
Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease

1
Faculty of Medicine, University of Coimbra, 3000-370 Coimbra, Portugal
2
CNC—Center for Neuroscience and Cell Biology, University of Coimbra, 3004-504 Coimbra, Portugal
3
CIBB—Center for Innovative Biomedicine and Biotechnology, University of Coimbra, 3004-504 Coimbra, Portugal
4
Institute of Physiology, Faculty of Medicine, University of Coimbra, 3000-370 Coimbra, Portugal
5
IIIUC—Institute for Interdisciplinary Research, University of Coimbra, 3030-789 Coimbra, Portugal
*
Author to whom correspondence should be addressed.
Biomedicines 2023, 11(9), 2331; https://doi.org/10.3390/biomedicines11092331
Submission received: 11 July 2023
/
Revised: 16 August 2023
/
Accepted: 18 August 2023
/
Published: 22 August 2023
(This article belongs to the Special Issue Alzheimer's Disease—115 Years after Its Discovery 2.0)
Abstract
:Alzheimer’s disease is the most prevalent neurodegenerative disorder and affects the lives not only of those who are diagnosed but also of their caregivers. Despite the enormous social, economic and political burden, AD remains a disease without an effective treatment and with several failed attempts to modify the disease course. The fact that AD clinical diagnosis is most often performed at a stage at which the underlying pathological events are in an advanced and conceivably irremediable state strongly hampers treatment attempts. This raises the awareness of the need to identify and characterize the early brain changes in AD, in order to identify possible novel therapeutic targets to circumvent AD’s cascade of events. One of the most auspicious targets is mitochondria, powerful organelles found in nearly all cells of the body. A vast body of literature has shown that mitochondria from AD patients and model organisms of the disease differ from their non-AD counterparts. In view of this evidence, preserving and/or restoring mitochondria’s health and function can represent the primary means to achieve advances to tackle AD. In this review, we will briefly assess and summarize the previous and latest evidence of mitochondria dysfunction in AD. A particular focus will be given to the recent updates and advances in the strategy options aimed to target faulty mitochondria in AD.
1. Introduction
Alzheimer’s disease (AD) is known to be the most common form of dementia among the elderly population, representing an estimated 60% to 80% of diagnosed dementia cases [1]. Although a small percentage of AD cases (approximately 1–5%) can be attributed to a genetically inherited predisposition and be diagnosed between 30 and 60 years old, the vast majority of AD cases are sporadic, being frequently diagnosed at over 65 years old [2]. The exact cause of the sporadic form of AD is not fully understood, but it is believed to have a multifactorial component involving both genetic and environmental factors that combine and move along a continuum until disease diagnosis [3]. In addition, it is also recognized that AD has a long asymptomatic pre-clinical phase, during which pathophysiological changes occur without noticeable cognitive symptoms. As the disease evolves, individuals start to perceive a gradual memory loss, cognitive decline, and behavioral changes that ultimately impair their daily routines [4]. Canonically, the pathological diagnosis of AD relies on the postmortem detection of the accumulation of abnormal structures, such as senile plaques resulting from the extracellular deposition of amyloid β protein (Aβ) and the intracellular accumulation of neurofibrillary tangles composed of hyperphosphorylated tau (pTau) protein, which could trigger the neurodegenerative cascade leading to the loss of neurons associated with higher brain function [4].
Despite the alarming numbers and the latest estimations suggesting that, by the year 2050, there will be more than 100 million people diagnosed with this disorder, AD continues to be a disease without an effective treatment. However, there are several ongoing attempts to modify the disease course, including the recently U.S. Food and Drug Administration (FDA)-approved drugs ducanumab and lecanema, both targeted to Aβ [5].
Considering the enormity of the problem, several countries have implemented a deadline to develop a cure or approved disease-modifying therapy to tackle AD [6,7]. In this scenario, recent reports highlight that the cognitive performance of individuals at older ages does not necessarily correlate with the accumulation of AD hallmarks [8], which highlights the need to move away from targeting the cardinal AD neuropathological hallmarks to identify and characterize the early brain changes in AD, in order to identify possible novel therapeutic targets to circumvent AD’s cascade of events. One of the most auspicious targets is mitochondria, conserved organelles indispensable in nearly all cells of the body [9]. Mitochondria are known as the cell’s powerhouses, responsible for generating the majority of the cell’s energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation. Beyond this role, mitochondria exert multiple essential functions in different cellular processes, e.g., the regulation of calcium homeostasis, reactive oxygen species formation (ROS), cell repair and cell death processes, intracellular signaling, and cell metabolism, among others [10]. In the brain, mitochondria acquire an even more important role, as the brain’s energy requirements, particularly of neuronal cells, are extremely high in order to support all of the complex processes and mechanisms necessary to ensure its adequate activity [11]. A vast body of literature has shown the occurrence of extensive mitochondria abnormalities in the continuum of AD-related brain events [12,13]. For instance, pivotal studies have revealed that during the transitional clinical stage, known as mild cognitive impairment (MCI), patients present a faulty cerebral energy metabolism, disturbances in the expression and/or activities of important regulatory mitochondrial energy-related enzymes (e.g., pyruvate dehydrogenase (PDH), isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase (α-KGDH), and cytochrome oxidase (COX)), a significant rise in oxidative stress markers, and alterations in mitochondria number, shape, and localization [14,15]. Recently, mitochondria deficits were identified as a key trigger for the loss of synaptic function observed in cortical tissue from post-mortem AD patients [16]. In further agreement, data from model organisms of the disease reinforce the notion that mitochondria dysfunction and metabolic alterations precede AD neuropathology [17,18,19]. According to these premises, preserving mitochondria’s health and function could represent the “holy grail” to address AD’s prevalence. In this review, we will assess and summarize the previous and latest evidence of mitochondria disturbances in AD. A particular emphasis will be placed on the recent advances and updates in the treatment options aimed to target faulty mitochondria in AD. To perform this narrative review, the search was conducted during March–June 2023 in the database PubMed. Relevant articles were searched using different combinations of the keywords “Alzheimer’s disease” (AND) “mitochondria” (AND) and (OR) “mitochondria-directed strategies” (OR) “mitochondria” (AND) “brain health” (AND) “energy metabolism”. We also used the database ClinicalTrials.gov to search for research that has advanced into clinical trials. Herein, the keywords used were “Alzheimer’s disease” (AND) “mitochondria”. The search performed was narrowed to articles written in English and was independently undertaken by two authors (Tiago Sousa and Susana Cardoso). The article selection was performed without year limitations but, since we aimed to provide an updated overview of the current literature available on the subject, preference was given to articles published during the last ten years.
2. Mitochondria (Dys)Function in Alzheimer’s Disease: A Brief Overview of the Main Mechanisms
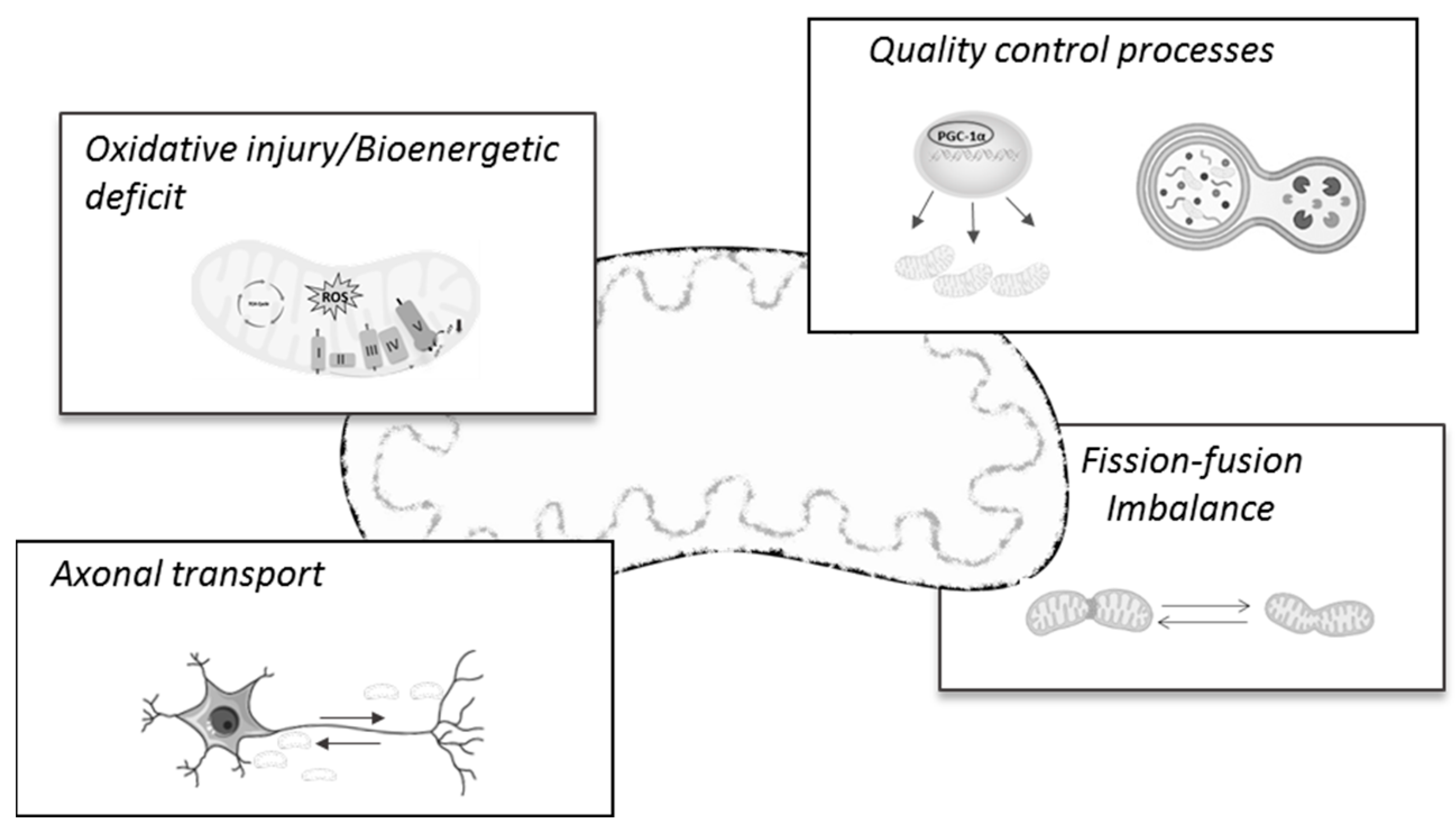
The first insights into mitochondria involvement in AD pathology emerged several years ago, when the electron microscopy analysis of the frontal cortex tissue of AD patients revealed the presence of abnormal mitochondria in apparent normal dendrites, but which were supposed to degenerate in later stages of the disease [20,21]. Since then, abundant research has demonstrated mitochondria and mitochondria-related mechanisms and processes as prominent features of the AD pathology and/or progression (for a review, see [22]). Notably, there is mounting evidence suggesting that many of these brain mitochondrial changes also occur in non-brain samples, such as peripheral blood lymphocytes [23], skin fibroblasts [24], peripheral blood mononuclear cells (PBMCs), and platelets [25]. In fact, a very recent work by Mahapatra and colleagues [25] confirmed that peripheral blood cells’ bioenergetics profiles closely correlated with subjects’ cognitive status across AD progression. In line with this, others report that the peripheral alterations of oxidative stress markers could represent feasible biomarkers for early AD diagnostics [26,27,28,29], thus highlighting and reinforcing the mitocentric view of AD. In the next subsections, we will provide a brief and up-to-date review of the main mitochondrial mechanisms found to be deregulated and/or impaired during the AD continuum, which will then be considered as possible targets for AD therapy (Figure 1).
2.1. Relevance of Oxidative Stress and Energy (Hypo)Metabolism in AD
Mitochondria are double-membrane-bound organelles with an outer membrane (OMM) facing the cytosol and a large folded inner mitochondrial membrane (IMM) with high selectivity capacity, which makes it impermeable to almost all molecules found in the matrix [10]. The oxidative phosphorylation system (OXPHOS) engrained in this membrane has the capacity to empower oxygen consumption with coupled ATP production through the coordination of five enzymatic complexes. Briefly, during cellular respiration, electrons flow through the mitochondrial electron transport chain (ETC) due to many redox reactions, forming an electrochemical gradient, which leads to ATP synthesis in complex V. If an electron escapes from the ETC, mainly in complexes I and III, it will subsequently bind oxygen (O2) and form superoxide (O2−), an anionic free radical (for a review, see [30]). Alongside O2−, hydroxyl radicals (·OH) and hydrogen peroxide (H2O2) constitute the reactive oxygen species (ROS) and are considered the main by-products of oxidative phosphorylation [31]. ROS are free radical and non-radical molecules that have the capacity to rapidly affect cell functionality and viability by damaging proteins, lipids, and nucleic acids, including mitochondrial structures that, due to the close production site, are not only the main producers but also the first targets of ROS-induced damage [31]. Importantly, it is also recognized that low/moderate concentrations of ROS are necessary for the regulation of different processes, such as cellular signaling, immune responses, and inflammation [32]. Thus, it is important to guarantee that ROS production and elimination exist in a highly controlled equilibrium (for a review, see [33]). To accomplish this, cells are equipped with a very efficient antioxidant system composed of enzymatic (e.g., superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase) and non-enzymatic defenses (e.g., glutathione, vitamins C and E) [31]. This becomes of particular importance regarding brain homeostasis, which, due to its inherent properties, e.g., high content of polyunsaturated fatty acids (PUFA), high oxygen consumption, and modest antioxidant defense levels, is an organ that is extremely vulnerable to the deleterious effects of oxidative injury [34]. As with AD, plentiful evidence has confirmed that oxidative stress has a pivotal role in the pathogenesis of the disease [35]. As previously reported, relative to non-diseased brains, AD brain tissue, especially brain regions with histopathological features of the disease, possesses high levels of oxidative stress markers [36]. In particular, protein oxidation markers (e.g., protein carbonyls and 3-nitrotyrosine) [37] and lipid peroxidation products (e.g., malondialdehyde (MDA) and 4-hydroxynonenal (HNE)) have been found to be elevated in the vulnerable regions of AD brains [38,39], whilst, in contrast, earlier studies show significant alterations in the antioxidant defense system in AD brains [40]. Further in vivo studies corroborate the relevant role of ROS in AD [41] and report a negative correlation between cerebral ROS levels and mitochondrial function and learning and memory in mAPP mice at an early age [41]. In fact, it has been observed that oxidative deregulation, particularly oxidative-associated damage on mitochondria, can represent a pivotal event in the transition from a normal state to cognitive impairment [42], which can precede other pathological features of AD [43,44]. This view is further supported by the work of Leuner and colleagues [45], which, by means of in vitro and in vivo models of AD, showed that mitochondria-derived ROS are able to trigger the amyloidogenic amyloid precursor protein (APP) processing, leading to Aβ formation, thus demonstrating the strong influence that mitochondria and Aβ can have on each other [46]. In this context, a previous study revealed a close relation between the degree of cognitive dysfunction observed in transgenic AD mice and the range of synaptic mitochondrial dysfunction and mitochondrial Aβ levels [47]. Consistent with these observations, earlier data indicated that extracellular Aβ can be internalized by cells and imported into mitochondria cristae via the translocase of the outer membrane (TOM) machinery [48]. Inside mitochondria, similar works demonstrate that Aβ interacts with mitochondrial 17β-hydroxysteroid dehydrogenase type 10 (HSD10), an enzyme present in neuronal mitochondria [17] and often referred to as Aβ-binding alcohol dehydrogenase (ABAD), and also with Presequence protease (PreP), a mitochondrial Aβ-degrading enzyme localized in the mitochondrial matrix [49]. However, whilst the interaction of Aβ with HSD10 causes a prompt increase in ROS levels, namely O2·− and H2O2, and a loss of mitochondria function [50], PreP activity was found to be decreased in AD brains and AD transgenic mouse brains, which also presented higher levels of oxidative products in their mitochondria [49]. It has been therefore proposed that modulating PreP activity could be a feasible strategy to control the mitochondrial Aβ content and mitochondrial oxidative stress [51,52]. Moreover, a recent in vitro study performed in iPSC-derived neurons and SY5Y cells transfected with an AβPP construct identified an undescribed correlation between the mitochondrial membrane potential, AβPP mitochondrial localization, and Aβ secretion [53]. In detail, the authors observed that depolarized mitochondria had more AβPP, secreted less Aβ, and consequently had higher intracellular Aβ levels; the opposite occurred when mitochondria were hyperpolarized (i.e., membrane potential was high), suggesting that Aβ secretion can represent a feasible biomarker of cell or tissue mitochondrial health [53]. In order to further explore the effects of the mitochondria–AD hallmark interaction, Rhein and colleagues [54] conducted a quantitative proteomic analysis and functional assays in four different strains of mice: the single transgenic pR5 mice to model tau pathology, the double transgenic APP/PS2 that model the Aβ plaque pathology, and the triple transgenic mice (pR5/APP/PS2) that combine the Aβ and tau pathologies. Among the main findings of their study, the authors observed a distinct pattern in Aβ and tau’s effects on OXPHOS components; for the same age, complex I activity was preferably affected in the tau mice model, whilst complex IV activity was firstly compromised in the APP/PS2 mice [54]. With advancing age, the triple transgenic strain presented the significant aggravation of the mitochondrial respiratory capacity, impaired mitochondrial enzyme activity, and lower ATP levels—alterations that were further associated with pronounced oxidative stress and, in line with other studies [55], disclosed the synergistic effects of both Aβ and tau on mitochondria. Following the same line of investigation, a recent proteomics study on AD post-mortem tissue observed a differentially altered pattern of mitochondrial electron transport chain components in age-dependent AD [56]. Specifically, the data obtained revealed the significant destabilization of complex I and the downregulation of subunits of complexes II, III, and V in the mitochondria of early-onset AD individuals, whilst complex IV subunits were only downregulated in AD aged mitochondria [56].
Previous research has shown that oxidative damage is closely interrelated with impairments in brain energy metabolism, particularly glucose metabolism [57,58]. Indeed, the use of proteomics analysis in MCI, in early and late AD brain tissue, made it possible to detect oxidative modifications in proteins involved in the glycolytic pathway [37,59], in the tricarboxylic acid (TCA) cycle [60], and in components of the respiratory chain [61], which, by causing proteins´ dysfunction, are believed to contribute to cellular energetic compromise and glucose hypometabolism [62]. In addition, evidence gathered from an in vitro study in which hippocampal and cortical primary neurons were exposed to increasing concentrations of Aβ25–35 suggested a strong link between the impairment of glucose transport and the neurotoxicity of the peptide [58]. In detail, the authors reported that the observed Aβ-mediated inhibition of glucose transport was causally related to Aβ-induced membrane lipid peroxidation and the generation of HNE [58]. Following these premises, and in agreement with prior neuroimaging studies using positron emission tomography (PET) imaging with 2-[18F] fluoro-2-deoxy-d-glucose ([18F]FDG) [63,64,65,66], a recent study has shown that brain glucose deregulation is undeniably an important early event in AD pathogenesis [67]. Using the well-characterized cohort of the Baltimore Longitudinal Study of Aging as an experimental group, An and colleagues [67] were able to observe a significant correlation between the brain tissue glucose concentration and the severity of AD hallmarks—an outcome that was associated with lower activity of the principal rate-controlling enzymes of glycolysis (hexokinase, phosphofructokinase, and pyruvate kinase) and lower expression levels of the neuronal glucose transporter 3 (GLUT3). In a complementary approach, Terada and colleagues, by means of updated neuroimaging techniques, disclosed the distinct contributions of glycolysis and mitochondrial OXPHOS activity to AD brain hypometabolism in the living brains of AD individuals [68]. The data obtained revealed an early decrease in mitochondrial oxidative activity in patients’ parahippocampi, detected using a specific PET probe for mitochondrial complex I, which was not accompanied by alterations in overall glucose metabolism, measured with the [18F]FDG tracer, thus suggesting that mitochondria-related energy failure may precede glycolysis-related hypometabolism in AD brains [68]. Consistent with this evidence, results obtained in the 5xFAD mouse model indicated that disturbances in mitochondria TCA cycle activity and mitochondria morphology in the hippocampi of young 5xFAD mice significantly impacted synaptic integrity during early amyloid pathology in this AD model [69]. Importantly, the authors’ experiments also disclosed an intricate interplay between hippocampal neurons and astrocytes’ metabolic function; herein, hippocampal astrocytes showed hampered glucose metabolism, which affected the glutamine supply to neuronal cells, impairing neuronal GABA synthesis and possibly the balance of synaptic excitation and inhibition in the AD brain, as previously reported [70].
Previous reports corroborate that, despite having a distinct metabolic profile (glycolytic for astrocytes and oxidative for neurons) characterized by cell-specific enzymatic features [71,72] and the organization of the mitochondrial respiratory chain [73], astrocyte–neuron coordination is of the utmost importance for the maintenance of brain function (for a review, see [74]). As an example, Vicente-Gutierrez and colleagues [75,76] demonstrated that astrocytic mitochondrial ROS production critically contributed to supporting brain energy metabolism, redox balance, neuronal function, and mice’s cognitive behavior. Concurrently, it has also been verified that an elevation in astrocytic aerobic glycolysis improves astrocytes’ support of neurons, thereby promoting neuronal survival and axon growth [77]. Aerobic glycolysis, i.e., the non-oxidative metabolism of glucose despite the presence of oxygen, has been considered a critical mechanism for neuroprotection, synaptic function, and rapid energy generation for cognitive task performance, etc., being associated with astrocytes’ glycolytic profiles [78] and often found to be decreased during the aging process [79]. In this line of investigation, recent findings identified an important association between the preservation of aerobic glycolysis and initial resilience to amyloid pathology, whilst, in contrast, the loss of this pattern correlated with cognitive impairment [80]. In this context, previous in vitro observations reveal that impaired astrocytic glycolysis results in the increased accumulation of Aβ within and around astrocytes and in the greater vulnerability of these cells to Aβ toxicity. In a similar manner, the impairment of glucose metabolism, mainly due to lower aerobic glycolysis, was linked with higher tau protein deposition in the vulnerable brain regions of pre-clinical and mildly symptomatic AD patients [81]. Overall, this evidence highlights the important impact that deregulated brain glucose metabolism can exert in the context of AD, suggesting new potential directions for AD treatment.
2.2. Mitochondrial Dynamics and Transport Alterations in AD
Mitochondria are described to be dynamic organelles with a striking ability to adapt their morphology, size, and location within the cell in order to maintain cellular requirements and proper function [82]. To successfully accomplish this task, the mitochondrial network relies on the balance between two opposing processes, fusion and fission, both regulated by several proteins belonging to the family of GTPases [83]. In a concise manner, under physiological conditions, mitochondrial fusion, regulated by mitofusins 1 and 2 (Mfn1 and Mfn2) located in the outer mitochondrial membrane and by optic dominant atrophy 1 (OPA1) present in the inner mitochondrial membrane, allows the cellular content and mitochondrial DNA to be combined to form a more resource-laden subcellular compartment. Unopposed fission, driven by the activity of the dynamin-related GTPase protein (Drp1) and mitochondrial fission 1 (Fis1), is essential to promote the conversion of damaged mitochondria into fragments, enabling their transport and selective degradation by mitophagy [84]. Altered mitochondria dynamics towards increased mitochondrial fission and consequent mitochondria fragmentation have been well established in AD [85]. This increased mitochondrial fragmentation was found to evolve as the disease progresses and was suggested to be related to the colocalization of Drp1 with Aβ monomers and oligomers in the postmortem brain tissue of AD patients [86]. Similarly, others reported increased mitochondrial fission in response to the increased production of Aβ in M17 cells and rat primary hippocampal neurons overexpressing APP [87]. Moreover, post-translational modifications of Drp1, such as increased S-nitrosylation [88] and phosphorylation at specific residues (e.g., ser616 and ser579) [89,90,91], were found to contribute to mitochondria dysfunction and AD pathology. More recently, a new mitochondrial phenotype characterized by elongated interconnected organelles was observed in the brain tissue of AD patients, animal models of AD, and wild-type aged mice [92]. This previously unknown morphology was designated as mitochondria-on-a-string (MOAS) and is believed to occur due to fission arrest at the final stages of the fission process as an attempt to preserve the remaining mitochondrial function in response to energetic stress [92]. Such a mitochondrial phenotype was later observed in the brains of OXYS rats, a model of sporadic AD [93], and in aged non-human primates [94]. Very recently, Panes and colleagues [95] reported that the MOAS morphology represented approximately 80% of the total mitochondria in the hippocampi of APP/PS1 mice. Moreover, they observed a significant interaction between MOAS and endoplasmic reticulum (ER) membranes, forming extensive mitochondria–ER contact sites (MERCS). Although the exact role of such an interaction remains unclear, the authors showed that reestablishing energy homeostasis reduced MOAS formation, MERCS coverage, and ER stress and enhanced the mitochondrial dynamics [95]. Alongside alterations in the CNS, previous studies refer to the existence of an abnormal mitochondrial distribution and morphology in fibroblasts from sporadic AD cases due to a reduction in Drp1 protein levels [96]. Additionally, others report slower mitochondria dynamics in sporadic AD fibroblasts due to the downregulation of both fission- and fusion-related proteins [97]. In further support, Drabik and colleagues [98] found that, in comparison with control patients, fibroblasts from sporadic AD individuals had lower levels of the fusion-regulating proteins and of Drp1, which were accompanied by a lower rate of mitochondrial fusion–fission. These alterations evoked the appearance of a less branched mitochondrial network characterized by less separated mitochondria that presented a smaller size. Although these findings suggest a less fragmented phenotype and oppose the alterations detected in brain tissue or in vitro models, they support the notion that mitochondrial dynamics have a prominent role in AD pathogenesis [99].
Intrinsically related to mitochondrial dynamics is the transport of mitochondria within neurons [100]. This motility is essential to provide energy in the form of ATP to neuronal synapses (anterograde transport driven by kinesin-1 (KIF5) motors) and to ensure the transport of aged or dysfunctional mitochondria back to the cell body, enabling its degradation (retrograde transport mediated by dynein motors) [101]. In AD, both in vivo and in vitro models demonstrate that the impairment of mitochondrial neuronal transport and the consequent energy deficit is one of the earliest events leading to the loss of axonal integrity and synaptic function [16,102,103,104]. For instance, it was reported that the lack of presynaptic mitochondria may underlie the selective regional loss of cortical synapses in the 5xFAD mouse model [105] and in AD brain tissue [106]. From a mechanistic point of view, earlier studies show that the combined overexpression of truncated tau with Aβ treatment in primary neurons promotes an impairment in mitochondrial transport by increasing the stationary mitochondrial population [107]. In further support, Vossel and collaborators found that the tau protein mediates Aβ-induced mitochondrial axonal transport impairment [108]. Using different experimental settings, the authors were able to verify that Aβ’s effects on mitochondrial transport involve glycogen synthase kinase 3beta (GSK3β) signaling [108], a kinase with proven involvement in Aβ-induced anterograde axonal transport deficits through its interaction with kinesin light chains [109]. More recently, it was demonstrated that the overexpression of tau P301L in primary neurons inhibited kinesin recruitment to mitochondria, thus inhibiting the anterograde mitochondria’s movement towards the axon terminals [110]. Notably, the retrograde transport of mitochondria was not affected by P301L [110]. In fact, several data suggest the increased vulnerability of the anterograde axonal mitochondrial transport in comparison with the opposite retrograde movement in AD-related conditions [103]. It was found that the expression content of KIF5A, a neuronal kinesin-1 isoform, and Miro1, an adaptor protein required for the axonal transport of mitochondria, was significantly decreased in the temporal lobes from postmortem AD brains, whilst dynein expression remained unchanged [111]. Similar findings were further observed in the neocortex in 5xFAD mice and in Aβ-treated primary neurons, whilst, in contrast, KIF5A overexpression in Aβ-treated neurons restored mitochondrial motility and attenuated neuronal oxidative stress and the loss of synaptic markers [111]. Consistently, others demonstrated that the knockdown of Miro1 or Milton, another adaptor protein, promoted the loss of axonal mitochondria in transgenic Drosophila expressing human tau [112]. As a result, the loss of mitochondria in axons was associated with the increased phosphorylation of the tau protein at AD-related residues, culminating in enhanced tau toxicity [112]. Following this line of investigation, Miro overexpression in AD transgenic flies was found to modulate mitochondria dynamics towards increased fusion, which correlated with increased ATP levels and decreased ROS levels [113], which supports the interlinked nature of mitochondrial quality control mechanisms.
2.3. Mitochondrial Biogenesis and Mitophagy
In concert with mitochondrial trafficking and dynamics, mitochondrial quality control relies on the balance between two complementary processes, the biogenesis of healthy and functional mitochondria and mitophagy, a selective mechanism where damaged or dysfunctional mitochondria are removed via autophagy [114]. In simple terms, mitochondria biogenesis is considered a self-renewal process that requires the participation of both mitochondrial and nuclear genomes, which interact in an organized multistep process [115]. The key components of mitochondrial biogenesis comprises peroxisome proliferator activated receptor-gamma (PPARα coactivator-1alpha (PGC-1α); two key nuclear transcription factors, nuclear respiratory factor 1 and 2 (NRF1 and NRF2); and the mitochondrial transcription factor A (Tfam). The sequential activation of each factor allows the integrative regulation of the nuclear-to-mitochondria proteins, thereby allowing the subunits of the respiratory chain to be encoded and contributing to the regulation of the antioxidant profile and mtDNA replication and transcription [116,117,118,119]. Previous and current studies demonstrate that compromised mitochondria biogenesis greatly contributes to mitochondrial impairment in AD [120,121]. Using postmortem brain samples from AD individuals and transgenic cells overexpressing APP mutations, Sheng and colleagues observed that the protein content of the critical regulators of mitochondrial biogenesis, PGC-1α, NRF1, NRF2, and TFAM, was significantly decreased in the AD milieu [120]. Additionally, the authors verified that the manipulation of PGC-1α expression in mutant M17 cells caused overt alterations in mitochondria function; if overexpressed, PGC-1α restored not only mitochondria biogenesis but also mitochondria ATP levels and complex IV activity, and opposite effects were noticed with PGC-1α silencing [120]. Concomitantly, data from human hippocampal brain samples of AD patients indicate that PGC-1α mRNA expression levels decrease as a function of the progression of clinical dementia [122]. Likewise, the PGC-1α protein content was inversely correlated with the Aβ peptide content and density of neuritic plaques [122]. In agreement with this, an in vivo study performed in 3xTg-AD mice at different ages revealed that the protein content and mRNA expression levels of mitochondrial biogenesis markers were already significantly decreased at 1 month of age, when mice did not present AD pathological hallmarks. Moreover, data obtained suggested that such earlier alterations were related to reduced mitochondria function at later ages, which prompted the authors to propose that disturbances in this quality control process precede the development and manifestation of AD pathology [121]. In further agreement, another study performed on the APP/PS1 mice model showed earlier changes affecting mitochondria biogenesis in the hippocampi of transgenic mice, prior to the formation of senile plaques and memory loss [123], thus reinforcing the idea that mitochondria malfunction, particularly in mitochondrial biogenesis, is an earlier event in the AD cascade. Accordingly, experimental evidence obtained from in vitro studies shows that Aβ1–42 peptide addition to neural stem cell cultures significantly compromises diverse mitochondrial parameters (e.g., ATP levels, mitochondrial ROS scavenging system, and mitochondrial dynamics), reduces mtDNA copy numbers, and downregulates the protein content of mitochondrial biogenesis regulators (PGC-1α, NRF1, and TFAM) [124]. In agreement with earlier works [125], the authors were able to observe that, when in culture, Aβ1–42 colocalizes with the mitochondrial network, suggesting a direct effect of Aβ on mitochondrial function. In addition, Dong and colleagues observed that hippocampal neurons exposed to Aβ25–35 experienced a significant decrease in the protein content of key mediators of mitochondria biogenesis and mtDNA levels, alongside the inhibition of the AMP-activated protein kinase (AMPK)–sirtuin 1 (SIRT1)–PGC-1α pathway [126]. Although the authors did not provide direct evidence of a causal link between AMPK–SIRT1–PGC-1α inhibition and Aβ25–35-induced mitochondrial biogenesis in hippocampal neurons, their findings support previous evidence about the important role that this signaling pathway has in rescuing mitochondrial defects by activating mitochondrial biogenesis in AD [126,127].
Alongside mitochondrial biogenesis, the other pillar of the mitochondrial quality control process relies on the identification of damaged or dysfunctional mitochondria for clearance [128]. This process, termed mitophagy, is a highly selective form of autophagy that begins with the recognition of depolarized and unhealthy mitochondria. This targeting step is regulated by PTEN-induced kinase 1 (PINK1), a protein kinase that accumulates and stabilizes on the OMM upon loss of mitochondrial membrane potential; this translocation leads to the phosphorylation of both ubiquitin and Parkin, an E3 ubiquitin ligase, at Ser(65), and Parkin recruitment to mitochondria [129,130]. Activated Parkin then initiates the ubiquitination of several substrates in the OMM, leading to the recruitment of autophagy adapter proteins such as p62, Optineurin (OPTN), and NDP52, which then initiates mitophagy. The clearance process proceeds with the formation of an autophagic membrane around the organelle (mitophagosome), which is later fused with a lysosome for degradation (mitophagolysosome). In this structure, lysosomal hydrolases digest mitochondria into small components that can then be recycled [131,132]. Besides PINK1–Parkin–ubiquitin-mediated mitophagy, the clearance of damaged mitochondria can also proceed with the involvement of receptor proteins. In mammalian cells, these receptors (e.g., NIP3-like protein X (NIX; also known as BNIP3L), FUN14 domain containing 1 (FUNDC1)) are transmembrane proteins attached to the OMM and must contain an LC3 motif, which allows the localization of the autophagosome with mitochondria [128]. Regardless of the mitophagy pathway, compelling studies have shown that this process plays a crucial role in regulating the machinery of the cell cycle, disposing of mitochondrial waste during mitosis, maintaining bioenergetic homeostasis, and preventing the accumulation of dysfunctional mitochondria that can lead to cellular degeneration. As such, mitophagy failure has been intrinsically related to a multitude of pathologies, being identified as a hallmark of age-related neurodegenerative disorders, including AD [132]. Indeed, different models of disease have consistently described the accumulation of damaged mitochondria as a fundamental event in AD pathogenesis [133,134]. In detail, immunohistochemical experiments allowed the observation of a significant reduction in the colocalization staining of TOMM20 (OMM protein) and LAMP2 (lysosomal protein), whilst electron microscopy images showed fewer mitophagy-like events in the postmortem hippocampal regions of AD patients relative to age-matched healthy controls, indicating mitophagy failure and the accumulation of compromised mitochondria in the hippocampi of AD patients [134]. An impairment in mitophagy was also observed in fibroblasts from sporadic AD individuals, as illustrated by a diminished number of autophagic vesicles (LC3) and inefficient Parkin translocation to the mitochondria, causing the accumulation of activated PINK1 [135]. Parallel in vivo and in vitro studies have contributed to better understanding the link between defective mitophagy and AD neuropathological hallmarks [135,136]. For instance, Cummins and colleagues found that tau protein expression directly inhibited mitophagy by aberrantly interacting with Parkin in cytosol and impeding its translocation to damaged mitochondria [136]. Notably, their experiments, which were performed in two different models, a neuroblastoma cell line and Caenorhabditis elegans neurons, showed that these effects occurred with both wild-type and frontotemporal dementia (P301L) mutant tau and did not require changes in mitochondrial membrane potential or the cytoskeleton [136]. Concurrently, others have described mitophagy deficits in HEK293 cells and primary hippocampal neurons overexpressing human tau, alongside reduced levels of PINK1/Parkin in the mitochondrial fraction [137]. However, differently from the previous study, these observations were reportedly linked to the allocation of the tau protein in the OMM, which caused increased mitochondrial membrane potential and blocked PINK1/Parkin mitochondrial location [137], highlighting the different possible mechanisms of tau-mediated mitochondrial toxicity. Besides the tau protein, two comparable studies highlighted a connection between the hippocampal accumulation of mutant APP and Aβ and reduced mitophagy in an animal model of AD and in an immortalized hippocampal transgenic cell line overexpressing mAPP [138,139]. More recently, Vaillant-Beuchot and co-workers uncovered that, independently of Aβ, APP-derived C-terminal fragment (APP-CTF) accumulation in mitochondria-enriched fractions can elicit basal mitophagy failure, apparently due to enhanced autophagy induction and the inefficient colocalization/targeting of mitochondria with/to lysosomes [140]. It was demonstrated that APP-CTF accumulation in the mitochondria of various AD study models caused the increased recruitment of PINK1/Parkin to mitochondria, the greater conversion of LC3, the accumulation of LC3-II, the non-degradation of the p62 substrate, enhanced levels of membrane and matrix mitochondrial proteins, and the deficient fusion of mitochondria with lysosomes [140]. Altogether, these findings point out the complex factors that can underlie mitophagy failure in AD pathology and pave the way for future interventions targeting this key process.
3. Mitochondria-Based Therapies for Alzheimer’s Disease
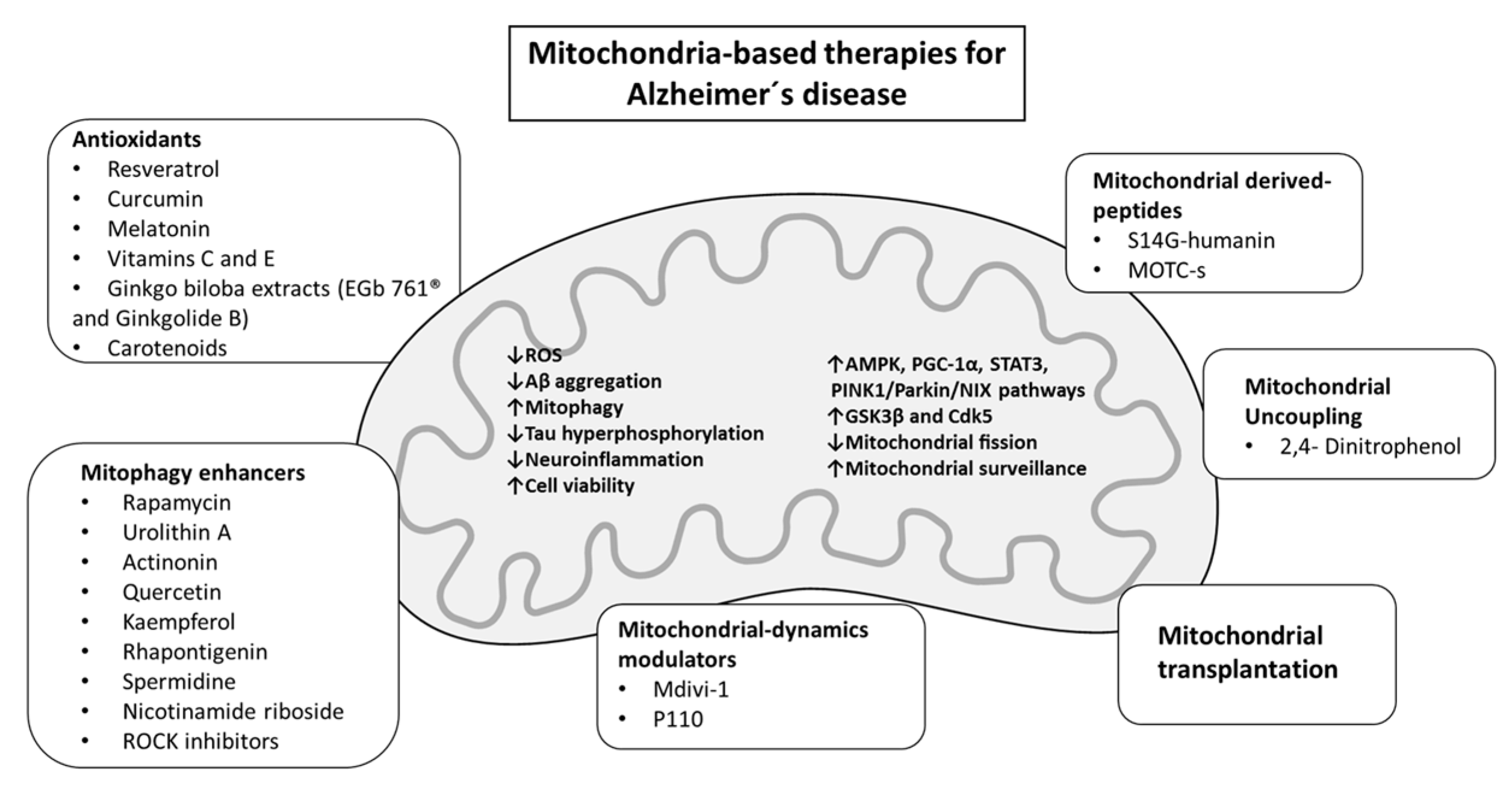
Due to the high complexity and multifactorial nature of the disease, AD investigation comprises a vast portfolio of attempted approaches that in general have been unsuccessful. With the goal of challenging the conventional scenario of targeting Aβ, research is now starting to focus on strategies directed at improving/restoring the multifaceted functions of mitochondria (Figure 2 and Table 1).
3.1. Mitochondrial Antioxidant Interventions
As aforementioned, an important hallmark of the AD pathology and one of the earliest changes in AD brains is the occurrence of a chronic imbalance between antioxidant defenses and the production and accumulation of ROS. Thus, it has been theorized that counterbalancing the levels of oxidative stress via antioxidant supplementation could be beneficial in the prevention or delay of the cascade of AD-induced neurodegeneration [141]. One well-known antioxidant is resveratrol, a polyphenol non-flavonoid present in several edible plants, such as grapes, peanuts, and berries, found to exert significant effects on mitochondria and to have immense therapeutic prospects, including in AD, being already evaluated in controlled clinical trials (NCT01504854) [142]. From a mechanistic perspective, earlier studies show that resveratrol is able to protect against Aβ-induced neurotoxicity through inhibiting Aβ aggregation [143], stimulating mitophagy [144], activating important metabolic and signaling pathways (e.g., AMPK, SIRT1, and PKC) [145,146,147], stimulating cellular antioxidant defenses [148], and reducing the inflammatory responses of APP/PS1 mice associated with Aβ-induced microglial activation [149]. However, despite the beneficial effects, the data obtained in clinical trials were not consistent and highlighted some limitations, mainly related to the low systemic bioavailability of the antioxidant [150], which can hamper its distribution to brain mitochondria. To address these issues, pre-clinical research is starting to investigate methods to improve resveratrol’s tissue targeting, bioavailability, and efficacy [147,151,152]. For instance, in a recent study, Han and colleagues conducted experiments using a nanostructured lipid particle system (NPs@RBCm) able to specifically deliver resveratrol to neuronal mitochondria. Their data showed that the resveratrol-loaded novel biomimetic nanosystem therapy improved the cognitive ability of APP/PS1 mice, decreased Aβ levels, mitigated brain inflammation, and improved mitochondrial oxidative stress in the hippocampi of AD mice [153]. Nevertheless, the literature highlights that resveratrol’s effects on mitochondria are highly dependent on the dose and redox status of the experimental model [154]. In detail, Gueguen and colleagues demonstrated that resveratrol directly binds to complex I of the respiratory chain, increasing its activity; when administered to aged mice, this event promoted an imbalance in the oxidative status towards increased oxidative stress [154]. In further agreement, others reinforce the idea that resveratrol-mediated mitochondrial effects strongly depend on the concentration administered [155].
Another natural product with antioxidant potential is curcumin, a phenolic compound extracted from the root of Curcuma longa [156]. Curcumin is described to have pleiotropic pharmacological effects and has been used in the treatment of several different pathologies (e.g., cardiac diseases, rheumatism, asthma, hepatic disorders, among others) [157]. In the context of AD, several lines of evidence have shown that curcumin has anti-amyloid properties, influencing Aβ aggregation [158] and cellular uptake [159], downregulating Aβ production [160], and reducing plaque deposition [161]. Others have also demonstrated that curcumin can interfere with the tau pathology, avoiding tau protein hyperphosphorylation and tangle formation [162], attenuating inflammatory processes [163], and improving mitochondrial function [164,165]. However, similarly to resveratrol, curcumin presents low bioavailability, which complicates its translation to clinical trials [166]. In fact, the first clinical studies evaluating the effects of curcumin in patients with AD did not show significant benefits with curcumin administration [167,168,169]. More recently, new curcumin formulations (Longvida® and Theracurmin) allowed for an improvement in the antioxidants´ bioavailability and achieved beneficial effects on attention and working memory tasks in a healthy older population [170] and in non-dementia adults that also presented decreases in Aβ and tau accumulation in brain regions modulating mood and memory [171]. In the same context, Gao and co-workers, using a neuronal mitochondria-targeted biomimetic engineered delivery nanosystem able to directly target curcumin to brain mitochondria, demonstrated positive outcomes upon intravenous administration in an AD mice model [172].
Concurrently with these observations, other considerable pre-clinical and clinical studies have demonstrated the potential of Ginkgo biloba extract EGb761® to treat age-related cognitive disorders, especially AD [173]. EGb 761®, an extract made from the dry leaves of Ginkgo biloba, was found to have strong free radical scavenging and mitochondrial-protective properties [174], mechanisms that seem to underlie the described protective effects, such as improved neuronal function [175] and neuroinflammation [176], a shift in APP metabolism towards non-amyloidogenic pathways [177], Aβ toxicity, and cognitive performance [178]. In this context, presently, the antioxidant and anti-inflammatory effects of EGb 761® are being evaluated in a Phase IV randomized, open-label clinical trial in MCI subjects to identify its possible clinical correlation with cognitive outcomes (NCT05594355) [179]. Complementing the existing data on to the neuroprotective effects of EGb 761®, recent studies have also investigated the effects of one of its main active components, Ginkgolide B, in the AD context. For example, evidence shows that Ginkgolide B is effective in reducing oxidative stress and protecting against Aβ toxicity [180], inactivating inflammasome formation [181], and modulating gut dysbiosis [182], allowing for improved cognitive function in animal models of AD [182,183]. Clinically, Ginkgolide B’s potential protective effects remain to be determined.
Alongside the potential of the abovementioned natural products, the latest findings suggest that melatonin can also have therapeutic effectiveness as a mitochondria-targeted antioxidant in AD [184]. Melatonin is widely known for its sleep–wake cycle regulatory properties. However, data confirm that melatonin exists inside mitochondria and modulates their function and acts as a free radical scavenger and antioxidant defense stimulator and as an anti-inflammatory agent at an organ and system level [185]. Earlier studies demonstrated that the melatonin concentration was decreased in the cerebrospinal fluid of AD patients, which seemed to parallel the pathophysiological progression of the disease [186,187]. Since then, considerable pre-clinical studies have supported the potentiality of melatonin as a treatment strategy to inhibit AD pathogenesis [188,189,190,191]. For instance, it was observed that melatonin supplementation in 5xFAD mice restored the mitophagy process, ameliorated mitochondrial energy metabolism, and inhibited Aβ formation, resulting in significant improvements in the cognitive function of transgenic mice [192]. Translating such outcomes to the clinic, the first therapeutic trials suggested that melatonin could be effective in ameliorating sleep efficacy and total sleep time in patients with dementia [193] and in improving mini-mental state examination (MMSE) scores in mild AD patients [194]. In a more recent clinical study, melatonin intake improved the sleep onset period and sleep cycle in early-to-moderate AD patients [195]. In the same context, the use of add-on prolonged-release melatonin showed positive effects on sleep maintenance in mild-to-moderate AD patients, especially in those with comorbid insomnia [196].
Beyond these novel mitochondrial-directed antioxidative supplements, considerable pre-clinical evidence exists demonstrating the beneficial effects of conventional antioxidants like vitamins C and E [197] and carotenoids (for a review, see [198]) on AD-related pathology. However, whist vitamins C and E’s clinical effectiveness remains inconclusive and more clinical studies must be conducted to demonstrate their efficacy in an AD context [199], carotenoid intake has been associated with a lower risk of MCI in the Chinese elderly population [200], with a higher cognitive score in the general population (NCT00272428) [201] and with a lower risk of AD neuropathology in a community-based cohort of older adults [202]. More recently, the administration of a combined micronutrient dietary supplement containing carotenoids, omega-3 fatty acids, and vitamin E achieved positive outcomes regarding the cognitive performance and mood of mild–moderate AD individuals [203], emphasizing the relevance of antioxidant therapy to AD.
3.2. Mitochondrial-Derived Peptides as Therapeutic Agents
The human mitochondrial DNA (mtDNA) has numerous short open reading frames (sORFs) that produce potential microproteins known as mitochondria-derived peptides (MDPs) [204]. The first MDP to be discovered was humanin. This 24-amino-acid peptide was first cloned from the resilient brain tissue of an AD patient by the group of Hashimoto et al., who identified it as an antagonizing factor against AD-associated neurotoxicity [205]. Later, Kelvin Yen and colleagues reported that humanin levels were decreased in the cerebrospinal fluid of AD patients, in comparison with controls, and a specific genetic variation in humanin sORF, m.2706A>G, was associated with more pronounced cognitive aging in African Americans [206]. Since then, multiple studies have been performed to assess the feasibility of humanin and its derivatives in the AD context. Using in vivo models of the disease, earlier evidence showed that the administration of S14G-HN, a more potent humanin derivative, ameliorated the cognitive performance of Aβ-injected [207] and 3xTg-AD [208] mice and reduced Aβ accumulation and neuroinflammation in the brains of middle-aged APP/PS1 mice [209]. Currently, it is known that the neuroprotective effects of humanin comprise its interaction with different cell surface receptors like the ciliary neurotrophic factor receptor (CNTFR) and the seven-transmembrane G-protein-coupled receptor formyl-peptide receptor-like-1 (FPRL1) [210]; the activation and modulation of the tyrosine kinase, JNK, AKT, ERK1/2, and STAT3 pathways as downstream effects [211]; and the prevention of mitochondria dysfunction and mitochondria-dependent apoptosis [212,213]. Complementing this evidence, it was also found that humanin can directly interact with Aβ, reducing its aggregation and oligomerization [214], and it is able to decrease the inactivation of phosphatase 2A that modulates tau pathology [206]. More recently, Han and colleagues showed that S14G-humanin-mediated protection in APP/PS1 transgenic mice entailed the regulation of autophagy, leading to the decreased accumulation of Aβ in the hippocampus and improved spatial learning and memory [215]. Beyond monotherapy, S14G-humanin has also been tested in a multifunctional hybrid peptide, HNSS, composed of S14G-humanin and the antioxidant peptide SS31, in 3xTg-AD mice [216]. It was observed that HNSS targeted mitochondria in the mouse brain, effectively rescuing mitochondria dysfunction via the PGC-1α and STAT3 pathways; neutralized AD-neurotoxic proteins, Aβ deposition, and tau hyperphosphorylation; and ameliorated memory defects and cholinergic neuronal damage in the 3xTg-AD mice [216]. Besides S14G-humanin, Colivelin has been identified as another potent humanin derivate able to improve the AD pathological damage and associated impairments in learning and memory that occurred in APP/PS1 mice [217] and to prevent Aβ25–35-induced deficits in spatial memory and synaptic plasticity in rats [218].
Alongside humanin, mitochondrial-derived peptides include MOTS-c (mitochondrial open reading frame of the 12S rRNA type-c) and six small humanin-like peptides (SHLP1-6) that differ in their expression patterns and ability to regulate cell viability and mitochondrial function [219]. Among these, SHLP2 was found to decline with age and, like humanin [206], was able to protect primary mouse cortical neurons from toxicity caused by Aβ1–42 [219]. Regarding MOTS-c, published evidence demonstrates that, under normal conditions, this 16-amino-acid peptide is found localized in mitochondria. However, in response to stress or exercise, it is rapidly translocated to the nucleus in a 5′-adenosine monophosphate-activated protein kinase (AMPK)/PGC1-α-dependent manner, where it promotes the expression of antioxidant and modifiable adaptive genes, improving redox homeostasis [220,221,222]. To further investigate the effect of MOTS-s on memory processes and the central inflammatory response under Aβ1–42 or LPS insults, an earlier study evaluated these outcomes using different routes of MOTS-c administration [223]. Their data showed that when administrated either by intrahippocampal CA1 microinjection or intracerebroventricular injection (icv), MOTS-s promoted the formation and consolidation of object and location recognition memory and attenuated Aβ1–42 or LPS-induced memory disability and the LPS-induced activation of microglia and astrocytes. In agreement with previous data, these effects were abolished when an inhibitor of the AMPK signaling pathway was administered prior to treatment. Importantly, the authors observed that MOTS-c did not cross the blood–brain barrier (BBB) and so, when administered peripherally, it was not able to exert neuroprotection. To circumvent such a drawback, a cell-penetrating MOTS analogue was designed and synthesized, demonstrating efficacy when intranasally injected in Aβ1–42 or LPS-icv-treated mice [223]. Notably, despite the favorable outcomes obtained with pre-clinical models, mitochondrial-derived peptides’ safety and efficacy have not been clinically tested in the context of AD. Presently, CB4211, a MOTS-c peptide analogue, is being evaluated in a Phase 1a double-blind, placebo-controlled clinical trial in obese subjects with non-alcoholic fatty liver disease (NAFLD) (NCT03998514).
3.3. Mitochondrial Dynamics and Mitophagy-Targeting Therapy in AD
As stated above, compelling research demonstrates that disrupted mitochondrial dynamics, biogenesis, and mitophagy are important and interconnected factors associated with the loss of neuronal cells’ homeostasis in AD. Such evidence opens up a new perspective on the beneficial effects that could stem from interventions that maintain and/or enhance mitophagy and mitochondrial structural and functional integrity in AD pathophysiology [224,225].
3.3.1. Mitochondria Dynamics Modulators
Under physiological conditions, the mitochondrial shape can quickly range from small round structures to elongated tubular networks due to the tightly coordinated action of mitochondrial fusion–fission proteins that rapidly react to challenges [84]. However, under pathological contexts, as is the case in AD, the mitochondrial dynamics’ equilibrium is disrupted, presenting a bias towards increased mitochondrial fragmentation due to increased levels of DRP1 [226]. In response to this, studies have been exploring the potentiality of targeting and inhibiting mitochondrial fission [227]. The most well-known compound is Mdivi-1. Discovered in the late 2000s as a potent inhibitor of DRP1 [228,229], Mdivi-1 treatment has been found to improve learning and memory processes, to rescue mitochondrial morphology, and to inhibit lipid peroxidation, BACE1 expression, and Aβ deposition in the brains of APP/PS1 mice [230]. Concurrently, others have reported the positive effects of Mdivi-1 in reverting Aβ-induced excessive mitochondrial fragmentation and synaptic toxicity in a mouse neuroblastoma (N2a) cell line [231].
Using the same rationale, P110 was developed as a new peptide able to inhibit mitochondrial fragmentation [232]. However, differently from Mdivi-1, P110 acts by disrupting the DRP1/Fis1 interaction, without affecting the integrity of the mitochondrial network in the basal state and the interaction of DRP1 with other adaptors [232]. In pre-clinical studies, P110 administration promoted neuroprotective outcomes such as the improvement of mitochondrial health, a reduction in Aβ levels, and the restoration of cognitive function in AD animal models [86]. Moreover, it was recently reported that P110, via the inhibition of DRP1/Fis1 interaction, was able to reduce the release of damaged mitochondria from neurotoxin-activated microglia and the consequent activation of the inflammatory response, protecting neuronal cells [233]. Nevertheless, despite the beneficial effects reported so far, there are no clinical data to document the efficacy of mitochondria dynamics modulators in AD.
3.3.2. Mitophagy Enhancers
One of the most well-known mitophagy-stimulating agents is rapamycin, an inhibitor of the mTOR pathway [234]. According to a study carried out on a mutant APP transgenic mouse model, the rapamycin-mediated inhibition of mTOR can ameliorate Aβ pathology and cognitive dysfunction and either slow down or halt the progression of AD [235]. Concurrently, others showed that rapamycin treatment could reverse AD-like pathology, mitochondrial abnormalities, and cognitive deficits via decreasing mTOR signaling in the hippocampus in a diabetic mice model [236]. Similarly, experimental evidence showed that rapamycin administration in young 3xTg-AD mice increased autophagy and decreased soluble Aβ and tau pathology [237]. Meanwhile, the same group observed that although rapamycin had beneficial effects when administered to young 3xTg-AD mice, in contrast, its administration in older mice had no effects on AD-like pathology and cognitive deficits [238]. In this regard, a very recent study by Shi and colleagues [239] demonstrated that rapamycin-induced mTOR pathway inhibition in 5XFAD mice significantly compromised microglia Aβ plaque clearance, corroborating the notion that rapamycin use in AD patients should be carefully analyzed [240]. At present, rapamycin’s safety and tolerability are being evaluated in a clinical trial in older adults with amnestic MCI (aMCI) and early-stage AD (NCT04629495).
Alongside rapamycin, research has been focused on evaluating the therapeutic potential of several pharmacological agents and natural supplements that could target mitophagy and improve mitochondrial function in the AD context [241]. In this regard, urolithin A (UA), a by-product of ellagitannin polyphenols occurring in pomegranate and walnuts, has demonstrated a significant ability to enhance cellular health by promoting mitochondrial function normalization and mitophagy and reducing detrimental inflammation in several pre-clinical models of aging and disease [242]. Regarding the clinical setting, the first clinical trial with UA showed that the regular oral consumption of UA in sedentary elderly individuals had a favorable safety profile (primary outcome) and led to an enhancement in mitochondrial and cellular molecular signs of fitness [243]. Corroborating data were obtained in a recent proof-of-concept investigation in which the benefits of the longer consumption of UA were assessed in middle-aged adults in terms of several physiological and biomarker endpoints [244]. Results obtained showed that UA promoted higher mitochondrial efficiency and mitophagy and reduced inflammation in the skeletal muscles of participants [244]. In cellular and animal models of AD, studies confirmed that UA lessened tau hyperphosphorylation in human neuronal cells and reversed memory impairments in transgenic nematodes and mice models [134,245]. Similar effects were observed with actinonin, an antibiotic able to trigger mitophagy in a PINK1/Parkin/NIX-dependent manner, and normalize mitochondria function and morphology in AD animal models [134]. Further, others report that UA’s beneficial effects can be enhanced when administered in combination with a green tea extract (epigallocatechin gallate, EGCG) [246]. Using the humanized Aβ knockin mice (hAbKI) as an experimental model, Kshirsagar and colleagues reported that the combined administration of UA+EGCG targeted multiple mechanisms of disease, such as inflammation, oxidative stress, synaptic loss, Aβ accumulation, cognitive deficits, and mitochondria homeostasis, thus suggesting the benefits of a combined approach instead of a monotherapy for AD [246]. In line with this, the combination of baicalein with memantine, one of the current U.S. FDA-approved drugs for AD, with an ability to enhance autophagic flux and the clearance of damaged mitochondria [247], showed enhanced therapeutic potential in Aβ-induced AD in Wistar rats [248] due to its ability to reach multi-therapeutic targets, demonstrating promising translational outcomes [248]. Previous studies show that flavonoids can have neuroprotective properties [249]. For instance, quercetin, a natural flavonoid that can be found in fruits, vegetables, berries, grapes, wine, various seeds, and nuts, has the ability to cross the BBB and modulate the expression and activity of PINK1/Parkin-mediated mitophagy and stimulate cellular defenses against oxidative stress, promoting positive effects in neurodegenerative contexts [250,251]. Compelling data from in vitro and in vivo models confirm the therapeutic efficacy of quercetin in modulating AD neuropathology in various experimental models of the disease [252,253,254]. More recently, Xie and co-workers [255], using combined advanced artificial intelligence with classical wet laboratory approaches, identified kaempferol, a natural flavonol, and rhapontigenin, a natural product and analog of resveratrol, as two potent mitophagy inducers that could be potential drug candidates for AD treatment. In their work, both compounds exhibited positive outcomes in one Aβ nematode model, two Tau nematode models, and the 3xTg-AD mouse model.
Spermidine is another versatile and multipurpose natural compound that can provide benefits for aging and age-related diseases like AD [256]. As shown in a recent pilot trial, spermidine supplementation for three months had a positive impact on memory performance in older adults at risk for the development of AD (NCT02755246) [257]. Recent evidence suggests that the maintenance of mitochondrial and autophagic function is the main mechanism of spermidine-mediated neuroprotection [258,259]. As noticed, spermidine was able to rescue the mitochondrial function of SH-SY5Y cells expressing a mutant form of the human tau protein (P301L tau mutation) [260]. As an autophagy inducer, spermidine modulates the mTOR and AMPK pathways [261] and activates Ataxia Telangiectasia mutated (ATM)-dependent PINK1/Parkin signaling [262].
Extensive studies using pre-clinical models of AD have demonstrated that increasing nicotinamide adenine dinucleotide (NAD+) levels through supplementation with NAD+ precursors (e.g., nicotinamide and its derivatives nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN)) stimulates mitophagy; mitigates Aβ, tau, and impaired energy metabolism; and modulates the inflammatory response, improving cognitive function [263,264,265,266]. Based on this, different clinical trials are currently being performed to evaluate the impact of NAD+ supplementation on brain function, cognition, oxidative stress, bioenergetics metabolism, and CSF pTau levels in MCI and AD patients (NCT03482167; NCT04430517; NCT04078178; NCT03061474). Following this rationale, the efficacy of combined metabolic activators (CMA) in AD patients is being evaluated in a Phase II clinical study (NCT04044131) [267]. CMA consists of the combination of L-serine, N-acetyl cysteine (NAC), nicotinamide riboside (NR), and L-carnitine tartrate (LCAT, the salt form of L-carnitine), and its oral administration to AD patients showed positive effects on cognitive function and markers of metabolic abnormalities, even in patients with severe AD [267].
Apart from the use of bioavailable neuronal mitophagy inducers, recent studies have identified a serine-threonine kinase (Rho-associated protein kinase 2; ROCK2) as a negative regulator of Parkin-dependent mitophagy [268]. ROCK2, which was firstly identified as a main regulator of the actin cytoskeleton and dendritic branches [269], is expressed in the cerebral cortex, hippocampal neurons, and cerebellar Purkinje cells, among other regions of the brain [270], in an age-dependent way. It was observed that brain sections from older human subjects possessed a higher number of ROCK2-positive cells as compared to younger controls [271]. In this regard, the application of ROCK inhibitors was found to improve synaptic processes and ameliorate rodents’ memory and cognitive behavior [272,273]. To investigate the applicability of ROCK inhibitors in AD, Hamano and colleagues [274] treated M1C cells (a human neuroblastoma cell line) overexpressing wild-type tau protein (4R0N) and a mouse model of tauopathy with three different ROCK inhibitors, H1152, Y-27632, and fasudil (HA-1077). The authors observed that ROCK inhibitors downregulated the activity of tau protein’s main kinases (GSK3β and Cdk5), reduced total tau levels, and upregulated autophagy [274]. In close agreement, ROCK knockdown in SH-SY5Y cells or its pharmacological inhibition in primary culture neurons contributed to a significant reduction in both tau mRNA and protein levels and to the decreased activation of mTOR, leading to the regulation of autophagy [275], thus supporting the clinical use of ROCK inhibitors as rational therapeutic approaches for AD and other tauopathies (NCT04734379).
Overall, these findings support the notion that strategies targeted at normalizing the balance between healthy and damaged mitochondria, by promoting the clearance of damaged and fragmented mitochondria, represent suitable candidates to provide neuronal protection in neurodegenerative diseases like AD.
3.4. Mitochondrial Uncoupling in AD
Alongside antioxidants, mitochondria are equipped with inner mitochondrial proteins able to counteract the production of free radicals [276]. These remarkable proteins, called mitochondrial uncoupling proteins (UCPs), exist in five isoforms depending on the organ in which they are expressed and serve the main purpose of transferring the protons produced during electron flow within the ETC against the concentration gradient. This, in turn, uncouples the process of electron transport from the synthesis of ATP through oxidative phosphorylation, allowing the regulation of the production of O2•− [277,278]. As reviewed elsewhere, whilst complete uncoupling would dissipate the cellular ATP pools, a small reduction in the mitochondrial membrane potential induced by mild uncoupling does not compromise the overall net ATP and has a significant effect in ameliorating ROS production [279]. Among the different isoforms, UCP2, UCP4, and UCP5 are the main UCPs detected in the brain [280]. Several studies suggest that brain UCPs play a key role against oxidative-stress-related brain pathologies, as is the case of AD, mainly through the modulation of ROS generation [281,282,283]. In fact, it has been shown that UCP2 protects hippocampal neurons against Aβ1–40-induced oxidative stress and toxicity [284]. Consistently, it was found that AD brain postmortem samples had lower expression levels of UCP2 and UCP4 when compared with samples from non-AD brains [285]. Furthermore, it has been reported that UCP2’s effects are not restricted to neurons but can also extend to other brain cells, such as microglia and astrocytes. For example, in LPS-stimulated microglia, UCP2 silencing caused an increase in M1 gene expression and an enhanced inflammatory response [286]. In line with this, Thangavel et al. demonstrated that AD brains presented increased expression of the proinflammatory protein glia maturation factor, which seemed to be related to the downregulation of both UCP2 and UCP4 expression and an increased proinflammatory environment in the parahippocampal gyrus in AD brains as compared to non-AD brains [285]. In this context, Rosenberg et al. demonstrated that the targeted overexpression of UCP4 in astrocytes’ mitochondria prevented hippocampal atrophy, neurons’ dendritic shrinkage, metabolic alterations, and spatial memory deterioration in a mouse model of AD [287]. These results suggest that modulating UCPs’ expression/activity in brain cells allows us to re-establish mitochondrial homeostasis and AD pathological outcomes. This may indicate that targeting the uncoupling mechanism is an approach worth investigating in AD treatment [288]. One well-known example of a pharmacological uncoupler able to mimic UCP-mediated brain effects is 2, 4-dinitrophenol (DNP). Described as a fast weight reduction agent, DNP is a chemical uncoupler that allows protons’ re-entrance to the mitochondrial matrix, thus interfering with ATP synthesis and mitochondrial membrane potential [289]. Compelling data suggest that DNP-mediated effects are dose-dependent [289]. This means that when used at high concentrations, DNP can evoke serious side effects, such as agranulocytosis, hyperthermia, skin reactions, and cataracts; in contrast, when used at low concentrations, it induces mild mitochondrial uncoupling that is beneficial in terms of reducing oxidative neuronal damage in different pathological conditions, including AD [290,291]. The first evidence came from the group of De Felice, who observed that DNP, at low concentrations, protected neurons against Aβ toxicity [292] and promoted neuritogenesis and neuronal differentiation in cortical and hippocampal neuronal cultures [293]. In this context, later studies showed that DNP was able to decrease the intracellular accumulation of APP in an immortalized cell line derived from the cerebral cortex of an animal model of Down’s syndrome, which presented a pathophysiology closely related to AD [294]. More recently, others observed that the peripheral administration of DNP resulted in the complex adaptive remodeling of the molecular pathways that regulate neuronal stress responses and synaptic plasticity [295]. In line with this, it was shown that DNP administration in APP/PS1 mice increased hippocampus-dependent spatial learning and memory [289]. Collectively, these data suggest the importance of examining the clinical potential of targeting uncoupling proteins and/or uncoupling agents as a disease-modifying strategy in AD pathophysiology.

{kind=link}
{kind=link}
Table 1.
Overview of mitochondria-based therapies for Alzheimer’s disease.
| Mitochondria-Based Therapeutics | Targets and Mechanism of Action | Benefits and Limitations | References | |
|---|---|---|---|---|
| Antioxidants | Resveratrol |
| Benefits (pre-clinical):
| [142,143,144,145,146,147,148,149,150,154,155] |
| Curcumin |
| Benefits (pre-clinical):
| [158,159,160,161,162,163,164,165,166,170,171] | |
| Melatonin |
| Benefits:
| [185,190,191,192,193,194,195,196] | |
| Vitamins C and E |
| Benefits:
| [197,199] | |
| a | Carotenoids |
| Benefits (clinical):
| [200,201,202] |
| Ginkgo biloba extracts (EGb761® and Ginkgolide B) |
| Benefits (pre-clinical):
| [175,176,177,178,179,180,181,182,183] | |
| Mitochondrial-derived peptides | S14G-humanin |
| Benefits (pre-clinical):
| [206,211,214,215] |
| Colivelin |
| Benefits (pre-clinical):
| [217,218] | |
| MOTC-s |
| Benefits (pre-clinical):
| [220,221,222,223] | |
| Mitochondrial dynamics modulators | Mdivi-1 |
| Benefits (pre-clinical):
| [230,231] |
| P110 |
| Benefits (pre-clinical):
| [85,233] | |
| Mitophagy enhancers | Rapamycin |
| Benefits (pre-clinical):
| [235,236,238,239] |
| Urolithin A |
| Benefits (pre-clinical):
| [134,242,243,244,245] | |
| Actinonin |
| Benefits (pre-clinical):
| [134] | |
| Memantine |
| Benefits (pre-clinical):
| [247] | |
| Quercetin |
| Benefits (pre-clinical):
| [250,251,252,253,254] | |
| Kaempferol and rhapontigenin |
| Benefits (pre-clinical):
| [255] | |
| Spermidine |
| Benefits (pre-clinical):
| [257,260,261,262] | |
| Nicotinamide adenine dinucleotide (NAD+) percursors |
| Benefits (pre-clinical):
| [263,264,265,266] | |
| ROCK inhibitors |
| Benefits (pre-clinical):
| [274,275] | |
| Combined metabolic activators (CMA) |
| Benefits (clinical):
| [267] | |
| Mitochondrial uncoupling | Mitochondrial uncoupling proteins(UCP2, UCP4) |
| Benefits (pre-clinical):
| [279,281,282,283,284,286,287] |
| 2,4-Dinitrophenol |
| Benefits (pre-clinical):
| [280,281,282,283,284,285] | |
4. Mitochondrial Transplantation: Could the Transfer of Healthy Mitochondria Be the Solution?
As further reported herein, a great deal of effort has been dedicated to achieving a successful mitochondria-based therapy able to tackle AD. However, to date, only a few approaches have been able to yield satisfactory results, revealing limited translatability. Thus, alongside therapeutic interventions, an alternative perspective would be to look to mitochondria as therapeutics themselves [296]. Based on the knowledge that mitochondria are extremely dynamic organelles that can move between different cells (for a review, see [297,298]), the transfer of functional mitochondria to replace disabled mitochondria is starting to be considered a feasible option as a basis for cell therapy in a plethora of neural and non-neural disorders [299,300,301,302]. One of the earliest pieces of evidence of intercellular mitochondrial transfer came from an in vitro study in which the authors observed that the co-culture of mtDNA mutated and depleted cells (A549 ρ° cells) with mesenchymal stem cells allowed them to rescue the mitochondrial function of A549 ρ° cells [298]. Subsequent studies provided evidence for the potential of transferring healthy mitochondria to cells with mitochondrial defects [303,304,305,306,307]. Nowadays, this process, referred to as mitochondrial transplantation, is commonly achieved by isolating viable mitochondria from healthy tissue or cell lines and transferring them into the compromised recipient, e.g., an organ, in vivo model, or in vitro system [296]. Once inside the diseased recipient cells, exogenous organelles are believed to rescue the cells’ viability and, consequently, to avert disease onset and/or progression [308]. As a proof-of-concept trial, the first clinical application of mitotherapy was conducted in pediatric patients who suffered from myocardial ischemia–reperfusion injury [309]. In this study, a single epicardial injection of freshly isolated autologous mitochondria from skeletal muscle promoted patients’ myocardial recovery and improved ventricular function within several days of treatment. Importantly, as the authors noted, mitochondria autotransplantation did not cause any adverse heart-related side effects or markers of a systemic inflammatory response [309]. Since this pioneering work, other studies have been registered at ClinicalTrials.gov to evaluate the efficacy of this unique therapeutic, mainly in ischemia-related conditions (NCT03639506, NCT02851758, NCT05669144, and NCT04998357).
Within the brain, previous studies demonstrate that mitochondria transfer can occur in a cell-to-cell manner between co-incubated astrocytes and neurons, as an endogenous strategy to safeguard neuronal survival after neurological damage [310,311]. More recently, others have observed that, alongside functional mitochondria, astrocytes also secrete the mitochondrial-genome-encoded small bioactive peptide humanin that is transported to microglia, reducing proinflammatory outcomes [312]. Notably, the efficacy of mitochondria transfer is intrinsically related to the age of the donor tissue/cell, as young mitochondria show better performance than older ones [313]. Corroborating these findings, mitotherapy with healthy young mitochondria obtained from the livers of young mice showed positive results in restoring the hippocampal mitochondrial bioenergetics of aged mice [314]. In a different setting, others demonstrated that the intravenous injection of hippocampus-derived isolated mitochondria in a status epilepticus (SE) disease model contributed to ameliorating hippocampal damage following SE and improved cognitive and mood dysfunction [315]. Their results showed that peripherally injected exogenous mitochondria could cross the BBB and revert SE-induced hippocampal injury by not only lessening SE-induced ROS production but also affecting the expression and metabolic pathways of important metabolites [315].
In the field of AD, mitochondrial transplantation as a therapeutic strategy is still in its infancy, but some recent pre-clinical research in experimental models of disease reports positive results. Using an in vitro model of disease established using okadaic acid (OA)-treated SH-SY5Y cells, Zhang and colleagues recently showed that a human-umbilical-cord-derived mesenchymal-stem-cell-conditioned medium (MSC-CM) exerted protective effects on AD through extracellular vesicles’ (EV) mitochondrial transfer [316]. The authors observed that treatment with MSC-CM significantly improved cell viability, apoptosis, and mitochondrial function in OA-treated SH-SY5Y cells via the donation of hucMSCs’ healthy mitochondria [316]. An additional study in an AD mice model achieved by the intracerebroventricular injection of the Aβ peptide showed that the intravenous injection of HeLa-cell-derived freshly isolated mitochondria contributed to ameliorating mice’s cognitive performance, hippocampal neuronal loss, and gliosis and reestablished mitochondrial functional outcomes [317]. More recently, a study conducted by Bobkova and collaborators verified that isolated mitochondria could be intranasally administered, leading to the recovery of spatial memory in olfactory bulbectomized (OBX) mice with AD-like degeneration [318]. Their data showed that functional isolated mitochondria from the brains of control mice, when intranasally injected, were detected in the hippocampus, neocortex, and olfactory bulbs in treated mice in a dose-dependent manner [318]. In a comparable manner, the same approach was evaluated in a mouse model of cisplatin-induced cognitive deficits. As the authors observed, nasally administered mitochondria isolated from human MSCs were able to reach brain meninges within 30 min of delivery, gaining access to the brain and reversing cisplatin-induced synaptic loss and hippocampal synaptosomal mitochondrial abnormalities [301], thus confirming the efficacy of mitochondria in crossing the BBB and restoring the homeostasis of brain cells. Overall, although some aspects of the efficacy, delivery, and safety concerns regarding mitochondria transplantation still need to be further explored [296], the remarkable outcomes reported in the literature strongly highlight the therapeutic prospects of mitotherapy in AD.
5. Final Remarks
Despite decades of investigation, the understanding of the AD pathophysiology remains incomplete. In this scenario, mitochondria, or the decline in mitochondrial function, appears as a common denominator in the majority of dedicated studies. As the world approaches a new milestone in the number of diagnosed AD cases, and developing new medicines is a challenging and slow process, the consideration of mitochondria as a target holds great promise in AD therapy. It is important to note, though, that the development/refinement of the presently reviewed mitochondria-based therapeutics will require a multidisciplinary approach, including the identification of specific mitochondrial targets, a deeper understanding of how the tested substances affect the interactions between mitochondria and AD hallmarks (e.g., Aβ and tau) and consequently their toxicity, the optimization of drug delivery, and the development of new imaging techniques to monitor the effectiveness of the therapy in order achieve its full potential [319]. The early occurrence of mitochondrial impairment in AD pathophysiology, the late diagnosis of the disease, and the existence of other comorbidities (e.g., diabetes) also stand as important factors contributing to the complexity of the exploration of mitochondrial targeting approaches in AD. In this regard, besides the therapeutics/strategies mentioned in this manuscript, other pharmacological agents have been considered as feasible therapeutic options for AD. For instance, metformin, which is a commonly prescribed anti-diabetic medication, has been suggested to improve cognitive performance in AD individuals [320] and to rescue human neural stem cells from Aβ-mediated mitochondrial deficiency [321]. Additionally, some compounds, like tricaprilin, intended to regulate metabolism/bioenergetics, have also demonstrated efficacy in the management of mild to moderate AD [322]. Being currently tested in clinical trials (NCT00142805), tricaprilin seems to serve as an alternative fuel source for glucose, inducing ketosis and improving patients’ metabolism [322].
Thus, despite being a complex field, the positive outcomes of pre-clinical studies and of some clinical trials suggest that the development of mitochondrial-targeted therapeutics for AD is a promising research area that has the potential to lay the groundwork for new treatment options for this debilitating disease. Finally, considering the importance of mitochondria, it is imperative to understand that, regardless of the targeted disease mechanism, if the drug candidate compromises mitochondrial function, its possible clinical application could be at risk.
Author Contributions
Conceptualization, S.C.; writing—original draft preparation, T.S. and S.C.; writing—review and editing, S.C. and P.I.M. All authors have read and agreed to the published version of the manuscript.
Funding
The authors’ work was supported by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Program, the COMPETE 2020—Operational Programme for Competitiveness, and national funds from the FCT—Foundation for Science and Technology under the project PEst-C/SAU/LA0001/2013-2014 and strategic projects UIDB/04539/2020, UIDP/04539/2020, and LA/P/0058/2020. Cardoso, S. has a Post-Doctoral Researcher Contract DL57/2016 (#SFRH/BPD/95770/2013) from the FCT.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-D.-C.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the Path to 2025: Understanding the Alzheimer’s Disease Continuum. Alzheimer’s Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s Disease Drug-Development Pipeline: 2023. Alzheimer’s Dement. 2023, 6, 37. [Google Scholar] [CrossRef]
- Vradenburg, G. A Pivotal Moment in Alzheimer’s Disease and Dementia: How Global Unity of Purpose and Action Can Beat the Disease by 2025. Expert Rev. Neurother. 2015, 15, 73–82. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug Development in Alzheimer’s Disease: The Path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef]
- Zhang, M.; Ganz, A.B.; Rohde, S.; Rozemuller, A.J.M.; Netherlands Brain Bank; Reinders, M.J.T.; Scheltens, P.; Hulsman, M.; Hoozemans, J.J.M.; Holstege, H. Resilience and Resistance to Alzheimer’s Disease-Associated Neuropathological Substrates in Centenarians: An Age-Continuous Perspective. Neurology 2022. preprint. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The Role of Mitochondrial Function and Cellular Bioenergetics in Ageing and Disease. Br. J. Dermatol. 2013, 169 (Suppl. 2), 1–8. [Google Scholar] [CrossRef]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz E Silva, O. Mitochondria, Energy, and Metabolism in Neuronal Health and Disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s Disease Is Associated with Reduced Expression of Energy Metabolism Genes in Posterior Cingulate Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [PubMed]
- Abyadeh, M.; Gupta, V.; Chitranshi, N.; Gupta, V.; Wu, Y.; Saks, D.; Wander Wall, R.; Fitzhenry, M.J.; Basavarajappa, D.; You, Y.; et al. Mitochondrial Dysfunction in Alzheimer’s Disease—A Proteomics Perspective. Expert Rev. Proteom. 2021, 18, 295–304. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial Dysfunction, Oxidative Stress, Neuroinflammation, and Metabolic Alterations in the Progression of Alzheimer’s Disease: A Meta-Analysis of in Vivo Magnetic Resonance Spectroscopy Studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Lu, Y.; Siedlak, S.L.; Fujioka, H.; Feng, H.; Perry, G.; Zhu, X. Damaged Mitochondria Coincide with Presynaptic Vesicle Loss and Abnormalities in Alzheimer’s Disease Brain. Acta Neuropathol. Commun. 2023, 11, 54. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial Bioenergetic Deficit Precedes Alzheimer’s Pathology in Female Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Fong, S.; Teo, E.; Ng, L.F.; Chen, C.-B.; Lakshmanan, L.N.; Tsoi, S.Y.; Moore, P.K.; Inoue, T.; Halliwell, B.; Gruber, J. Energy Crisis Precedes Global Metabolic Failure in a Novel Caenorhabditis Elegans Alzheimer Disease Model. Sci. Rep. 2016, 6, 33781. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W.E. Mitochondrial Dysfunction: An Early Event in Alzheimer Pathology Accumulates with Age in AD Transgenic Mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Saraiva, A.A.; Borges, M.M.; Madeira, M.D.; Tavares, M.A.; Paula-Barbosa, M.M. Mitochondrial Abnormalities in Cortical Dendrites from Patients with Alzheimer’s Disease. J. Submicrosc. Cytol. 1985, 17, 459–464. [Google Scholar]
- Paula-Barbosa, M.M.; Cardoso, R.M.; Guimaraes, M.L.; Cruz, C. Dendritic Degeneration and Regrowth in the Cerebral Cortex of Patients with Alzheimer’s Disease. J. Neurol. Sci. 1980, 45, 129–134. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, J.; Tan, M.; Albers, K.M.; Jia, J. Mitochondrial Fission Proteins in Peripheral Blood Lymphocytes Are Potential Biomarkers for Alzheimer’s Disease. Eur. J. Neurol. 2012, 19, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ponce, D.P.; Osorio-Fuentealba, C.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Bioenergetics Is Altered in Fibroblasts from Patients with Sporadic Alzheimer’s Disease. Front. Neurosci. 2017, 11, 553. [Google Scholar] [CrossRef]
- Mahapatra, G.; Gao, Z.; Bateman, J.R.; Lockhart, S.N.; Bergstrom, J.; DeWitt, A.R.; Piloso, J.E.; Kramer, P.A.; Gonzalez-Armenta, J.L.; Amick, K.A.; et al. Blood-Based Bioenergetic Profiling Reveals Differences in Mitochondrial Function Associated with Cognitive Performance and Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, P.; Martín-Aragón, S.; Benedí, J.; Susín, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Peripheral Levels of Glutathione and Protein Oxidation as Markers in the Development of Alzheimer’s Disease from Mild Cognitive Impairment. Free Radic. Res. 2008, 42, 162–170. [Google Scholar] [CrossRef]
- Padurariu, M.; Ciobica, A.; Hritcu, L.; Stoica, B.; Bild, W.; Stefanescu, C. Changes of Some Oxidative Stress Markers in the Serum of Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Neurosci. Lett. 2010, 469, 6–10. [Google Scholar] [CrossRef]
- Torres, L.L.; Quaglio, N.B.; de Souza, G.T.; Garcia, R.T.; Dati, L.M.M.; Moreira, W.L.; Loureiro, A.P.d.M.; de Souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral Oxidative Stress Biomarkers in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef]
- López, N.; Tormo, C.; De Blas, I.; Llinares, I.; Alom, J. Oxidative Stress in Alzheimer’s Disease and Mild Cognitive Impairment with High Sensitivity and Specificity. J. Alzheimer’s Dis. 2013, 33, 823–829. [Google Scholar] [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox Metabolism: ROS as Specific Molecular Regulators of Cell Signaling and Function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 Reasons Why the Brain Is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B. Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M. Brain Regional Correspondence between Alzheimer’s Disease Histopathology and Biomarkers of Protein Oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Pierce, W.M., Jr.; Turner, D.M.; Markesbery, W.R.; Butterfield, A.D. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J. Cell. Mol. Med. 2009, 13, 2019–2029. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-Hydroxynonenal, a Product of Lipid Peroxidation, Is Increased in the Brain in Alzheimer’s Disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the Lipid Peroxidation Product, HNE, in the Pathogenesis and Progression of Alzheimer’s Disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased Peroxidation and Reduced Antioxidant Enzyme Activity in Alzheimer’s Disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Fang, D.; Zhang, Z.; Li, H.; Yu, Q.; Douglas, J.T.; Bratasz, A.; Kuppusamy, P.; Yan, S.S. Increased Electron Paramagnetic Resonance Signal Correlates with Mitochondrial Dysfunction and Oxidative Stress in an Alzheimer’s Disease Mouse Brain. J. Alzheimer’s Dis. 2016, 51, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Tamaoki, T.; Motohashi, N.; Nakamura, M.; McKeel, D.W.; Tabaton, M.; Lee, H.-G.; Smith, M.A.; Perry, G.; Zhu, X. The Earliest Stage of Cognitive Impairment in Transition from Normal Aging to Alzheimer Disease Is Marked by Prominent RNA Oxidation in Vulnerable Neurons. J. Neuropathol. Exp. Neurol. 2012, 71, 233–241. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Oxidatively Modified RNA in Mild Cognitive Impairment. Neurobiol. Dis. 2008, 29, 169–175. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial Abnormalities in Alzheimer’s Disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-Derived Reactive Oxygen Species Lead to Enhanced Amyloid Beta Formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef]
- Wilkins, H.M. Interactions between Amyloid, Amyloid Precursor Protein, and Mitochondria. Biochem. Soc. Trans. 2023, 51, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Mamcarz, M.; Zhu, Y.; Buzzeo, R.; Tan, J.; Arendash, G.W.; Bradshaw, P.C. Mitochondrial Amyloid-Beta Levels Are Associated with the Extent of Mitochondrial Dysfunction in Different Brain Regions and the Degree of Cognitive Impairment in Alzheimer’s Transgenic Mice. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S535–S550. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The Amyloid Beta-Peptide Is Imported into Mitochondria via the TOM Import Machinery and Localized to Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Alikhani, N.; Guo, L.; Yan, S.; Du, H.; Pinho, C.M.; Chen, J.X.; Glaser, E.; Yan, S.S. Decreased Proteolytic Activity of the Mitochondrial Amyloid-β Degrading Enzyme, PreP Peptidasome, in Alzheimer’s Disease Brain Mitochondria. J. Alzheimer’s Dis. 2011, 27, 75–87. [Google Scholar] [CrossRef]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.-C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD Enhances Abeta-Induced Cell Stress via Mitochondrial Dysfunction. FASEB J. 2005, 19, 597–598. [Google Scholar] [CrossRef]
- Fang, D.; Wang, Y.; Zhang, Z.; Du, H.; Yan, S.; Sun, Q.; Zhong, C.; Wu, L.; Vangavaragu, J.R.; Yan, S.; et al. Increased Neuronal PreP Activity Reduces Aβ Accumulation, Attenuates Neuroinflammation and Improves Mitochondrial and Synaptic Function in Alzheimer Disease’s Mouse Model. Hum. Mol. Genet. 2015, 24, 5198–5210. [Google Scholar] [CrossRef]
- Dou, Y.; Tan, Y. Presequence Protease Reverses Mitochondria-Specific Amyloid-β-Induced Mitophagy to Protect Mitochondria. FASEB J. 2023, 37, e22890. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Troutwine, B.R.; Menta, B.W.; Manley, S.J.; Strope, T.A.; Lysaker, C.R.; Swerdlow, R.H. Mitochondrial Membrane Potential Influences Amyloid-β Protein Precursor Localization and Amyloid-β Secretion. J. Alzheimer’s Dis. 2022, 85, 381–394. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging. 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain. 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.G.M.; Koopman, W.J.H.; Grefte, S. Interactions between Mitochondrial Reactive Oxygen Species and Cellular Glucose Metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid Beta-Peptide Impairs Glucose Transport in Hippocampal and Cortical Neurons: Involvement of Membrane Lipid Peroxidation. J. Neurosci. 1997, 17, 1046–1054. [Google Scholar] [CrossRef]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of Nitrated Proteins in Alzheimer’s Disease Brain Using a Redox Proteomics Approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef]
- Reed, T.T.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic Identification of HNE-Bound Proteins in Early Alzheimer Disease: Insights into the Role of Lipid Peroxidation in the Progression of AD. Brain Res. 2009, 1274, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox Proteomics Identification of 4-Hydroxynonenal-Modified Brain Proteins in Alzheimer’s Disease: Role of Lipid Peroxidation in Alzheimer’s Disease Pathogenesis. Proteom. Clin. Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Redox Proteomics and Amyloid β-Peptide: Insights into Alzheimer Disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET Changes in Brain Glucose Metabolism from Normal Cognition to Pathologically Verified Alzheimer’s Disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal Hypometabolism Predicts Cognitive Decline from Normal Aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Brain Glucose Metabolism in the Early and Specific Diagnosis of Alzheimer’s Disease. FDG-PET Studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional Brain Abnormalities in Young Adults at Genetic Risk for Late-Onset Alzheimer’s Dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for Brain Glucose Dysregulation in Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In Vivo Mitochondrial and Glycolytic Impairments in Patients with Alzheimer Disease. Neurology 2020, 94, e1592–e1604. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Skotte, N.H.; Christensen, S.K.; Polli, F.S.; Shabani, M.; Markussen, K.H.; Haukedal, H.; Westi, E.W.; Diaz-delCastillo, M.; Sun, R.C.; et al. Hippocampal Disruptions of Synaptic and Astrocyte Metabolism Are Primary Events of Early Amyloid Pathology in the 5xFAD Mouse Model of Alzheimer’s Disease. Cell Death Dis. 2021, 12, 954. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient Astrocyte Metabolism Impairs Glutamine Synthesis and Neurotransmitter Homeostasis in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef]
- Halim, N.D.; Mcfate, T.; Mohyeldin, A.; Okagaki, P.; Korotchkina, L.G.; Patel, M.S.; Jeoung, N.H.; Harris, R.A.; Schell, M.J.; Verma, A. Phosphorylation Status of Pyruvate Dehydrogenase Distinguishes Metabolic Phenotypes of Cultured Rat Brain Astrocytes and Neurons. Glia 2010, 58, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Laughton, J.D.; Bittar, P.; Charnay, Y.; Pellerin, L.; Kovari, E.; Magistretti, P.J.; Bouras, C. Metabolic Compartmentalization in the Human Cortex and Hippocampus: Evidence for a Cell- and Region-Specific Localization of Lactate Dehydrogenase 5 and Pyruvate Dehydrogenase. BMC Neurosci. 2007, 8, 35. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef]
- Bouzier-Sore, A.-K.; Merle, M.; Magistretti, P.J.; Pellerin, L. Feeding Active Neurons: (Re)Emergence of a Nursing Role for Astrocytes. J. Physiol. Paris 2002, 96, 273–282. [Google Scholar] [CrossRef]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic Mitochondrial ROS Modulate Brain Metabolism and Mouse Behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Gutierrez, C.; Bonora, N.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Bates, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Abrogating Mitochondrial ROS in Neurons or Astrocytes Reveals Cell-Specific Impact on Mouse Behaviour. Redox Biol. 2021, 41, 101917. [Google Scholar] [CrossRef]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 Improves the Supportive Ability of Astrocytes to Neurons by Promoting Aerobic Glycolysis in Alzheimer’s Disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.-A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e8. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.-S.; Morris, J.C.; Raichle, M.E. Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 2017, 26, 353–360.e3. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Blazey, T.; Metcalf, N.V.; McAvoy, M.P.; Strain, J.F.; Rahmani, M.; Durbin, T.J.; Xiong, C.; Benzinger, T.L.-S.; Morris, J.C.; et al. Brain Aerobic Glycolysis and Resilience in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2212256120. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Gordon, B.A.; Goyal, M.S.; Su, Y.; Blazey, T.M.; Durbin, T.J.; Couture, L.E.; Christensen, J.J.; Jafri, H.; Morris, J.C.; et al. Aerobic Glycolysis and Tau Deposition in Preclinical Alzheimer’s Disease. Neurobiol. Aging 2018, 67, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal Interaction between the Mitochondrial Fission Protein Drp1 and Hyperphosphorylated Tau in Alzheimer’s Disease Neurons: Implications for Mitochondrial Dysfunction and Neuronal Damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 Interaction Mediates Mitochondrial Dysfunction, Bioenergetic Failure and Cognitive Decline in Alzheimer’s Disease. Oncotarget 2018, 9, 6128–6143. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-Beta Overproduction Causes Abnormal Mitochondrial Dynamics via Differential Modulation of Mitochondrial Fission/Fusion Proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates Beta-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Espino de la Fuente-Muñoz, C.; Rosas-Lemus, M.; Moreno-Castilla, P.; Bermúdez-Rattoni, F.; Uribe-Carvajal, S.; Arias, C. Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 8727. [Google Scholar] [CrossRef]
- Guo, M.-Y.; Shang, L.; Hu, Y.-Y.; Jiang, L.-P.; Wan, Y.-Y.; Zhou, Q.-Q.; Zhang, K.; Liao, H.-F.; Yi, J.-L.; Han, X.-J. The Role of Cdk5-Mediated Drp1 Phosphorylation in Aβ1-42 Induced Mitochondrial Fission and Neuronal Apoptosis. J. Cell. Biochem. 2018, 119, 4815–4825. [Google Scholar] [CrossRef]
- Xu, D.; Yang, P.; Yang, Z.-J.; Li, Q.-G.; Ouyang, Y.-T.; Yu, T.; Shangguan, J.-H.; Wan, Y.-Y.; Jiang, L.-P.; Qu, X.-H.; et al. Blockage of Drp1 Phosphorylation at Ser579 Protects Neurons against Aβ1-42-induced Degeneration. Mol. Med. Rep. 2021, 24, 657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered Brain Energetics Induces Mitochondrial Fission Arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef] [PubMed]
- Tyumentsev, M.A.; Stefanova, N.A.; Kiseleva, E.V.; Kolosova, N.G. Mitochondria with Morphology Characteristic for Alzheimer’s Disease Patients Are Found in the Brain of OXYS Rats. Biochem. Biokhimiia 2018, 83, 1083–1088. [Google Scholar] [CrossRef]
- Morozov, Y.M.; Datta, D.; Paspalas, C.D.; Arnsten, A.F.T. Ultrastructural Evidence for Impaired Mitochondrial Fission in the Aged Rhesus Monkey Dorsolateral Prefrontal Cortex. Neurobiol. Aging 2017, 51, 9–18. [Google Scholar] [CrossRef]
- Panes, J.; Nguyen, T.K.O.; Gao, H.; Christensen, T.A.; Stojakovic, A.; Trushin, S.; Salisbury, J.L.; Fuentealba, J.; Trushina, E. Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice. Cells 2023, 12, 1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like Protein 1 Reduction Underlies Mitochondrial Morphology and Distribution Abnormalities in Fibroblasts from Sporadic Alzheimer’s Disease Patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; García, E.; Perry, G.; Avila, J.; García-Escudero, V. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9302761. [Google Scholar] [CrossRef]
- Drabik, K.; Piecyk, K.; Wolny, A.; Szulc-Dąbrowska, L.; Dębska-Vielhaber, G.; Vielhaber, S.; Duszyński, J.; Malińska, D.; Szczepanowska, J. Adaptation of Mitochondrial Network Dynamics and Velocity of Mitochondrial Movement to Chronic Stress Present in Fibroblasts Derived from Patients with Sporadic Form of Alzheimer’s Disease. FASEB J. 2021, 35, e21586. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial Dynamics and Transport in Alzheimer’s Disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Vevea, J.D.; Chapman, E.R. Mitofusin 2 Sustains the Axonal Mitochondrial Network to Support Presynaptic Ca2+ Homeostasis and the Synaptic Vesicle Cycle in Rat Hippocampal Axons. J. Neurosci. 2023, 43, 3421–3438. [Google Scholar] [CrossRef]
- Lin, M.-Y.; Sheng, Z.-H. Regulation of Mitochondrial Transport in Neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef]
- Calkins, M.J.; Reddy, P.H. Amyloid Beta Impairs Mitochondrial Anterograde Transport and Degenerates Synapses in Alzheimer’s Disease Neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired Mitochondrial Biogenesis, Defective Axonal Transport of Mitochondria, Abnormal Mitochondrial Dynamics and Synaptic Degeneration in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Seo, N.-Y.; Kim, G.H.; Noh, J.E.; Shin, J.W.; Lee, C.H.; Lee, K.J. Selective Regional Loss of Cortical Synapses Lacking Presynaptic Mitochondria in the 5xFAD Mouse Model. Front. Neuroanat. 2021, 15, 690168. [Google Scholar] [CrossRef]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.-A.; King, D.; Smith, C.; Gillingwater, T.H.; Henstridge, C.M.; Spires-Jones, T.L. Region-Specific Depletion of Synaptic Mitochondria in the Brains of Patients with Alzheimer’s Disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Dolan, P.J.; Jin, Y.N.; Johnson, G.V.W. Truncated Tau and Aβ Cooperatively Impair Mitochondria in Primary Neurons. Neurobiol. Aging 2012, 33, e25–e35. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Xu, J.C.; Fomenko, V.; Miyamoto, T.; Suberbielle, E.; Knox, J.A.; Ho, K.; Kim, D.H.; Yu, G.-Q.; Mucke, L. Tau Reduction Prevents Aβ-Induced Axonal Transport Deficits by Blocking Activation of GSK3β. J. Cell Biol. 2015, 209, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Szebenyi, G.; Elluru, R.; Ratner, N.; Brady, S.T. Glycogen Synthase Kinase 3 Phosphorylates Kinesin Light Chains and Negatively Regulates Kinesin-Based Motility. EMBO J. 2002, 21, 281–293. [Google Scholar] [CrossRef]
- Sabui, A.; Biswas, M.; Somvanshi, P.R.; Kandagiri, P.; Gorla, M.; Mohammed, F.; Tammineni, P. Decreased Anterograde Transport Coupled with Sustained Retrograde Transport Contributes to Reduced Axonal Mitochondrial Density in Tauopathy Neurons. Front. Mol. Neurosci. 2022, 15, 927195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tian, J.; Chen, H.; Du, H.; Guo, L. Amyloid Beta-Mediated KIF5A Deficiency Disrupts Anterograde Axonal Mitochondrial Movement. Neurobiol. Dis. 2019, 127, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Iijima-Ando, K.; Sekiya, M.; Maruko-Otake, A.; Ohtake, Y.; Suzuki, E.; Lu, B.; Iijima, K.M. Loss of Axonal Mitochondria Promotes Tau-Mediated Neurodegeneration and Alzheimer’s Disease-Related Tau Phosphorylation via PAR-1. PLoS Genet. 2012, 8, e1002918. [Google Scholar] [CrossRef] [PubMed]
- Panchal, K.; Tiwari, A.K. Miro, a Rho GTPase Genetically Interacts with Alzheimer’s Disease-Associated Genes (Tau, Aβ42 and Appl) in Drosophila Melanogaster. Biol. Open 2020, 9, bio049569. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial Biogenesis and Clearance: A Balancing Act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.-D. Mitochondrial Biogenesis: An Update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional Activators and Coactivators in the Nuclear Control of Mitochondrial Function in Mammalian Cells. Gene 2002, 286, 81–89. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial Biogenesis in Neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.; Casadesus, G.; Perry, G.; Zhu, X. Impaired Mitochondrial Biogenesis Contributes to Mitochondrial Dysfunction in Alzheimer’s Disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Singulani, M.P.; Pereira, C.P.M.; Ferreira, A.F.F.; Garcia, P.C.; Ferrari, G.D.; Alberici, L.C.; Britto, L.R. Impairment of PGC-1α-Mediated Mitochondrial Biogenesis Precedes Mitochondrial Dysfunction and Alzheimer’s Pathology in the 3xTg Mouse Model of Alzheimer’s Disease. Exp. Gerontol. 2020, 133, 110882. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha Expression Decreases in the Alzheimer Disease Brain as a Function of Dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Pedrós, I.; Petrov, D.; Allgaier, M.; Sureda, F.; Barroso, E.; Beas-Zarate, C.; Auladell, C.; Pallàs, M.; Vázquez-Carrera, M.; Casadesús, G.; et al. Early Alterations in Energy Metabolism in the Hippocampus of APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.F.; Genebra, T.; Rego, A.C.; Rodrigues, C.M.P.; Solá, S. Amyloid β Peptide Compromises Neural Stem Cell Fate by Irreversibly Disturbing Mitochondrial Oxidative State and Blocking Mitochondrial Biogenesis and Dynamics. Mol. Neurobiol. 2019, 56, 3922–3936. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-Beta-Induced Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Dong, W.; Wang, F.; Guo, W.; Zheng, X.; Chen, Y.; Zhang, W.; Shi, H. Aβ25-35 Suppresses Mitochondrial Biogenesis in Primary Hippocampal Neurons. Cell. Mol. Neurobiol. 2016, 36, 83–91. [Google Scholar] [CrossRef]
- Jia, J.; Yin, J.; Zhang, Y.; Xu, G.; Wang, M.; Jiang, H.; Li, L.; Zeng, X.; Zhu, D. Thioredoxin-1 Promotes Mitochondrial Biogenesis Through Regulating AMPK/Sirt1/PGC1α Pathway in Alzheimer’s Disease. ASN Neuro 2023, 15, 17590914231159226. [Google Scholar] [CrossRef]
- Onishi, M.; Okamoto, K. Mitochondrial Clearance: Mechanisms and Roles in Cellular Fitness. FEBS Lett. 2021, 595, 1239–1263. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin Is Activated by PINK1-Dependent Phosphorylation of Ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 Enhancement Is Able to Compensate Mitophagy Alterations Found in Sporadic Alzheimer’s Disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-Associated Tau Impairs Mitophagy by Inhibiting Parkin Translocation to Mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal Mutant APP and Amyloid Beta-Induced Cognitive Decline, Dendritic Spine Loss, Defective Autophagy, Mitophagy and Mitochondrial Abnormalities in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef]
- Hu, Y.; Li, X.-C.; Wang, Z.; Luo, Y.; Zhang, X.; Liu, X.-P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau Accumulation Impairs Mitophagy via Increasing Mitochondrial Membrane Potential and Reducing Mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and Amyloid Beta-Induced Defective Autophagy, Mitophagy, Mitochondrial Structural and Functional Changes and Synaptic Damage in Hippocampal Neurons from Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of Amyloid Precursor Protein C-Terminal Fragments Triggers Mitochondrial Structure, Function, and Mitophagy Defects in Alzheimer’s Disease Models and Human Brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Drygalski, K.; Fereniec, E.; Koryciński, K.; Chomentowski, A.; Kiełczewska, A.; Odrzygóźdź, C.; Modzelewska, B. Resveratrol and Alzheimer’s Disease. From Molecular Pathophysiology to Clinical Trials. Exp. Gerontol. 2018, 113, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Rivière, C.; Richard, T.; Vitrac, X.; Mérillon, J.-M.; Valls, J.; Monti, J.-P. New Polyphenols Active on Beta-Amyloid Aggregation. Bioorg. Med. Chem. Lett. 2008, 18, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, T.; Li, W.; Gao, N.; Zhang, T. Resveratrol Attenuates Oxidative Damage through Activating Mitophagy in an in Vitro Model of Alzheimer’s Disease. Toxicol. Lett. 2018, 282, 100–108. [Google Scholar] [CrossRef]
- Han, Y.-S.; Zheng, W.-H.; Bastianetto, S.; Chabot, J.-G.; Quirion, R. Neuroprotective Effects of Resveratrol against Beta-Amyloid-Induced Neurotoxicity in Rat Hippocampal Neurons: Involvement of Protein Kinase C. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.-C.; Nicol, C.J.; Cheng, Y.-C. Resveratrol Activation of AMPK-Dependent Pathways Is Neuroprotective in Human Neural Stem Cells against Amyloid-Beta-Induced Inflammation and Oxidative Stress. Neurochem. Int. 2018, 115, 1–10. [Google Scholar] [CrossRef]
- Abozaid, O.A.R.; Sallam, M.W.; El-Sonbaty, S.; Aziza, S.; Emad, B.; Ahmed, E.S.A. Resveratrol-Selenium Nanoparticles Alleviate Neuroinflammation and Neurotoxicity in a Rat Model of Alzheimer’s Disease by Regulating Sirt1/MiRNA-134/GSK3β Expression. Biol. Trace Elem. Res. 2022, 200, 5104–5114. [Google Scholar] [CrossRef]
- Savaskan, E.; Olivieri, G.; Meier, F.; Seifritz, E.; Wirz-Justice, A.; Müller-Spahn, F. Red Wine Ingredient Resveratrol Protects from Beta-Amyloid Neurotoxicity. Gerontology 2003, 49, 380–383. [Google Scholar] [CrossRef]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol Mitigates Lipopolysaccharide- and Aβ-Mediated Microglial Inflammation by Inhibiting the TLR4/NF-ΚB/STAT Signaling Cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E.; Walle, U.K. High Absorption but Very Low Bioavailability of Oral Resveratrol in Humans. Drug Metab. Dispos. Biol. Fate Chem. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Walia, V.; Kaushik, D.; Mittal, V.; Kumar, K.; Verma, R.; Parashar, J.; Akter, R.; Rahman, M.H.; Bhatia, S.; Al-Harrasi, A.; et al. Delineation of Neuroprotective Effects and Possible Benefits of AntioxidantsTherapy for the Treatment of Alzheimer’s Diseases by Targeting Mitochondrial-Derived Reactive Oxygen Species: Bench to Bedside. Mol. Neurobiol. 2022, 59, 657–680. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Z.; Lei, H.; Zhang, D. Recent Progress in Nanotechnology-Based Drug Carriers for Resveratrol Delivery. Drug Deliv. 2023, 30, 2174206. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chu, X.; Cui, L.; Fu, S.; Gao, C.; Li, Y.; Sun, B. Neuronal Mitochondria-Targeted Therapy for Alzheimer’s Disease by Systemic Delivery of Resveratrol Using Dual-Modified Novel Biomimetic Nanosystems. Drug Deliv. 2020, 27, 502–518. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, N.; Desquiret-Dumas, V.; Leman, G.; Chupin, S.; Baron, S.; Nivet-Antoine, V.; Vessières, E.; Ayer, A.; Henrion, D.; Lenaers, G.; et al. Resveratrol Directly Binds to Mitochondrial Complex I and Increases Oxidative Stress in Brain Mitochondria of Aged Mice. PLoS ONE 2015, 10, e0144290. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Graier, W.F. Dosis Facit Sanitatem-Concentration-Dependent Effects of Resveratrol on Mitochondria. Nutrients 2017, 9, 1117. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Zinjarde, S.; Bhargava, S.; Rajamohanan, P.R.; Ravikumar, A. Discovering Bisdemethoxycurcumin from Curcuma Longa Rhizome as a Potent Small Molecule Inhibitor of Human Pancreatic α-Amylase, a Target for Type-2 Diabetes. Food Chem. 2012, 135, 2638–2642. [Google Scholar] [CrossRef]
- Sivani, B.M.; Azzeh, M.; Patnaik, R.; Pantea Stoian, A.; Rizzo, M.; Banerjee, Y. Reconnoitering the Therapeutic Role of Curcumin in Disease Prevention and Treatment: Lessons Learnt and Future Directions. Metabolites 2022, 12, 639. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin Has Potent Anti-Amyloidogenic Effects for Alzheimer’s Beta-Amyloid Fibrils in Vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef]
- Zhang, L.; Fiala, M.; Cashman, J.; Sayre, J.; Espinosa, A.; Mahanian, M.; Zaghi, J.; Badmaev, V.; Graves, M.C.; Bernard, G.; et al. Curcuminoids Enhance Amyloid-Beta Uptake by Macrophages of Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2006, 10, 1–7. [Google Scholar] [CrossRef]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Epigallocatechin-3-Gallate and Curcumin Suppress Amyloid Beta-Induced Beta-Site APP Cleaving Enzyme-1 Upregulation. Neuroreport 2008, 19, 1329–1333. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin Labels Amyloid Pathology in Vivo, Disrupts Existing Plaques, and Partially Restores Distorted Neurites in an Alzheimer Mouse Model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in Vitro. J. Alzheimer’s Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- ELBini-Dhouib, I.; Doghri, R.; Ellefi, A.; Degrach, I.; Srairi-Abid, N.; Gati, A. Curcumin Attenuated Neurotoxicity in Sporadic Animal Model of Alzheimer’s Disease. Molecules 2021, 26, 3011. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Kandimalla, R.; Kuruva, C.S. Protective Effects of a Natural Product, Curcumin, against Amyloid β Induced Mitochondrial and Synaptic Toxicities in Alzheimer’s Disease. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2016, 64, 1220–1234. [Google Scholar] [CrossRef]
- Song, X.-J.; Zhou, H.-Y.; Sun, Y.-X.; Huang, H.-C. Inhibitory Effects of Curcumin on H2O2-Induced Cell Damage and APP Expression and Processing in SH-SY5Y Cells Transfected with APP Gene with Swedish Mutation. Mol. Biol. Rep. 2020, 47, 2047–2059. [Google Scholar] [CrossRef]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A Review of Its Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-Month Randomized, Placebo-Controlled, Double-Blind, Pilot Clinical Trial of Curcumin in Patients with Alzheimer Disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral Curcumin for Alzheimer’s Disease: Tolerability and Efficacy in a 24-Week Randomized, Double Blind, Placebo-Controlled Study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef]
- Huang, H.-C.; Xu, K.; Jiang, Z.-F. Curcumin-Mediated Neuroprotection against Amyloid-β-Induced Mitochondrial Dysfunction Involves the Inhibition of GSK-3β. J. Alzheimer’s Dis. 2012, 32, 981–996. [Google Scholar] [CrossRef]
- Cox, K.H.M.; Pipingas, A.; Scholey, A.B. Investigation of the Effects of Solid Lipid Curcumin on Cognition and Mood in a Healthy Older Population. J. Psychopharmacol. Oxf. Engl. 2015, 29, 642–651. [Google Scholar] [CrossRef]
- Small, G.W.; Siddarth, P.; Li, Z.; Miller, K.J.; Ercoli, L.; Emerson, N.D.; Martinez, J.; Wong, K.-P.; Liu, J.; Merrill, D.A.; et al. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am. J. Geriatr. Psychiatry 2018, 26, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, Y.; Sun, J.; Han, Y.; Gong, W.; Li, Y.; Feng, Y.; Wang, H.; Yang, M.; Li, Z.; et al. Neuronal Mitochondria-Targeted Delivery of Curcumin by Biomimetic Engineered Nanosystems in Alzheimer’s Disease Mice. Acta Biomater. 2020, 108, 285–299. [Google Scholar] [CrossRef]
- Kandiah, N.; Ong, P.A.; Yuda, T.; Ng, L.-L.; Mamun, K.; Merchant, R.A.; Chen, C.; Dominguez, J.; Marasigan, S.; Ampil, E.; et al. Treatment of dementia and mild cognitive impairment with or without cerebrovascular disease: Expert consensus on the use of Ginkgo biloba extract, EGb 761®. CNS Neurosci. Ther. 2019, 25, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.; Eckert, A.; Eckert, G.P.; Fink, H.; Friedland, K.; Gauthier, S.; Hoerr, R.; Ihl, R.; Kasper, S.; Möller, H.-J. Therapeutic efficacy of the Ginkgo special extract EGb761® within the framework of the mitochondrial cascade hypothesis of Alzheimer’s disease. World J. Biol. Psychiatry 2019, 20, 173–189. [Google Scholar] [CrossRef]
- Tchantchou, F.; Xu, Y.; Wu, Y.; Christen, Y.; Luo, Y. EGb 761 enhances adult hippocampal neurogenesis and phosphorylation of CREB in transgenic mouse model of Alzheimer’s disease. FASEB J. 2007, 21, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Zhang, C.; Danielsen, M.; Li, Q.; Chen, W.; Chan, Y.; Li, Y. EGb761 improves cognitive function and regulates inflammatory responses in the APP/PS1 mouse. Exp. Gerontol. 2016, 81, 92–100. [Google Scholar] [CrossRef]
- Colciaghi, F.; Borroni, B.; Zimmermann, M.; Bellone, C.; Longhi, A.; Padovani, A.; Cattabeni, F.; Christen, Y.; Di Luca, M. Amyloid precursor protein metabolism is regulated toward alpha-secretase pathway by Ginkgo biloba extracts. Neurobiol. Dis. 2004, 16, 454–460. [Google Scholar] [CrossRef]
- Ge, W.; Ren, C.; Xing, L.; Guan, L.; Zhang, C.; Sun, X.; Wang, G.; Niu, H.; Qun, S. Ginkgo biloba extract improves cognitive function and increases neurogenesis by reducing Aβ pathology in 5×FAD mice. Am. J. Transl. Res. 2021, 13, 1471–1482. [Google Scholar]
- Morató, X.; Marquié, M.; Tartari, J.P.; Lafuente, A.; Abdelnour, C.; Alegret, M.; Jofresa, S.; Buendía, M.; Pancho, A.; Aguilera, N.; et al. A randomized, open-label clinical trial in mild cognitive impairment with EGb 761 examining blood markers of inflammation and oxidative stress. Sci. Rep. 2023, 13, 5406. [Google Scholar] [CrossRef]
- Gill, I.; Kaur, S.; Kaur, N.; Dhiman, M.; Mantha, A.K. Phytochemical Ginkgolide B Attenuates Amyloid-β1-42 Induced Oxidative Damage and Altered Cellular Responses in Human Neuroblastoma SH-SY5Y Cells. J. Alzheimer’s Dis. 2017, 60 (Suppl. 1), S25–S40. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B inactivates the NLRP3 inflammasome by promoting autophagic degradation to improve learning and memory impairment in Alzheimer’s disease. Metab. Brain Dis. 2022, 37, 329–341. [Google Scholar] [CrossRef]
- Liu, J.; Ye, T.; Zhang, Y.; Zhang, R.; Kong, Y.; Zhang, Y.; Sun, J. Protective Effect of Ginkgolide B against Cognitive Impairment in Mice via Regulation of Gut Microbiota. J. Agric. Food Chem. 2021, 69, 12230–12240. [Google Scholar] [CrossRef]
- Shao, L.; Dong, C.; Geng, D.; He, Q.; Shi, Y. Ginkgolide B protects against cognitive impairment in senescence-accelerated P8 mice by mitigating oxidative stress, inflammation and ferroptosis. Biochem. Biophys. Res. Commun. 2021, 572, 7–14. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Zhou, X.J.; Xu, B. Mitochondria: Central Organelles for Melatonin’s Antioxidant and Anti-Aging Actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [PubMed]
- Chitimus, D.M.; Popescu, M.R.; Voiculescu, S.E.; Panaitescu, A.M.; Pavel, B.; Zagrean, L.; Zagrean, A.-M. Melatonin’s Impact on Antioxidative and Anti-Inflammatory Reprogramming in Homeostasis and Disease. Biomolecules 2020, 10, 1211. [Google Scholar] [CrossRef]
- Maurizi, C.P. Loss of Intraventricular Fluid Melatonin Can Explain the Neuropathology of Alzheimer’s Disease. Med. Hypotheses 1997, 49, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-N.; Liu, R.-Y.; Kamphorst, W.; Hofman, M.A.; Swaab, D.F. Early Neuropathological Alzheimer’s Changes in Aged Individuals Are Accompanied by Decreased Cerebrospinal Fluid Melatonin Levels. J. Pineal. Res. 2003, 35, 125–130. [Google Scholar] [CrossRef]
- Feng, Z.; Chang, Y.; Cheng, Y.; Zhang, B.; Qu, Z.; Qin, C.; Zhang, J. Melatonin Alleviates Behavioral Deficits Associated with Apoptosis and Cholinergic System Dysfunction in the APP 695 Transgenic Mouse Model of Alzheimer’s Disease. J. Pineal. Res. 2004, 37, 129–136. [Google Scholar] [CrossRef]
- Olcese, J.M.; Cao, C.; Mori, T.; Mamcarz, M.B.; Maxwell, A.; Runfeldt, M.J.; Wang, L.; Zhang, C.; Lin, X.; Zhang, G.; et al. Protection against Cognitive Deficits and Markers of Neurodegeneration by Long-Term Oral Administration of Melatonin in a Transgenic Model of Alzheimer Disease. J. Pineal. Res. 2009, 47, 82–96. [Google Scholar] [CrossRef]
- Cheng, Y.; Feng, Z.; Zhang, Q.; Zhang, J. Beneficial Effects of Melatonin in Experimental Models of Alzheimer Disease. Acta Pharmacol. Sin. 2006, 27, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Qin, C.; Chang, Y.; Zhang, J. Early Melatonin Supplementation Alleviates Oxidative Stress in a Transgenic Mouse Model of Alzheimer’s Disease. Free Radic. Biol. Med. 2006, 40, 101–109. [Google Scholar] [CrossRef]
- Chen, C.; Yang, C.; Wang, J.; Huang, X.; Yu, H.; Li, S.; Li, S.; Zhang, Z.; Liu, J.; Yang, X.; et al. Melatonin Ameliorates Cognitive Deficits through Improving Mitophagy in a Mouse Model of Alzheimer’s Disease. J. Pineal. Res. 2021, 71, e12774. [Google Scholar] [CrossRef]
- Xu, J.; Wang, L.-L.; Dammer, E.B.; Li, C.-B.; Xu, G.; Chen, S.-D.; Wang, G. Melatonin for Sleep Disorders and Cognition in Dementia: A Meta-Analysis of Randomized Controlled Trials. Am. J. Alzheimer’s Dis. Other Demen. 2015, 30, 439–447. [Google Scholar] [CrossRef]
- Sumsuzzman, D.M.; Choi, J.; Jin, Y.; Hong, Y. Neurocognitive Effects of Melatonin Treatment in Healthy Adults and Individuals with Alzheimer’s Disease and Insomnia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Neurosci. Biobehav. Rev. 2021, 127, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguilar, M.A.; Ramírez-Salado, I.; Guevara, M.A.; Hernández-González, M.; Benitez-King, G. Melatonin Effects on EEG Activity During Sleep Onset in Mild-to-Moderate Alzheimer’s Disease: A Pilot Study. J. Alzheimer’s Dis. Rep. 2018, 2, 55–65. [Google Scholar] [CrossRef]
- Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Laudon, M.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on Prolonged-Release Melatonin for Cognitive Function and Sleep in Mild to Moderate Alzheimer’s Disease: A 6-Month, Randomized, Placebo-Controlled, Multicenter Trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Nelson, V.A.; Davila, H.; Ratner, E.; Fink, H.A.; Hemmy, L.S.; McCarten, J.R.; Barclay, T.R.; Brasure, M.; Kane, R.L. Over-the-Counter Supplement Interventions to Prevent Cognitive Decline, Mild Cognitive Impairment, and Clinical Alzheimer-Type Dementia: A Systematic Review. Ann. Intern. Med. 2018, 168, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Manochkumar, J.; Doss, C.G.P.; El-Seedi, H.R.; Efferth, T.; Ramamoorthy, S. The neuroprotective potential of carotenoids in vitro and in vivo. Phytomedicine 2021, 91, 153676. [Google Scholar] [CrossRef]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin E and Alzheimer’s Disease: What Do We Know so Far? Clin. Interv. Aging 2019, 14, 1303–1317. [Google Scholar] [CrossRef]
- Lu, Y.; An, Y.; Guo, J.; Zhang, X.; Wang, H.; Rong, H.; Xiao, R. Dietary Intake of Nutrients and Lifestyle Affect the Risk of Mild Cognitive Impairment in the Chinese Elderly Population: A Cross-Sectional Study. Front. Behav. Neurosci. 2016, 10, 229. [Google Scholar] [CrossRef]
- Kesse-Guyot, E.; Andreeva, V.A.; Ducros, V.; Jeandel, C.; Julia, C.; Hercberg, S.; Galan, P. Carotenoid-rich dietary patterns during midlife and subsequent cognitive function. Br. J. Nutr. 2014, 111, 915–923. [Google Scholar] [CrossRef]
- Yuan, C.; Chen, H.; Wang, Y.; Schneider, J.A.; Willett, W.C.; Morris, M.C. Dietary carotenoids related to risk of incident Alzheimer dementia (AD) and brain AD neuropathology: A community-based cohort of older adults. Am. J. Clin. Nutr. 2021, 113, 200–208. [Google Scholar] [CrossRef]
- Nolan, J.M.; Power, R.; Mulcahy, R. Supplementation With Carotenoids, Omega-3 Fatty Acids, and Vitamin E Has a Positive Effect on the Symptoms and Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 90, 233–249. [Google Scholar] [CrossRef]
- Miller, B.; Kim, S.-J.; Kumagai, H.; Yen, K.; Cohen, P. Mitochondria-Derived Peptides in Aging and Healthspan. J. Clin. Investig. 2022, 132, e158449. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A Rescue Factor Abolishing Neuronal Cell Death by a Wide Spectrum of Familial Alzheimer’s Disease Genes and Abeta. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef]
- Yen, K.; Wan, J.; Mehta, H.H.; Miller, B.; Christensen, A.; Levine, M.E.; Salomon, M.P.; Brandhorst, S.; Xiao, J.; Kim, S.-J.; et al. Humanin Prevents Age-Related Cognitive Decline in Mice and Is Associated with Improved Cognitive Age in Humans. Sci. Rep. 2018, 8, 14212. [Google Scholar] [CrossRef]
- Tajima, H.; Kawasumi, M.; Chiba, T.; Yamada, M.; Yamashita, K.; Nawa, M.; Kita, Y.; Kouyama, K.; Aiso, S.; Matsuoka, M.; et al. A Humanin Derivative, S14G-HN, Prevents Amyloid-Beta-Induced Memory Impairment in Mice. J. Neurosci. Res. 2005, 79, 714–723. [Google Scholar] [CrossRef]
- Niikura, T.; Sidahmed, E.; Hirata-Fukae, C.; Aisen, P.S.; Matsuoka, Y. A Humanin Derivative Reduces Amyloid Beta Accumulation and Ameliorates Memory Deficit in Triple Transgenic Mice. PLoS ONE 2011, 6, e16259. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, W.; Li, Z.; Hao, J.; Zhang, Z.; Liu, L.; Mao, N.; Miao, J.; Zhang, L. S14G-Humanin Improves Cognitive Deficits and Reduces Amyloid Pathology in the Middle-Aged APPswe/PS1dE9 Mice. Pharmacol. Biochem. Behav. 2012, 100, 361–369. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin Inhibits Neuronal Cell Death by Interacting with a Cytokine Receptor Complex or Complexes Involving CNTF Receptor Alpha/WSX-1/Gp130. Mol. Biol. Cell 2009, 20, 2864–2873. [Google Scholar] [CrossRef]
- Kim, S.-J.; Guerrero, N.; Wassef, G.; Xiao, J.; Mehta, H.H.; Cohen, P.; Yen, K. The Mitochondrial-Derived Peptide Humanin Activates the ERK1/2, AKT, and STAT3 Signaling Pathways and Has Age-Dependent Signaling Differences in the Hippocampus. Oncotarget 2016, 7, 46899–46912. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, T.; Wang, W.-X.; Xu, J.-H.; Yang, P.-B.; Lu, H.-X.; Sun, Q.-R.; Hu, H.-T. Protective Effects of [Gly14]-Humanin on Beta-Amyloid-Induced PC12 Cell Death by Preventing Mitochondrial Dysfunction. Neurochem. Int. 2010, 56, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-W.; Liu, D.-X. Humanin Decreases Mitochondrial Membrane Permeability by Inhibiting the Membrane Association and Oligomerization of Bax and Bid Proteins. Acta Pharmacol. Sin. 2018, 39, 1012–1021. [Google Scholar] [CrossRef]
- Romeo, M.; Stravalaci, M.; Beeg, M.; Rossi, A.; Fiordaliso, F.; Corbelli, A.; Salmona, M.; Gobbi, M.; Cagnotto, A.; Diomede, L. Humanin Specifically Interacts with Amyloid-β Oligomers and Counteracts Their in Vivo Toxicity. J. Alzheimer’s Dis. 2017, 57, 857–871. [Google Scholar] [CrossRef]
- Han, K.; Jia, N.; Zhong, Y.; Shang, X. S14G-Humanin Alleviates Insulin Resistance and Increases Autophagy in Neurons of APP/PS1 Transgenic Mouse. J. Cell. Biochem. 2018, 119, 3111–3117. [Google Scholar] [CrossRef]
- Qian, K.; Bao, X.; Li, Y.; Wang, P.; Guo, Q.; Yang, P.; Xu, S.; Yu, F.; Meng, R.; Cheng, Y.; et al. Cholinergic Neuron Targeting Nanosystem Delivering Hybrid Peptide for Combinatorial Mitochondrial Therapy in Alzheimer’s Disease. ACS Nano 2022, 16, 11455–11472. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Shi, H.; He, Y.; Yuan, L.; Qu, X.; Zhang, J.; Wang, Z.; Cai, H.; Qi, J. Colivelin Ameliorates Impairments in Cognitive Behaviors and Synaptic Plasticity in APP/PS1 Transgenic Mice. J. Alzheimer’s Dis. 2017, 59, 1067–1078. [Google Scholar] [CrossRef]
- Wu, M.-N.; Zhou, L.-W.; Wang, Z.-J.; Han, W.-N.; Zhang, J.; Liu, X.-J.; Tong, J.-Q.; Qi, J.-S. Colivelin Ameliorates Amyloid β Peptide-Induced Impairments in Spatial Memory, Synaptic Plasticity, and Calcium Homeostasis in Rats. Hippocampus 2015, 25, 363–372. [Google Scholar] [CrossRef]
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally Occurring Mitochondrial-Derived Peptides Are Age-Dependent Regulators of Apoptosis, Insulin Sensitivity, and Inflammatory Markers. Aging 2016, 8, 796–809. [Google Scholar] [CrossRef]
- Wan, W.; Zhang, L.; Lin, Y.; Rao, X.; Wang, X.; Hua, F.; Ying, J. Mitochondria-Derived Peptide MOTS-c: Effects and Mechanisms Related to Stress, Metabolism and Aging. J. Transl. Med. 2023, 21, 36. [Google Scholar] [CrossRef]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 2018, 28, 516–524.e7. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.X.; Finkel, T. Mitochondria as Intracellular Signaling Platforms in Health and Disease. J. Cell Biol. 2020, 219, e202002179. [Google Scholar] [CrossRef]
- Jiang, J.; Chang, X.; Nie, Y.; Shen, Y.; Liang, X.; Peng, Y.; Chang, M. Peripheral Administration of a Cell-Penetrating MOTS-c Analogue Enhances Memory and Attenuates Aβ1-42- or LPS-Induced Memory Impairment through Inhibiting Neuroinflammation. ACS Chem. Neurosci. 2021, 12, 1506–1518. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s Disease Pathogenesis: Therapeutic Potential and Future Perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Protective Effects of Mitophagy Enhancers against Amyloid Beta-Induced Mitochondrial and Synaptic Toxicities in Alzheimer Disease. Hum. Mol. Genet. 2022, 31, 423–439. [Google Scholar] [CrossRef]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal Mitochondrial Dynamics in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S253–S262. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective Effects of Reduced Dynamin-Related Protein 1 against Amyloid Beta-Induced Mitochondrial Dysfunction and Synaptic Damage in Alzheimer’s Disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Hemachandra Reddy, P. Corrigendum: Mitochondrial Division Inhibitor 1 Reduces Dynamin-Related Protein 1 and Mitochondrial Fission Activity. Hum. Mol. Genet. 2019, 28, 875–876. [Google Scholar] [CrossRef]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.-H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-β Induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 Inhibitor Diminishes Aberrant Mitochondrial Fission and Neurotoxicity. J. Cell Sci. 2013, 126 Pt 3, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W. Inhibition of the Mechanistic Target of Rapamycin (MTOR)-Rapamycin and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6, a025924. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of MTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-Beta Levels in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, H.; Cen, M.; Tao, Y.; Lai, C.; Tang, Z. Rapamycin Ameliorates Cognitive Impairments and Alzheimer’s Disease-Like Pathology with Restoring Mitochondrial Abnormality in the Hippocampus of Streptozotocin-Induced Diabetic Mice. Neurochem. Res. 2021, 46, 265–275. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular Interplay between Mammalian Target of Rapamycin (MTOR), Amyloid-Beta, and Tau: Effects on Cognitive Impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing Autophagy by Rapamycin before, but Not after, the Formation of Plaques and Tangles Ameliorates Cognitive Deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef]
- Shi, Q.; Chang, C.; Saliba, A.; Bhat, M.A. Microglial MTOR Activation Upregulates Trem2 and Enhances β-Amyloid Plaque Clearance in the 5XFAD Alzheimer’s Disease Model. J. Neurosci. 2022, 42, 5294–5313. [Google Scholar] [CrossRef]
- Carosi, J.M.; Sargeant, T.J. Rapamycin and Alzheimer Disease: A Double-Edged Sword? Autophagy 2019, 15, 1460–1462. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Hindle, A.; Kshirsagar, S.; Reddy, P.H. Are Mitophagy Enhancers Therapeutic Targets for Alzheimer’s Disease? Biomed. Pharmacother. 2022, 149, 112918. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The Mitophagy Activator Urolithin A Is Safe and Induces a Molecular Signature of Improved Mitochondrial and Cellular Health in Humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; D’Amico, D.; Andreux, P.A.; Fouassier, A.M.; Blanco-Bose, W.; Evans, M.; Aebischer, P.; Auwerx, J.; Rinsch, C. Urolithin A Improves Muscle Strength, Exercise Performance, and Biomarkers of Mitochondrial Health in a Randomized Trial in Middle-Aged Adults. Cell Rep. Med. 2022, 3, 100633. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A Attenuates Memory Impairment and Neuroinflammation in APP/PS1 Mice. J. Neuroinflammation 2019, 16, 62. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Alvir, R.V.; Pradeepkiran, J.A.; Hindle, A.; Vijayan, M.; Ramasubramaniam, B.; Kumar, S.; Reddy, A.P.; Reddy, P.H. A Combination Therapy of Urolithin A+EGCG Has Stronger Protective Effects than Single Drug Urolithin A in a Humanized Amyloid Beta Knockin Mice for Late-Onset Alzheimer’s Disease. Cells 2022, 11, 2660. [Google Scholar] [CrossRef]
- Hirano, K.; Fujimaki, M.; Sasazawa, Y.; Yamaguchi, A.; Ishikawa, K.-I.; Miyamoto, K.; Souma, S.; Furuya, N.; Imamichi, Y.; Yamada, D.; et al. Neuroprotective Effects of Memantine via Enhancement of Autophagy. Biochem. Biophys. Res. Commun. 2019, 518, 161–170. [Google Scholar] [CrossRef]
- Jadhav, R.; Kulkarni, Y.A. The Combination of Baicalein and Memantine Reduces Oxidative Stress and Protects against β-Amyloid-Induced Alzheimer’s Disease in Rat Model. Antioxidants 2023, 12, 707. [Google Scholar] [CrossRef]
- Sowndhararajan, K.; Deepa, P.; Kim, M.; Park, S.J.; Kim, S. Neuroprotective and Cognitive Enhancement Potentials of Baicalin: A Review. Brain Sci. 2018, 8, 104. [Google Scholar] [CrossRef]
- Wang, W.-W.; Han, R.; He, H.-J.; Li, J.; Chen, S.-Y.; Gu, Y.; Xie, C. Administration of Quercetin Improves Mitochondria Quality Control and Protects the Neurons in 6-OHDA-Lesioned Parkinson’s Disease Models. Aging 2021, 13, 11738–11751. [Google Scholar] [CrossRef]
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxid. Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [PubMed]
- Paula, P.-C.; Angelica Maria, S.-G.; Luis, C.-H.; Gloria Patricia, C.-G. Preventive Effect of Quercetin in a Triple Transgenic Alzheimer’s Disease Mice Model. Molecules 2019, 24, 2287. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Ohta, K. Quercetin Regulates the Integrated Stress Response to Improve Memory. Int. J. Mol. Sci. 2019, 20, 2761. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Y.; Mu, X. Effect of Quercetin on PC12 Alzheimer’s Disease Cell Model Induced by Aβ 25-35 and Its Mechanism Based on Sirtuin1/Nrf2/HO-1 Pathway. BioMed Res. Int. 2020, 2020, 8210578. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Zhuang, X.-X.; Niu, Z.; Ai, R.; Lautrup, S.; Zheng, S.; Jiang, Y.; Han, R.; Gupta, T.S.; Cao, S.; et al. Amelioration of Alzheimer’s Disease Pathology by Mitophagy Inducers Identified via Machine Learning and a Cross-Species Workflow. Nat. Biomed. Eng. 2022, 6, 76–93. [Google Scholar] [CrossRef]
- Ni, Y.-Q.; Liu, Y.-S. New Insights into the Roles and Mechanisms of Spermidine in Aging and Age-Related Diseases. Aging Dis. 2021, 12, 1948–1963. [Google Scholar] [CrossRef]
- Wirth, M.; Benson, G.; Schwarz, C.; Köbe, T.; Grittner, U.; Schmitz, D.; Sigrist, S.J.; Bohlken, J.; Stekovic, S.; Madeo, F.; et al. The Effect of Spermidine on Memory Performance in Older Adults at Risk for Dementia: A Randomized Controlled Trial. Cortex J. Devoted Study Nerv. Syst. Behav. 2018, 109, 181–188. [Google Scholar] [CrossRef]
- Schroeder, S.; Hofer, S.J.; Zimmermann, A.; Pechlaner, R.; Dammbrueck, C.; Pendl, T.; Marcello, G.M.; Pogatschnigg, V.; Bergmann, M.; Müller, M.; et al. Dietary Spermidine Improves Cognitive Function. Cell Rep. 2021, 35, 108985. [Google Scholar] [CrossRef]
- Jing, Y.-H.; Yan, J.-L.; Wang, Q.-J.; Chen, H.-C.; Ma, X.-Z.; Yin, J.; Gao, L.-P. Spermidine Ameliorates the Neuronal Aging by Improving the Mitochondrial Function in Vitro. Exp. Gerontol. 2018, 108, 77–86. [Google Scholar] [CrossRef]
- Fairley, L.H.; Lejri, I.; Grimm, A.; Eckert, A. Spermidine Rescues Bioenergetic and Mitophagy Deficits Induced by Disease-Associated Tau Protein. Int. J. Mol. Sci. 2023, 24, 5297. [Google Scholar] [CrossRef]
- Makarov, M.; Korkotian, E. Differential Role of Active Compounds in Mitophagy and Related Neurodegenerative Diseases. Toxins 2023, 15, 202. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Qiu, Q.; Gu, X.; Tian, Y.; Zhang, Y. ATM Mediates Spermidine-Induced Mitophagy via PINK1 and Parkin Regulation in Human Fibroblasts. Sci. Rep. 2016, 6, 24700. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ Supplementation Normalizes Key Alzheimer’s Features and DNA Damage Responses in a New AD Mouse Model with Introduced DNA Repair Deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ Supplementation Reduces Neuroinflammation and Cell Senescence in a Transgenic Mouse Model of Alzheimer’s Disease via CGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- Xie, X.; Gao, Y.; Zeng, M.; Wang, Y.; Wei, T.-F.; Lu, Y.-B.; Zhang, W.-P. Nicotinamide Ribose Ameliorates Cognitive Impairment of Aged and Alzheimer’s Disease Model Mice. Metab. Brain Dis. 2019, 34, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide Riboside Restores Cognition through an Upregulation of Proliferator-Activated Receptor-γ Coactivator 1α Regulated β-Secretase 1 Degradation and Mitochondrial Gene Expression in Alzheimer’s Mouse Models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef]
- Yulug, B.; Altay, O.; Li, X.; Hanoglu, L.; Cankaya, S.; Lam, S.; Velioglu, H.A.; Yang, H.; Coskun, E.; Idil, E.; et al. Combined Metabolic Activators Improve Cognitive Functions in Alzheimer’s Disease Patients: A Randomised, Double-Blinded, Placebo-Controlled Phase-II Trial. Transl. Neurodegener. 2023, 12, 4. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK Inhibitors Upregulate the Neuroprotective Parkin-Mediated Mitophagy Pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef]
- Koch, J.C.; Tönges, L.; Barski, E.; Michel, U.; Bähr, M.; Lingor, P. ROCK2 Is a Major Regulator of Axonal Degeneration, Neuronal Death and Axonal Regeneration in the CNS. Cell Death Dis. 2014, 5, e1225. [Google Scholar] [CrossRef]
- Liu, J.; Gao, H.-Y.; Wang, X.-F. The Role of the Rho/ROCK Signaling Pathway in Inhibiting Axonal Regeneration in the Central Nervous System. Neural Regen. Res. 2015, 10, 1892–1896. [Google Scholar] [CrossRef]
- Saal, K.-A.; Galter, D.; Roeber, S.; Bähr, M.; Tönges, L.; Lingor, P. Altered Expression of Growth Associated Protein-43 and Rho Kinase in Human Patients with Parkinson’s Disease. Brain Pathol. 2017, 27, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.C.; Tatenhorst, L.; Roser, A.-E.; Saal, K.-A.; Tönges, L.; Lingor, P. ROCK Inhibition in Models of Neurodegeneration and Its Potential for Clinical Translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Huentelman, M.J.; Stephan, D.A.; Talboom, J.; Corneveaux, J.J.; Reiman, D.M.; Gerber, J.D.; Barnes, C.A.; Alexander, G.E.; Reiman, E.M.; Bimonte-Nelson, H.A. Peripheral Delivery of a ROCK Inhibitor Improves Learning and Working Memory. Behav. Neurosci. 2009, 123, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Shirafuji, N.; Yen, S.-H.; Yoshida, H.; Kanaan, N.M.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Fujita, Y.; Kuriyama, M.; et al. Rho-Kinase ROCK Inhibitors Reduce Oligomeric Tau Protein. Neurobiol. Aging 2020, 89, 41–54. [Google Scholar] [CrossRef]
- Gentry, E.G.; Henderson, B.W.; Arrant, A.E.; Gearing, M.; Feng, Y.; Riddle, N.C.; Herskowitz, J.H. Rho Kinase Inhibition as a Therapeutic for Progressive Supranuclear Palsy and Corticobasal Degeneration. J. Neurosci. 2016, 36, 1316–1323. [Google Scholar] [CrossRef]
- Hass, D.T.; Barnstable, C.J. Uncoupling Proteins in the Mitochondrial Defense against Oxidative Stress. Prog. Retin. Eye Res. 2021, 83, 100941. [Google Scholar] [CrossRef]
- Kumar, R.; T, A.; Singothu, S.; Singh, S.B.; Bhandari, V. Uncoupling Proteins as a Therapeutic Target for the Development of New Era Drugs against Neurodegenerative Disorder. Biomed. Pharmacother. 2022, 147, 112656. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef]
- Cunha, F.M.; Caldeira da Silva, C.C.; Cerqueira, F.M.; Kowaltowski, A.J. Mild Mitochondrial Uncoupling as a Therapeutic Strategy. Curr. Drug Targets 2011, 12, 783–789. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Diano, S.; Horvath, T.L. Mitochondrial Uncoupling Proteins in the CNS: In Support of Function and Survival. Nat. Rev. Neurosci. 2005, 6, 829–840. [Google Scholar] [CrossRef]
- Mehta, S.L.; Li, P.A. Neuroprotective Role of Mitochondrial Uncoupling Protein 2 in Cerebral Stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.B.; Ho, P.W.-L.; Ho, J.W.-M.; Liu, H.-F.; So, D.H.-F.; Tse, H.-M.; Chan, K.-H.; Ho, S.-L. Human Neuronal Uncoupling Proteins 4 and 5 (UCP4 and UCP5): Structural Properties, Regulation, and Physiological Role in Protection against Oxidative Stress and Mitochondrial Dysfunction. Brain Behav. 2012, 2, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Seiça, R.M.; Moreira, P.I. Uncoupling Protein 2 Inhibition Exacerbates Glucose Fluctuation-Mediated Neuronal Effects. Neurotox. Res. 2018, 33, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Jun, Z.; Ibrahim, M.M.; Dezheng, G.; Bo, Y.; Qiong, W.; Yuan, Z. UCP2 Protects against Amyloid Beta Toxicity and Oxidative Stress in Primary Neuronal Culture. Biomed. Pharmacother. 2015, 74, 211–214. [Google Scholar] [CrossRef]
- Thangavel, R.; Kempuraj, D.; Zaheer, S.; Raikwar, S.; Ahmed, M.E.; Selvakumar, G.P.; Iyer, S.S.; Zaheer, A. Glia Maturation Factor and Mitochondrial Uncoupling Proteins 2 and 4 Expression in the Temporal Cortex of Alzheimer’s Disease Brain. Front. Aging Neurosci. 2017, 9, 150. [Google Scholar] [CrossRef]
- De Simone, R.; Ajmone-Cat, M.A.; Pandolfi, M.; Bernardo, A.; De Nuccio, C.; Minghetti, L.; Visentin, S. The Mitochondrial Uncoupling Protein-2 Is a Master Regulator of Both M1 and M2 Microglial Responses. J. Neurochem. 2015, 135, 147–156. [Google Scholar] [CrossRef]
- Rosenberg, N.; Reva, M.; Binda, F.; Restivo, L.; Depierre, P.; Puyal, J.; Briquet, M.; Bernardinelli, Y.; Rocher, A.-B.; Markram, H.; et al. Overexpression of UCP4 in Astrocytic Mitochondria Prevents Multilevel Dysfunctions in a Mouse Model of Alzheimer’s Disease. Glia 2023, 71, 957–973. [Google Scholar] [CrossRef]
- Zorov, D.B.; Andrianova, N.V.; Babenko, V.A.; Pevzner, I.B.; Popkov, V.A.; Zorov, S.D.; Zorova, L.D.; Plotnikov, E.Y.; Sukhikh, G.T.; Silachev, D.N. Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons. Brain Sci. 2021, 11, 1050. [Google Scholar] [CrossRef]
- Geisler, J.G.; Marosi, K.; Halpern, J.; Mattson, M.P. DNP, Mitochondrial Uncoupling, and Neuroprotection: A Little Dab’ll Do Ya. Alzheimer’s Dement. 2017, 13, 582–591. [Google Scholar] [CrossRef]
- Aribal, P.; Alver, E.N.; Kaltalioglu, K.; Balabanli, B.; Ebegil, M.; Coskun-Cevher, S. The Relationship between Experimental 2,4-Dinitrophenol Administration and Neurological Oxidative Stress: In Terms of Dose, Time and Gender Differences. Mol. Cell. Biochem. 2023, 478, 1161–1168. [Google Scholar] [CrossRef]
- Geisler, J.G. 2,4 Dinitrophenol as Medicine. Cells 2019, 8, 280. [Google Scholar] [CrossRef]
- De Felice, F.G.; Houzel, J.C.; Garcia-Abreu, J.; Louzada, P.R.; Afonso, R.C.; Meirelles, M.N.; Lent, R.; Neto, V.M.; Ferreira, S.T. Inhibition of Alzheimer’s Disease Beta-Amyloid Aggregation, Neurotoxicity, and in Vivo Deposition by Nitrophenols: Implications for Alzheimer’s Therapy. FASEB J. 2001, 15, 1297–1299. [Google Scholar] [CrossRef]
- Wasilewska-Sampaio, A.P.; Silveira, M.S.; Holub, O.; Goecking, R.; Gomes, F.C.A.; Neto, V.M.; Linden, R.; Ferreira, S.T.; De Felice, F.G. Neuritogenesis and Neuronal Differentiation Promoted by 2,4-Dinitrophenol, a Novel Anti-Amyloidogenic Compound. FASEB J. 2005, 19, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Paula Lima, A.C.; Arriagada, C.; Toro, R.; Cárdenas, A.M.; Caviedes, R.; Ferreira, S.T.; Caviedes, P. Small-Molecule Aggregation Inhibitors Reduce Excess Amyloid in a Trisomy 16 Mouse Cortical Cell Line. Biol. Res. 2008, 41, 129–136. [Google Scholar] [PubMed]
- Liu, D.; Zhang, Y.; Gharavi, R.; Park, H.R.; Lee, J.; Siddiqui, S.; Telljohann, R.; Nassar, M.R.; Cutler, R.G.; Becker, K.G.; et al. The Mitochondrial Uncoupler DNP Triggers Brain Cell MTOR Signaling Network Reprogramming and CREB Pathway Up-Regulation. J. Neurochem. 2015, 134, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, S.; Ali Pour, P.; Kheradvar, A. Prospects of Mitochondrial Transplantation in Clinical Medicine: Aspirations and Challenges. Mitochondrion 2022, 65, 33–44. [Google Scholar] [CrossRef]
- Berridge, M.V.; McConnell, M.J.; Grasso, C.; Bajzikova, M.; Kovarova, J.; Neuzil, J. Horizontal Transfer of Mitochondria between Mammalian Cells: Beyond Co-Culture Approaches. Curr. Opin. Genet. Dev. 2016, 38, 75–82. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial Transfer between Cells Can Rescue Aerobic Respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Hayashida, K.; Takegawa, R.; Endo, Y.; Yin, T.; Choudhary, R.C.; Aoki, T.; Nishikimi, M.; Murao, A.; Nakamura, E.; Shoaib, M.; et al. Exogenous Mitochondrial Transplantation Improves Survival and Neurological Outcomes after Resuscitation from Cardiac Arrest. BMC Med. 2023, 21, 56. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef]
- Alexander, J.F.; Seua, A.V.; Arroyo, L.D.; Ray, P.R.; Wangzhou, A.; Heiβ-Lückemann, L.; Schedlowski, M.; Price, T.J.; Kavelaars, A.; Heijnen, C.J. Nasal Administration of Mitochondria Reverses Chemotherapy-Induced Cognitive Deficits. Theranostics 2021, 11, 3109–3130. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D. Promising Treatment for Multiple Sclerosis: Mitochondrial Transplantation. Int. J. Mol. Sci. 2022, 23, 2245. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Liou, C.-W.; Chen, S.-D.; Hsu, T.-Y.; Chuang, J.-H.; Wang, P.-W.; Huang, S.-T.; Tiao, M.-M.; Chen, J.-B.; Lin, T.-K.; et al. Mitochondrial Transfer from Wharton’s Jelly-Derived Mesenchymal Stem Cells to Mitochondria-Defective Cells Recaptures Impaired Mitochondrial Function. Mitochondrion 2015, 22, 31–44. [Google Scholar] [CrossRef]
- Shi, X.; Bai, H.; Zhao, M.; Li, X.; Sun, X.; Jiang, H.; Fu, A. Treatment of Acetaminophen-Induced Liver Injury with Exogenous Mitochondria in Mice. Transl. Res. J. Lab. Clin. Med. 2018, 196, 31–41. [Google Scholar] [CrossRef]
- Su, Y.; Zhu, L.; Yu, X.; Cai, L.; Lu, Y.; Zhang, J.; Li, T.; Li, J.; Xia, J.; Xu, F.; et al. Erratum: Mitochondrial Transplantation Attenuates Airway Hyperresponsiveness by Inhibition of Cholinergic Hyperactivity: Erratum. Theranostics 2019, 9, 1385–1386. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.-F.; Mak, J.C.W.; Lian, Q. Mitochondrial Transfer of Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells to Airway Epithelial Cells Attenuates Cigarette Smoke-Induced Damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Nakamura, Y.; Park, J.-H.; Hayakawa, K. Therapeutic Use of Extracellular Mitochondria in CNS Injury and Disease. Exp. Neurol. 2020, 324, 113114. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial Transfer/Transplantation: An Emerging Therapeutic Approach for Multiple Diseases. Cell Biosci. 2022, 12, 66. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous Mitochondrial Transplantation for Dysfunction after Ischemia-Reperfusion Injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Corrigendum: Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 539, 123. [Google Scholar] [CrossRef]
- English, K.; Shepherd, A.; Uzor, N.-E.; Trinh, R.; Kavelaars, A.; Heijnen, C.J. Astrocytes Rescue Neuronal Health after Cisplatin Treatment through Mitochondrial Transfer. Acta Neuropathol. Commun. 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Sun, G.; Bautista Garrido, J.; Obertas, L.; Mobley, A.S.; Ting, S.-M.; Zhao, X.; Aronowski, J. The Mitochondria-Derived Peptide Humanin Improves Recovery from Intracerebral Hemorrhage: Implication of Mitochondria Transfer and Microglia Phenotype Change. J. Neurosci. 2020, 40, 2154–2165. [Google Scholar] [CrossRef]
- Tashiro, R.; Ozaki, D.; Bautista-Garrido, J.; Sun, G.; Obertas, L.; Mobley, A.S.; Kim, G.S.; Aronowski, J.; Jung, J.E. Young Astrocytic Mitochondria Attenuate the Elevated Level of CCL11 in the Aged Mice, Contributing to Cognitive Function Improvement. Int. J. Mol. Sci. 2023, 24, 5187. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Benson, T.; Albensi, B.C. Mitochondrial Transfusion Improves Mitochondrial Function Through Up-Regulation of Mitochondrial Complex II Protein Subunit SDHB in the Hippocampus of Aged Mice. Mol. Neurobiol. 2022, 59, 6009–6017. [Google Scholar] [CrossRef]
- Jia, X.; Wang, Q.; Ji, J.; Lu, W.; Liu, Z.; Tian, H.; Guo, L.; Wang, Y. Mitochondrial Transplantation Ameliorates Hippocampal Damage Following Status Epilepticus. Anim. Models Exp. Med. 2023, 6, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sheng, H.; Liao, L.; Xu, C.; Zhang, A.; Yang, Y.; Zhao, L.; Duan, L.; Chen, H.; Zhang, B. Mesenchymal Stem Cell-Conditioned Medium Improves Mitochondrial Dysfunction and Suppresses Apoptosis in Okadaic Acid-Treated SH-SY5Y Cells by Extracellular Vesicle Mitochondrial Transfer. J. Alzheimer’s Dis. 2020, 78, 1161–1176. [Google Scholar] [CrossRef]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef]
- Bobkova, N.V.; Zhdanova, D.Y.; Belosludtseva, N.V.; Penkov, N.V.; Mironova, G.D. Intranasal Administration of Mitochondria Improves Spatial Memory in Olfactory Bulbectomized Mice. Exp. Biol. Med. 2022, 247, 416–425. [Google Scholar] [CrossRef]
- Khan, T.; Waseem, R.; Zehra, Z.; Aiman, A.; Bhardwaj, P.; Ansari, J.; Hassan, M.I.; Islam, A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics 2022, 14, 2657. [Google Scholar] [CrossRef]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data from a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Chiang, M.C.; Cheng, Y.C.; Chen, S.J.; Yen, C.H.; Huang, R.N. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp. Cell Res. 2016, 347, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bemis, M.; Desilets, A.R. Role of Medium Chain Triglycerides (Axona®) in the Treatment of Mild to Moderate Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Demen. 2014, 29, 409–414. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic illustration of the main mitochondrial-related processes disturbed in Alzheimer´s disease. As is widely documented, Alzheimer’s disease (AD) (brain) is characterized by the loss of mitochondria homeostasis. This is mainly manifested by disturbances in the interconnected mechanisms of mitochondrial quality control (autophagy and mitochondrial biogenesis), mitochondrial axonal transport, fission/fusion processes and bioenergetics (energy production), and reactive oxygen species production (ROS). I—Complex I of the mitochondrial respiratory chain; II—Complex II of the mitochondrial respiratory chain; III—Complex III of the mitochondrial respiratory chain; IV—Complex IV of the mitochondrial respiratory chain; V—Complex V of the mitochondrial respiratory chain.
Figure 1.
A schematic illustration of the main mitochondrial-related processes disturbed in Alzheimer´s disease. As is widely documented, Alzheimer’s disease (AD) (brain) is characterized by the loss of mitochondria homeostasis. This is mainly manifested by disturbances in the interconnected mechanisms of mitochondrial quality control (autophagy and mitochondrial biogenesis), mitochondrial axonal transport, fission/fusion processes and bioenergetics (energy production), and reactive oxygen species production (ROS). I—Complex I of the mitochondrial respiratory chain; II—Complex II of the mitochondrial respiratory chain; III—Complex III of the mitochondrial respiratory chain; IV—Complex IV of the mitochondrial respiratory chain; V—Complex V of the mitochondrial respiratory chain.

Figure 2.
Simplistic representation of current strategies aimed at targeting Alzheimer´s disease faulty mitochondria. Regarding the Alzheimer´s disease (AD) pathophysiology, mitochondria decline arises as a common denominator in the majority of dedicated studies. In view of this evidence, preserving and/or restoring mitochondria’s health and function can represent the primary means to achieve advances to tackle AD. Thus far, a wealth of research has been dedicated to developing mitochondria-targeted therapeutics that could strengthen/restore the multifaceted functions of mitochondria to circumvent AD’s cascade of events. Herein, we show mitochondrial-directed antioxidants, mitochondrial-derived peptides, mitochondrial dynamics and mitophagy-targeting compounds, mitochondrial uncoupling, and mitochondrial transplantation as disease-modifying strategies in AD pathophysiology. Aβ—abeta peptide; AMPK—AMP-activated protein kinase; GSK3β—glycogen synthase kinase 3beta; PGC-1α—peroxisome proliferator activated receptor-gamma (PPARγ) coactivator-1alpha; PINK1—PTEN-induced kinase 1; ROCK—Rho-associated protein kinase; ROS—reactive oxygen species; STAT3—signal transducer and activator of transcription 3. ↑ Increase; ↓ Decrease.
Figure 2.
Simplistic representation of current strategies aimed at targeting Alzheimer´s disease faulty mitochondria. Regarding the Alzheimer´s disease (AD) pathophysiology, mitochondria decline arises as a common denominator in the majority of dedicated studies. In view of this evidence, preserving and/or restoring mitochondria’s health and function can represent the primary means to achieve advances to tackle AD. Thus far, a wealth of research has been dedicated to developing mitochondria-targeted therapeutics that could strengthen/restore the multifaceted functions of mitochondria to circumvent AD’s cascade of events. Herein, we show mitochondrial-directed antioxidants, mitochondrial-derived peptides, mitochondrial dynamics and mitophagy-targeting compounds, mitochondrial uncoupling, and mitochondrial transplantation as disease-modifying strategies in AD pathophysiology. Aβ—abeta peptide; AMPK—AMP-activated protein kinase; GSK3β—glycogen synthase kinase 3beta; PGC-1α—peroxisome proliferator activated receptor-gamma (PPARγ) coactivator-1alpha; PINK1—PTEN-induced kinase 1; ROCK—Rho-associated protein kinase; ROS—reactive oxygen species; STAT3—signal transducer and activator of transcription 3. ↑ Increase; ↓ Decrease.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sousa, T.; Moreira, P.I.; Cardoso, S. Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease. Biomedicines 2023, 11, 2331. https://doi.org/10.3390/biomedicines11092331
AMA Style
Sousa T, Moreira PI, Cardoso S. Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease. Biomedicines. 2023; 11(9):2331. https://doi.org/10.3390/biomedicines11092331
Chicago/Turabian StyleSousa, Tiago, Paula I. Moreira, and Susana Cardoso. 2023. "Current Advances in Mitochondrial Targeted Interventions in Alzheimer’s Disease" Biomedicines 11, no. 9: 2331. https://doi.org/10.3390/biomedicines11092331
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.