Quinpirole-Mediated Regulation of Dopamine D2 Receptors Inhibits Glial Cell-Induced Neuroinflammation in Cortex and Striatum after Brain Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Quinpirole Treatment for Mice
2.3. Stab Wound Cortical Injury
2.4. Protein Extraction
2.5. Western Blot Analysis
2.6. Brain Tissue Collection and Sample Preparation
2.7. Immunofluorescence Staining
2.8. Assessment of Brain Lesion Volume
2.9. Nissl Staining
2.10. Cell Culture and Treatment
2.11. Microglial Conditioned Media
2.12. Statistical Analysis
3. Results
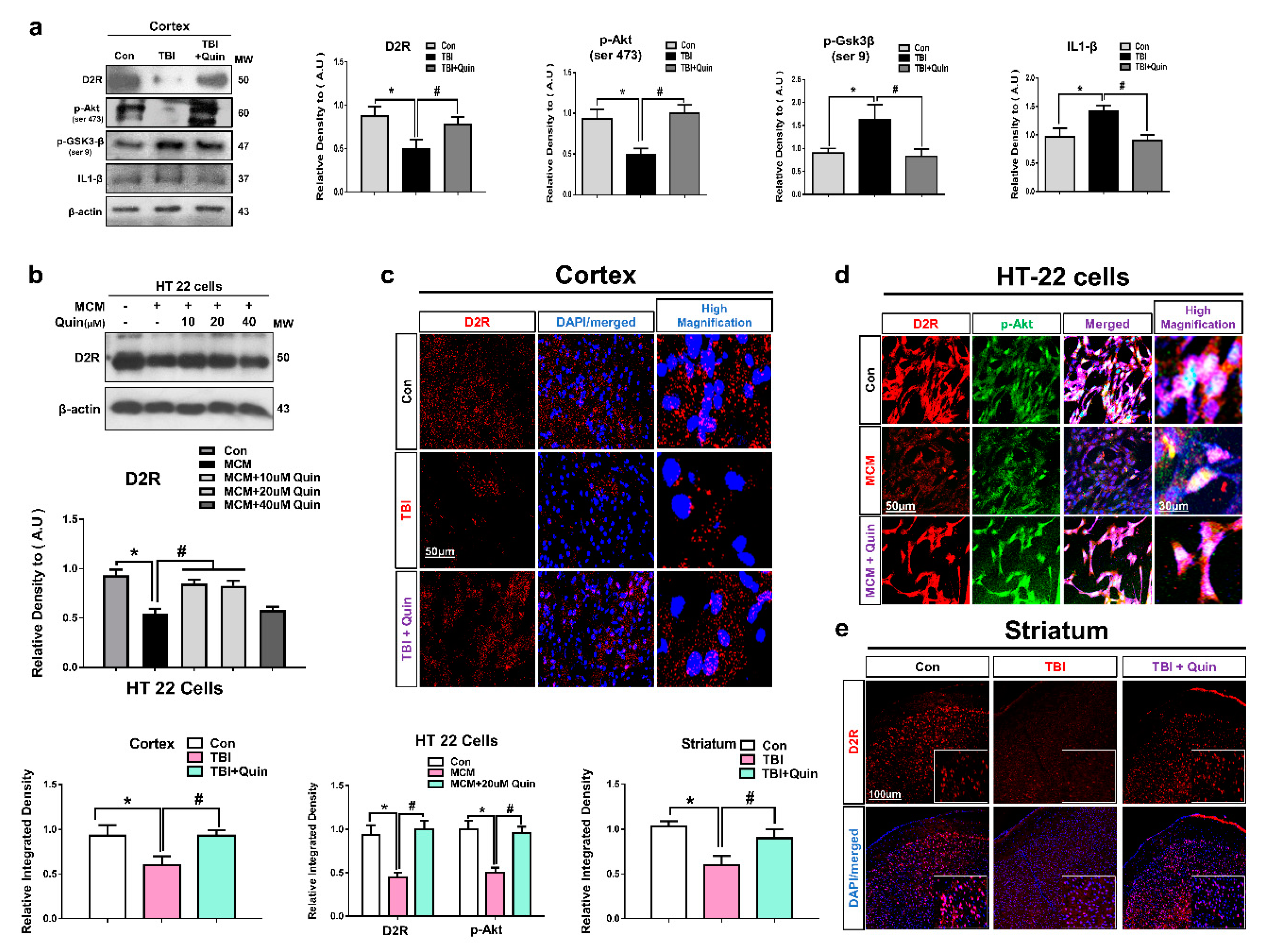
3.1. Quinpirole Regulated the D2R Expression Level in the Injured Brain and HT22 Cells
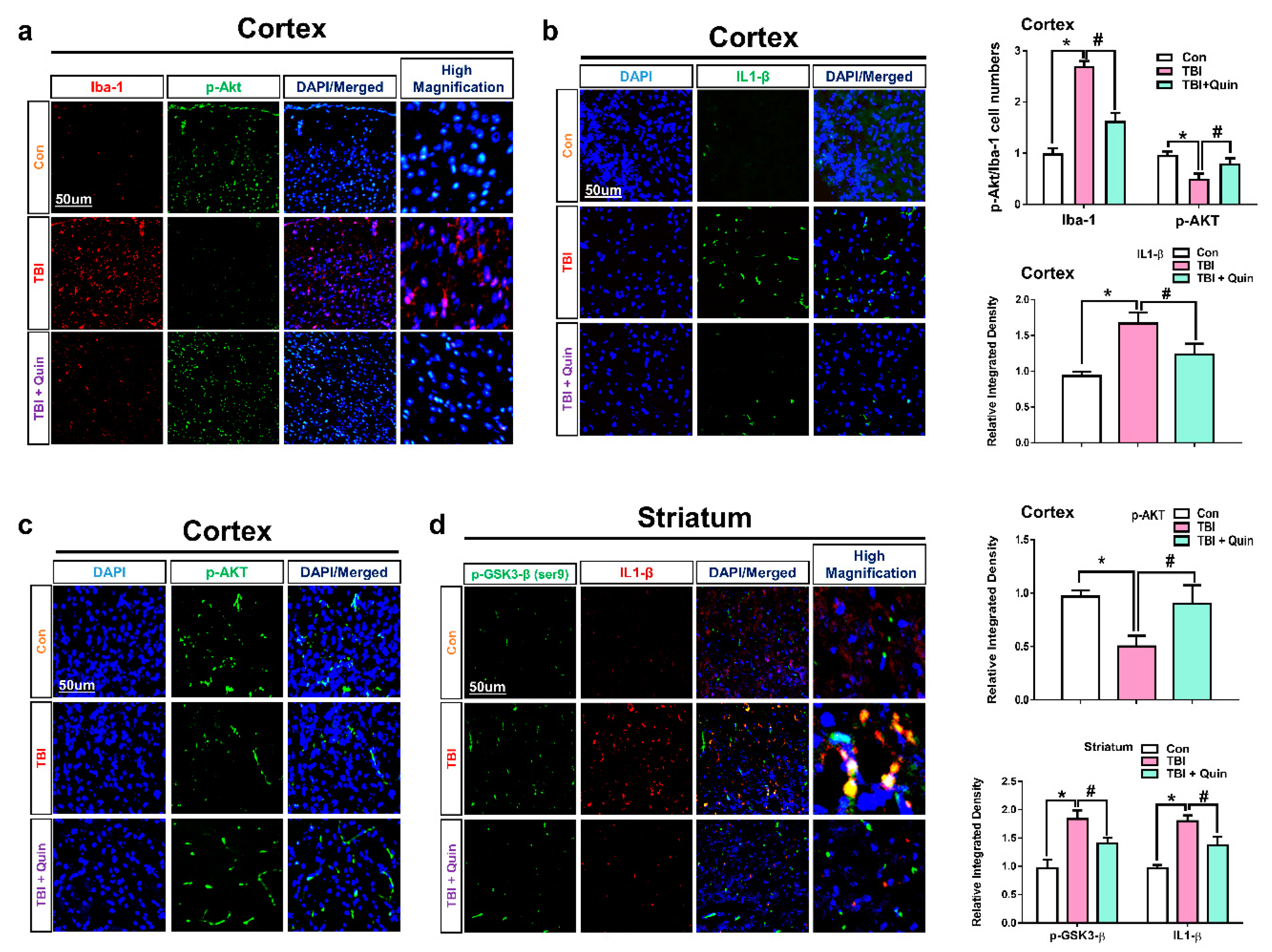
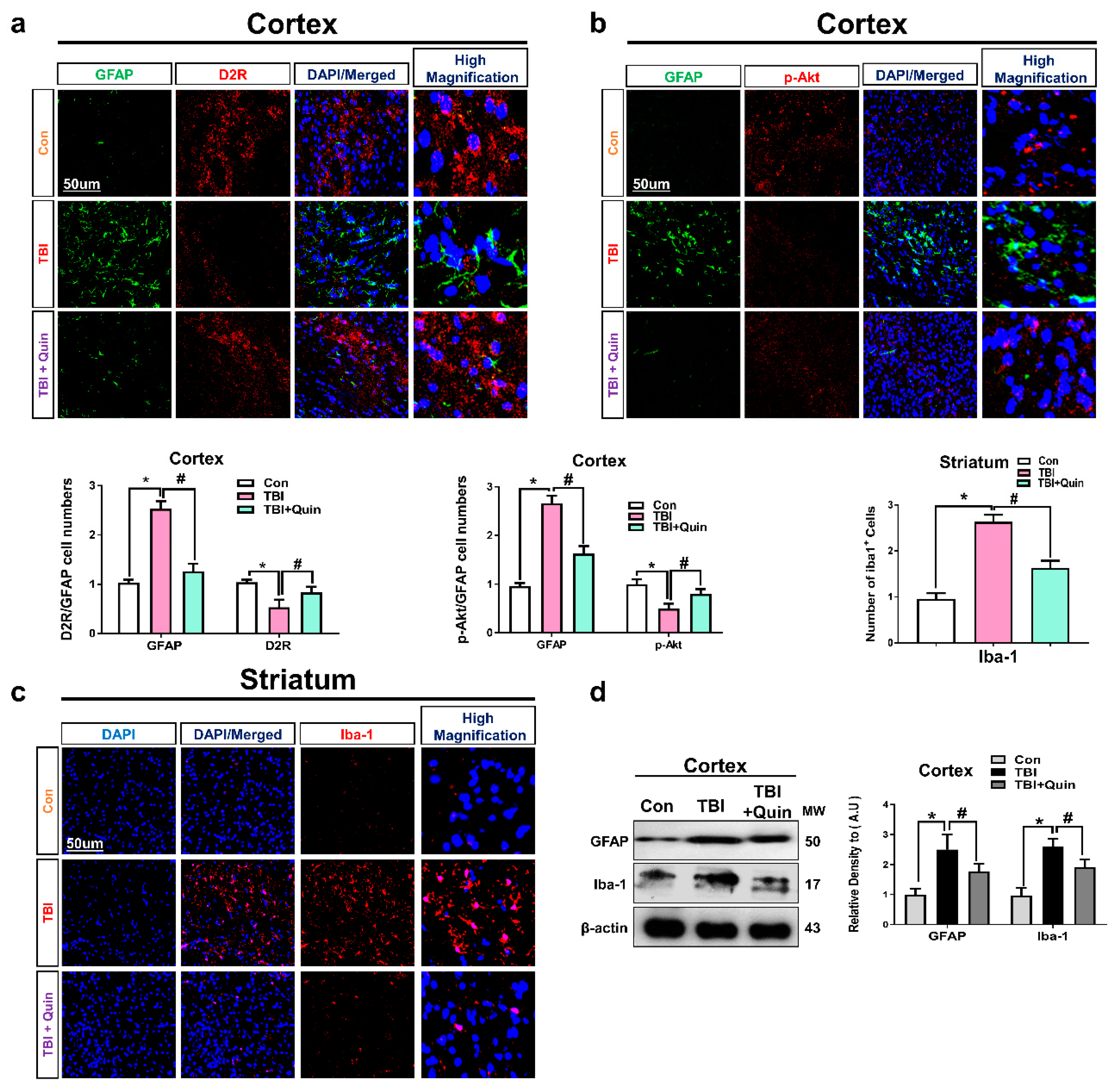
3.2. Quinpirole Reduced Gloisis and Atttenates D2R/Akt Level after Brain Injury
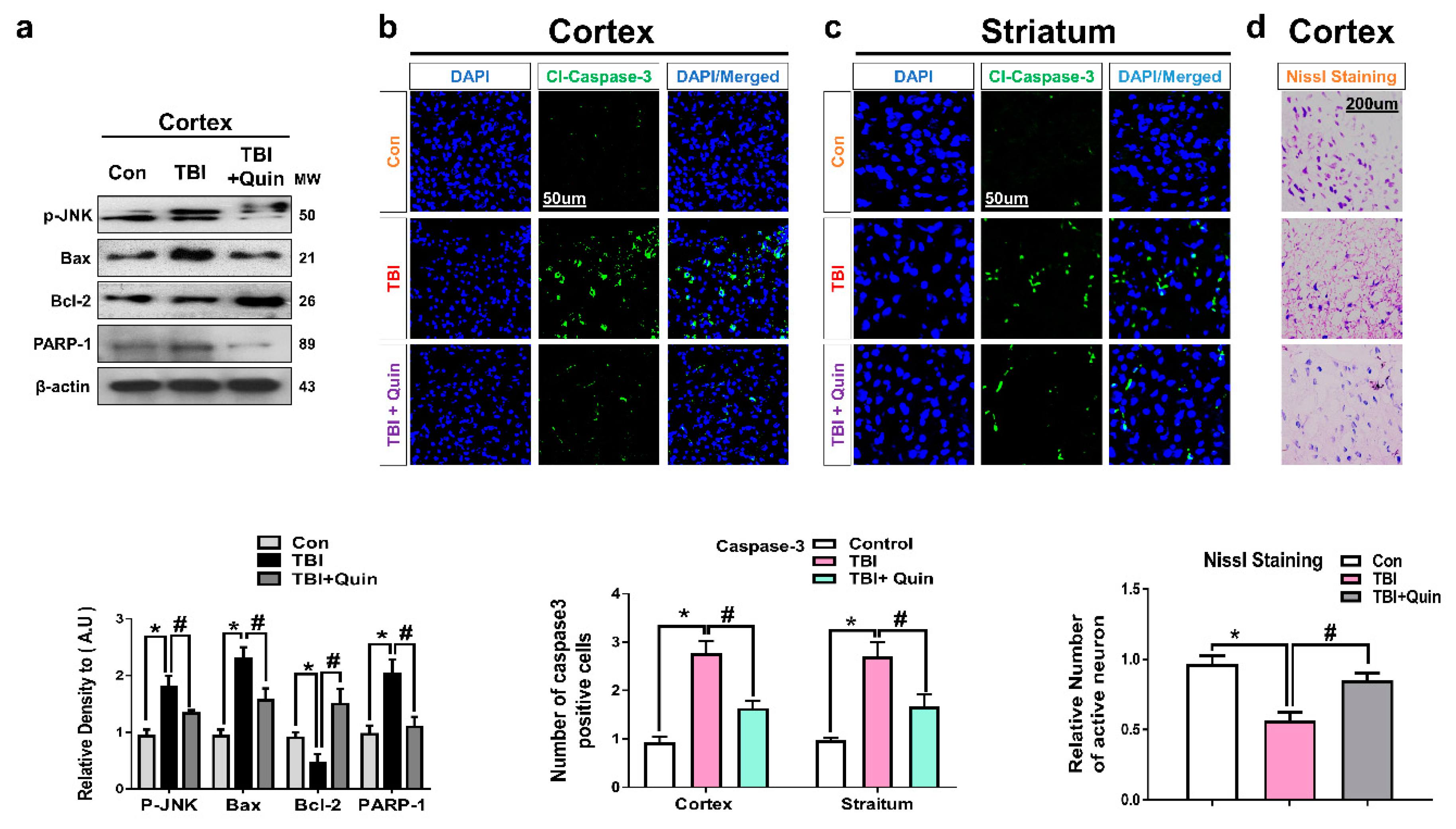
3.3. Quinpirole Reduced Neuronal Apoptosis after Brain Injury
3.4. Quinpirole-Induced Restoration of Blood–Brain Barrier Disruption and Lesion Volume after Brain Injury
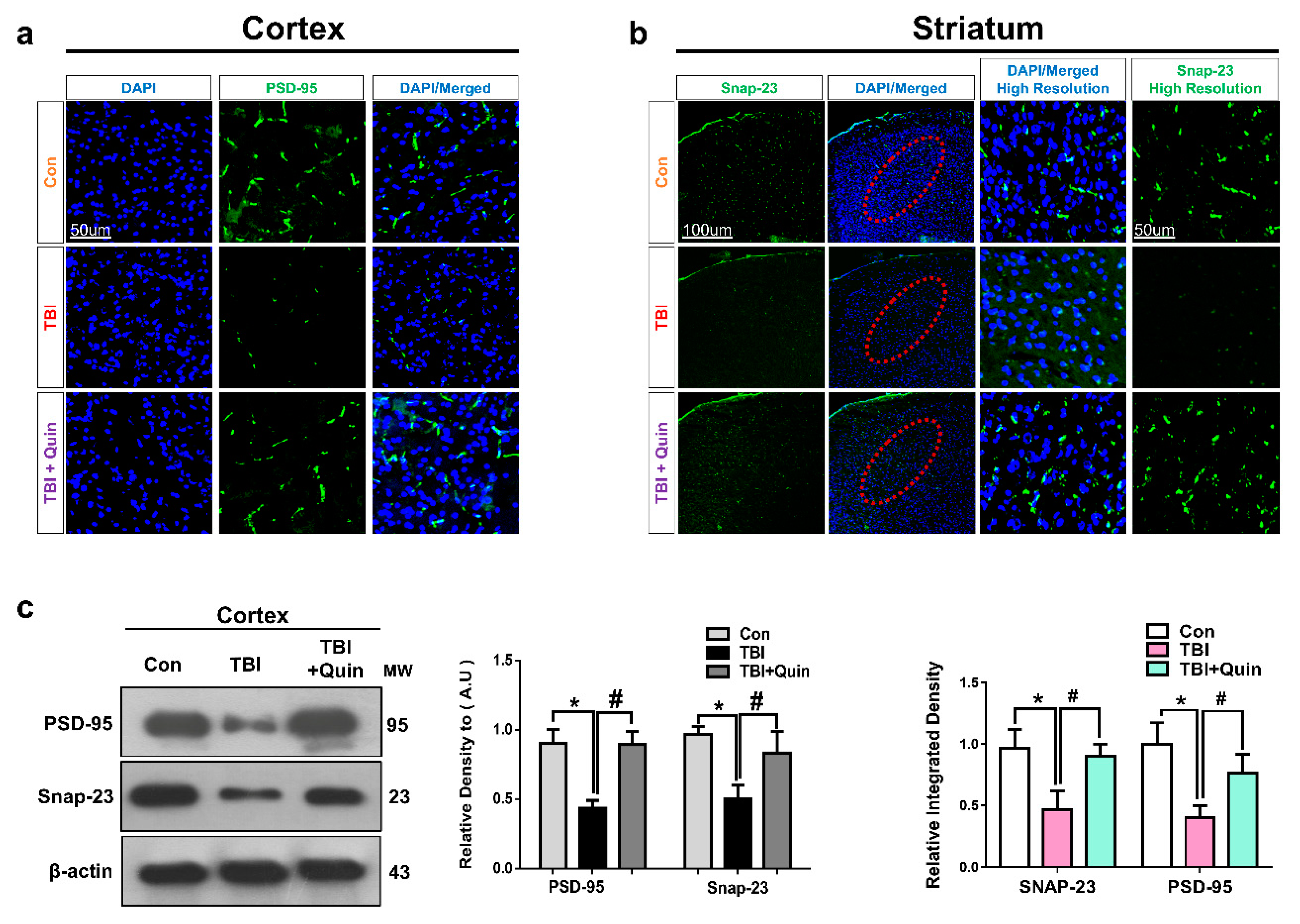
3.5. Quinpirole Attenuated Synaptic Dysfunction after Brain Injury
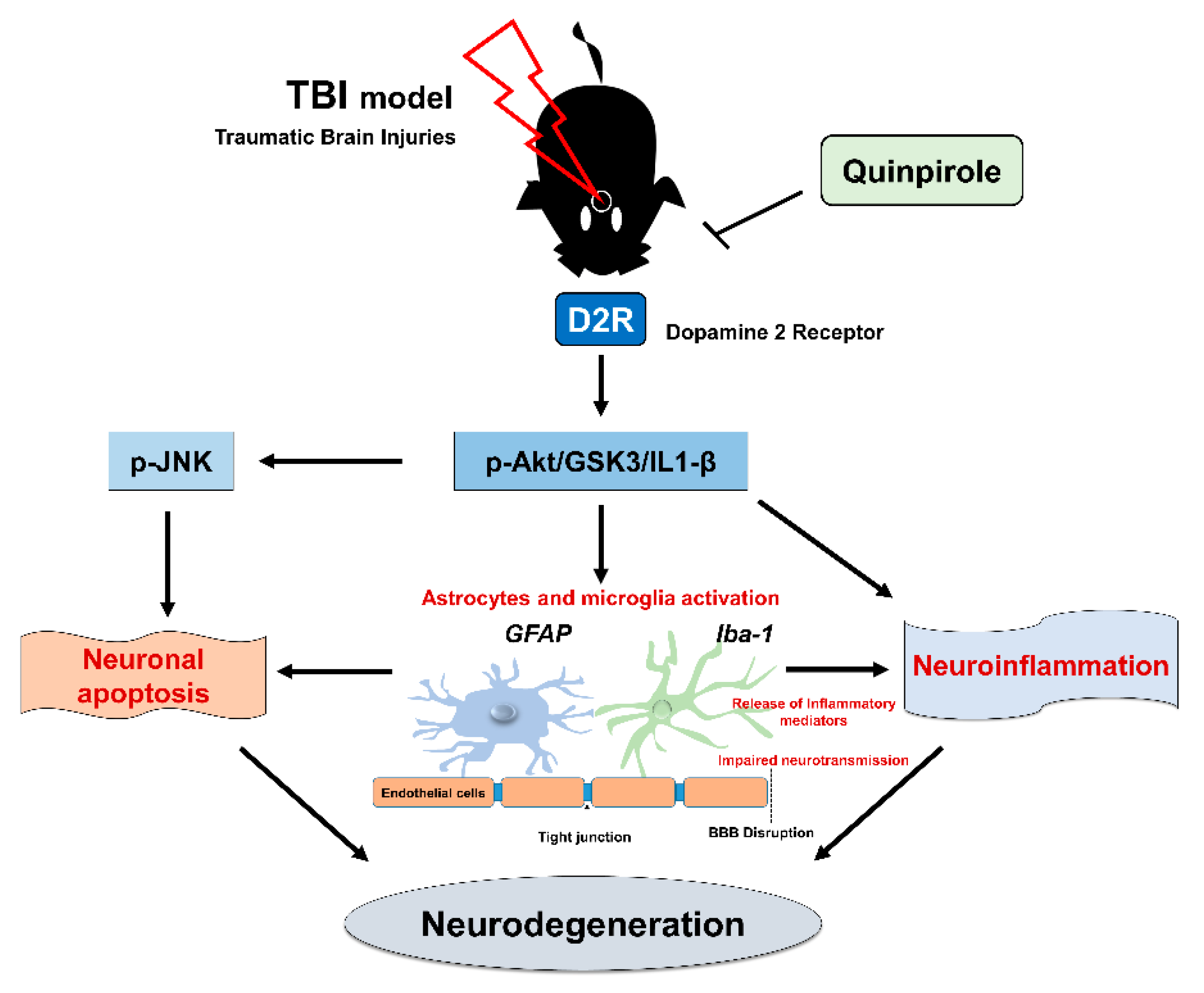
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delic, V.; Beck, K.D.; Pang, K.C.H.; Citron, B.A. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.S.; Sharp, D.J. Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1221–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallenquist, U.; Holmqvist, K.; Hanell, A.; Marklund, N.; Hillered, L.; Forsberg-Nilsson, K. Ibuprofen attenuates the inflammatory response and allows formation of migratory neuroblasts from grafted stem cells after traumatic brain injury. Restor. Neurol. Neurosci. 2012, 30, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Campos-Pires, R.; Hirnet, T.; Valeo, F.; Ong, B.E.; Radyushkin, K.; Aldhoun, J.; Saville, J.; Edge, C.J.; Franks, N.P.; Thal, S.C.; et al. Xenon improves long-term cognitive function, reduces neuronal loss and chronic neuroinflammation, and improves survival after traumatic brain injury in mice. Br. J. Anaesth. 2019, 123, 60–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajiri, N.; Hernandez, D.; Acosta, S.; Shinozuka, K.; Ishikawa, H.; Ehrhart, J.; Diamandis, T.; Gonzales-Portillo, C.; Borlongan, M.C.; Tan, J.; et al. Suppressed cytokine expression immediatey following traumatic brain injury in neonatal rats indicates an expeditious endogenous anti-inflammatory response. Brain Res. 2014, 1559, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, E.; Somera-Molina, K.; Van Eldik, L.J.; Watterson, D.M.; Wainwright, M.S. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J. Neuroinflamm. 2008, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Becker, A.; Grecksch, G.; Thiemann, W.; Hollt, V. Pentylenetetrazol-kindling modulates stimulated dopamine release in the nucleus accumbens of rats. Pharmacol. Biochem. Behav. 2000, 66, 425–428. [Google Scholar] [CrossRef]
- Sellnow, R.C.; Newman, J.H.; Chambers, N.; West, A.R.; Steece-Collier, K.; Sandoval, I.M.; Benskey, M.J.; Bishop, C.; Manfredsson, F.P. Regulation of dopamine neurotransmission from serotonergic neurons by ectopic expression of the dopamine D2 autoreceptor blocks levodopa-induced dyskinesia. Acta Neuropathol. Commun. 2019, 7, 8. [Google Scholar] [CrossRef]
- Bibb, J.A. Decoding dopamine signaling. Cell 2005, 122, 153–155. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhang, X.; Yang, D.; Du, Y.; Li, L.; Li, X.; Ming, M.; Le, W. D2/D3 receptor agonist ropinirole protects dopaminergic cell line against rotenone-induced apoptosis through inhibition of caspase-and JNK-dependent pathways. FEBS Lett. 2008, 582, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, W.; Zhang, S.Z.; Tang, M.; Zhang, X.H.; Zhou, Z.; Yin, Y.Q.; Zhou, Q.B.; Huang, Y.Y.; Liu, Y.J.; Wawrousek, E.; et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature 2013, 494, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kato, M.; Takano, H.; Arakawa, R.; Okumura, M.; Otsuka, T.; Kodaka, F.; Hayashi, M.; Okubo, Y.; Ito, H. Differential contributions of prefrontal and hippocampal dopamine D1 and D2 receptors in human cognitive functions. J. Neurosci. 2008, 28, 12032–12038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, C.; Basu, B.; Chakroborty, D.; Dasgupta, P.S.; Basu, S. The immunoregulatory role of dopamine: An update. Brain Behav. Immun. 2010, 24, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Campo, N.; Chamberlain, S.R.; Sahakian, B.J.; Robbins, T.W. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol. Psychiatry 2011, 69, e145–e157. [Google Scholar] [CrossRef]
- Russell, V.A.; Sagvolden, T.; Johansen, E.B. Animal models of attention-deficit hyperactivity disorder. Behav. Brain Funct. 2005, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Khlghatyan, J.; Quintana, C.; Parent, M.; Beaulieu, J.M. High Sensitivity Mapping of Cortical Dopamine D2 Receptor Expressing Neurons. Cereb. Cortex 2018, 29, 3813–3827. [Google Scholar] [CrossRef]
- Kuric, E.; Wieloch, T.; Ruscher, K. Dopamine receptor activation increases glial cell line-derived neurotrophic factor in experimental stroke. Exp. Neurol. 2013, 247, 202–208. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Wu, J.; Manaenko, A.; Yang, P.; Tang, J.; Fu, W.; Zhang, J.H. Activation of Dopamine D2 Receptor Suppresses Neuroinflammation Through alphaB-Crystalline by Inhibition of NF-kappaB Nuclear Translocation in Experimental ICH Mice Model. Stroke 2015, 46, 2637–2646. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.J.; Yen, C.T.; Liu, T.L.; Tsao, H.W.; Hsu, J.W.; Tsai, M.L. Effects of dopamine D2 agonist quinpirole on neuronal activity of anterior cingulate cortex and striatum in rats. Psychopharmacology 2013, 227, 459–466. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Tirotta, E.; Sotnikova, T.D.; Masri, B.; Salahpour, A.; Gainetdinov, R.R.; Borrelli, E.; Caron, M.G. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J. Neurosci. 2007, 27, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koller, W.; Herbster, G.; Anderson, D.; Wack, R.; Gordon, J. Quinpirole hydrochloride, a potential anti-parkinsonism drug. Neuropharmacology 1987, 26, 1031–1036. [Google Scholar] [CrossRef]
- Alam, S.I.; Rehman, S.U.; Kim, M.O. Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-kappaB Signaling Pathway after Brain Injury. J. Clin. Med. 2019, 8, 271. [Google Scholar] [CrossRef] [Green Version]
- Rehman, S.U.; Ali, T.; Alam, S.I.; Ullah, R.; Zeb, A.; Lee, K.W.; Rutten, B.P.F.; Kim, M.O. Ferulic Acid Rescues LPS-Induced Neurotoxicity via Modulation of the TLR4 Receptor in the Mouse Hippocampus. Mol. Neurobiol. 2019, 56, 2774–2790. [Google Scholar] [CrossRef]
- Ahmad, R.; Khan, A.; Lee, H.J.; Ur Rehman, I.; Khan, I.; Alam, S.I.; Kim, M.O. Lupeol, a Plant-Derived Triterpenoid, Protects Mice Brains against Abeta-Induced Oxidative Stress and Neurodegeneration. Biomedicines 2020, 8, 380. [Google Scholar] [CrossRef]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Khan, M.; Rutten, B.P.F.; Kim, M.O. MST1 Regulates Neuronal Cell Death via JNK/Casp3 Signaling Pathway in HFD Mouse Brain and HT22 Cells. Int. J. Mol. Sci. 2019, 20, 2504. [Google Scholar] [CrossRef] [Green Version]
- Ullah, R.; Jo, M.H.; Riaz, M.; Alam, S.I.; Saeed, K.; Ali, W.; Rehman, I.U.; Ikram, M.; Kim, M.O. Glycine, the smallest amino acid, confers neuroprotection against D-galactose-induced neurodegeneration and memory impairment by regulating c-Jun N-terminal kinase in the mouse brain. J. Neuroinflamm. 2020, 17, 303. [Google Scholar] [CrossRef]
- Han, X.; Li, B.; Ye, X.; Mulatibieke, T.; Wu, J.; Dai, J.; Wu, D.; Ni, J.; Zhang, R.; Xue, J.; et al. Dopamine D2 receptor signalling controls inflammation in acute pancreatitis via a PP2A-dependent Akt/NF-kappaB signalling pathway. Br. J. Pharmacol. 2017, 174, 4751–4770. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Yan, Z.; Tao, K.; Li, Y.; Li, Y.; Li, J.; Dong, Y.; Feng, D.; Chen, H. Sinomenine activates astrocytic dopamine D2 receptors and alleviates neuroinflammatory injury via the CRYAB/STAT3 pathway after ischemic stroke in mice. J. Neuroinflamm. 2016, 13, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maixner, D.W.; Weng, H.-R. The role of glycogen synthase kinase 3 beta in neuroinflammation and pain. J. Pharm. Pharmacol. 2013, 1, 001. [Google Scholar]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic acid attenuates Abeta1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017, 7, 40753. [Google Scholar] [CrossRef] [PubMed]
- Clement, T.; Lee, J.B.; Ichkova, A.; Rodriguez-Grande, B.; Fournier, M.L.; Aussudre, J.; Ogier, M.; Haddad, E.; Canini, F.; Koehl, M.; et al. Juvenile mild traumatic brain injury elicits distinct spatiotemporal astrocyte responses. Glia 2020, 68, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Balarezo, M.G.; Affram, K.; Saavedra, J.M.; Symes, A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 2015, 138, 3299–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younger, D.; Murugan, M.; Rama Rao, K.V.; Wu, L.J.; Chandra, N. Microglia Receptors in Animal Models of Traumatic Brain Injury. Mol. Neurobiol. 2019, 56, 5202–5228. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kanno, T.; Yanagisawa, Y.; Yasutake, K.; Hadano, S.; Yoshii, F.; Ikeda, J.E. Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 232, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Lee, J.H.; Park, S.K.; Yang, W.M.; Jeon, G.S.; Lee, Y.H.; Chung, C.K.; Cho, S.S. Astrocytic expressions of phosphorylated Akt, GSK3beta and CREB following an excitotoxic lesion in the mouse hippocampus. Neurochem. Res. 2007, 32, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Gerbatin, R.D.R.; Cassol, G.; Dobrachinski, F.; Ferreira, A.P.O.; Quines, C.B.; Pace, I.D.D.; Busanello, G.L.; Gutierres, J.M.; Nogueira, C.W.; Oliveira, M.S.; et al. Guanosine Protects Against Traumatic Brain Injury-Induced Functional Impairments and Neuronal Loss by Modulating Excitotoxicity, Mitochondrial Dysfunction, and Inflammation. Mol. Neurobiol. 2017, 54, 7585–7596. [Google Scholar] [CrossRef]
- Villapol, S.; Byrnes, K.R.; Symes, A.J. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front. Neurol. 2014, 5, 82. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Zhao, Z.; Yu, G.; Zhou, Z.; Zhou, Y.; Hu, T.; Jiang, R.; Zhang, J. VEGI attenuates the inflammatory injury and disruption of blood–brain barrier partly by suppressing the TLR4/NF-kappaB signaling pathway in experimental traumatic brain injury. Brain Res. 2015, 1622, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Bales, J.W.; Yan, H.Q.; Ma, X.; Li, Y.; Samarasinghe, R.; Dixon, C.E. The dopamine and cAMP regulated phosphoprotein, 32 kDa (DARPP-32) signaling pathway: A novel therapeutic target in traumatic brain injury. Exp. Neurol. 2011, 229, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.S.; Bray, E.R.; Dixon, C.E. Effects of nicotine administration on striatal dopamine signaling after traumatic brain injury in rats. J. Neurotrauma 2012, 29, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.N.; Yang, L.Y.; Greig, N.H.; Wang, Y.C.; Lai, C.C.; Wang, J.Y. Neuroprotective effects of pifithrin-alpha against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep. 2018, 8, 2368. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Webster, S.J.; Goulding, D.S.; Morton, J.E.; Watterson, D.M.; Van Eldik, L.J. Attenuation of traumatic brain injury-induced cognitive impairment in mice by targeting increased cytokine levels with a small molecule experimental therapeutic. J. Neuroinflamm. 2015, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Webster, S.J.; Van Eldik, L.J.; Watterson, D.M.; Bachstetter, A.D. Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J. Neurosci. 2015, 35, 6554–6569. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Endo, H.; Nito, C.; Kamada, H.; Yu, F.; Chan, P.H. Akt/GSK3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke 2006, 37, 2140–2146. [Google Scholar] [CrossRef] [Green Version]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393. [Google Scholar] [CrossRef] [Green Version]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of c-Jun N-Terminal Kinase Protects Against Brain Damage and Improves Learning and Memory After Traumatic Brain Injury in Adult Mice. Cereb. Cortex 2018, 28, 2854–2872. [Google Scholar] [CrossRef] [PubMed]
- De Beaumont, L.; Tremblay, S.; Poirier, J.; Lassonde, M.; Theoret, H. Altered bidirectional plasticity and reduced implicit motor learning in concussed athletes. Cereb. Cortex 2012, 22, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Lutton, E.M.; Razmpour, R.; Andrews, A.M.; Cannella, L.A.; Son, Y.-J.; Shuvaev, V.V.; Muzykantov, V.R.; Ramirez, S.H. Acute administration of catalase targeted to ICAM-1 attenuates neuropathology in experimental traumatic brain injury. Sci. Rep. 2017, 7, 3846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Chen, Y.; Meng, J.; Wu, M.; Bi, F.; Chang, C.; Li, H.; Zhang, L. Ablation of caspase-1 protects against TBI-induced pyroptosis in vitro and in vivo. J. Neuroinflamm. 2018, 15, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OLANOW, C.W.; Sealfon, S.C. Activation of phosphoinositide 3-kinase by D2 receptor prevents apoptosis in dopaminergic cell lines. Biochem. J. 2003, 373, 25–32. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, S.I.; Jo, M.G.; Park, T.J.; Ullah, R.; Ahmad, S.; Rehman, S.U.; Kim, M.O. Quinpirole-Mediated Regulation of Dopamine D2 Receptors Inhibits Glial Cell-Induced Neuroinflammation in Cortex and Striatum after Brain Injury. Biomedicines 2021, 9, 47. https://doi.org/10.3390/biomedicines9010047
Alam SI, Jo MG, Park TJ, Ullah R, Ahmad S, Rehman SU, Kim MO. Quinpirole-Mediated Regulation of Dopamine D2 Receptors Inhibits Glial Cell-Induced Neuroinflammation in Cortex and Striatum after Brain Injury. Biomedicines. 2021; 9(1):47. https://doi.org/10.3390/biomedicines9010047
Chicago/Turabian StyleAlam, Sayed Ibrar, Min Gi Jo, Tae Ju Park, Rahat Ullah, Sareer Ahmad, Shafiq Ur Rehman, and Myeong Ok Kim. 2021. "Quinpirole-Mediated Regulation of Dopamine D2 Receptors Inhibits Glial Cell-Induced Neuroinflammation in Cortex and Striatum after Brain Injury" Biomedicines 9, no. 1: 47. https://doi.org/10.3390/biomedicines9010047