population [82,83]. Nevertheless, several ongoing trials are focusing on the important roles of TRL with respect to the residual ASCVD risk in statin users. The results of these ongoing clinical trials and upcoming evidence regarding omega-3 fatty acids (high-dose icosapent ethyl) [78], and the selective peroxisome proliferator-activated receptor modulator pemafibrate may help to clarify which population will benefit from a reduced risk of ASCVD by lowering TRL levels [84,85]. The development of molecular technologies has provided more detailed information on the pathways underlying TRL modulation. Several emerging therapeutic molecules have been targeted, including inhibitors of angiopoietin-like protein 3 (evinacumab; allele-specific oligonucleotide IONISANGPTL3-LRx) (Table 1) [86], and inhibitors of intestinal diacylglycerol acyltransferase (pradigastat) [87], as well as those targeting apoC-II and A-V and angiopoietin-like protein 4 [88]. TRL modification strategies in specific patients can be expected to become a crucial part of lipid-directed treatment in the near future.
6. Apolipoprotein C-III
ApoC-III is a glycoprotein with 79 amino acids that is principally synthesized in the liver and is associated with TRLs [96]. ApoC-III increases the plasma level of TGs by facilitating the assembly and secretion of TRLs in the liver, inhibiting lipoprotein lipase activity, and interrupting the binding of apoE to its hepatic receptors, thus resulting in impaired clearance of TRL remnants [97]. The accumulation of apoC-III on HDL leads to conformation changes of HDL, including decreased apoA-I content, impaired insulin sensitivity, and reduced cholesterol efflux capacity [98,99]. ApoC-III also enhances the expression of adhesion molecules in endothelial cells via the NF-κB pathway and promotes the chemoattraction of monocytes [100]. Besides its role in atherosclerosis, apoC-III has been proposed to induce the apoptosis of insulin-secreting pancreatic β-cells [101].
There is ample evidence showing the strong link between increased apoC-III levels and ASCVD risk [102]. Genome-wide association studies and Mendelian randomization studies have demonstrated a direct relationship between loss-of-function mutations of the APOC3 gene, lower TG levels, and reduced ASCVD risk [103,104,105,106] Certain genetic polymorphisms have been associated with the upregulation of APOC3 gene expression and mRNA stability, such the SstI gene polymorphism [107]. Other studies have demonstrated a 4% decrease in the risk of ASCVD with every 1 mg/dl decrease in plasma apoC-III level. However, currently available lipid-modifying therapies are not able to target apoC-III level specifically [108,109,110,111].
Antisense oligonucleotide (ASO) targeting the messenger RNA of APOC3 gene was developed in 2014 to interrupt the transcription of APOC3 and thus reduce the level of TGs [112]. After showing promising efficacy in reducing the level of TG, apoC-III, and non-HDL cholesterol in phase 1 and phase 2 clinical trials, volanesorsen has been evaluated in two major phase 3 clinical trials. The APPROACH trial was a randomized double-blinded placebo-controlled trial that enrolled 66 patients with familial chylomicronemia syndrome (FCS), a rare genetic disease caused by an inactivating mutation of both alleles of LPL genes and genes encoding other proteins required for LPL activity [91]. After three months, volanesorsen resulted in a 76.5% reduction in the plasma level of TGs, equivalent to a reduction of 750–880 mg/dl, and 84.2% reduction in the plasma level of apoC-III. Other effects on the lipid profile included a significant increase in the levels of HDL, apoA1, and LDL, and decreased levels of chylomicron TGs, VLDL-C, and non-HDL-C. However, thrombocytopenia occurred in 76% of the subjects receiving volanesorsen compared to 24% in the placebo group. The COMPASS trial was a randomized, double-blinded, placebo-controlled trial that enrolled 114 patients with severe hypertriglyceridemia (fasting TG ≥ 500 mg/dl) [92]. Similar to the results of the APPROACH trial, the level of TGs was decreased by 73% after three months of volanesorsen treatment. No serious adverse effects regarding platelet count were reported. Another phase 2 randomized double-blinded placebo-controlled trial enrolled patients with hypertriglyceridemia (TG > 200 mg/dL and < 500 mg/dL) and uncontrolled type 2 diabetes mellitus (HbA1c > 7.5%) to test whether Volanesorsen was able to improve insulin sensitivity [113]. In the volanesorsen group, insulin sensitivity improved by 50% and the level of HbA1c decreased by 0.44%. Since insulin resistance is associated with an increased risk of ASCVD, the effect of improved insulin sensitivity and reduced apoC-III may be translated to a reduction in the risk of ASCVD. However, the Food and Drug Administration refused to approve volanesorsen for the treatment of FCS due to safety concerns, and thus a large cardiovascular outcome trial will not be conducted. Structurally, volanesorsen is a 2ʹ-O-(2-methoxyethyl)-modified ASO which has demonstrated good metabolic stability and RNA binding affinity. Despite this modification, less than 15% of the drug is distributed to hepatocytes [114].
A third generation of ASO modified with triantennary N-acetyl galactosamine (GalNAc3) to target the asialoglycoprotein receptors on hepatocytes has been developed. This modification enhances the affinity of ASO to hepatocytes and allows similar efficacy to untargeted ASO with 20–30-fold lower dosing, thus minimizing systemic exposure [115,116,117]. In a phase 1/2a double-blind, randomized, placebo-controlled, dose-escalation study, the novel GalNAc3-modified ASO, AKCEA-ApoCIII-LRx, was administered to healthy volunteers (ages 18–65 years) with elevated TG levels, with predefined doses ranging from 10 mg to 120 mg and varying schedules. After single doses of AKCEA-ApoCIII-LRx of 10, 30, 60, 90, and 120 mg, the median reductions in apoC-III were 0, −42%, −73%, −81%, and −92%, and the mean reductions in TG were −12%, −7%, −42%, −73%, and −77%, respectively. Significant reductions in total cholesterol, apoB, non-HDL-C, VLDL-C, and increases in HDL-C were also noted after multiple doses. The safety of AKCEA-ApoCIII-LRx was demonstrated in this study as no significant effects on the liver or kidney function were noted, and most importantly, no thrombocytopenia events occurred. Recently, a phase 2 trial (NCT03385239) enrolling patients with hypertriglyceridemia and established ASCVD or those at a high risk of ASCVD was completed. It is very interesting to see what effects AKCEA-ApoCIII-LRx may have on the risk of ASCVD.
7. Apolipoprotein A-I
ApoA-I contains ten consecutive helical regions, which are critical for its amphipathic properties to facilitate the dissolution of lipids in an aqueous environment [118]. The conformation of apoA-I is highly dynamic and readily accommodates changes in size and composition of HDL particles [119,120,121,122]. After transporting cholesterol to the liver, apoA-I dissociates from HDL particles and continues to initiate de novo RCT and HDL formation. The degree of release of apoA-I from HDL has been associated with the efficiency of cholesterol efflux [123]. Besides promoting RCT, apoA-I inhibits pro-oxidative and pro-inflammatory processes, induces vasodilation, and inhibits the activation of platelets. The cholesterol efflux capacity of apoA-I can be compromised in specific conditions [124]. For example, point mutations of APOAI gene may result in decreased HDL lipoprotein formation, stability [125], and lipid-binding affinity [126,127,128,129,130,131,132,133,134]. The A164S point mutation has been shown to result in a series of functional impairments and increased cardiovascular mortality without reducing HDL level [135]. Oxidative modifications of apoA-I by MPO have also been shown to suppress its anti-inflammatory and antiapoptotic activities on endothelial cells in vitro and to activate pro-inflammatory pathways [136]. Certain mutations of the APOA1 gene may produce MPO-resistant apoA-I forms that are antiatherosclerotic [137].
Previous studies have demonstrated a strong association between low apoA-I levels and increased risk of ASCVD [138,139,140,141]. In a cohort study of subjects suspected of having coronary artery disease who received coronary computed tomography angiography, the serum oxidized HDL/apoA-I ratio was significantly correlated with the prevalence of high-risk plaque and severe luminal narrowing [142]. In patients with type 2 diabetes mellitus, the prevalence and incidence of peripheral artery disease have also been reported to be significantly higher in subjects with low apoA-I levels [143]. On the other hand, a good cholesterol efflux capacity has been shown to promote angiogenesis and the development of collateral circulation in patients with chronic total coronary occlusion [144].
The roles of a well-function apoA-I and HDL have gained increased attention in the past decades as most therapies targeting an increase in HDL concentration have failed to reduce ASCVD risk [48,49,145,146,147,148]. The only CETP inhibitor to demonstrate a beneficial effect is anacetrapib. In the REVEAL trial, patients with ASCVD receiving anacetrapib had fewer coronary events compared to those receiving placebo. However, it took around two years for the full beneficial effects of anacetrapib to emerge, and the rate ratio was only 0.91. In addition, anacetrapib injection was associated with a reduction in non-HDL cholesterol that could be translated into a 10% relative reduction in coronary events. Therefore, it is difficult to independently evaluate the impacts of increased HDL and increased apoA-I on ASCVD risk.
CSL112 is a plasma-derived apoA-I reconstituted into disk-shaped lipoproteins with phosphatidylcholine and stabilized with sucrose [149]. In the phase 2b AEGIS-I trial, the favorable safety profile of CSL112 infusion was established, as no hepatic or renal adverse effects were noted, and the total and ABCA1-dependent cholesterol efflux increased significantly [94,150,151]. Based on the safety profile, the AEGIS-II study has been designed to assess the efficacy of CSL112 in MACE reduction in high-risk patients with acute MI, and it is currently enrolling subjects. The study protocol has been published elsewhere [152]. In brief, patients with acute MI, multivessel disease, or left main disease, and established risk factors defined as diabetes mellitus or at least two of an age ≥ 65 years, prior history of MI, or peripheral arterial disease will be enrolled. The primary outcome is the time to the first occurrence of a MACE composite of cardiovascular death, MI, or stroke from the time of randomization through 90 days, during which most recurrent cardiovascular events occur. So far, none of the HDL-raising therapies have been demonstrated to decrease the risk of cardiovascular events despite a significant increase in the level of HDL-C by up to 130% or apoA-I by up to 50% [49]. In the AEGIS-I trial, weekly infusions of CSL112 increased the level of apoA-I by nearly 100%. With the results of the AEGIS-II trial, the impact of this potent cholesterol efflux enhancer on the reduction of residual cardiovascular risk may be seen.
8. Lipoprotein(a)
Lp(a) is composed of LDL particles and glycoprotein apo(a) covalently bound to the apoB component of LDL [153,154,155]. It is synthesized in the liver and catabolized in the liver and kidneys. In patients with chronic kidney disease, the level of Lp(a) is elevated due to decreased excretion [156,157,158,159] Physiologically, Lp(a) participates in thrombogenesis, wound healing, tissue repair, and vascular remodeling [154,160]. Part of its thrombogenicity comes from apo(a), which is a structural homolog to plasminogen. Apo(a) inhibits the conversion of plasminogen to plasmin and may result in reduced fibrinolysis [161]. Lp(a) affects platelet activation, aggregation, increases the synthesis of plasminogen activator inhibitor-1 and inhibits synthesis of the tissue factor pathway inhibitor [162]. It is also one of the most important carriers of oxidized phospholipid (OxPL), which stimulates inflammatory responses in endothelial cells, and promotes proinflammatory and apoptotic genes expression in macrophages [163,164,165].
The associations between the Lp(a) level and an increased risk of atherosclerosis in different vascular beds, large artery atherosclerotic stroke, and calcification of the aortic valve have been well-demonstrated in large-scale studies [166,167,168,169,170,171,172]. These associations apply to different ethnicities and may be independent of LDL-C. In the Brisighella Heart Study, the Lp(a) level has been shown to predict long-term cardiovascular mortality in the intermediate-risk female and high-risk subjects without a history of ASCVD [173]. Another recently published biobank study showed a linear correlation between the Lp(a) level and incident ASCVD, with a hazard ratio of 1.11 (95% CI, 1.10–1.12) per 50 nmol/L increments [174]. The causal relationship between Lp(a) and the risk of ASCVD has been established by genome-wide association studies and Mendelian randomization studies [175,176,177,178]. Through genetic studies, two single nucleotide polymorphisms, rs10455872 and rs3798220, have been strongly associated with small apo(a) isoforms and high Lp(a) concentration [179,180]. The association between rs10455872 and an increased risk of MI was further demonstrated in a meta-analysis [181]. The combination of rs10455872 and rs3798220 single nucleotide polymorphism in the LPA gene have been causally associated with aortic valve stenosis [182].
Evidence has consistently shown that Lp(a) contributes to residual ASCVD risk in statin-treated patients. The residual risk of ASCVD has been shown to increase gradually as the Lp(a) concentration rises above 30 mg/dL, and sharply as the concentration rises above 50 mg/dL [166,183,184]. In a recent statement by the National Lipid Association, an Lp(a) level of 50 mg/dl or 100 nmol/L was used as a threshold [185], and an estimated 20% of the world’s population was estimated to have an Lp(a) level above this [186]. According to the 2018 ACC/AHA guidelines, measuring Lp(a) should be considered for subjects with a past or family history of ASCVD [3]. The 2019 ESC/EAS guidelines also recommended measuring Lp(a) to identify subjects with a very high Lp(a) level (≥180mg/dL or ≥ 430mmol/L), especially in those with established ASCVD and recurrent events, familial hypercholesterolemia, premature ASCVD, or a first-degree family member with premature ASCVD [7]. Lp(a)-lowering is a promising method to eliminate residual ASCVD risk [187,188], and an analysis of the ODYSSEY OUTCOMES trial revealed that a 1 mg/dl reduction in Lp(a) by alirocumab was associated with a hazard ratio of 0.994 [189,190]. However, no approved medications can specifically reduce the level of Lp(a).
Statin treatment does not lower but may even increase the level of Lp(a) by 10-20% [191,192,193]. The PCSK9 inhibitors evolocumab and alirocumab have been shown to reduce the level of Lp(a), however, the effects were only modest [194]. Apheresis has been demonstrated to result in a 60 –70% reduction in Lp(a) level and ASCVD risk [195,196,197,198], however, it is reserved for very high-risk patients in a few countries due to its time-consuming process. Recent pharmaceutical research has revealed that subcutaneous injections of ASO against hepatic apo(a) mRNA with PELACARSEN, previously known as ISIS 681257, IONIS APO(a)-LRx, AKCEA-Apo(a)-LRX, or TQJ230, significantly reduced the levels of Lp(a) and OxPL [95,116,199]. In a phase 2 trial, 286 patients with established ASCVD and Lp(a) levels above 60 mg/dl or 150 nmol/L were enrolled to receive PELACARSEN. At 6 months of treatment, the subjects receiving PELACARSEN demonstrated an 80% reduction in the Lp(a) level, compared to a 6% reduction in the placebo group and a 25% reduction with PCSK9 inhibitors. In addition, 98% of the subjects receiving PELACARSEN achieved the prespecified Lp(a) target level of 50 mg/dl or 125 nmol/L. Moreover, PELACARSEN decreased oxidized phospholipids by up to 90%. A favorable safety profile was also demonstrated as no significant change in platelet count, liver function, and renal function were noted. PELACARSEN was structurally modified from its prototype, IONIS-Apo(a), with the addition of a covalently attached GalNAC3 complex in order to target the ASGP receptors on the surface of hepatocytes more efficiently [200,201]. The ongoing Lp(a)HORIZON trial (NCT04023552) is a phase 3 randomized placebo-controlled trial that plans to enroll 7,680 subjects with an Lp(a) level above 150 nmol/L and established ASCVD, including prior MI, prior CVA, and significant symptomatic peripheral artery disease to receive PELACARSEN or placebo. The primary outcome is trial committee-confirmed expanded MACEs that include cardiovascular death, non-fatal MI, non-fatal stroke, and urgent coronary re-vascularization requiring hospitalization. The enrollment will be completed by 2024. This trial will provide more essential insights to answer the question of whether targeting Lp(a) reduction can specifically further reduce residual cardiovascular risk [202].
9. Conclusions
The novel evidence reported in this study provides deep insights into dyslipidemia, and the changing viewpoint from simply treating the concentration to potentially modifying lipoprotein functions. LDL may undergo structural change by oxidation, glycation, and peroxidation, and these modifications result in progressive atherosclerosis. HDL may undergo post-translational modifications such as oxidation, carbamylation, and glycation, and alteration of its cargo molecules such as SAA, LCAT, apoM, S1P, and PON1.
These modifications could decrease the atheroprotective function of HDL. Currently, no pharmacological approach has been developed to block or reverse these modifications. Research on how these modifications are modulated is still needed and will uncover more potential therapeutic targets. Pharmacotherapies targeting a reduction in TGs, apoC-III, and Lp(a), and an increase in apoA-1, are currently under investigation, and promising preliminary results have been reported. Clinical trials that assess the effects of these therapies on cardiovascular events are underway. These targets may further shape the landscape of dyslipidemia treatment and decrease the residual risk of cardiovascular events.
Author Contributions
Conceptualization, W.-K.W., J.-Y.C., and M.-S.W.; writing—original draft preparation, C.-K.L., C.-W.L., S.-W.M., J.-Y.C.; writing—review and editing, C.-K.L., C.-W.L., W.-K.W., J.-Y.C., M.-S.W.; visualization, J.-Y.C.; supervision, M.-S.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mensah, G.A.; Wei, G.S.; Sorlie, P.D.; Fine, L.J.; Rosenberg, Y.; Kaufmann, P.G.; Mussolino, M.E.; Hsu, L.; Addou, E.; Engelgau, M.M.; et al. Decline in Cardiovascular Mortality: Possible Causes and Implications. Circ. Res. 2017, 120, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Amini, M.; Zayeri, F.; Salehi, M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: Results from global burden of disease study 2017. BMC Public Health 2021, 21, 1–12. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guideline. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef]
- Banach, M.; Duell, P.B.; Gotto, A.M., Jr.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Flaim, J.; Ye, Z.; Catapano, A.L. Association of Bempedoic Acid Administration with Atherogenic Lipid Levels in Phase 3 Randomized Clinical Trials of Patients with Hypercholesterolemia. JAMA Cardiol. 2020, 5, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Pontremoli, R.; Fogacci, F.; Viazzi, F.; Borghi, C. Effect of Bempedoic Acid on Serum Uric Acid and Related Outcomes: A Systematic Review and Meta-analysis of the available Phase 2 and Phase 3 Clinical Studies. Drug Saf. 2020, 43, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef]
- Ruscica, M.; Macchi, C.; Fogacci, F.; Ferri, N.; Grandi, E.; Rizzoli, E.; D’Addato, S.; Borghi, C.; Cicero, A.F. Angiopoietin-like 3 and subclinical peripheral arterial disease: Evidence from the Brisighella Heart Study. Eur. J. Prev. Cardiol. 2020, 27, 2251–2254. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, E.; Kobayashi, K.; Tabuchi, M.; Lopez, L.R. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog. Lipid Res. 2006, 45, 466–486. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Leitinger, N.; Ley, K. Effects of Native and Modified Low-Density Lipoproteins on Monocyte Recruitment in Atherosclerosis. Hypertension 2007, 50, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Quehenberger, O.; Kondratenko, N.; Green, S.; Steinberg, D. Minimally Oxidized Low-Density Lipoprotein Increases Expression of Scavenger Receptor A, CD36, and Macrosialin in Resident Mouse Peritoneal Macrophages. Arter. Thromb. Vasc. Biol. 1998, 18, 794–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, D. Oxidized low density lipoprotein--an extreme example of lipoprotein heterogeneity. Isr. J. Med Sci. 1996, 32, 469–472. [Google Scholar] [PubMed]
- Navab, M.; Berliner, J.A.; Watson, A.D.; Hama, S.Y.; Territo, M.C.; Lusis, A.J.; Shih, D.M.; Van Lenten, B.J.; Frank, J.S.; Demer, L.; et al. The Yin and Yang of Oxidation in the Development of the Fatty Streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arter. Thromb. Vasc. Biol. 1996, 16, 831–842. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger Receptors Class A-I/II and CD36 Are the Principal Receptors Responsible for the Uptake of Modified Low Density Lipoprotein Leading to Lipid Loading in Macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef] [Green Version]
- Oka, K.; Sawamura, T.; Kikuta, K.-I.; Itokawa, S.; Kume, N.; Kita, T.; Masaki, T. Lectin-like oxidized low-density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9535–9540. [Google Scholar] [CrossRef] [Green Version]
- Shimaoka, T.; Kume, N.; Minami, M.; Hayashida, K.; Kataoka, H.; Kita, T.; Yonehara, S. Molecular Cloning of a Novel Scavenger Receptor for Oxidized Low Density Lipoprotein, SR-PSOX, on Macrophages. J. Biol. Chem. 2000, 275, 40663–40666. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.; Leake, D. Oxidation of low density lipoprotein by iron or copper at acidic pH. J. Lipid Res. 1995, 36, 2504–2512. [Google Scholar] [CrossRef]
- Lamb, D.J.; Leake, D.S. The effect of EDTA on the oxidation of low density lipoprotein. Atherosclerosis 1992, 94, 35–42. [Google Scholar] [CrossRef]
- Stadler, N.; Lindner, R.A.; Davies, M. Direct Detection and Quantification of Transition Metal Ions in Human Atherosclerotic Plaques: Evidence for the Presence of Elevated Levels of Iron and Copper. Arter. Thromb. Vasc. Biol. 2004, 24, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Gaut, J.P.; Heinecke, J.W. Mechanisms for oxidizing low-density lipoprotein. Insights from patterns of oxidation products in the artery wall and from mouse models of atherosclerosis. Trends Cardiovasc. Med. 2001, 11, 103–112. [Google Scholar] [CrossRef]
- Ascherio, A.; Willett, W.C. Are Body Iron Stores Related to the Risk of Coronary Heart Disease? N. Engl. J. Med. 1994, 330, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S. Mammalian lipoxygenases: Molecular structures and functions. Biochim. Biophys. Acta 1992, 1128, 117–131. [Google Scholar] [CrossRef]
- Heydeck, D.; Upston, J.M.; Viita, H.; Ylä-Herttuala, S.; Stocker, R. Oxidation of LDL by rabbit and human 15-lipoxygenase: Prevalence of nonenzymatic reactions. J. Lipid Res. 2001, 42, 1082–1088. [Google Scholar] [CrossRef]
- Yla-Herttuala, S.; Rosenfeld, M.E.; Parthasarathy, S.; Sigal, E.; Särkioja, T.; Witztum, J.L.; Steinberg, D. Gene expression in macrophage-rich human atherosclerotic lesions. 15-lipoxygenase and acetyl low density lipoprotein receptor messenger RNA colocalize with oxidation specific lipid-protein adducts. J. Clin. Investig. 1991, 87, 1146–1152. [Google Scholar] [CrossRef]
- Steinberg, D. The LDL modification hypothesis of atherogenesis: An update. J. Lipid Res. 2009, 50, S376–S381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Schindhelm, R.K.; Van Der Zwan, L.P.; Teerlink, T.; Scheffer, P.G. Myeloperoxidase: A Useful Biomarker for Cardiovascular Disease Risk Stratification? Clin. Chem. 2009, 55, 1462–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younis, N.; Sharma, R.; Soran, H.; Charlton-Menys, V.; Elseweidy, M.; Durrington, P.N. Glycation as an atherogenic modification of LDL. Curr. Opin. Lipidol. 2008, 19, 378–384. [Google Scholar] [CrossRef]
- Menzel, E.J.; Sobal, G.; Staudinger, A. The role of oxidative stress in the long-term glycation of LDL. BioFactors 1997, 6, 111–124. [Google Scholar] [CrossRef]
- Bucala, R.; Makita, Z.; Koschinsky, T.; Cerami, A.; Vlassara, H. Lipid advanced glycosylation: Pathway for lipid oxidation in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 6434–6438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiserich, J.P.; Baldus, S.; Brennan, M.-L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a Leukocyte-Derived Vascular NO Oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef]
- Berliner, J.A.; Heinecke, J.W. The role of oxidized lipoproteins in atherogenesis. Free. Radic. Biol. Med. 1996, 20, 707–727. [Google Scholar] [CrossRef]
- Mora, S.; Caulfield, M.P.; Wohlgemuth, J.; Chen, Z.; Superko, H.R.; Rowland, C.M.; Glynn, R.J.; Ridker, P.M.; Krauss, R.M. Atherogenic Lipoprotein Subfractions Determined by Ion Mobility and First Cardiovascular Events After Random Allocation to High-Intensity Statin or Placebo: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) Trial. Circulation 2015, 132, 2220–2229. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.-J.; Krauss, R.M. Apolipoprotein CIII bound to apoB-containing lipoproteins is associated with small, dense LDL independent of plasma triglyceride levels in healthy men. Atherosclerosis 2010, 211, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Chancharme, L.; Thérond, P.; Nigon, F.; Lepage, S.; Couturier, M.; Chapman, M.J. Cholesteryl Ester Hydroperoxide Lability Is a Key Feature of the Oxidative Susceptibility of Small, Dense LDL. Arter. Thromb. Vasc. Biol. 1999, 19, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thongtang, N.; Diffenderfer, M.R.; Ooi, E.M.M.; Barrett, P.H.R.; Turner, S.M.; Le, N.-A.; Brown, W.V.; Schaefer, E.J. Metabolism and proteomics of large and small dense LDL in combined hyperlipidemia: Effects of rosuvastatin. J. Lipid Res. 2017, 58, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Koplev, S.; Fisher, E.A.; Tabas, I.; Björkegren, J.L.M.; Doran, A.C.; Kovacic, J.C. Macrophage Trafficking, Inflammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series (Part 2). J. Am. Coll. Cardiol. 2018, 72, 2181–2197. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Witztum, J.L.; Lichtman, A.H. The Influence of Innate and Adaptive Immune Responses on Atherosclerosis. Annu. Rev. Pathol. 2014, 9, 73–102. [Google Scholar] [CrossRef] [Green Version]
- Kovanen, P.T.; Bot, I. Mast cells in atherosclerotic cardiovascular disease—Activators and actions. Eur. J. Pharmacol. 2017, 816, 37–46. [Google Scholar] [CrossRef]
- Castelli, W.P.; Garrison, R.J.; Wilson, P.W.F.; Abbott, R.D.; Kalousdian, S.; Kannel, W.B. Incidence of Coronary Heart Disease and Lipoprotein Cholesterol Levels: The Framingham Study. JAMA 1986, 256, 2835–2838. [Google Scholar] [CrossRef] [PubMed]
- Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; Packard, C.J.; et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Keene, D.; Price, C.; Shun-Shin, M.J.; Francis, D.P. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: Meta-analysis of randomised controlled trials including 117 411 patients. BMJ 2014, 349, g4379. [Google Scholar] [CrossRef] [Green Version]
- Riaz, H.; Khan, S.U.; Rahman, H.; Shah, N.P.; Kaluski, E.; Lincoff, A.M.; Nissen, S.E. Effects of high-density lipoprotein targeting treatments on cardiovascular outcomes: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2019, 26, 533–543. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle Subclasses and Molecular Components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonacina, F.; Pirillo, A.; Catapano, A.; Norata, G. HDL in Immune-Inflammatory Responses: Implications beyond Cardiovascular Diseases. Cells 2021, 10, 1061. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L.; Norata, G.D. Biological Consequences of Dysfunctional HDL. Curr. Med. Chem. 2019, 26, 1644–1664. [Google Scholar] [CrossRef] [PubMed]
- Márquez, A.B.; Nazir, S.; Van Der Vorst, E.P. High-Density Lipoprotein Modifications: A Pathological Consequence or Cause of Disease Progression? Biomedicines 2020, 8, 549. [Google Scholar] [CrossRef]
- Chiesa, S.T.; Charakida, M. High-Density Lipoprotein Function and Dysfunction in Health and Disease. Cardiovasc. Drugs Ther. 2019, 33, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Ertek, S. High-density Lipoprotein (HDL) Dysfunction and the Future of HDL. Curr. Vasc. Pharmacol. 2018, 16, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol. Cell. Biochem. 2017, 440, 167–187. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Sheng, H.; Li, H.; Wang, R. Acute phase reactant serum amyloid A in inflammation and other diseases. Advances in Clinical Chemistry 2019, 90, 25–80. [Google Scholar] [CrossRef]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nofer, J.-R.; Van Der Giet, M.; Tolle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Efrat, M.; Aviram, M. Paraoxonase 1 Interactions with HDL, Antioxidants and Macrophages Regulate Atherogenesis—A Protective Role for HDL Phospholipids. Adv. Exp. Med. Biol. 2009, 660, 153–166. [Google Scholar] [CrossRef]
- Hafiane, A.; Jabor, B.; Ruel, I.; Ling, J.; Genest, J. High-Density Lipoprotein Mediated Cellular Cholesterol Efflux in Acute Coronary Syndromes. Am. J. Cardiol. 2014, 113, 249–255. [Google Scholar] [CrossRef]
- Nobeécourt, E.; Tabet, F.; Lambert, G.; Puranik, R.; Bao, S.; Yan, L.; Davies, M.J.; Brown, B.E.; Jenkins, A.J.; Dusting, G.J.; et al. Nonenzymatic Glycation Impairs the Antiinflammatory Properties of Apolipoprotein A-I. Arter. Thromb. Vasc. Biol. 2010, 30, 766–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Yancey, P.G.; Ikizler, T.A.; Jerome, W.G.; Kaseda, R.; Cox, B.; Bian, A.; Shintani, A.; Fogo, A.B.; Linton, M.F.; et al. Dysfunctional High-Density Lipoprotein in Patients on Chronic Hemodialysis. J. Am. Coll. Cardiol. 2012, 60, 2372–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dron, J.S.; Wang, J.; Cao, H.; McIntyre, A.D.; Iacocca, M.A.; Menard, J.R.; Movsesyan, I.; Malloy, M.J.; Pullinger, C.R.; Kane, J.P.; et al. Severe hypertriglyceridemia is primarily polygenic. J. Clin. Lipidol. 2019, 13, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Truthmann, J.; Schienkiewitz, A.; Busch, M.A.; Mensink, G.B.M.; Du, Y.; Bosy-Westphal, A.; Knopf, H.; Scheidt-Nave, C. Changes in mean serum lipids among adults in Germany: Results from National Health Surveys 1997-99 and 2008-11. BMC Public Health 2016, 16, 240. [Google Scholar] [CrossRef] [Green Version]
- Hegele, R.A.; Ginsberg, H.N.; Chapman, M.J.; Nordestgaard, B.G.; Kuivenhoven, J.A.; Averna, M.; Borén, J.; Bruckert, E.; Catapano, A.L.; Descamps, O.S.; et al. The polygenic nature of hypertriglyceridaemia: Implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014, 2, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Dash, S.; Morgantini, C.; Hegele, R.A.; Lewis, G.F. Pharmacological Targeting of the Atherogenic Dyslipidemia Complex: The Next Frontier in CVD Prevention Beyond Lowering LDL Cholesterol. Diabetes 2016, 65, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Stahel, P.; Xiao, C.; Hegele, R.A.; Lewis, G.F. The Atherogenic Dyslipidemia Complex and Novel Approaches to Cardiovascular Disease Prevention in Diabetes. Can. J. Cardiol. 2018, 34, 595–604. [Google Scholar] [CrossRef]
- Ganda, O.P.; Bhatt, D.L.; Mason, R.P.; Miller, M.; Boden, W.E. Unmet Need for Adjunctive Dyslipidemia Therapy in Hypertriglyceridemia Management. J. Am. Coll. Cardiol. 2018, 72, 330–343. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Unmet need for primary prevention in individuals with hypertriglyceridaemia not eligible for statin therapy according to European Society of Cardiology/European Atherosclerosis Society guidelines: A contemporary population-based study. Eur. Heart J. 2018, 39, 610–619. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Abt, M.; Bao, W.; DeMicco, D.; Kallend, D.; Miller, M.; Mundl, H.; Olsson, A.G. Fasting Triglycerides Predict Recurrent Ischemic Events in Patients with Acute Coronary Syndrome Treated with Statins. J. Am. Coll. Cardiol. 2015, 65, 2267–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarwar, N.; Sandhu, M.S.; Ricketts, S.L.; Butterworth, A.S.; Di Angelantonio, E.; Boekholdt, M.; Ouwehand, W.; Watkins, H.; Samani, N.J.; Saleheen, D.; et al. Triglyceride-mediated pathways and coronary disease: Collaborative analysis of 101 studies. Lancet 2010, 375, 1634–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dron, J.S.; Hegele, R.A. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr. Opin. Lipidol. 2017, 28, 161–169. [Google Scholar] [CrossRef]
- Ference, B.A.; Kastelein, J.J.P.; Ray, K.K.; Ginsberg, H.N.; Chapman, M.J.; Packard, C.J.; Laufs, U.; Oliver-Williams, C.; Wood, A.M.; Butterworth, A.S.; et al. Association of Triglyceride-Lowering LPL Variants and LDL-C–Lowering LDLR Variants with Risk of Coronary Heart Disease. JAMA 2019, 321, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, N.; Danesh, J.; Eiriksdottir, G.; Sigurdsson, G.; Wareham, N.; Bingham, S.; Boekholdt, S.M.; Khaw, K.-T.; Gudnason, V. Triglycerides and the Risk of Coronary Heart Disease. Circulation 2007, 115, 450–458. [Google Scholar] [CrossRef]
- Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.J.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; Saleheen, D.; et al. Lipid-Related Markers and Cardiovascular Disease Prediction. JAMA 2012, 307, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Laufs, U.; Parhofer, K.G.; Ginsberg, H.N.; Hegele, R.A. Clinical review on triglycerides. Eur. Heart J. 2020, 41, 99–109c. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels: A Systematic Review and Meta-analysis. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef] [Green Version]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Ference, B.A.; Yoo, W.; Alesh, I.; Mahajan, N.; Mirowska, K.K.; Mewada, A.; Kahn, J.; Afonso, L.; Williams, K.A.; Flack, J.M. Effect of Long-Term Exposure to Lower Low-Density Lipoprotein Cholesterol Beginning Early in Life on the Risk of Coronary Heart Disease: A Mendelian Randomization Analysis. J. Am. Coll. Cardiol. 2012, 60, 2631–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 2015, 36, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razi, F.; Forouzanfar, K.; Bandarian, F.; Nasli-Esfahani, E. LDL-cholesterol measurement in diabetic type 2 patients: A comparison between direct assay and popular equations. J. Diabetes Metab. Disord. 2017, 16, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlois, M.R.; Descamps, O.S.; van der Laarse, A.; Weykamp, C.; Baum, H.; Pulkki, K.; von Eckardstein, A.; De Bacquer, D.; Borén, J.; Wiklund, O.; et al. Clinical impact of direct HDLc and LDLc method bias in hypertriglyceridemia. A simulation study of the EAS-EFLM Collaborative Project Group. Atherosclerosis 2014, 233, 83–90. [Google Scholar] [CrossRef]
- Baca, A.M.; Warnick, G.R. Estimation of LDL-Associated Apolipoprotein B from Measurements of Triglycerides and Total Apolipoprotein B. Clin. Chem. 2008, 54, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.P.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.-C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef]
- Kuehn, B.M. Evinacumab Approval Adds a New Option for Homozygous Familial Hypercholesterolemia with a Hefty Price Tag. Circulation 2021, 143, 2494–2496. [Google Scholar] [CrossRef]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’Dea, L.S.L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef]
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796. [Google Scholar] [CrossRef]
- Michael Gibson, C.; Korjian, S.; Tricoci, P.; Daaboul, Y.; Yee, M.; Jain, P.; Alexander, J.H.; Steg, P.G.; Lincoff, A.M.; Kastelein, J.J.; et al. Safety and Tolerability of CSL112, a Reconstituted, Infusible, Plasma-Derived Apolipoprotein A-I, After Acute Myocardial Infarction: The AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation 2016, 134, 1918–1930. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Norata, G.D.; Tsimikas, S.; Pirillo, A.; Catapano, A.L. Apolipoprotein C-III: From Pathophysiology to Pharmacology. Trends Pharmacol. Sci. 2015, 36, 675–687. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Gallo, A.; Di Costanzo, A.; Bruckert, E.; Arca, M. Evaluation of efficacy and safety of antisense inhibition of apolipoprotein C-III with volanesorsen in patients with severe hypertriglyceridemia. Expert Opin. Pharmacother. 2020, 21, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Jensen, M.K.; Aroner, S.; Furtado, J.D.; Rosner, B.; Hu, F.B.; Balkau, B.; Natali, A.; Ferrannini, E.; Baldi, S.; et al. HDL Containing Apolipoprotein C-III is Associated with Insulin Sensitivity: A Multicenter Cohort Study. J. Clin. Endocrinol. Metab. 2021, e2928–e2940. [Google Scholar] [CrossRef]
- Zvintzou, E.; Lhomme, M.; Chasapi, S.; Filou, S.; Theodoropoulos, V.; Xapapadaki, E.; Kontush, A.; Spyroulias, G.; Tellis, C.C.; Tselepis, A.D.; et al. Pleiotropic effects of apolipoprotein C3 on HDL functionality and adipose tissue metabolic activity. J. Lipid Res. 2017, 58, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, A.; Aikawa, M.; Alcaide, P.; Luscinskas, F.W.; Libby, P.; Sacks, F.M. Apolipoprotein CIII Induces Expression of Vascular Cell Adhesion Molecule-1 in Vascular Endothelial Cells and Increases Adhesion of Monocytic Cells. Circulation 2006, 114, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Valladolid-Acebes, I.; Berggren, P.-O.; Juntti-Berggren, L. Apolipoprotein CIII Is an Important Piece in the Type-1 Diabetes Jigsaw Puzzle. Int. J. Mol. Sci. 2021, 22, 932. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.B.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Loss-of-Function Mutations inAPOC3and Risk of Ischemic Vascular Disease. N. Engl. J. Med. 2014, 371, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Wulff, A.B.; Nordestgaard, B.G.; Tybjærg-Hansen, A. APOC3 Loss-of-Function Mutations, Remnant Cholesterol, Low-Density Lipoprotein Cholesterol, and Cardiovascular Risk: Mediation- and Meta-Analyses of 137 895 Individuals. Arter. Thromb. Vasc. Biol. 2018, 38, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, P.; Kohli, P.; Baber, U.; Nguyen, K.-D.H.; Sartori, S.; Reilly, D.F.; Mehran, R.; Muntendam, P.; Fuster, V.; Rader, D.J.; et al. Association of APOC3 Loss-of-Function Mutations with Plasma Lipids and Subclinical Atherosclerosis: The Multi-Ethnic BioImage Study. J. Am. Coll. Cardiol. 2015, 66, 2053–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosby, J.; Peloso, G.M.; Auer, P.L.; Crosslin, D.R.; Stitziel, N.; Lange, L.A.; Lu, Y.; Tang, Z.Z.; Zhang, H.; Hindy, G.; et al. Loss-of-Function Mutations in APOC3, Triglycerides, and Coronary Disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleheen, D.; Natarajan, P.; Armean, I.; Zhao, W.; Rasheed, A.; Khetarpal, S.A.; Won, H.-H.; Karczewski, K.J.; O’Donnell-Luria, A.; Samocha, K.; et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 2017, 544, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.N.; Raffield, L.M.; Freedman, B.I.; Langefeld, C.D.; Ng, M.C.; Carr, J.J.; Cox, A.J.; Bowden, N.W. Analysis of common and coding variants with cardiovascular disease in the diabetes heart study. Cardiovasc. Diabetol. 2014, 13, 77. [Google Scholar] [CrossRef] [Green Version]
- Maki, K.C.; Bays, H.E.; Dicklin, M.R.; Johnson, S.L.; Shabbout, M. Effects of prescription omega-3-acid ethyl esters, coadministered with atorvastatin, on circulating levels of lipoprotein particles, apolipoprotein CIII, and lipoprotein-associated phospholipase A2 mass in men and women with mixed dyslipidemia. J. Clin. Lipidol. 2011, 5, 483–492. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Ooi, E.M.; Ji, J.; Johnson, A.G.; Barrett, P.H. Atorvastatin and Fenofibrate Have Comparable Effects on VLDL-Apolipoprotein C-III Kinetics in Men With the Metabolic Syndrome. Arter. Thromb. Vasc. Biol. 2008, 28, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Ooi, E.; Watts, G.; Chan, D.; Chen, M.M.; Nestel, P.J.; Sviridov, D.; Barrett, P.H.R. Dose-Dependent Effect of Rosuvastatin on VLDL-Apolipoprotein C-III Kinetics in the Metabolic Syndrome. Diabetes Care 2008, 31, 1656–1661. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, K.; Lopez, C.; Donovan, D.; Ngai, C.; Fontanez, N.; Bensadoun, A.; Fruchart-Najib, J.; Holleran, S.; Cohn, J.S.; Ramakrishnan, R.; et al. Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J. Clin. Investig. 2005, 115, 1323–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogacci, F.; Norata, G.D.; Toth, P.P.; Arca, M.; Cicero, A.F.G. Efficacy and Safety of Volanesorsen (ISIS 304801): The Evidence from Phase 2 and 3 Clinical Trials. Curr. Atheroscler. Rep. 2020, 22, 18. [Google Scholar] [CrossRef]
- Digenio, A.; Dunbar, R.L.; Alexander, V.J.; Hompesch, M.; Morrow, L.; Lee, R.G.; Graham, M.J.; Hughes, S.G.; Yu, R.; Singleton, W.; et al. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care 2016, 39, 1408–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, M.J.; Crooke, S.T.; Lemonidis, K.M.; Gaus, H.J.; Templin, M.V.; Crooke, R.M. Hepatic distribution of a phosphorothioate oligodeoxynucleotide within rodents following intravenous administration. Biochem. Pharmacol. 2001, 62, 297–306. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Yu, R.Z.; Graham, M.J.; Post, N.; Riney, S.; Zanardi, T.; Hall, S.; Burkey, J.; Shemesh, C.S.; Prakash, T.P.; Seth, P.P.; et al. Disposition and Pharmacology of a GalNAc3-conjugated ASO Targeting Human Lipoprotein (a) in Mice. Mol. Ther. Nucleic Acids 2016, 5, e317. [Google Scholar] [CrossRef] [Green Version]
- Gillotte, K.L.; Zaiou, M.; Lund-Katz, S.; Anantharamaiah, G.; Holvoet, P.; Dhoest, A.; Palgunachari, M.N.; Segrest, J.P.; Weisgraber, K.H.; Rothblat, G.H.; et al. Apolipoprotein-mediated Plasma Membrane Microsolubilization. Role of lipid affinity and membrane penetration in the efflux of cellular cholesterol and phospholipid. J. Biol. Chem. 1999, 274, 2021–2028. [Google Scholar] [CrossRef] [Green Version]
- Okuhira, K.-I.; Tsujita, M.; Yamauchi, Y.; Abe-Dohmae, S.; Kato, K.; Handa, T.; Yokoyama, S. Potential involvement of dissociated apoA-I in the ABCA1-dependent cellular lipid release by HDL. J. Lipid Res. 2004, 45, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtiss, L.K.; Valenta, D.T.; Hime, N.J.; Rye, K.-A. What Is So Special About Apolipoprotein AI in Reverse Cholesterol Transport? Arter. Thromb. Vasc. Biol. 2006, 26, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Duong, P.T.; Weibel, G.L.; Lund-Katz, S.; Rothblat, G.H.; Phillips, M.C. Characterization and properties of preβ-HDL particles formed by ABCA1-mediated cellular lipid efflux to apoA-I. J. Lipid Res. 2008, 49, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenson, R.S.; Brewer, H.B., Jr.; Chapman, M.J.; Fazio, S.; Hussain, M.M.; Kontush, A.; Krauss, R.M.; Otvos, J.D.; Remaley, A.T.; Schaefer, E.J. HDL Measures, Particle Heterogeneity, Proposed Nomenclature, and Relation to Atherosclerotic Cardiovascular Events. Clin. Chem. 2011, 57, 392–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dergunov, A.D.; Litvinov, D.Y.; Malkov, A.A.; Baserova, V.B.; Nosova, E.V.; Dergunova, L.V. Denaturation of human plasma high-density lipoproteins by urea studied by apolipoprotein A-I dissociation. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2021, 1866, 158814. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.S.; DiDonato, J.A.; et al. Effects of Native and Myeloperoxidase-Modified Apolipoprotein A-I on Reverse Cholesterol Transport and Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Wilson, C.J.; Mei, X.; Wales, T.E.; Engen, J.R.; Gursky, O. Structural Stability and Local Dynamics in Disease-Causing Mutants of Human Apolipoprotein A-I: What Makes the Protein Amyloidogenic? J. Mol. Biol. 2016, 428, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Petrlova, J.; Duong, T.; Cochran, M.C.; Axelsson, A.; Mörgelin, M.; Roberts, L.M.; Lagerstedt, J.O. The fibrillogenic L178H variant of apolipoprotein A-I forms helical fibrils. J. Lipid Res. 2012, 53, 390–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosu, S.A.; Rimoldi, O.J.; Prieto, E.D.; Curto, L.M.; Delfino, J.M.; Ramella, N.A.; Tricerri, M.A. Amyloidogenic Propensity of a Natural Variant of Human Apolipoprotein A-I: Stability and Interaction with Ligands. PLoS ONE 2015, 10, e0124946. [Google Scholar] [CrossRef] [Green Version]
- Dalla-Riva, J.; Lagerstedt, J.O.; Petrlova, J. Structural and Functional Analysis of the ApolipoproteinA-I A164S Variant. PLoS ONE 2015, 10, e0143915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sviridov, D.; Hoang, A.; Huang, W.; Sasaki, J. Structure-function studies of apoA-I variants: Site-directed mutagenesis and natural mutations. J. Lipid Res. 2002, 43, 1283–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chroni, A.; Kan, H.-Y.; Kypreos, K.E.; Gorshkova, I.N.; Shkodrani, A.; Zannis, V.I. Substitutions of Glutamate 110 and 111 in the Middle Helix 4 of Human Apolipoprotein A-I (apoA-I) by Alanine Affect the Structure and In Vitro Functions of apoA-I and Induce Severe Hypertriglyceridemia in apoA-I-Deficient Mice. Biochemistry 2004, 43, 10442–10457. [Google Scholar] [CrossRef] [PubMed]
- Kateifides, A.K.; Gorshkova, I.; Duka, A.; Chroni, A.; Kardassis, D.; Zannis, V.I. Alteration of negatively charged residues in the 89 to 99 domain of apoA-I affects lipid homeostasis and maturation of HDL. J. Lipid Res. 2011, 52, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniil, G.; Zannis, V.I.; Chroni, A. Effect of apoA-I Mutations in the Capacity of Reconstituted HDL to Promote ABCG1-Mediated Cholesterol Efflux. PLoS ONE 2013, 8, e67993. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.H.; Jonas, A. A key point mutation (V156E) affects the structure and functions of human apolipoprotein A-I. J. Biol. Chem. 2000, 275, 26821–26827. [Google Scholar] [CrossRef]
- Tian, S.; Jonas, A. Structural and functional properties of apolipoprotein A-I mutants containing disulfide-linked cysteines at positions 124 or 232. Biochim. Biophys. Acta 2002, 1599, 56–64. [Google Scholar] [CrossRef]
- Gkolfinopoulou, C.; Soukou, F.; Dafnis, I.; Kellici, T.; Sanoudou, D.; Mavromoustakos, T.; Stratikos, E.; Chroni, A. Structure–function analysis of naturally occurring apolipoprotein A-I L144R, A164S and L178P mutants provides insight on their role on HDL levels and cardiovascular risk. Cell. Mol. Life Sci. 2021, 78, 1523–1544. [Google Scholar] [CrossRef]
- Undurti, A.; Huang, Y.; Lupica, J.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of High Density Lipoprotein by Myeloperoxidase Generates a Pro-inflammatory Particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Zhou, L.; Tian, R.; Lu, N. Myeloperoxidase Targets Apolipoprotein A-I for Site-Specific Tyrosine Chlorination in Atherosclerotic Lesions and Generates Dysfunctional High-Density Lipoprotein. Chem. Res. Toxicol. 2021, 34, 1672–1680. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; De La Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; De Lemos, J.A.; et al. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühnast, S.; Fiocco, M.; van der Hoorn, J.W.; Princen, H.M.; Jukema, J.W. Innovative pharmaceutical interventions in cardiovascular disease: Focusing on the contribution of non-HDL-C/LDL-C-lowering versus HDL-C-raisingA systematic review and meta-analysis of relevant preclinical studies and clinical trials. Eur. J. Pharmacol. 2015, 763, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Hovingh, G.K.; Smits, L.P.; Stefanutti, C.; Soran, H.; Kwok, S.; de Graaf, J.; Gaudet, D.; Keyserling, C.H.; Klepp, H.; Frick, J.; et al. The effect of an apolipoprotein A-I–containing high-density lipoprotein–mimetic particle (CER-001) on carotid artery wall thickness in patients with homozygous familial hypercholesterolemia. Am. Heart J. 2015, 169, 736–742. [Google Scholar] [CrossRef]
- Suruga, K.; Miyoshi, T.; Kotani, K.; Ichikawa, K.; Miki, T.; Osawa, K.; Ejiri, K.; Toda, H.; Nakamura, K.; Morita, H.; et al. Higher oxidized high-density lipoprotein to apolipoprotein A-I ratio is associated with high-risk coronary plaque characteristics determined by CT angiography. Int. J. Cardiol. 2021, 324, 193–198. [Google Scholar] [CrossRef]
- Bertrand, C.; Saulnier, P.-J.; Potier, L.; Croyal, M.; Blanchard, V.; Gand, E.; Ragot, S.; Schneider, F.; Bocock, O.; Baillet-Blanco, L.; et al. Plasma concentrations of lipoproteins and risk of lower-limb peripheral artery disease in people with type 2 diabetes: The SURDIAGENE study. Diabetol. 2021, 64, 668–680. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.M.; Ann, S.; Kang, M.; Cheon, E.J.; An, D.B.; Choi, Y.R.; Lee, C.J.; Oh, J.; Park, S.; et al. Cholesterol Efflux and Collateral Circulation in Chronic Total Coronary Occlusion: Effect-Circ Study. J. Am. Heart Assoc. 2021, 10, e019060. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Kiss, D.; Rader, D. HDL-cholesterol and cardiovascular disease: Rethinking our approach. Curr. Opin. Cardiol. 2015, 30, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.; Rohatgi, A. HDL Cholesterol Efflux Capacity: Cardiovascular Risk Factor and Potential Therapeutic Target. Curr. Atheroscler. Rep. 2015, 18, 1–8. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [Green Version]
- Diditchenko, S.; Gille, A.; Pragst, I.; Stadler, D.; Waelchli, M.; Hamilton, R.; Leis, A.; Wright, S.D. Novel Formulation of a Reconstituted High-Density Lipoprotein (CSL112) Dramatically Enhances ABCA1-Dependent Cholesterol Efflux. Arter. Thromb. Vasc. Biol. 2013, 33, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Tricoci, P.; D’Andrea, D.M.; Gurbel, P.A.; Yao, Z.; Cuchel, M.; Winston, B.; Schott, R.; Weiss, R.; Blazing, M.A.; Cannon, L.; et al. Infusion of Reconstituted High-Density Lipoprotein, CSL112, in Patients with Atherosclerosis: Safety and Pharmacokinetic Results from a Phase 2a Randomized Clinical Trial. J. Am. Heart Assoc. 2015, 4, e002171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, E.; Pragst, I.; Waelchli, M.; Gille, A.; Schenk, S.; Mueller-Cohrs, J.; Diditchenko, S.; Zanoni, P.; Cuchel, M.; Seubert, A.; et al. Reconstituted high-density lipoprotein can elevate plasma alanine aminotransferase by transient depletion of hepatic cholesterol: Role of the phospholipid component. J. Appl. Toxicol. 2016, 36, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Kastelein, J.J.; Phillips, A.T.; Aylward, P.E.; Yee, M.K.; Tendera, M.; Nicholls, S.J.; Pocock, S.; Goodman, S.G.; Alexander, J.H.; et al. Rationale and design of ApoA-I Event Reducing in Ischemic Syndromes II (AEGIS-II): A phase 3, multicenter, double-blind, randomized, placebo-controlled, parallel-group study to investigate the efficacy and safety of CSL112 in subjects after acute myocardial infarction. Am. Heart J. 2021, 231, 121–127. [Google Scholar] [CrossRef]
- Berman, A.N.; Blankstein, R. Current and future role of lipoprotein(a) in preventive cardiology. Curr. Opin. Cardiol. 2019, 34, 514–518. [Google Scholar] [CrossRef]
- Orsó, E.; Schmitz, G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin. Res. Cardiol. Suppl. 2017, 12, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Tada, H.; Takamura, M.; Kawashiri, M.-A. Lipoprotein(a) as an Old and New Causal Risk Factor of Atherosclerotic Cardiovascular Disease. J. Atheroscler. Thromb. 2019, 26, 583–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopewell, J.C.; Haynes, R.; Baigent, C. The role of lipoprotein (a) in chronic kidney disease. J. Lipid Res. 2018, 59, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Bermúdez-López, M.; Forne, C.; Amigo, N.; Bozic, M.; Arroyo, D.; Bretones, T.; Alonso, N.; Cambray, S.; Del Pino, M.D.; Mauricio, D.; et al. An in-depth analysis shows a hidden atherogenic lipoprotein profile in non-diabetic chronic kidney disease patients. Expert Opin. Ther. Targets 2019, 23, 619–630. [Google Scholar] [CrossRef]
- Agrawal, S.; Zaritsky, J.J.; Fornoni, A.; Smoyer, W.E. Dyslipidaemia in nephrotic syndrome: Mechanisms and treatment. Nat. Rev. Nephrol. 2018, 14, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Prado, R.; Perez-Gomez, M.V.; Ortiz, A. Pelacarsen for lowering lipoprotein(a): Implications for patients with chronic kidney disease. Clin. Kidney J. 2020, 13, 753–757. [Google Scholar] [CrossRef]
- Stulnig, T.M.; Morozzi, C.; Reindl-Schwaighofer, R.; Stefanutti, C. Looking at Lp(a) and Related Cardiovascular Risk: From Scientific Evidence and Clinical Practice. Curr. Atheroscler. Rep. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Tang, X.-F.; Song, Y.; Zhang, Y.; Gao, L.-J.; Gao, Z.; Chen, J.; Yang, Y.-J.; Gao, R.-L.; Xu, B.; et al. Association of lipoprotein(a) with platelet aggregation and thrombogenicity in patients undergoing percutaneous coronary intervention. Platelets 2021, 32, 684–689. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T.; Johnston, T.P.; Banach, M.; Pirro, M.; Sahebkar, A. Lipoprotein(a): A missing culprit in the management of athero-thrombosis? J. Cell. Physiol. 2017, 233, 2966–2981. [Google Scholar] [CrossRef]
- Kadl, A.; Sharma, P.R.; Chen, W.; Agrawal, R.; Meher, A.K.; Rudraiah, S.; Grubbs, N.; Sharma, R.; Leitinger, N. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free. Radic. Biol. Med. 2011, 51, 1903–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; Van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circ. 2016, 134, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Tselepis, A.D. Oxidized phospholipids and lipoprotein-associated phospholipase A2 as important determinants of Lp(a) functionality and pathophysiological role. J. Biomed. Res. 2016, 32, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme Lipoprotein(a) Levels and Improved Cardiovascular Risk Prediction. J. Am. Coll. Cardiol. 2013, 61, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Andres, E.; Mata, N.; Fuentes-Jiménez, F.; Badimón, L.; Lopez-Miranda, J.; Padro, T.; Muñiz, O.; Díaz, J.L.D.; Mauri, M.; et al. Lipoprotein(a) Levels in Familial Hypercholesterolemia: An important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J. Am. Coll. Cardiol. 2014, 63, 1982–1989. [Google Scholar] [CrossRef] [Green Version]
- Langsted, A.; Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: A prospective cohort study. Lancet Diabetes Endocrinol. 2016, 4, 577–587. [Google Scholar] [CrossRef]
- Ong, K.L.; McClelland, R.L.; Allison, M.A.; Cushman, M.; Garg, P.K.; Tsai, M.Y.; Rye, K.-A.; Tabet, F. Lipoprotein (a) and coronary artery calcification: Prospective study assessing interactions with other risk factors. Metab. Clin. Exp. 2021, 116, 154706. [Google Scholar] [CrossRef]
- Jun, J.E.; Kang, H.; Hwang, Y.-C.; Ahn, K.J.; Chung, H.-Y.; Jeong, I.-K. The association between lipoprotein (a) and carotid atherosclerosis in patients with type 2 diabetes without pre-existing cardiovascular disease: A cross-sectional study. Diabetes Res. Clin. Pr. 2021, 171, 108622. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Schweizer, J.; Nakas, C.T.; Schütz, V.; Westphal, L.P.; Inauen, C.; Pokorny, T.; Luft, A.; Leichtle, A.; Arnold, M.; et al. Lipoprotein(a) is associated with large artery atherosclerosis stroke aetiology and stroke recurrence among patients below the age of 60 years: Results from the BIOSIGNAL study. Eur. Heart J. 2021, 42, 2186–2196. [Google Scholar] [CrossRef]
- De Isla, L.P.; Watts, G.F.; Alonso, R.; Díaz-Díaz, J.L.; Muñiz-Grijalvo, O.; Zambón, D.; Fuentes, F.; De Andrés, R.; Padró, T.; López-Miranda, J.; et al. Lipoprotein(a), LDL-cholesterol, and hypertension: Predictors of the need for aortic valve replacement in familial hypercholesterolaemia. Eur. Heart J. 2021, 42, 2201–2211. [Google Scholar] [CrossRef]
- Fogacci, F.; Cicero, A.F.G.; D’Addato, S.; D’Agostini, L.; Rosticci, M.; Giovannini, M.; Bertagnin, E.; Borghi, C. Serum lipoprotein(a) level as long-term predictor of cardiovascular mortality in a large sample of subjects in primary cardiovascular prevention: Data from the Brisighella Heart Study. Eur. J. Intern. Med. 2017, 37, 49–55. [Google Scholar] [CrossRef]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights from a Large National Biobank. Arter. Thromb. Vasc. Biol. 2020, 41, 465–474. [Google Scholar] [CrossRef]
- Kronenberg, F.; Utermann, G. Lipoprotein(a): Resurrected by genetics. J. Intern. Med. 2013, 273, 6–30. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, F. Human Genetics and the Causal Role of Lipoprotein(a) for Various Diseases. Cardiovasc. Drugs Ther. 2016, 30, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Vongpromek, R.; Bos, S.; Kate, G.-J.R.T.; Yahya, R.; Verhoeven, A.J.M.; De Feyter, P.J.; Kronenberg, F.; Van Lennep, J.E.R.; Sijbrands, E.; Mulder, M.T. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J. Intern. Med. 2015, 278, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjærg -Hansen, A.; Nordestgaard, B.G. Genetic Evidence That Lipoprotein(a) Associates with Atherosclerotic Stenosis Rather Than Venous Thrombosis. Arter. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, M.A.; Yeung, M.W.; van de Vegte, Y.J.; Benjamins, J.W.; Dullaart, R.P.; Ruotsalainen, S.; Ripatti, S.; Natarajan, P.; Juarez-Orozco, L.E.; Verweij, N.; et al. Genome-Wide Association Study and Identification of a Protective Missense Variant on Lipoprotein(a) Concentration: Protective Missense Variant on Lipoprotein(a) Concentration-Brief Report. Arter. Thromb. Vasc. Biol. 2021, 41, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Nordestgaard, B.G. Lipoprotein(a) concentrations, isoform size, and risk of type 2 diabetes: A Mendelian randomisation study. Lancet Diabetes Endocrinol. 2013, 1, 220–227. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated Lipoprotein(a) and Risk of Aortic Valve Stenosis in the General Population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; Danesh, J. Lipoprotein(a) Concentration and the Risk of Coronary Heart Disease, Stroke, and Nonvascular Mortality. JAMA 2009, 302, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, N.; Joshi, P.H. Lp(a): Addressing a Target for Cardiovascular Disease Prevention. Curr. Cardiol. Rep. 2019, 21, 102. [Google Scholar] [CrossRef]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Utermann, G.; Hoppichler, F.; Dieplinger, H.; Seed, M.; Thompson, G.; Boerwinkle, E. Defects in the low density lipoprotein receptor gene affect lipoprotein (a) levels: Multiplicative interaction of two gene loci associated with premature atherosclerosis. Proc. Natl. Acad. Sci. USA 1989, 86, 4171–4174. [Google Scholar] [CrossRef] [Green Version]
- Marcovina, S.M.; Koschinsky, M.; Albers, J.J.; Skarlatos, S. Report of the National Heart, Lung, and Blood Institute Workshop on Lipoprotein(a) and Cardiovascular Disease: Recent Advances and Future Directions. Clin. Chem. 2003, 49, 1785–1796. [Google Scholar] [CrossRef] [Green Version]
- Marcovina, S.M.; Albers, J.J. Lipoprotein (a) measurements for clinical application. J. Lipid Res. 2016, 57, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Bittner, V.A.; Szarek, M.; Aylward, P.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Szarek, M.; Bittner, V.A.; Aylward, P.; Baccara-Dinet, M.; Bhatt, D.L.; Diaz, R.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Harrington, R.A.; et al. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur. Heart J. 2020, 41, 4245–4255. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statins and increases in Lp(a): An inconvenient truth that needs attention. Eur. Heart J. 2019, 41, 192–193. [Google Scholar] [CrossRef]
- Cao, Y.-X.; Liu, H.-H.; Li, S.; Li, J.-J. A Meta-Analysis of the Effect of PCSK9-Monoclonal Antibodies on Circulating Lipoprotein (a) Levels. Am. J. Cardiovasc. Drugs 2019, 19, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, E.; Parhofer, K.G. Lipoprotein apheresis to treat elevated lipoprotein (a). J. Lipid Res. 2016, 57, 1751–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Lehmacher, W.; Kamstrup, P.R.; Nordestgaard, B.G.; et al. Lipoprotein Apheresis for Lipoprotein(a)-Associated Cardiovascular Disease: Prospective 5 Years of Follow-Up and Apolipoprotein(a) Characterization. Arter. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef] [Green Version]
- Safarova, M.S.; Ezhov, M.V.; Afanasieva, O.; Matchin, Y.G.; Atanesyan, R.V.; Adamova, I.Y.; Utkina, E.; Konovalov, G.A.; Pokrovsky, S. Effect of specific lipoprotein(a) apheresis on coronary atherosclerosis regression assessed by quantitative coronary angiography. Atheroscler. Suppl. 2013, 14, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Stefanutti, C.; Pisciotta, L.; Favari, E.; Di Giacomo, S.; Vacondio, F.; Zenti, M.G.; Morozzi, C.; Berretti, D.; Mesce, D.; Vitale, M.; et al. Lipoprotein(a) concentration, genetic variants, apo(a) isoform size, and cellular cholesterol efflux in patients with elevated Lp(a) and coronary heart disease submitted or not to lipoprotein apheresis: An Italian case-control multicenter study on Lp(a). J. Clin. Lipidol. 2020, 14, 487–497. [Google Scholar] [CrossRef]
- Schreml, J.; Gouni-Berthold, I. Apolipoprotein(a) Antisense Oligonucleotides: A New Treatment Option for Lowering Elevated Lipoprotein(a)? Curr. Pharm. Des. 2017, 23, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assessing the Impact of Lipoprotein (a) Lowering with TQJ230 on Major Cardiovascular Events in Patients With CVD. Available online: https://ClinicalTrials.gov/show/NCT04023552 (accessed on 7 July 2021).
Figure 1.
Schematic diagram showing the essential modifications of low-density lipoprotein (LDL) and high-density lipoprotein (HDL) and potential therapeutic targets. HDL promotes cholesterol efflux from cells within atherosclerotic plaques through reverse cholesterol transport (RCT) and transports excess cholesterol from peripheral tissues to the liver for excretion. Post-translational modifications including oxidation, carbamylation, glycation, and alterations of its lipidomic and proteomic structure result in dysfunctional HDL. Infusion of reconstituted apolipoprotein A-I, CSL112, enhances RCT. Lipoprotein(a) (Lp(a)) promotes atherosclerosis through its proinflammatory and antifibrinolytic effects. The production of Lp(a) in the liver can be reduced by the novel antisense oligonucleotide, PELACARSEN. Angiopoietin-like protein 3 (ANGPTL3) produced in the liver inhibits lipoprotein lipase (LPL)-induced lipolysis, resulting in increased circulating triglycerides carried by triglyceride-rich lipoproteins (TRLs), and accelerated atherosclerosis [9]. This process can be blocked by the ANGPTL3 inhibitor, evinacumab. Native LDL is modified by oxidation, glycation, peroxidation, and apolipoprotein C-III (apoC-III) adhesion and becomes more atherogenic. The expression of apoC-III can be suppressed by another novel antisense oligonucleotide, AKCEA-ApoCIII-LRx.
Figure 1.
Schematic diagram showing the essential modifications of low-density lipoprotein (LDL) and high-density lipoprotein (HDL) and potential therapeutic targets. HDL promotes cholesterol efflux from cells within atherosclerotic plaques through reverse cholesterol transport (RCT) and transports excess cholesterol from peripheral tissues to the liver for excretion. Post-translational modifications including oxidation, carbamylation, glycation, and alterations of its lipidomic and proteomic structure result in dysfunctional HDL. Infusion of reconstituted apolipoprotein A-I, CSL112, enhances RCT. Lipoprotein(a) (Lp(a)) promotes atherosclerosis through its proinflammatory and antifibrinolytic effects. The production of Lp(a) in the liver can be reduced by the novel antisense oligonucleotide, PELACARSEN. Angiopoietin-like protein 3 (ANGPTL3) produced in the liver inhibits lipoprotein lipase (LPL)-induced lipolysis, resulting in increased circulating triglycerides carried by triglyceride-rich lipoproteins (TRLs), and accelerated atherosclerosis [9]. This process can be blocked by the ANGPTL3 inhibitor, evinacumab. Native LDL is modified by oxidation, glycation, peroxidation, and apolipoprotein C-III (apoC-III) adhesion and becomes more atherogenic. The expression of apoC-III can be suppressed by another novel antisense oligonucleotide, AKCEA-ApoCIII-LRx.


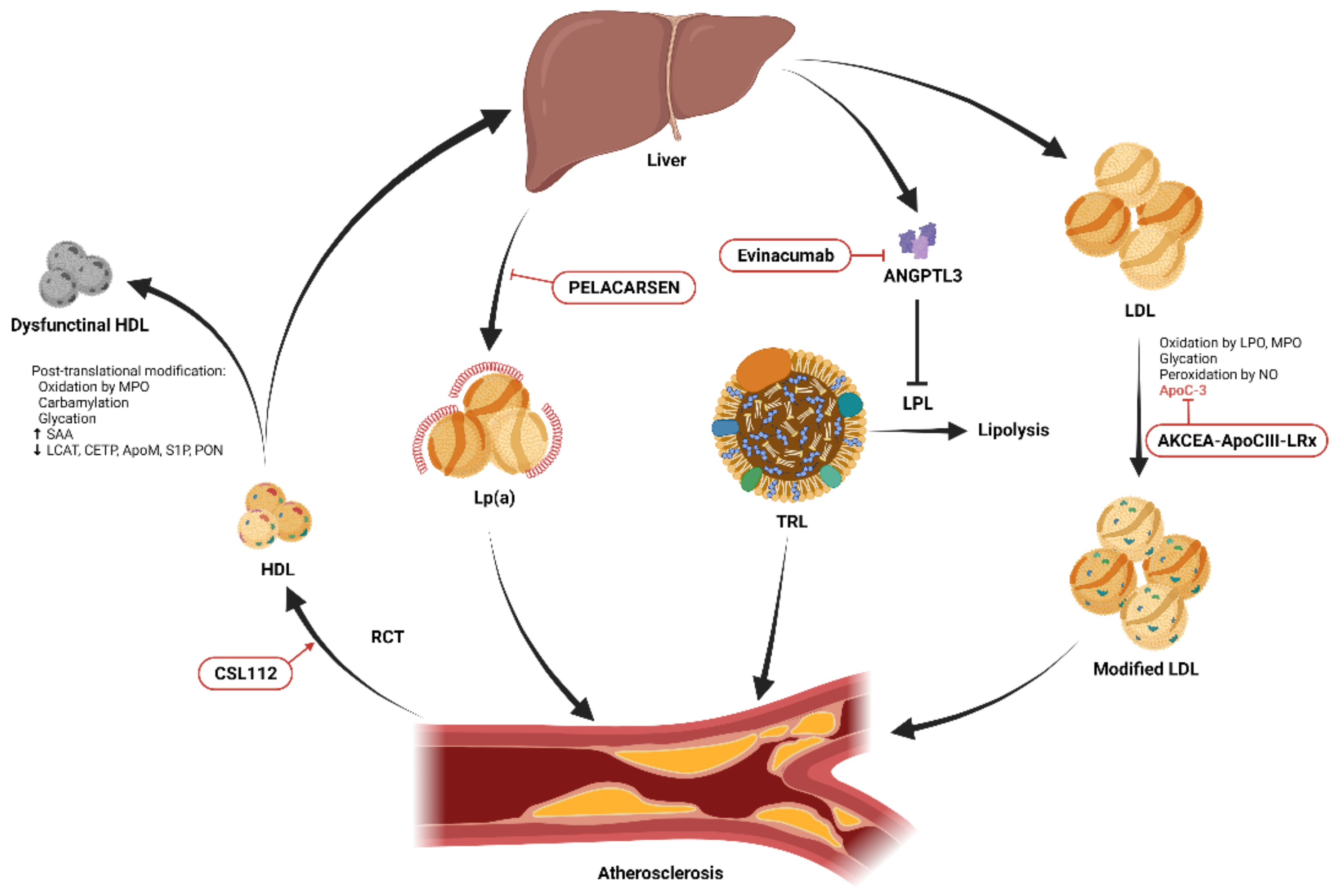{kind=link}
Table 1.
Potential therapeutic targets and emerging pharmacological lipid-lowering approaches.
| Potential Therapeutic Target | Pharmacological Approach | Published Clinical Trials | Subjects | Pros | Cons | Ongoing Trials and The Aims of The Trials |
|---|---|---|---|---|---|---|
| Angiopoietin-like protein 3 | Evinacumab, a recombinant human monoclonal antibody that inhibits angiopoietin-like protein 3 | ELIPSE HoFH (phase 3) [89] | Patients with homozygous familial hypercholesterolemia |
|
|
|
| ApoC-III | Volanesorsen, a 2ʹ-O-(2-methoxyethyl)-modified antisense oligonucleotide | APPROACH (phase 3) [91] | Patients with familial chylomicronemia syndrome |
|
| |
| COMPASS (phase 3) [92] | Patients with severe hypertriglyceridemia |
| ||||
| AKCEA-ApoCIII-LRx, a GalNAc3 modified antisense oligonucleotide | Phase 1/2a trial [93] | Healthy volunteers |
|
| ||
| ApoA-I | CSL112, a plasma-derived reconstituted apoA-I | Phase 2b AEGIS-I trial [94] | Patients with myocardial infarction |
|
| |
| Apolipoprotein(a) | PELACARSEN, an GalNAC3 modified antisense oligonucleotide | Phase 2 trial [95] | Patients with established ASCVD |
|
|
ABCA1, ATP binding cassette subfamily A member 1, ASCVD, atherosclerotic cardiovascular disease; GalNAc3, triantennary N-acetyl galactosamine; LDL-C, low-density lipoprotein cholesterol; TG, triglyceride; Lp(a), lipoprotein(a).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, C.-K.; Liao, C.-W.; Meng, S.-W.; Wu, W.-K.; Chiang, J.-Y.; Wu, M.-S. Lipids and Lipoproteins in Health and Disease: Focus on Targeting Atherosclerosis. Biomedicines 2021, 9, 985. https://doi.org/10.3390/biomedicines9080985
AMA Style
Lee C-K, Liao C-W, Meng S-W, Wu W-K, Chiang J-Y, Wu M-S. Lipids and Lipoproteins in Health and Disease: Focus on Targeting Atherosclerosis. Biomedicines. 2021; 9(8):985. https://doi.org/10.3390/biomedicines9080985
Chicago/Turabian StyleLee, Chih-Kuo, Che-Wei Liao, Shih-Wei Meng, Wei-Kai Wu, Jiun-Yang Chiang, and Ming-Shiang Wu. 2021. "Lipids and Lipoproteins in Health and Disease: Focus on Targeting Atherosclerosis" Biomedicines 9, no. 8: 985. https://doi.org/10.3390/biomedicines9080985
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.