Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Construction and Characterization of PD-L1 Blocker Peptide
2.3. Recombinant Virus Construction
2.4. Cytotoxicity Assay
2.5. Replication Kinetics of Mutant Viruses
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. IL-2 Assay
2.8. In Vitro Quantification of the Combinatorial Effect between IL-12, IL-15/IL-15RA and PD-L1 Blocker
2.9. In Vivo Tumor Models
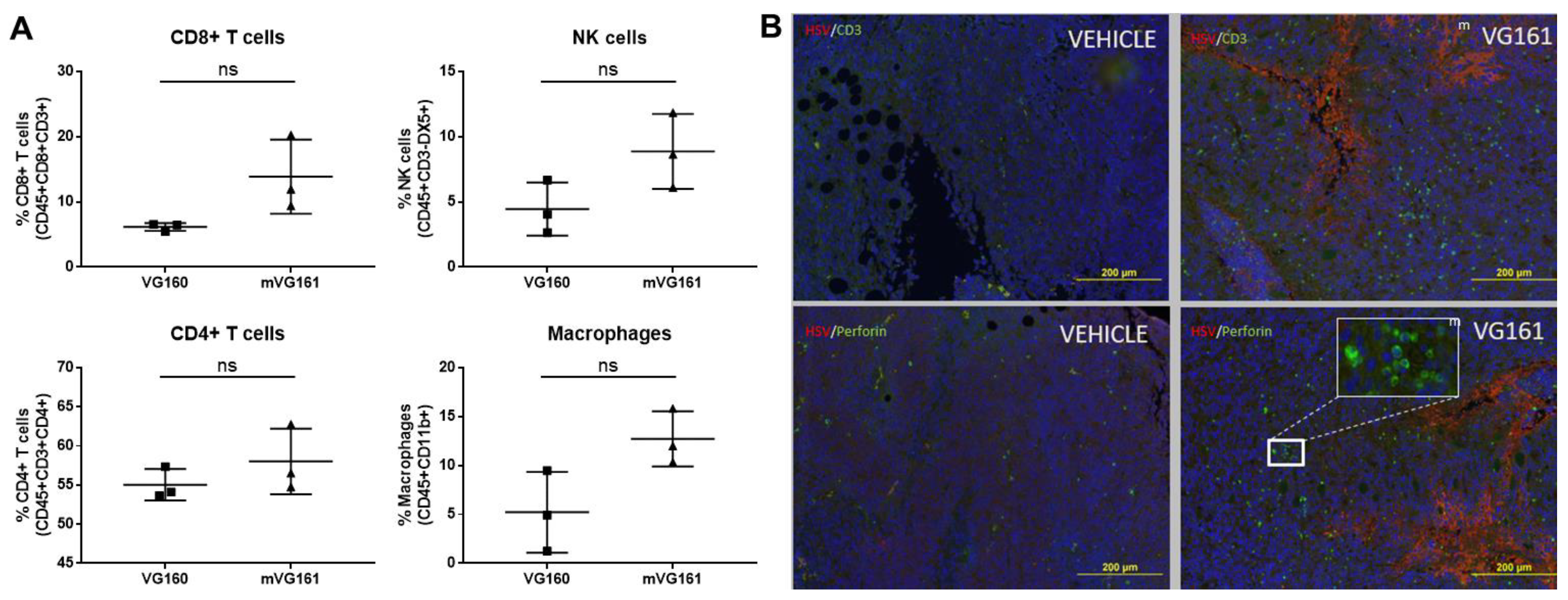
2.10. Flow Cytometric Analysis
2.11. Detection of Tumor Infiltrating Lymphocytes
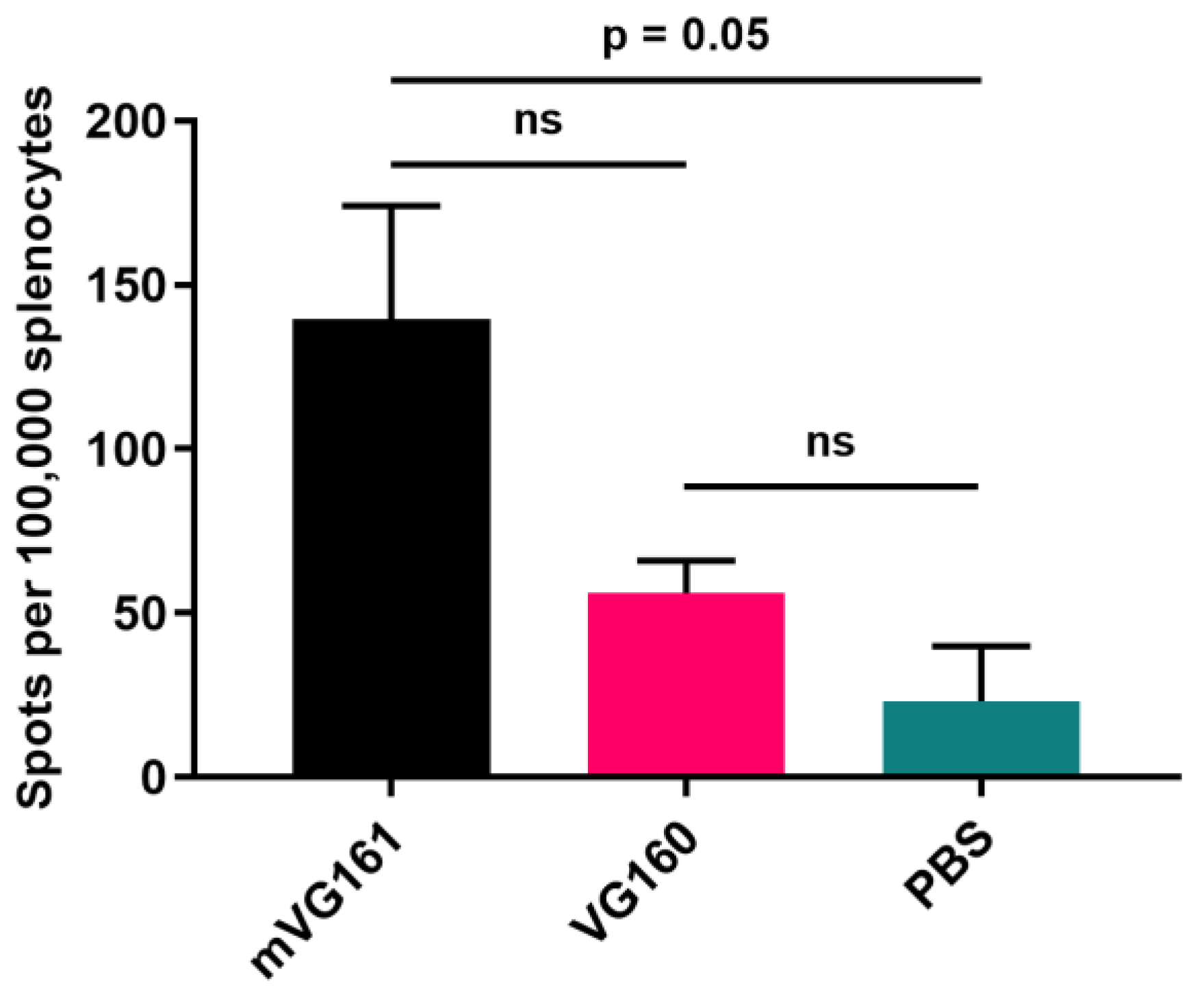
2.12. ELISpot T Cell Activity Assay
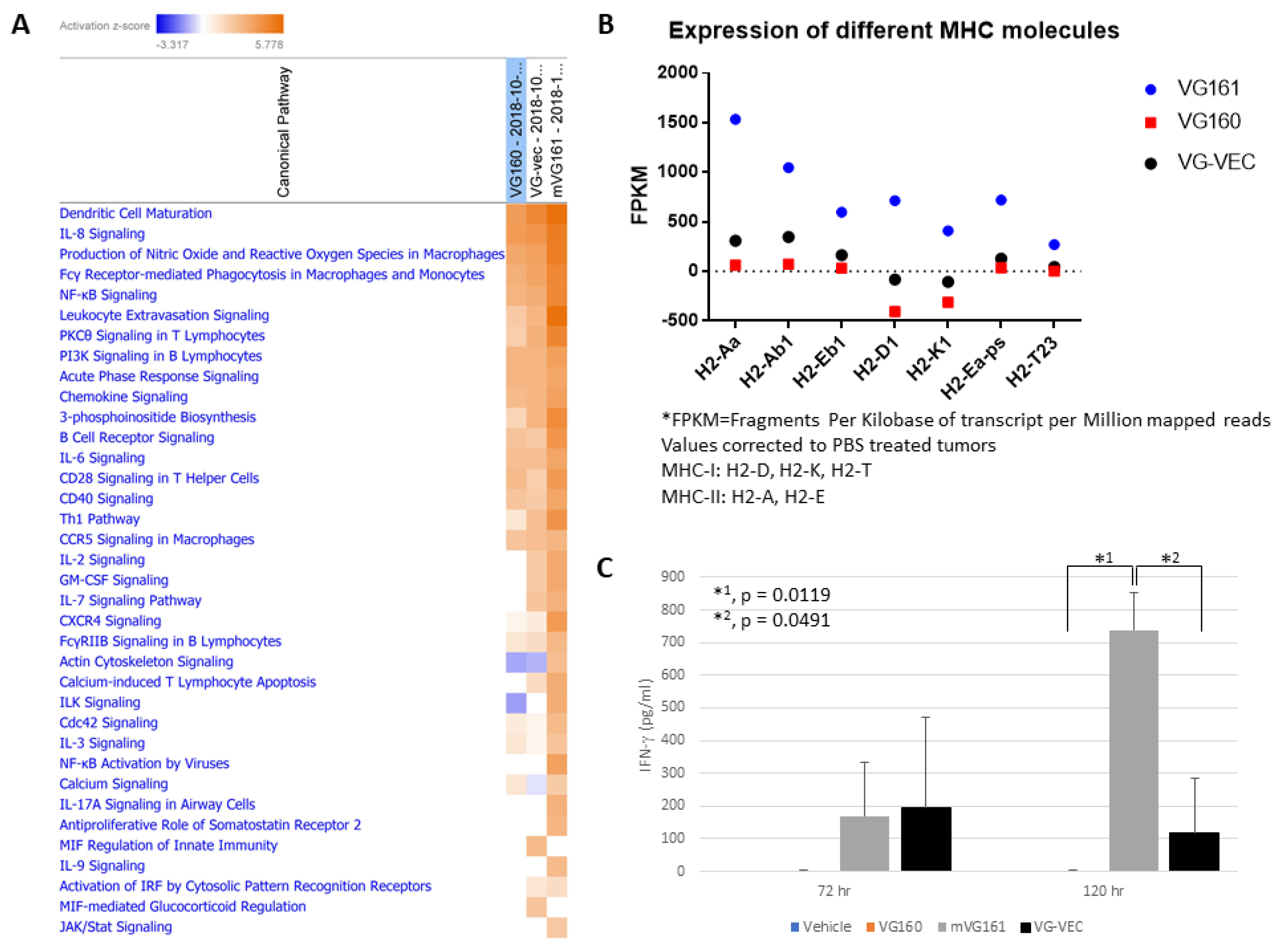
2.13. Gene Expression Analysis
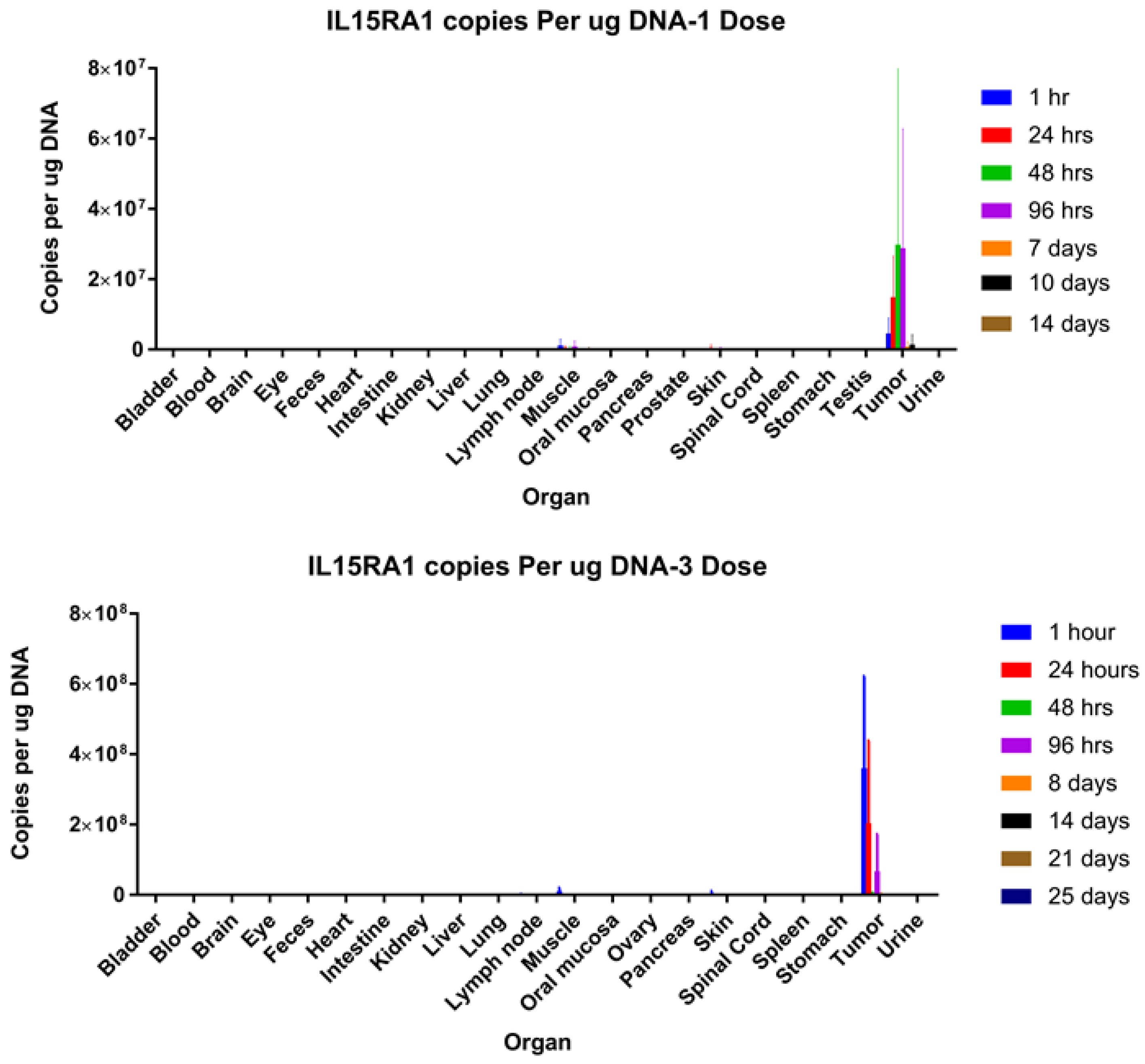
2.14. Biodistribution Analysis
2.15. Primate Toxicity Studies
2.16. Statistical Tests Employed in the Study
3. Results
3.1. Virus Characterization and In Vitro Validation of Oncolytic Activity
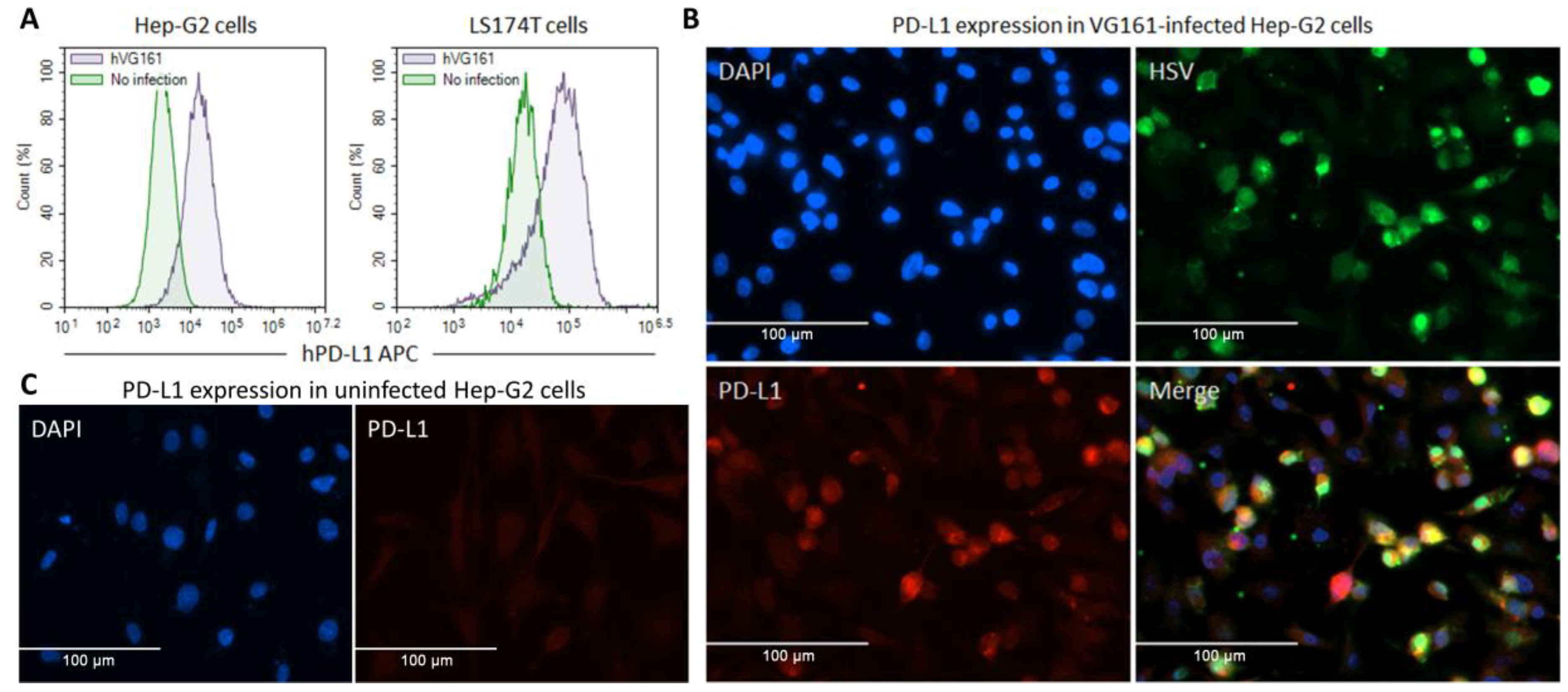
3.2. In Vitro Transgene Expression and Characterization
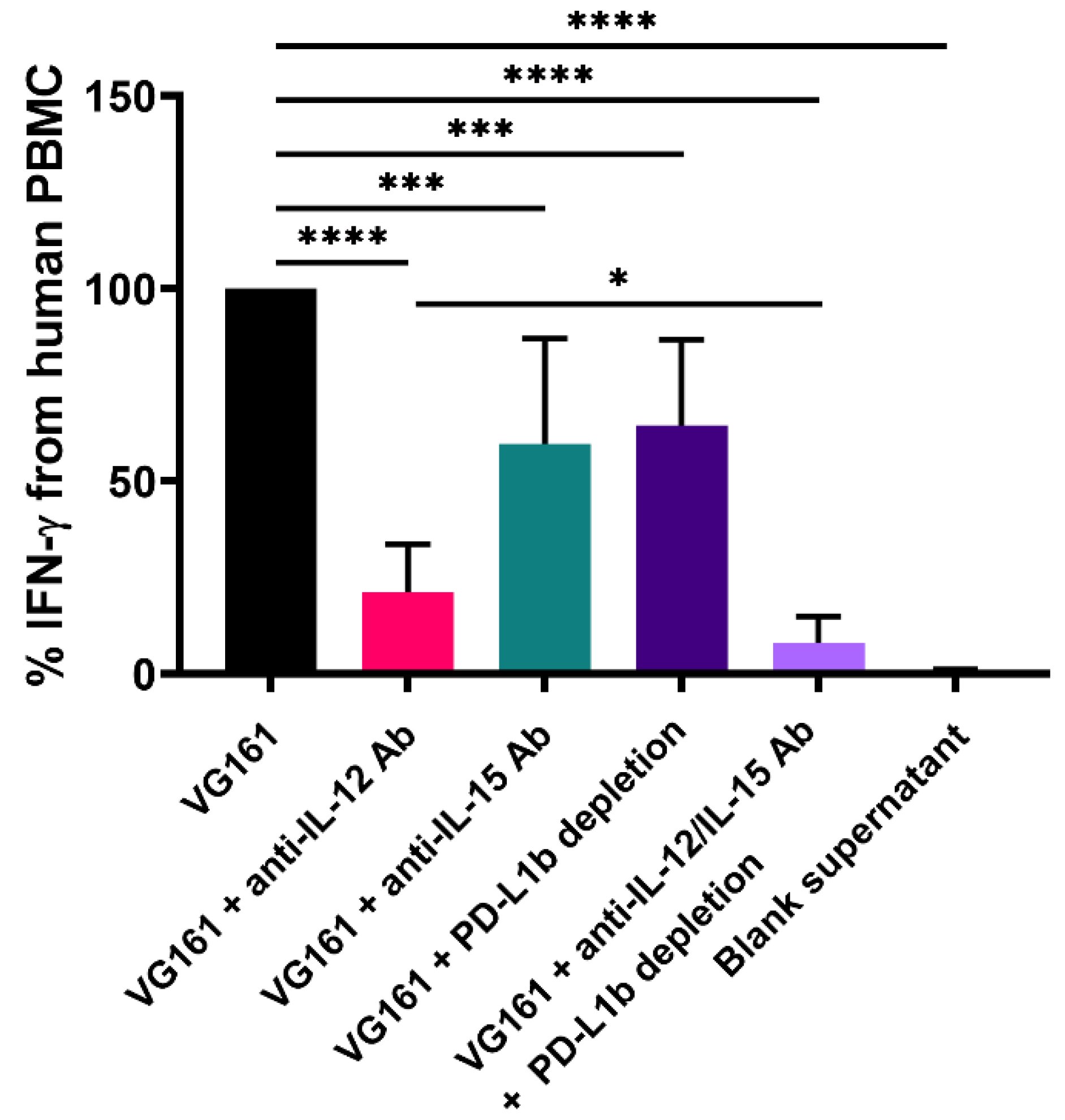
3.3. Immune Activation by Virally Encoded Transgenes In Vitro
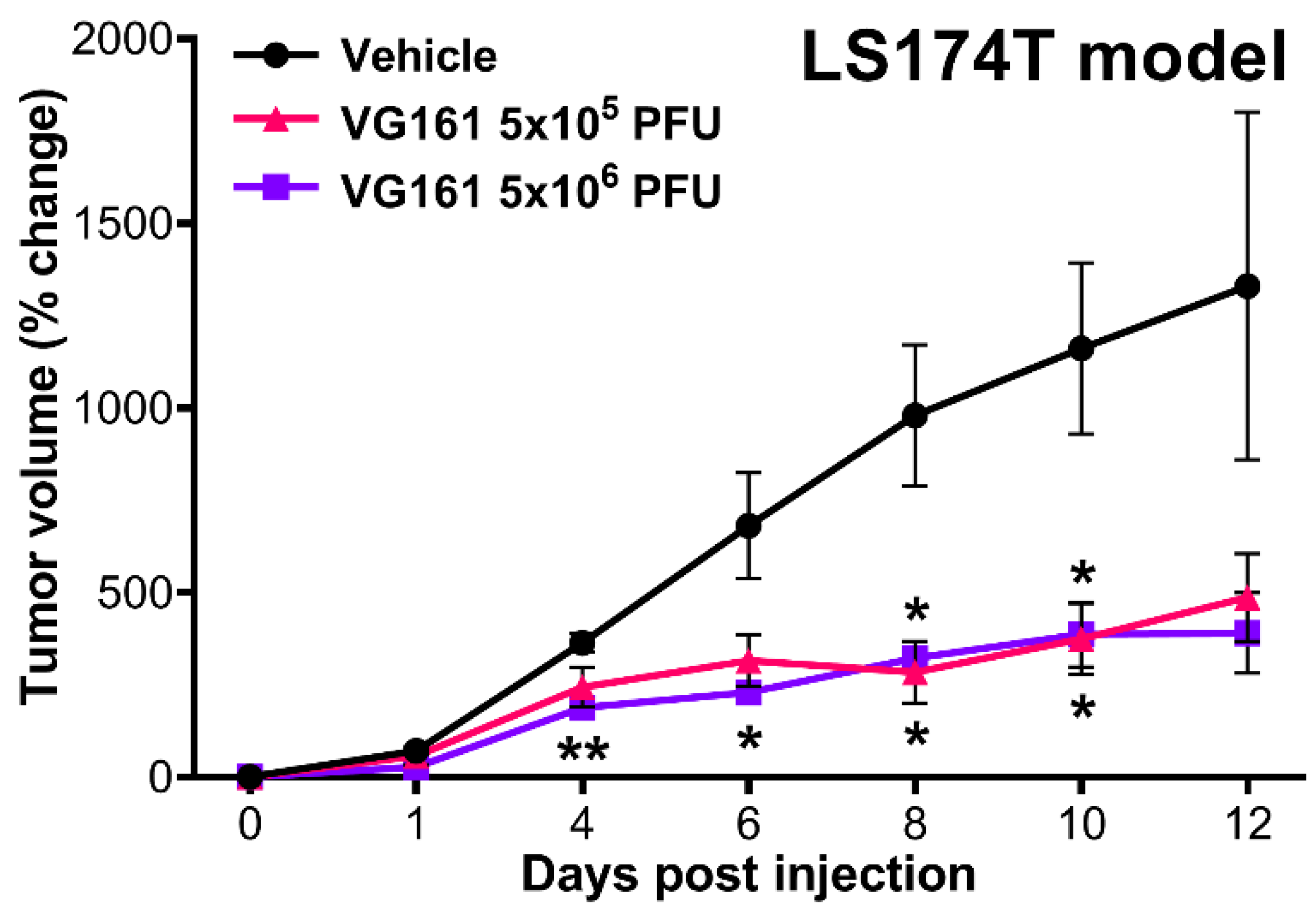
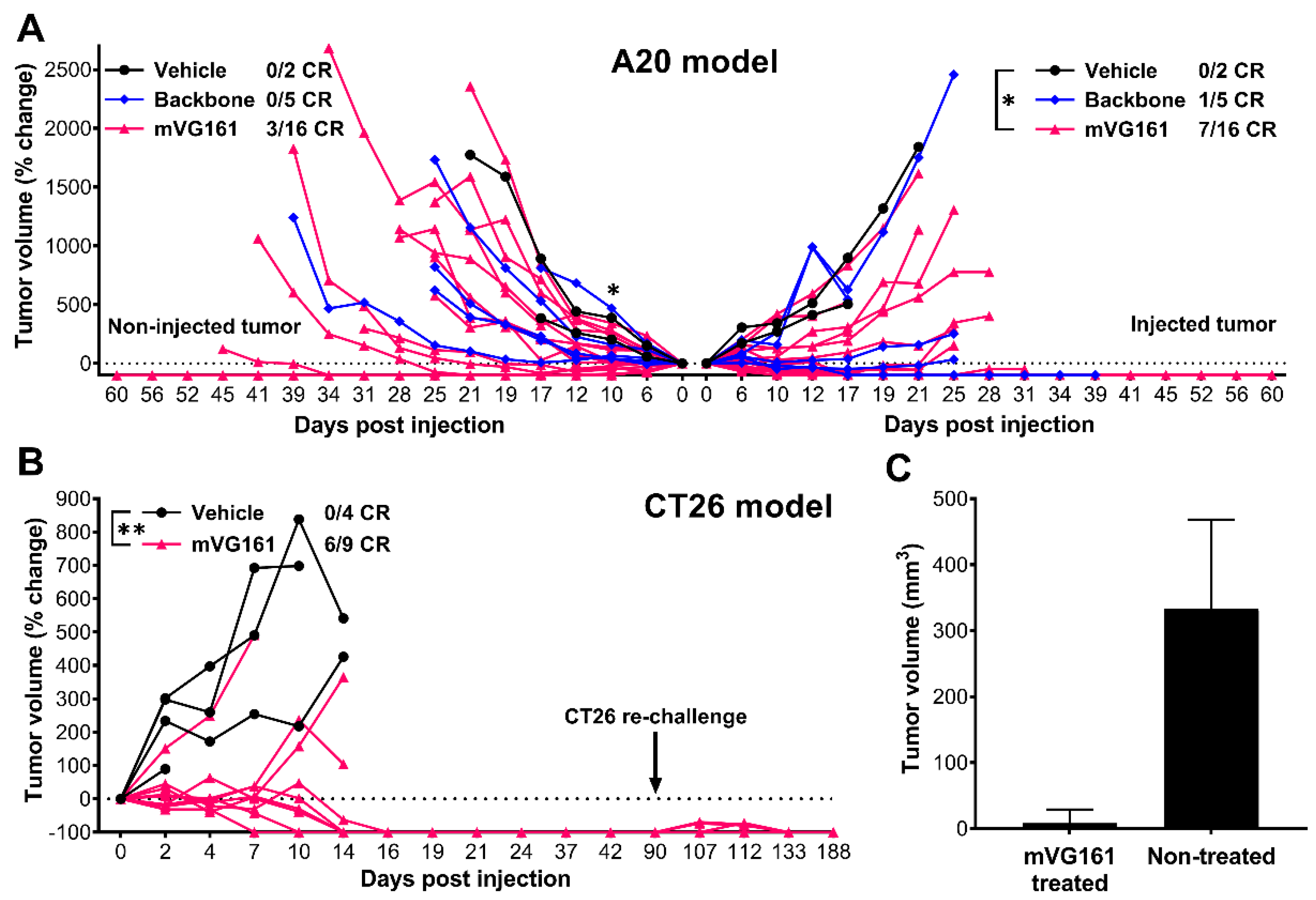
3.4. VG161 Promotes Efficient Tumor Clearance In Vivo
3.5. mVG161 Induces a Potent Anti-Tumor Immune Response In Vivo
3.6. mVG161 Alters the Tumor Microenvironment (TME)
3.7. VG161 Displays a Robust Safety Profile in Primates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.-C.; Cho, C.-F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.L.; Robinson, M.; Han, Z.-Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Awasthi, S.; Curti, B.; Hutchins, L.; Linette, G.; Triozzi, P.; Tan, M.C.B.; Brown, R.E.; Nemunaitis, J.; Whitman, E.; et al. Phase IIIb safety results from an expanded-access protocol of talimogene laherparepvec for patients with unresected, stage IIIB-IVM1c melanoma. Melanoma Res. 2018, 28, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Bu, X.; Lee, I.-F. Compositions and Methods for Inhibiting CD279 Interactions. U.S. Patent No. 10,590,169, 17 March 2020. [Google Scholar]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamarin, D.; Ricca, J.M.; Sadekova, S.; Oseledchyk, A.; Yu, Y.; Blumenschein, W.M.; Wong, J.; Gigoux, M.; Merghoub, T.; Wolchok, J.D. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J. Clin. Invest. 2018, 128, 1413–1428. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.P.; Trinchieri, G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 155–168. [Google Scholar] [CrossRef]
- Wigginton, J.M.; Wiltrout, R.H. IL-12/IL-2 combination cytokine therapy for solid tumours: Translation from bench to bedside. Expert Opin. Biol. Ther. 2002, 2, 513–524. [Google Scholar] [CrossRef]
- Heinzerling, L.; Burg, G.; Dummer, R.; Maier, T.; Oberholzer, P.A.; Schultz, J.; Elzaouk, L.; Pavlovic, J.; Moelling, K. Intratumoral injection of DNA encoding human interleukin 12 into patients with metastatic melanoma: Clinical efficacy. Hum. Gene Ther. 2005, 16, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom Berg, J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef] [Green Version]
- Lasek, W.; Golab, J.; Maśliński, W.; Switaj, T.; Bałkowiec, E.Z.; Stokłosa, T.; Giermasz, A.; Malejczyk, M.; Jakóbisiak, M. Subtherapeutic doses of interleukin-15 augment the antitumor effect of interleukin-12 in a B16F10 melanoma model in mice. Eur. Cytokine Netw. 1999, 10, 345–356. [Google Scholar]
- Robinson, T.O.; Schluns, K.S. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Hofs, E.; Cho, B.; Li, M.; Bolduc, K.; Bu, L.; Liu, G.; Lam, V.; Rennie, P.; Jia, W.; et al. UV Light-inactivated HSV-1 Stimulates Natural Killer Cell-induced Killing of Prostate Cancer Cells. J. Immunother. 2019, 42, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Rezvani, K.; Shaim, H.; Hofs, E.; Ngom, M.; Bu, L.; Liu, G.; Lee, J.T.C.; Imren, S.; Lam, V.; et al. UV-inactivated HSV-1 potently activates NK cell killing of leukemic cells. Blood 2016, 127, 2575–2586. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Luan, L.; Patil, N.K.; Sherwood, E.R. Immunobiology of the IL-15/IL-15Rα complex as an antitumor and antiviral agent. Cytokine Growth Factor Rev. 2017, 38, 10–21. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front. Immunol. 2017, 8, 1597. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.-H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha}. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar] [CrossRef] [Green Version]
- Van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Halford, W.P.; Balliet, J.W.; Gebhardt, B.M. Re-evaluating natural resistance to herpes simplex virus type 1. J. Virol. 2004, 78, 10086–10095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Jaggi, U.; Ghiasi, H. CCR2+ migratory macrophages with M1 status are the early-responders in the cornea of HSV-1 infected mice. PLoS ONE 2019, 14, e0215727. [Google Scholar] [CrossRef]
- Givechian, K.B.; Wnuk, K.; Garner, C.; Benz, S.; Garban, H.; Rabizadeh, S.; Niazi, K.; Soon-Shiong, P. Identification of an immune gene expression signature associated with favorable clinical features in Treg-enriched patient tumor samples. NPJ Genom. Med. 2018, 3, 14. [Google Scholar] [CrossRef]
- Niemann, J.; Woller, N.; Brooks, J.; Fleischmann-Mundt, B.; Martin, N.T.; Kloos, A.; Knocke, S.; Ernst, A.M.; Manns, M.P.; Kubicka, S.; et al. Molecular retargeting of antibodies converts immune defense against oncolytic viruses into cancer immunotherapy. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, K.J.; Wong, J.; Fong, Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin. Investig. Drugs 2008, 17, 1105–1113. [Google Scholar] [CrossRef]
- Kuryk, L.; Møller, A.-S.W.; Jaderberg, M. Abscopal effect when combining oncolytic adenovirus and checkpoint inhibitor in a humanized NOG mouse model of melanoma. J. Med. Virol. 2019, 91, 1702–1706. [Google Scholar] [CrossRef] [Green Version]
- Broman, K.K.; Zager, J.S. An evaluation of talimogene laherparepvec for the treatment of melanoma. Expert Opin. Biol. Ther. 2020, 20, 9–14. [Google Scholar] [CrossRef]
- Chou, J.; Chen, J.J.; Gross, M.; Roizman, B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 1995, 92, 10516–10520. [Google Scholar] [PubMed] [Green Version]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar]
- Car, B.D.; Eng, V.M.; Lipman, J.M.; Anderson, T.D. The toxicology of interleukin-12: A review. Toxicol. Pathol. 1999, 27, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Fernandez Gomez, A.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef]
- Morales, J.K.; Kmieciak, M.; Knutson, K.L.; Bear, H.D.; Manjili, M.H. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res. Treat. 2010, 123, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Parmiani, G.; Castelli, C.; Pilla, L.; Santinami, M.; Colombo, M.P.; Rivoltini, L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann. Oncol. 2007, 18, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks-Konczalik, J.; Dubois, S.; Losi, J.M.; Sabzevari, H.; Yamada, N.; Feigenbaum, L.; Waldmann, T.A.; Tagaya, Y. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11445–11450. [Google Scholar] [CrossRef] [Green Version]
- Leong, J.W.; Chase, J.M.; Romee, R.; Schneider, S.E.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol. Blood Marrow Transplant. 2014, 20, 463–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusty, E.; Poznanski, S.M.; Kwofie, K.; Mandur, T.S.; Lee, D.A.; Richards, C.D.; Ashkar, A.A. IL-18/IL-15/IL-12 synergy induces elevated and prolonged IFN-γ production by ex vivo expanded NK cells which is not due to enhanced STAT4 activation. Mol. Immunol. 2017, 88, 138–147. [Google Scholar] [CrossRef]
- Chertova, E.; Bergamaschi, C.; Chertov, O.; Sowder, R.; Bear, J.; Roser, J.D.; Beach, R.K.; Lifson, J.D.; Felber, B.K.; Pavlakis, G.N. Characterization and favorable in vivo properties of heterodimeric soluble IL-15·IL-15Rα cytokine compared to IL-15 monomer. J. Biol. Chem. 2013, 288, 18093–18103. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A. Technology evaluation: Ipilimumab, Medarex/Bristol-Myers Squibb. Curr. Opin. Mol. Ther. 2005, 7, 588–597. [Google Scholar]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.-J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Mühlbauer, M.; Fleck, M.; Schütz, C.; Weiss, T.; Froh, M.; Blank, C.; Schölmerich, J.; Hellerbrand, C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J. Hepatol. 2006, 45, 520–528. [Google Scholar] [CrossRef]
- Woller, N.; Gürlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Sharon, E.; Streicher, H.; Goncalves, P.; Chen, H.X. Immune checkpoint inhibitors in clinical trials. Chin. J. Cancer 2014, 33, 434–444. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Vom Berg, J.; Vrohlings, M.; Haller, S.; Haimovici, A.; Kulig, P.; Sledzinska, A.; Weller, M.; Becher, B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 2013, 210, 2803–2811. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Wang, P.-Y.; Hutzen, B.; Sprague, L.; Swain, H.M.; Love, J.K.; Stanek, J.R.; Boon, L.; Conner, J.; Cripe, T.P. Cooperation of Oncolytic Herpes Virotherapy and PD-1 Blockade in Murine Rhabdomyosarcoma Models. Sci. Rep. 2017, 7, 2396. [Google Scholar] [CrossRef]
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, H.; Ren, J.; Liu, W.; Chen, L.; Chen, H.; Ye, J.; Dai, E.; Ma, C.; Ju, S.; et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Backhaus, P.S.; Veinalde, R.; Hartmann, L.; Dunder, J.E.; Jeworowski, L.M.; Albert, J.; Hoyler, B.; Poth, T.; Jäger, D.; Ungerechts, G.; et al. Immunological Effects and Viral Gene Expression Determine the Efficacy of Oncolytic Measles Vaccines Encoding IL-12 or IL-15 Agonists. Viruses 2019, 11, 914. [Google Scholar] [CrossRef] [Green Version]
- Gaston, D.C.; Odom, C.I.; Li, L.; Markert, J.M.; Roth, J.C.; Cassady, K.A.; Whitley, R.J.; Parker, J.N. Production of bioactive soluble interleukin-15 in complex with interleukin-15 receptor alpha from a conditionally-replicating oncolytic HSV-1. PLoS ONE 2013, 8, e81768. [Google Scholar] [CrossRef]
- Wu, C.; Wu, M.; Liang, M.; Xiong, S.; Dong, C. A novel oncolytic virus engineered with PD-L1 scFv effectively inhibits tumor growth in a mouse model. Cell. Mol. Immunol. 2019, 16, 780–782. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chouljenko, D.V.; Ding, J.; Lee, I.-F.; Murad, Y.M.; Bu, X.; Liu, G.; Delwar, Z.; Sun, Y.; Yu, S.; Samudio, I.; et al. Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes. Biomedicines 2020, 8, 484. https://doi.org/10.3390/biomedicines8110484
Chouljenko DV, Ding J, Lee I-F, Murad YM, Bu X, Liu G, Delwar Z, Sun Y, Yu S, Samudio I, et al. Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes. Biomedicines. 2020; 8(11):484. https://doi.org/10.3390/biomedicines8110484
Chicago/Turabian StyleChouljenko, Dmitry V., Jun Ding, I-Fang Lee, Yanal M. Murad, Xuexian Bu, Guoyu Liu, Zahid Delwar, Yi Sun, Sheng Yu, Ismael Samudio, and et al. 2020. "Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes" Biomedicines 8, no. 11: 484. https://doi.org/10.3390/biomedicines8110484