Nrf2 Lowers the Risk of Lung Injury via Modulating the Airway Innate Immune Response Induced by Diesel Exhaust in Mice

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. DE Exposure
2.3. Study Design
2.4. Electron Microscopy Analysis
2.5. BAL and Cell Count in BALF
2.6. Quantitation of Cytokine Protein Levels in BALF
2.7. Quantitation of Cytokine Protein Levels in Lung Tissue Supernatants
2.8. Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction
2.9. Statistical Analysis
3. Results
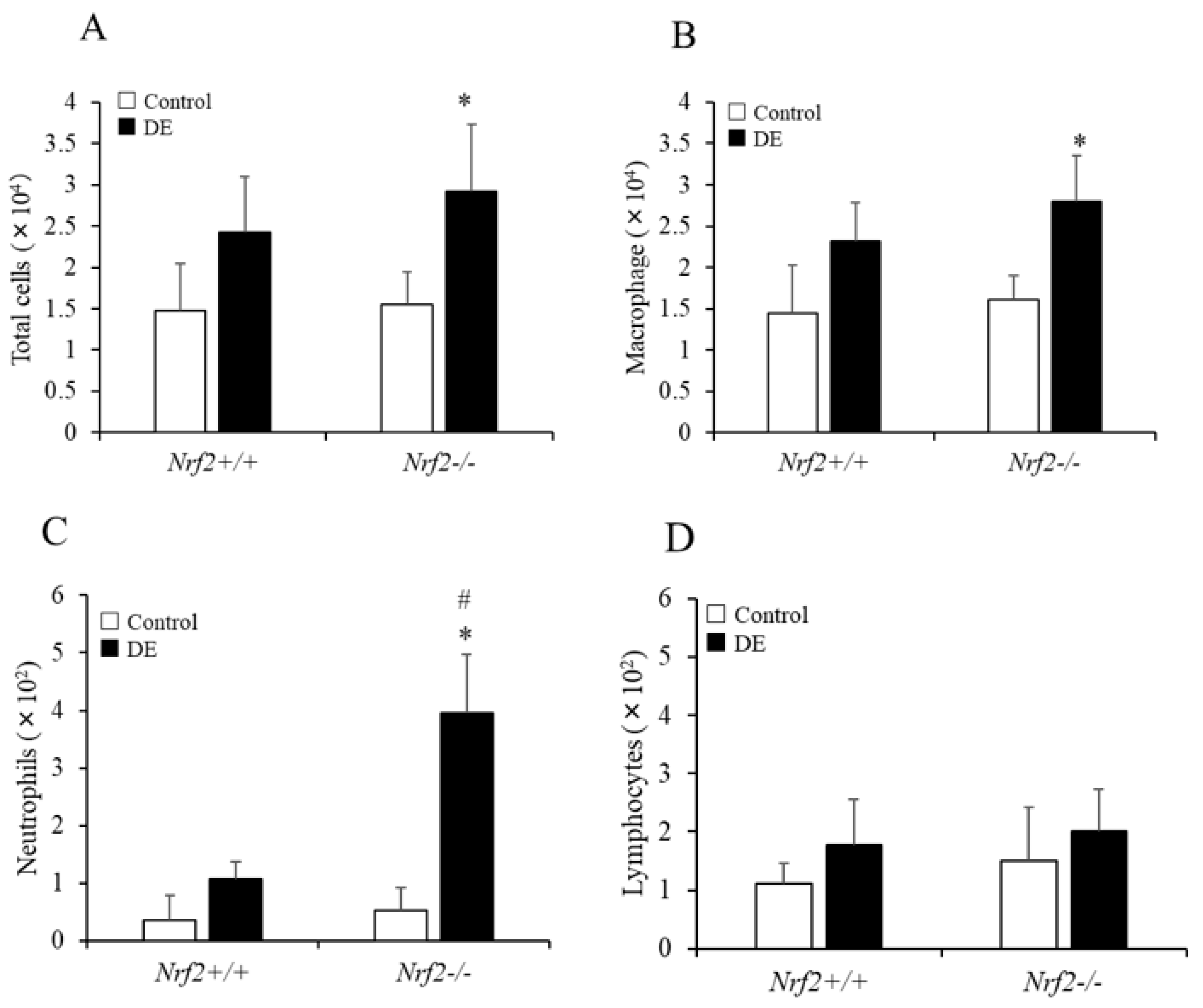
3.1. Differential Cell Counts in BALF
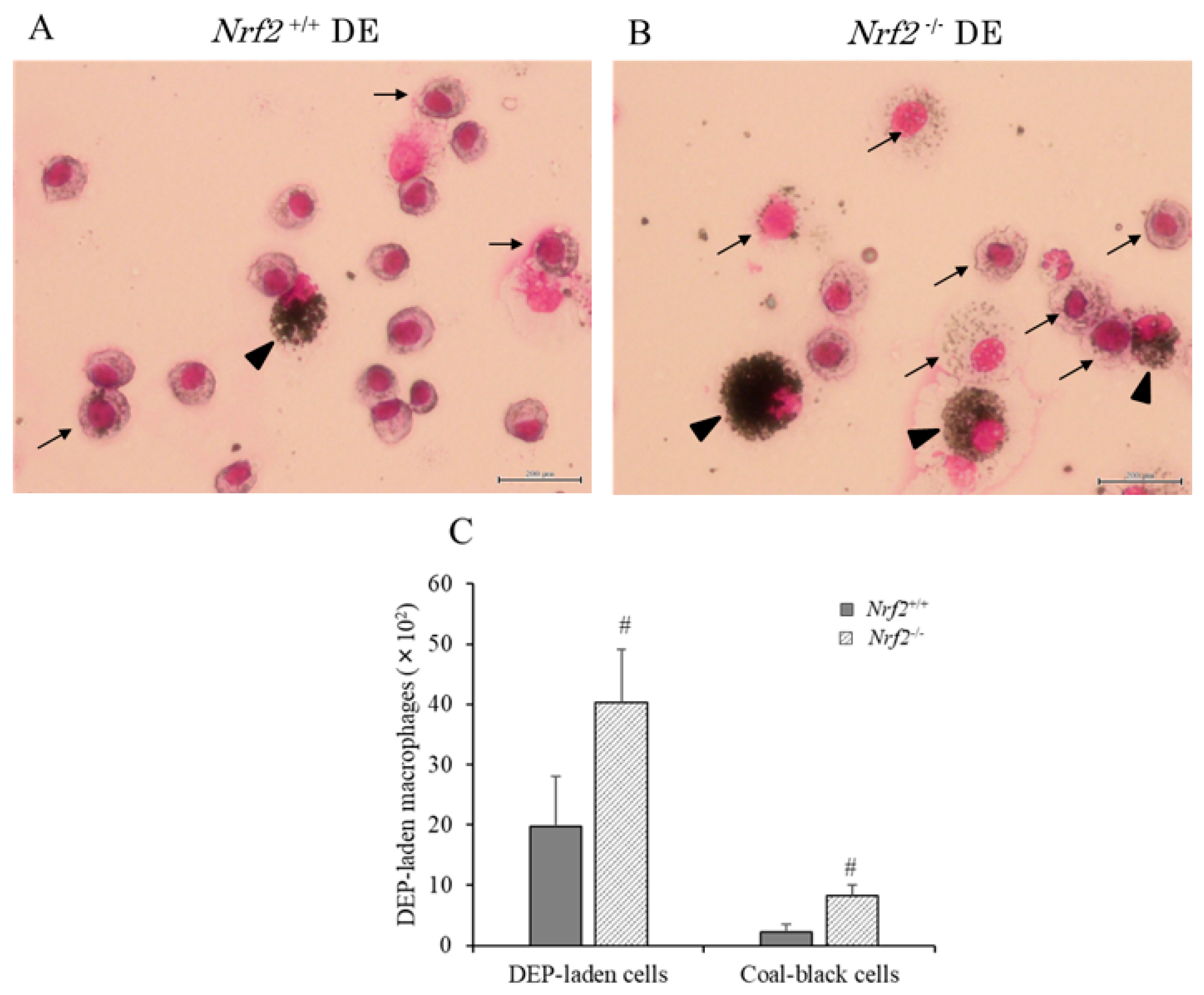
3.2. DEP-Laden Alveolar Macrophages in BALF
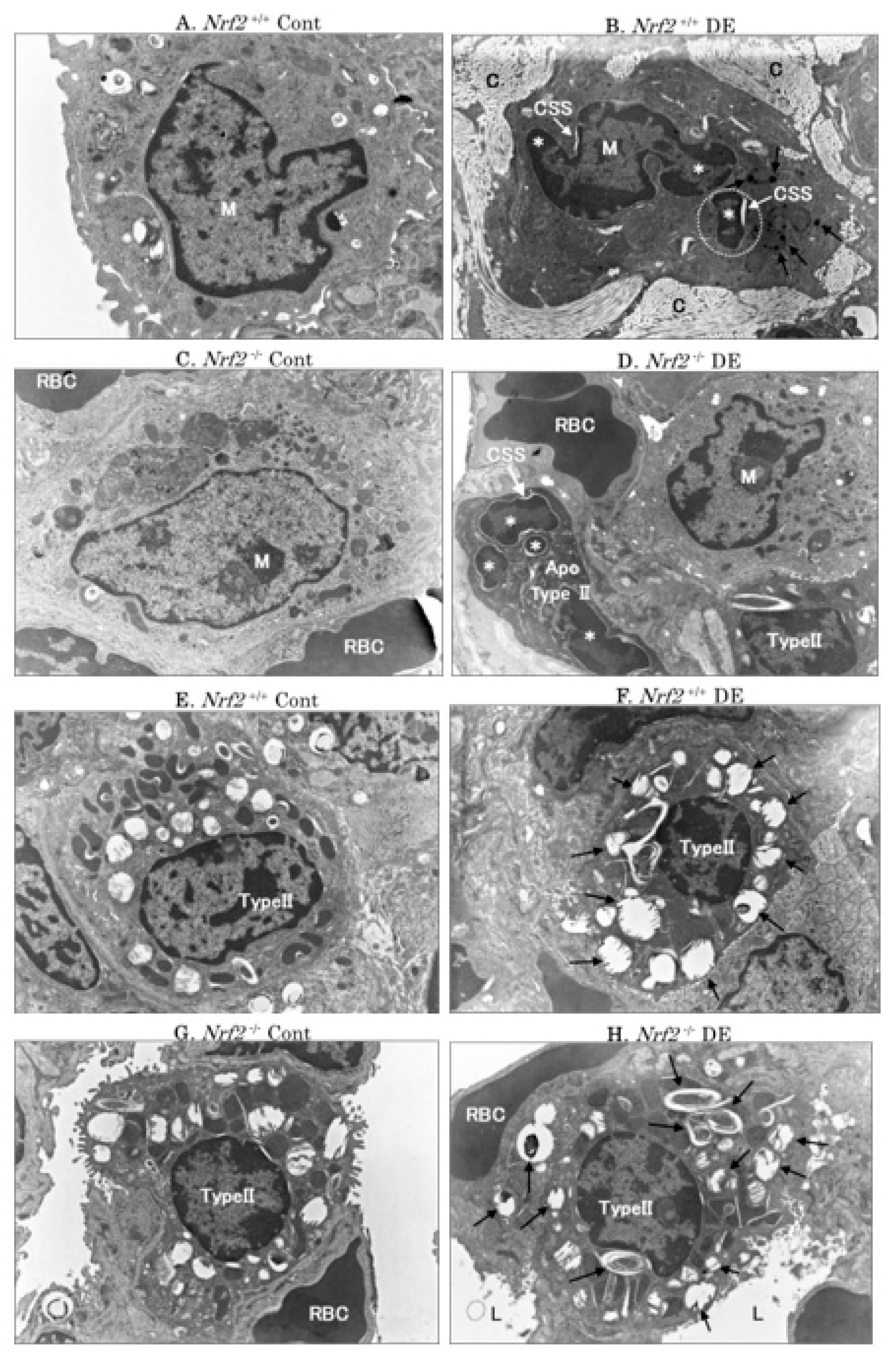
3.3. Electron Microscopic Analysis
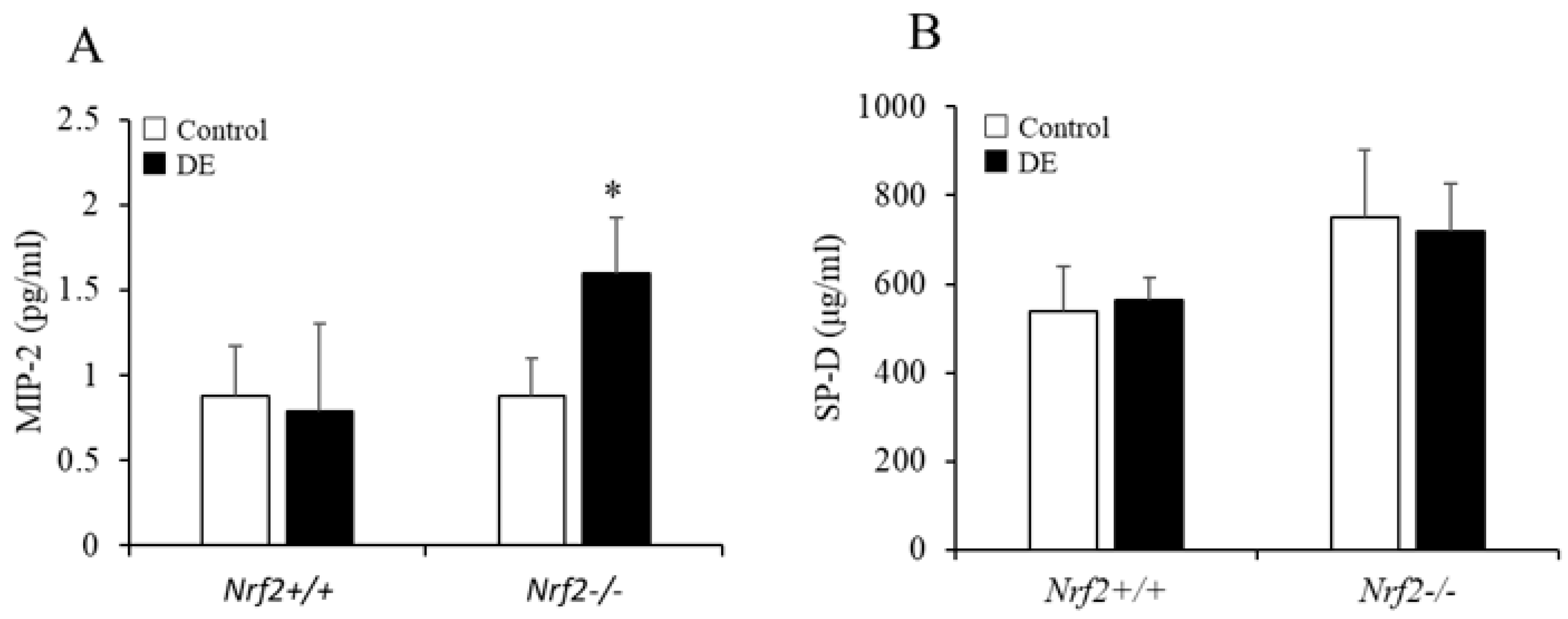
3.4. MIP-2 and SP-D Levels in BALF
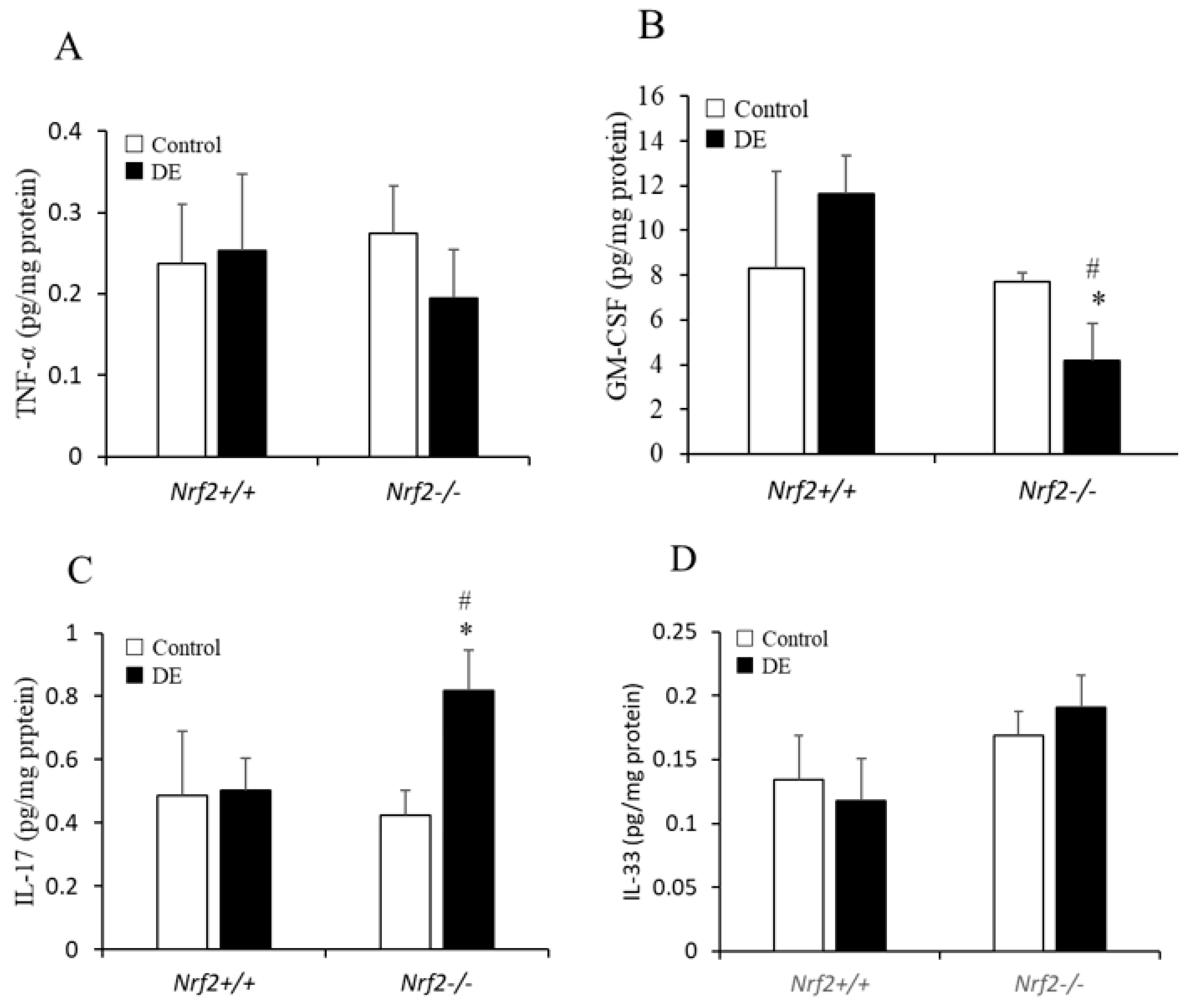
3.5. TNF-α, GM-CSF, IL-17, and IL-33 Levels in Lung Tissue
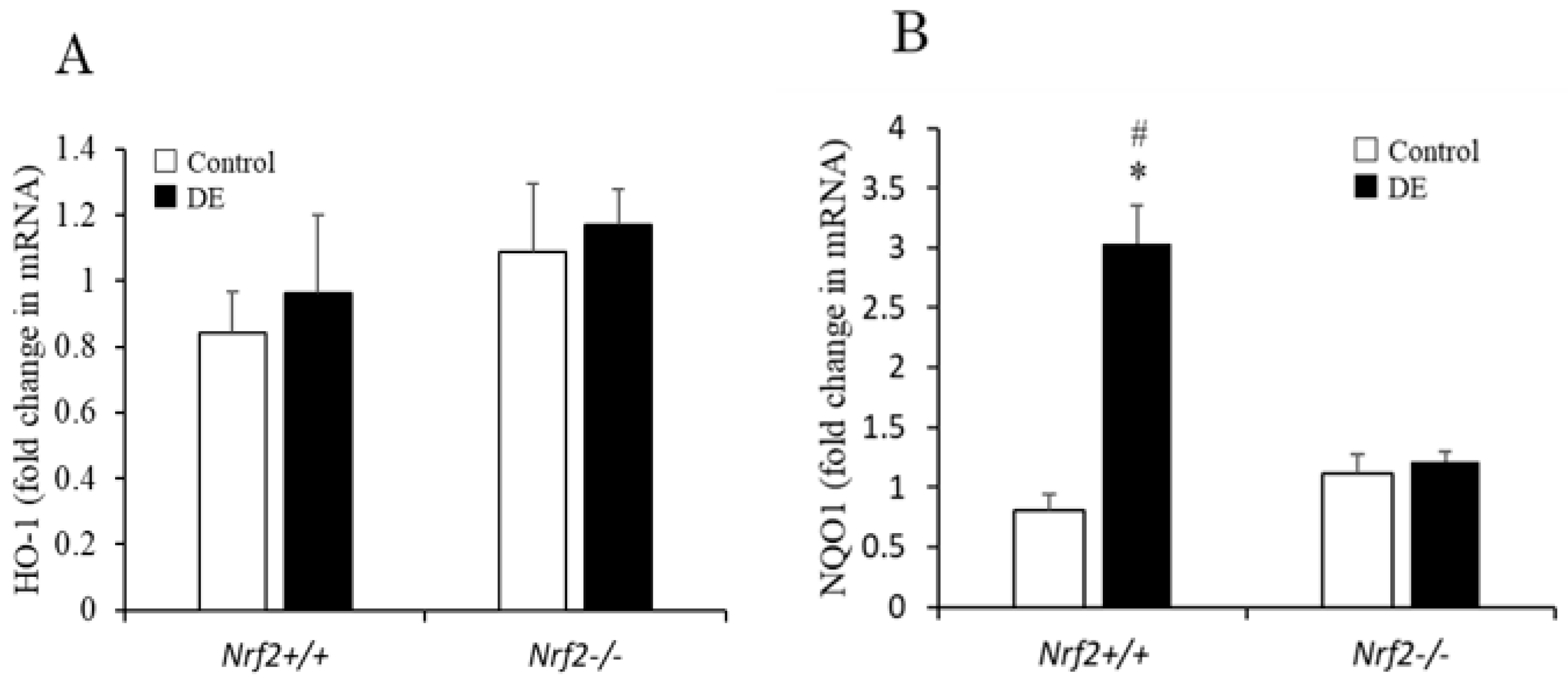
3.6. Induction of Antioxidant Enzyme mRNA Expression in the Lung Tissues
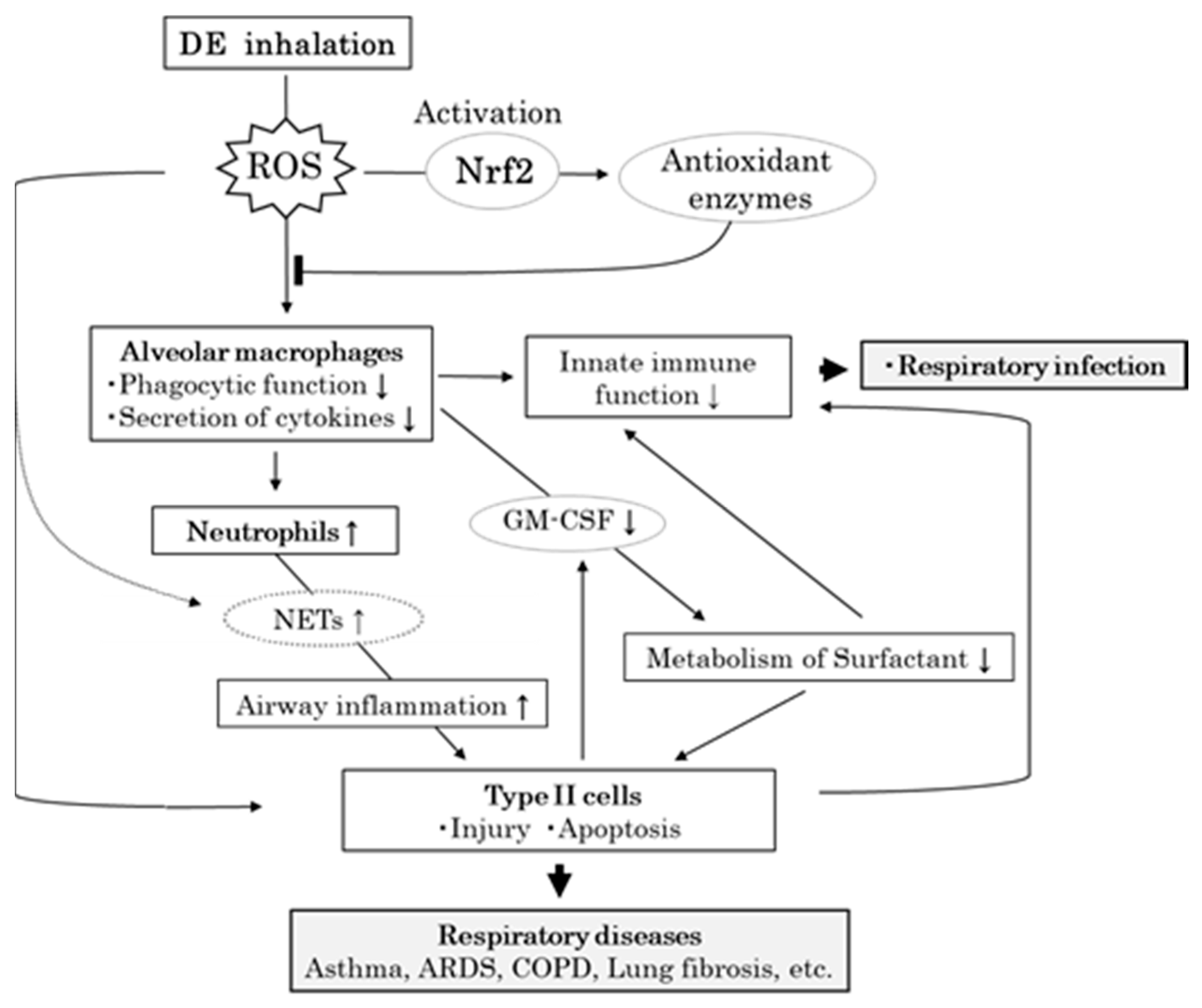
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dockery, D.W.; Pope, C.A.; Xu, X.; Spengler, J.D.; Ware, J.H.; Fay, M.E.; Ferris, B.G.; Speizer, F.E. Anassociation between air pollution and mortality in six USA cities. N. Engl. J. Med. 1993, 329, 1753–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, G.; Brunekreef, B.; Goldbohm, S.; Fischer, P.; van den Brandt, P.A. Association between mortality and indicators of traffic-related air pollution in The Netherlands: A cohort study. Lancet 2002, 360, 1203–1209. [Google Scholar] [CrossRef] [Green Version]
- Araujo, J.A.; Nel, A.E. Particulate matter and atherosclerosis: Role of particle size, composition and oxidative stress. Part. Fibre. Toxicol. 2009, 6, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 2015, 525, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Umezawa, M.; Takizawa, H.; Takeda, K.; Kawada, T. PM2.5: Role of Oxidative Stress in Health Effects and Prevention Strategy; Nova Science Publishers: New York, NY, USA, 2015. [Google Scholar]
- Takizawa, H. Diesel exhaust particles and their effect on induced cytokine expression in human bronchial epithelial cells. Curr. Opin. Allergy Clin. Immunol. 2004, 4, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Abe, S.; Okazaki, H.; Kohyama, T.; Sugawara, I.; Saito, Y.; Ohtoshi, T.; Kawasaki, S.; Desaki, M.; Nakahara, K.; et al. Diesel Exhaust Particles Upregulate Eotaxin Gene Expression in Human Bronchial Epithelial Cells via Nuclear Factor-Kappa B-Dependent Pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L1055–L1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, S.; Gon, Y.; Takeshita, I.; Matsumoto, K.; Jibiki, I.; Takizawa, H.; Kudoh, S.; Horie, T. Diesel Exhaust particles activate p38 MAP kinase to produce interleukin 8 and RANTES by human bronchial epithelial cells and N-acetylcysteine attenuates p38 MAP kinase activation. Am. J. Respir. Crit. Care Med. 2000, 161, 280–285. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.G.; Wang, M.; Li, N.; Loo, J.A.; Nel, A.E. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J. Biol. Chem. 2003, 278, 50781–50790. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Venkatesan, M.I.; Miguel, A.; Kaplan, R.; Gujuluva, C.; Alam, J.; Nel, A. Induction of heme oxygenase-1 expression in macrophages by diesel exhaust particle chemicals and quinones via the antioxidant-responsive element. J. Immunol. 2000, 165, 3393–3401. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Alam, J.; Venkatesan, M.I.; Eiguren-Fernandez, A.; Schmitz, D.; Di Stefano, E.; Slaughter, N.; Killeen, E.; Wang, X.; Huang, A.; et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: Protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 2004, 173, 3467–3481. [Google Scholar] [CrossRef]
- Li, Y.J.; Kawada, T.; Matsumoto, A.; Azuma, A.; Kudoh, S.; Takizawa, H.; Sugawara, I. Airway inflammatory responses to oxidative stress induced by low-dose diesel exhaust particle exposure differ between mouse strains. Exp. Lung. Res. 2007, 33, 227–244. [Google Scholar] [CrossRef]
- Li, Y.J.; Kawada, T.; Takizawa, H.; Azuma, A.; Kudoh, S.; Sugawara, I.; Yamauchi, Y.; Kohyama, T. Airway inflammatory responses to oxidative stress induced by prolonged low-dose diesel exhaust particle exposure from birth differ between mouse BALB/c and C57BL/6 strains. Exp. Lung. Res. 2008, 34, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Sato, S.; Nishimura, N.; Takahashi, S.; Itoh, K.; Yamamoto, M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol. Appl. Pharmacol. 2001, 173, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Takizawa, H.; Azuma, A.; Kohyama, T.; Yamauchi, Y.; Takahashi, S.; Masayuki, Y.; Kawada, T.; Kudoh, S.; Sugawara, I. Disruption of Nrf2 enhances susceptibility to airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clin. Immunol. 2008, 128, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Takizawa, H.; Azuma, A.; Kohyama, T.; Yamauchi, Y.; Takahashi, S.; Yamamoto, M.; Kawada, T.; Kudoh, S.; Sugawara, I. Nrf2 is closely related to allergic airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clin. Immunol. 2010, 137, 234–241. [Google Scholar] [CrossRef]
- Li, Y.J.; Shimizu, T.; Shinkai, Y.; Hirata, Y.; Inagaki, H.; Takeda, K.; Azuma, A.; Yamamoto, M.; Kawada, T. Nrf2 Regulates the Risk of a Diesel Exhaust Inhalation-Induced Immune Response during Bleomycin Lung Injury and Fibrosis in Mice. Int. J. Mol. Sci. 2017, 18, 649. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, J.; Sun, J.; Li, W.; Yang, L.; Wen, L.; Wang, W.; Wang, X.; Collett, J.L., Jr.; Shi, Y.; et al. Severe haze episodes and seriously polluted fog water in Ji’nan. China Sci. Total. Environ. 2014, 493, 133–137. [Google Scholar] [CrossRef]
- Saito, Y.; Azuma, A.; Kudo, S.; Takizawa, H.; Sugawara, I. Long-term inhalation of diesel exhaust affects cytokine expression in murine lung tissues: Comparison between low—And high-dose diesel exhaust exposure. Exp. Lung. Res. 2002, 28, 493–506. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Azuma, A.; Kudoh, S.; Desaki, M.; Takizawa, H.; Sugawara, I. Inhalation of Diesel Exhaust for Three Months Affects Major Cytokine Expression and Induces Bronchus-Associated Lymphoid Tissue Formation in Murine Lungs. Exp. Lung. Res. 2003, 29, 607–622. [Google Scholar] [CrossRef]
- Sakai, M.; Yamashita, K.; Takemoto, N.; Ohshima, Y.; Tsukimoto, M.; Shinkai, Y.; Takeda, K.; Oshio, S.; Kojima, S. Diesel exhaust (DE) aggravates pathology of delayed-type hypersensitivity (DTH) induced by methyl-bovine serum albumin (mBSA) in mice. J. Toxicol. Sci. 2009, 34, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Umezawa, M.; Sakata, C.; Tanaka, N.; Tabata, M.; Takeda, K.; Ihara, T.; Sugamata, M. Pathological study for the effects of in utero and postnatal exposure to diesel exhaust on a rat endometriosis model. J. Toxicol. Sci. 2011, 36, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umezawa, M.; Sekita, K.; Suzuki, K.; Kubo-Irie, M.; Niki, R.; Ihara, T.; Sugamata, M.; Takeda, K. Effect of aerosol particles generated by ultrasonic humidifiers on the lung in mouse. Part. Fibre. Toxicol. 2013, 10, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.R.; Frevert, C.W. Innate Immunity in the Lungs. Proc. Am. Thorac. Soc. 2005, 2, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Y.; Ma, J.K. The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J. Environ. Sci. Health Part C 2002, 20, 117–147. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.J.; Dong, C.C.; Ma, J.Y.; Roberts, J.R.; Antonini, J.M.; Ma, J.K. Suppression of phagocytic and bactericidal functions of rat alveolar macrophages by the organic component of diesel exhaust particles. J. Toxicol. Environ. Health A 2007, 70, 820–828. [Google Scholar] [CrossRef]
- Yin, X.J.; Schafer, R.; Ma, J.Y.; Antonini, J.M.; Weissman, D.D.; Siegel, P.D.; Barger, M.W.; Roberts, J.R.; Ma, J.K. Alteration of pulmonary immunity to Listeria monocytogenes by diesel exhaust particles (DEPs) I. Effects of DEPs on early pulmonary responses. Environ. Health Perspect. 2002, 110, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Cattani-Cavalieri, I.; Valenca, S.S.; Lanzetti, M.; Carvalho, G.M.C.; Zin, W.A.; Monte-Alto-Costa, A.; Porto, L.C.; Romana-Souza, B. Acute Exposure to Diesel-Biodiesel Particulate Matter Promotes Murine Lung Oxidative Stress by Nrf2/HO-1 and Inflammation Through the NF-kB/TNF-α Pathways. Inflammation 2019, 42, 526–537. [Google Scholar] [CrossRef]
- Huffman, J.A.; Hull, W.M.; Dranoff, G.; Mulligan, R.C.; Whitsett, J.A. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J. Clin. Investig. 1996, 97, 649–655. [Google Scholar] [CrossRef]
- Berclaz, P.Y.; Shibata, Y.; Whitsett, J.A.; Trapnell, B.C. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood 2002, 100, 4193–4200. [Google Scholar] [CrossRef]
- Staitieh, B.S.; Egea, E.E.; Fan, X.; Azih, N.; Neveu, W.; Guidot, D.M. Activation of alveolar macrophages with interferon-γ promotes antioxidant defenses via the Nrf2-ARE pathway. J. Clin. Cell Immunol. 2015, 6, 365. [Google Scholar]
- Ihara, T.; Yamamoto, T.; Sugamata, M.; Okumura, H.; Ueno, Y. The process of ultrastructural changes from nuclei to apoptotic body. Virchows Arch. 1998, 433, 443–447. [Google Scholar] [CrossRef]
- Sugamata, M.; Ihara, T.; Takano, H.; Oshio, S.; Takeda, K. Maternal Diesel Exhaust Exposure Damages Newborn Murine Brains. J. Health Sci. 2006, 52, 82–84. [Google Scholar] [CrossRef] [Green Version]
- Salvi, S.; Blomberg, A.; Rudell, B.; Kelly, F.; Sandstrom, T.; Holgate, S.T.; Frew, A. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am. J. Respir. Crit. Care. Med. 1999, 159, 702–709. [Google Scholar] [CrossRef]
- Stenfors, N.; Nordenh¨all, C.; Salvi, S.S.; Mudway, I.; Söderberg, M.; Blomberg, A.; Helleday, R.; Levin, J.O.; Holgate, S.T.; Kelly, F.J.; et al. Different airway inflammatory responses in asthmatic and healthy humans exposed to diesel. Eur. Respir. J. 2004, 23, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Lou, N.; Lennard Richard, M.L.; Yu, J.; Kindy, M.; Zhang, X.K. The Fli-1 transcription factor is a critical regulator for controlling the expression of chemokine C-X-C motif ligand 2 (CXCL2). Mol. Immunol. 2016, 81, 59–66. [Google Scholar]
- Lindén, A.; Laan, M.; Anderson, G.P. Neutrophils, interleukin-17A and lung disease. Eur. Respir. J. 2005, 25, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Chen, H.; Ding, Q.; Xu, X.; Yu, B.; Huang, Z. Nuclear Factor Erythroid 2-related Factor 2 Deficiency Exacerbates Lupus Nephritis in B6/lpr mice by Regulating Th17 Cell Function. Sci. Rep. 2016, 6, 38619. [Google Scholar] [CrossRef] [Green Version]
- Dworski, R.; Simon, H.U.; Hoskins, A.; Yousefi, S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J. Allergy Clin. Immunol. 2011, 127, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- McCarthy, P.C.; Phair, I.R.; Greger, C.; Pardali, K.; McGuire, V.A.; Clark, A.R.; Gaestel, M.; Arthur, J.S.C. IL-33 regulates cytokine production and neutrophil recruitment via the p38 MAPK-activated kinases MK2/3. Immunol. Cell Biol. 2019, 97, 54–71. [Google Scholar] [CrossRef]
- Uchida, M.; Anderson, E.L.; Squillace, D.L.; Patil, N.; Maniak, P.J.; Iijima, K.; Kita, H.; O’Grady, S.M. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy 2017, 72, 1521–1531. [Google Scholar] [CrossRef]
- Schürch, S.; Lee, M.; Gehr, P. Pulmonary surfactant: Surface properties and function of alveolar and airway surfactant. Pure Appl. Chem. 1992, 64, 1745–1750. [Google Scholar] [CrossRef]
- Chander, A.; Fisher, A.B. Regulation of lung surfactant secretion. Am. J. Physiol. 1990, 258, L241–L253. [Google Scholar] [CrossRef]
- McIntosh, J.C.; Swyers, A.H.; Fisher, J.H.; Wright, J.R. Surfactant proteins A and D increase in response to intratracheal lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 1996, 15, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.R. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005, 5, 58–68. [Google Scholar] [CrossRef]
- Cross, P.C.; Mercer, K.L. Cell and Tissue Ultrastructure; W.H. Freeman and Company: New York, NY, USA, 1993; pp. 314–315. [Google Scholar]
- Mason, R.J.; Lewis, M.C.; Edeen, K.E.; McCormick-Shannon, K.; Nielsen, L.D.; Shannon, J.M. Maintenance of surfactant protein A and D secretion by rat alveolar type II cells in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L249–L258. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.Y.; Wang, J.S.; Chao, M.W. Causation by diesel exhaust particles of endothelial dysfunctions in cytotoxicity, pro-inflammation, permeability, and apoptosis induced by ROS generation. Cardiovasc. Toxicol. 2017, 17, 384–392. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chamber | CO (ppm) | SO2 (ppb) | NO (ppm) | NO2 (ppm) | NOx (ppm) | DEP (mg/m3) | DEP (#/cc) |
|---|---|---|---|---|---|---|---|
| Clean | 0.44 ± 0.17 | 0.64 ± 0.50 | 0.00 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.01 ± 0.01 | 3 ± 1 |
| DE | 10.26 ± 2.72 | 21.03 ± 5.50 | 3.65 ± 0.84 | 1.91 ± 0.45 | 5.55 ± 1.26 | 1.02 ± 0.29 | 343,700 ± 2900 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-J.; Shimizu, T.; Shinkai, Y.; Ihara, T.; Sugamata, M.; Kato, K.; Kobayashi, M.; Hirata, Y.; Inagaki, H.; Uzuki, M.; et al. Nrf2 Lowers the Risk of Lung Injury via Modulating the Airway Innate Immune Response Induced by Diesel Exhaust in Mice. Biomedicines 2020, 8, 443. https://doi.org/10.3390/biomedicines8100443
Li Y-J, Shimizu T, Shinkai Y, Ihara T, Sugamata M, Kato K, Kobayashi M, Hirata Y, Inagaki H, Uzuki M, et al. Nrf2 Lowers the Risk of Lung Injury via Modulating the Airway Innate Immune Response Induced by Diesel Exhaust in Mice. Biomedicines. 2020; 8(10):443. https://doi.org/10.3390/biomedicines8100443
Chicago/Turabian StyleLi, Ying-Ji, Takako Shimizu, Yusuke Shinkai, Tomomi Ihara, Masao Sugamata, Katsuhito Kato, Maiko Kobayashi, Yukiyo Hirata, Hirofumi Inagaki, Makoto Uzuki, and et al. 2020. "Nrf2 Lowers the Risk of Lung Injury via Modulating the Airway Innate Immune Response Induced by Diesel Exhaust in Mice" Biomedicines 8, no. 10: 443. https://doi.org/10.3390/biomedicines8100443