
A Protein Microarray-Based Respiratory Viral Antigen Testing Platform for COVID-19 Surveillance
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Recombinant Proteins and Antibodies
2.2. Protein Biotinylation
2.3. Inactivated SARS-CoV-2 Samples and SARS-CoV-2 Clinical Samples
2.4. Influenza A Virus Culture and Virus Titer Measurement
2.5. Protein Microarray Printing
2.6. Fluorescence Signal Development and Reagent Screening
2.7. Antibody Pair Screening for Sandwich Immunoassays on Protein Microarrays
2.8. Clinical SARS-CoV-2 Samples and Multiplex Viral Antigen Detection
2.9. Protein Microarray Imaging and Data Analysis
2.10. Statistical Methods
3. Results
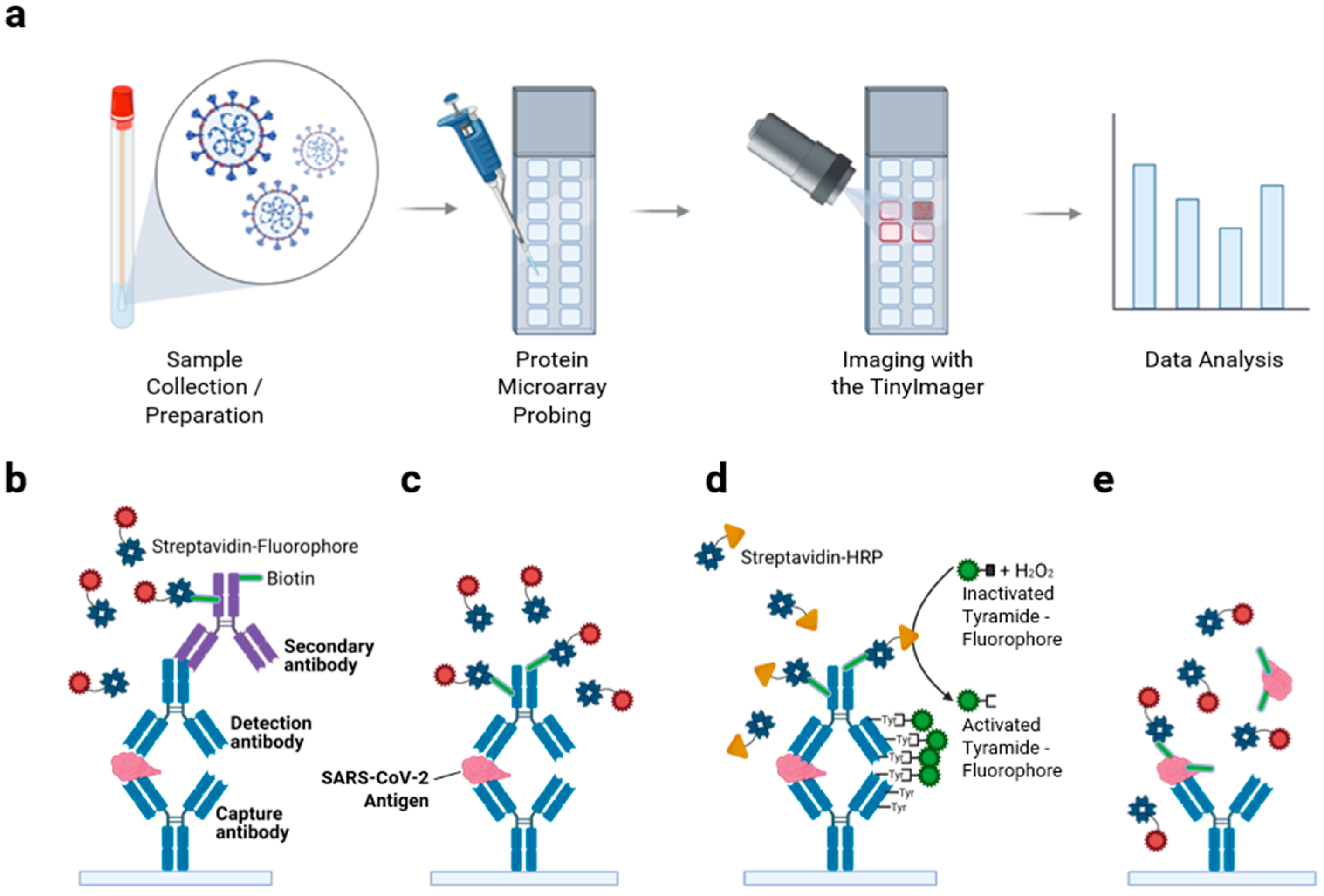
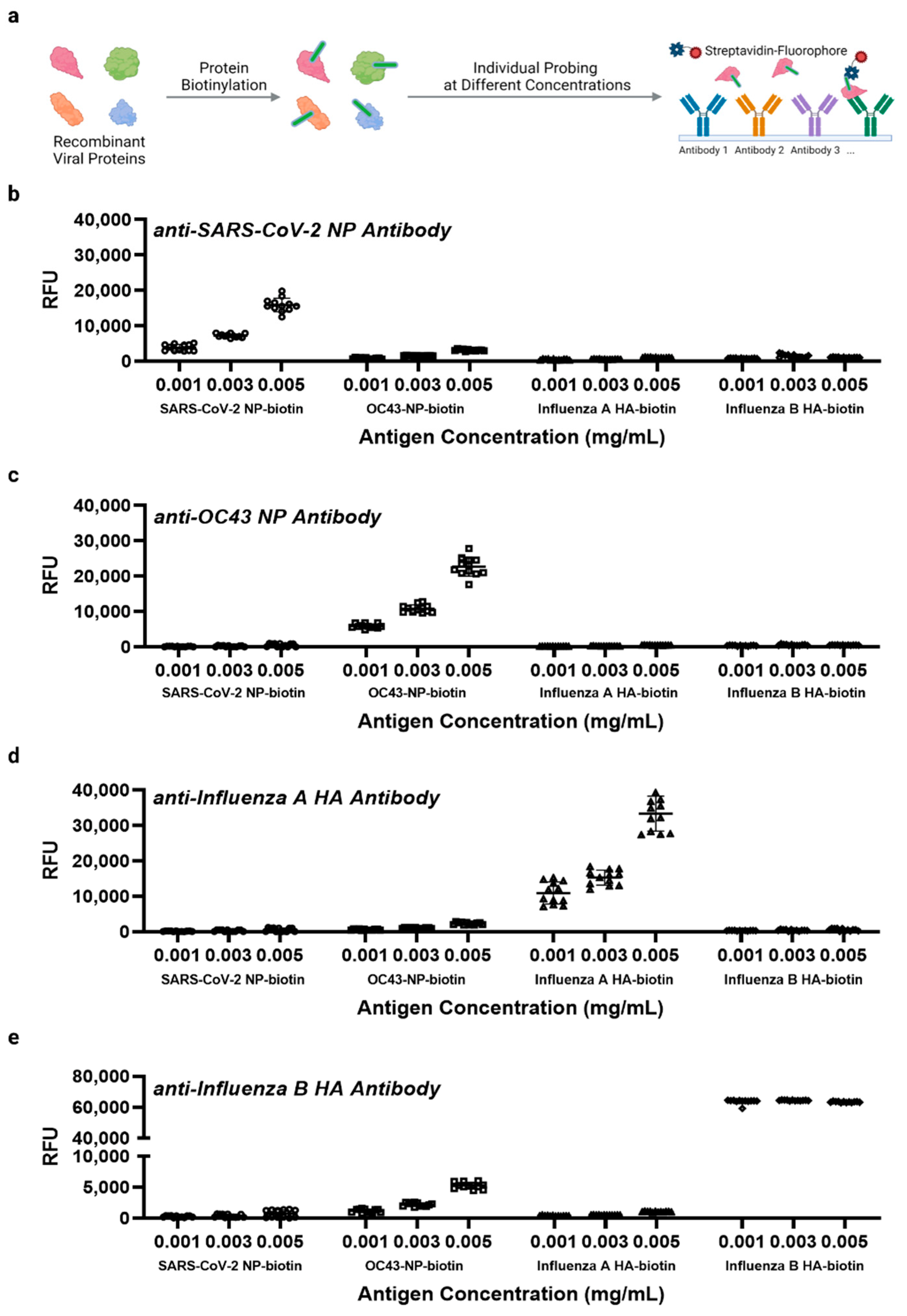
3.1. Protein Microarray Design Considerations and Assay Workflow for Viral Antigen Detection

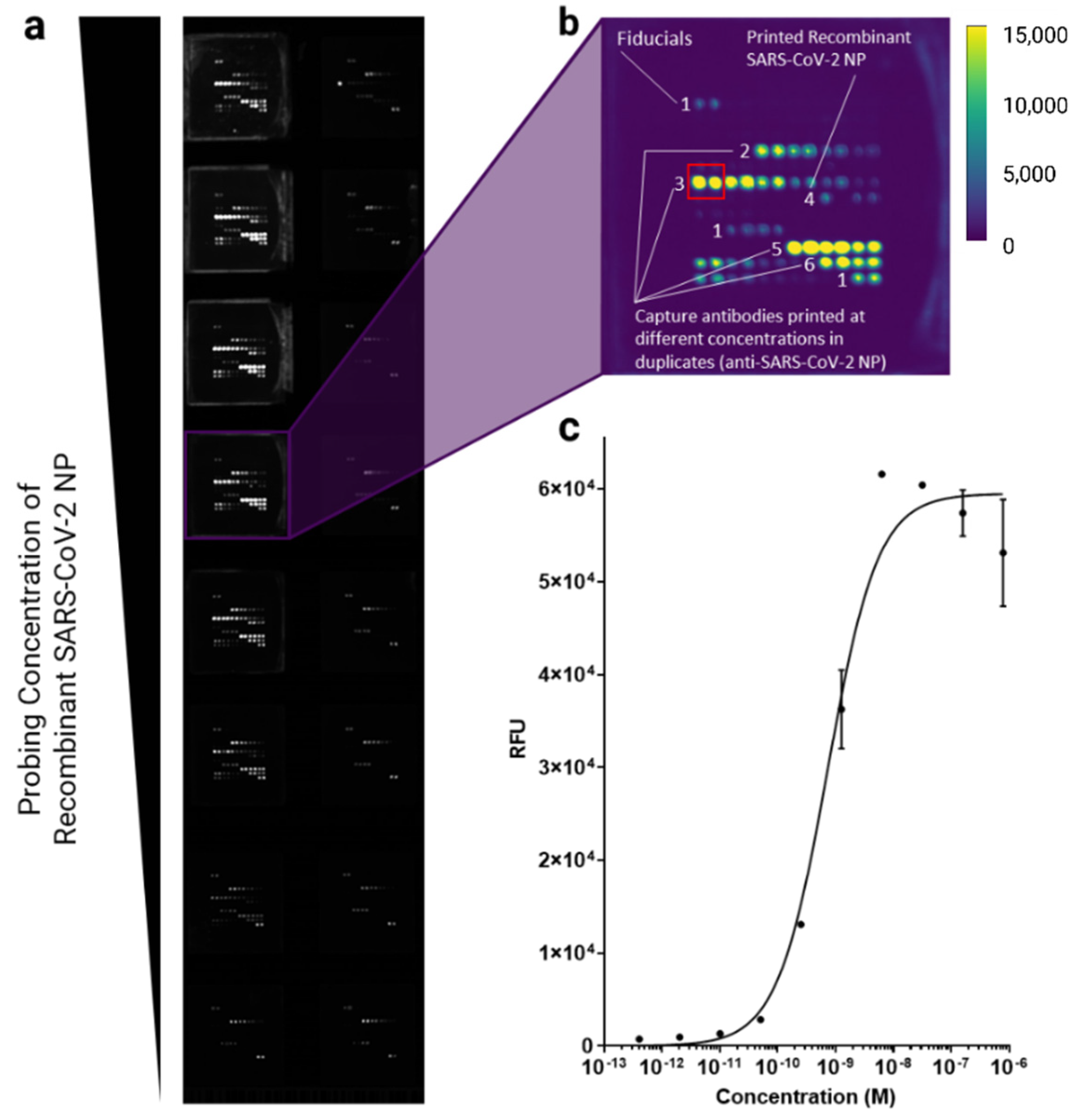
3.2. Screening for Optimal Anti-SARS-CoV-2 Antibodies
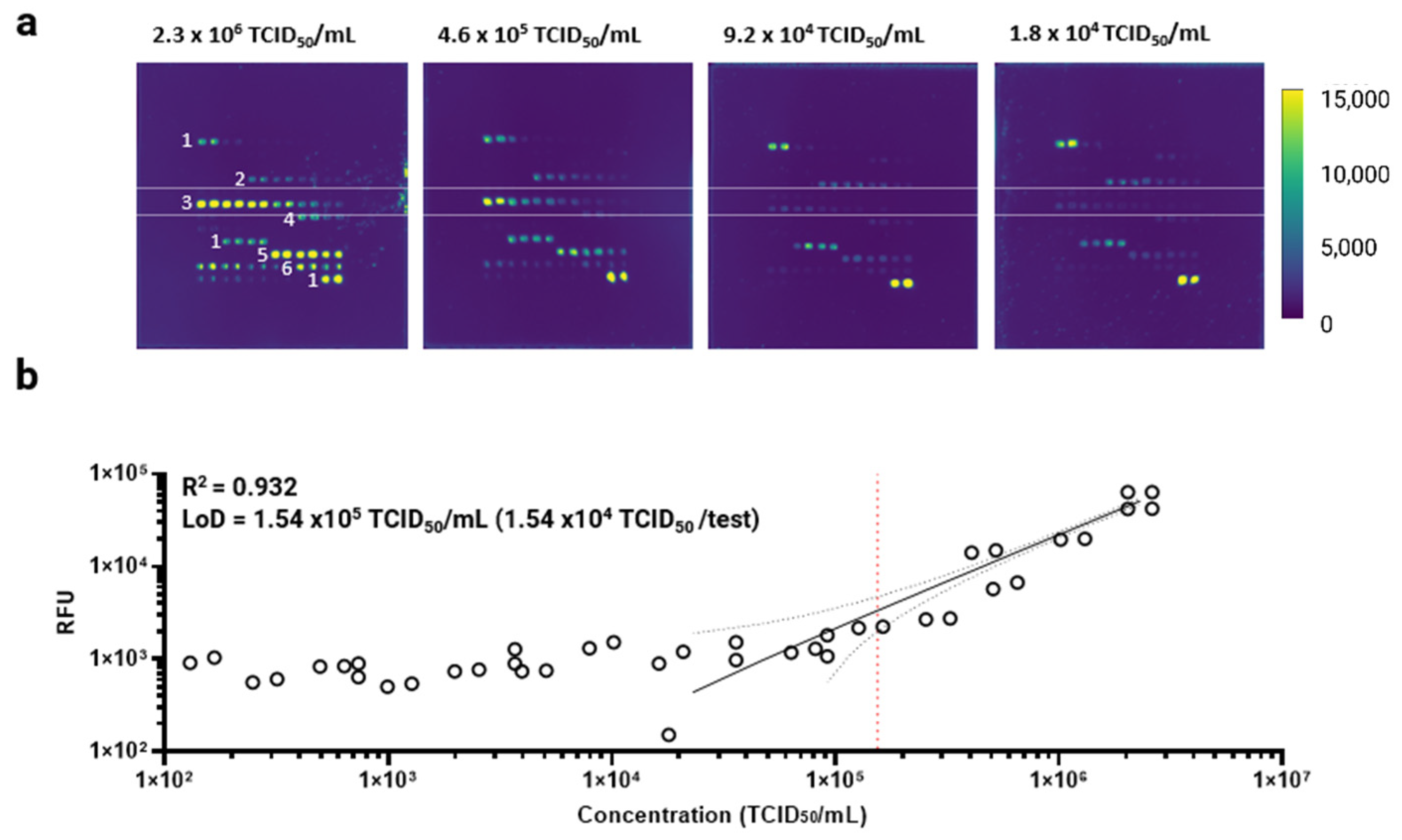
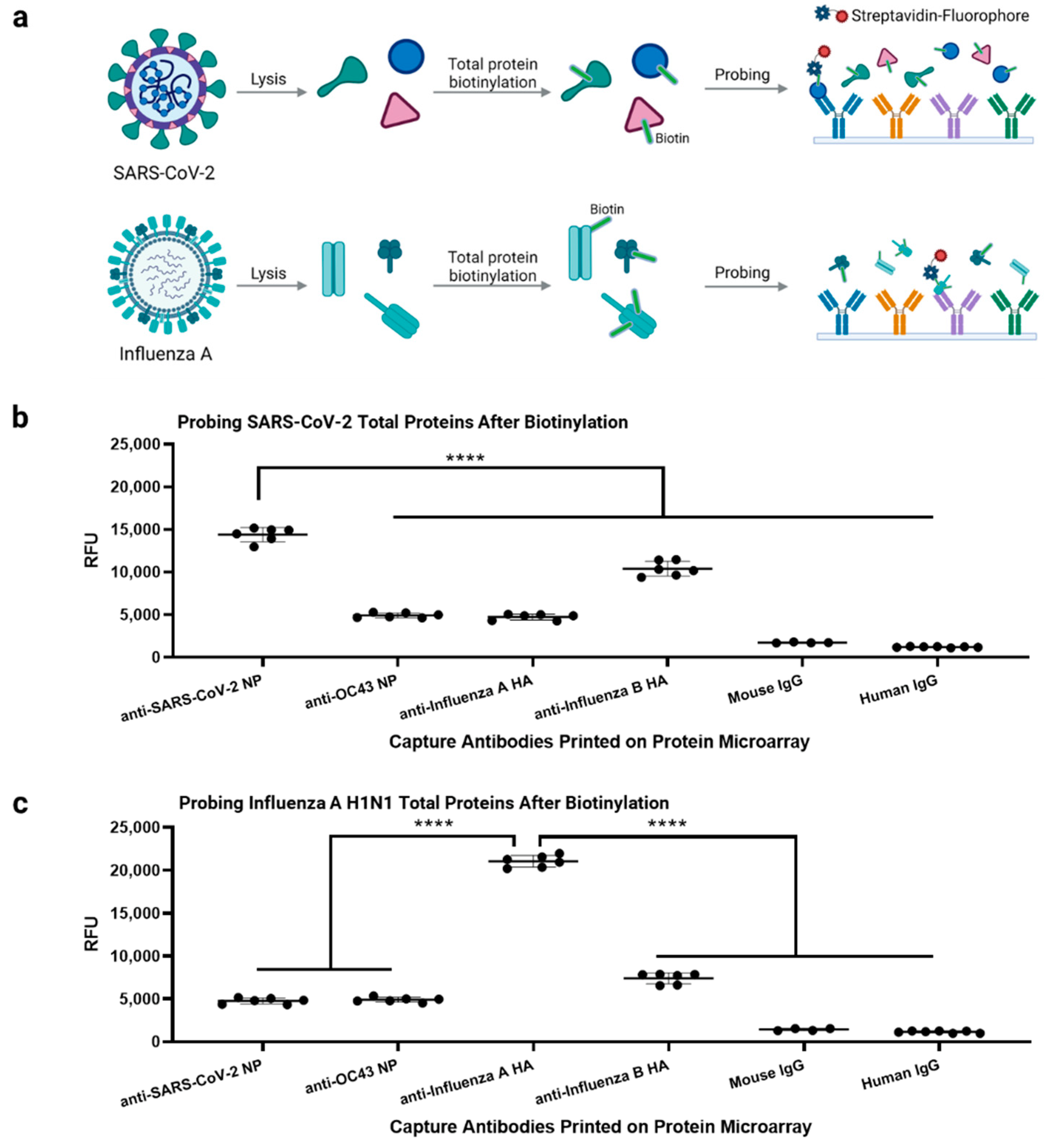
3.3. Antibody Microarray Characterization Using Inactivated SARS-CoV-2 Samples

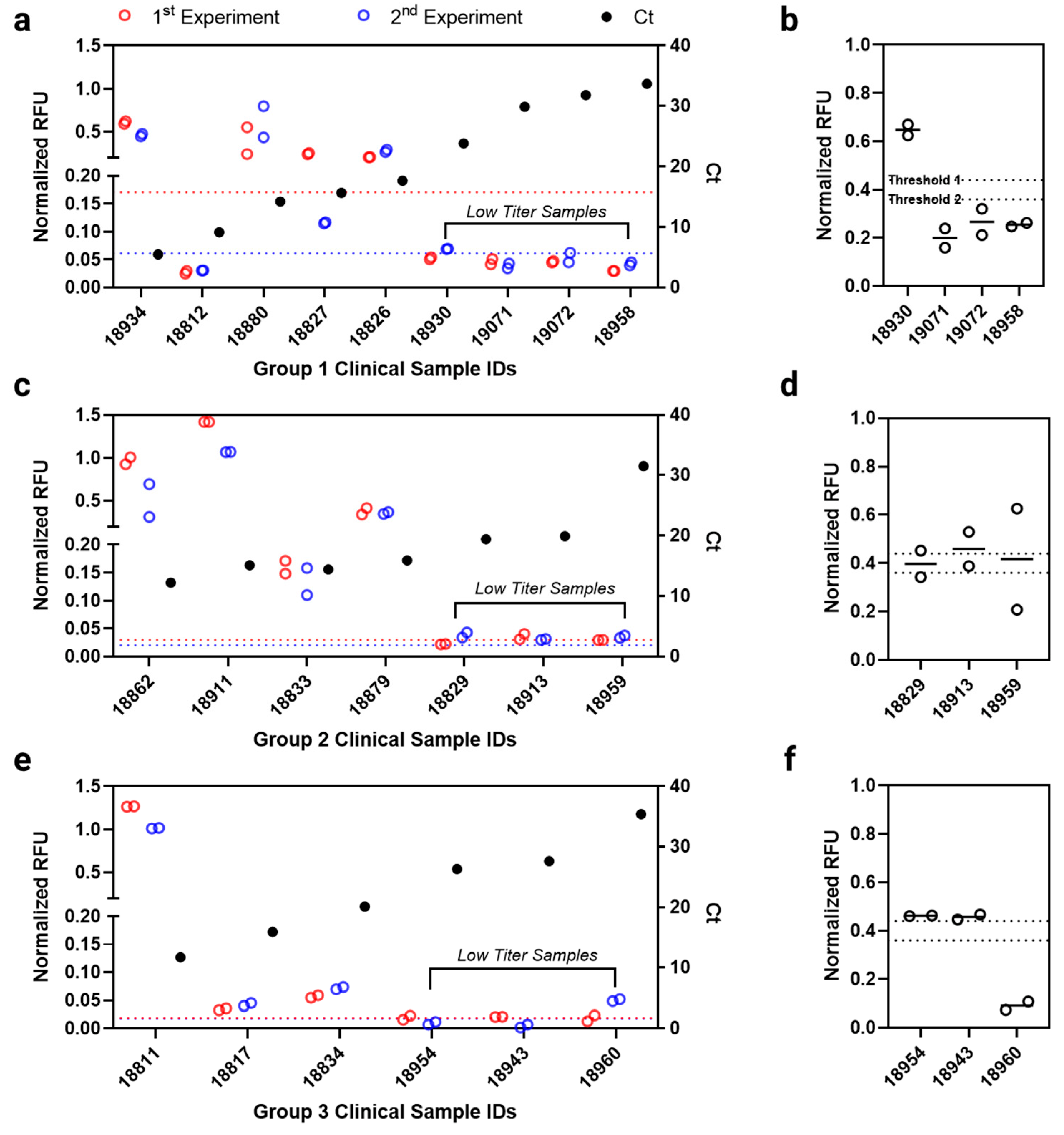
3.4. Antibody Microarray Test Evaluation with Clinical Samples

3.5. Multiplexed Antibody Microarray Test for Respiratory Viruses


4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kevadiya, B.D.; Machhi, J.; Herskovitz, J.; Oleynikov, M.D.; Blomberg, W.R.; Bajwa, N.; Soni, D.; Das, S.; Hasan, M.; Patel, M. Diagnostics for SARS-CoV-2 infections. Nat. Mater. 2021, 20, 593–605. [Google Scholar] [CrossRef]
- Yuan, X.; Yang, C.; He, Q.; Chen, J.; Yu, D.; Li, J.; Zhai, S.; Qin, Z.; Du, K.; Chu, Z. Current and perspective diagnostic techniques for COVID-19. ACS Infect. Dis. 2020, 6, 1998–2016. [Google Scholar] [CrossRef]
- Rabiee, N.; Akhavan, O.; Fatahi, Y.; Ghadiri, A.M.; Kiani, M.; Makvandi, P.; Rabiee, M.; Nicknam, M.H.; Saeb, M.R.; Varma, R.S. CaZnO-based nanoghosts for the detection of ssDNA, pCRISPR and recombinant SARS-CoV-2 spike antigen and targeted delivery of doxorubicin. Chemosphere 2022, 306, 135578. [Google Scholar] [CrossRef]
- Afshari, R.; Akhavan, O.; Hamblin, M.R.; Varma, R.S. Review of oxygenation with nanobubbles: Possible treatment for hypoxic COVID-19 patients. ACS Appl. Nano Mater. 2021, 4, 11386–11412. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Asadi, M.; Akhavan, O. Graphene-based nanomaterials in fighting the most challenging viruses and immunogenic disorders. ACS Biomater. Sci. Eng. 2021, 8, 54–81. [Google Scholar] [CrossRef]
- FDA. In Vitro Diagnostics EUAs-Antigen Diagnostic Tests for SARS-CoV-2. Available online: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-antigen-diagnostic-tests-sars-cov-2#IndividualEUAs (accessed on 15 February 2022).
- Dinnes, J.; Sharma, P.; Berhane, S.; van Wyk, S.S.; Nyaaba, N.; Domen, J.; Taylor, M.; Cunningham, J.; Davenport, C.; Dittrich, S. Rapid, point-of-care antigen tests for diagnosis of SARS-CoV-2 infection. Cochrane Database Syst. Rev. 2022, CD013705. [Google Scholar]
- Giberti, I.; Costa, E.; Domnich, A.; Ricucci, V.; De Pace, V.; Garzillo, G.; Guarona, G.; Icardi, G. High Diagnostic Accuracy of a Novel Lateral Flow Assay for the Point-of-Care Detection of SARS-CoV-2. Biomedicines 2022, 10, 1558. [Google Scholar] [CrossRef]
- Thapa, D.; Samadi, N.; Baker, A.; Santos, C.d.; Trahtemberg, U.; Tabatabaei, N. Rapid and Low-Cost Detection and Quantification of SARS-CoV-2 Antibody Titers of ICU Patients with Respiratory Deterioration Using a Handheld Thermo-Photonic Device. Biomedicines 2022, 10, 1424. [Google Scholar] [CrossRef]
- Mokhtary, P.; Pourhashem, Z.; Mehrizi, A.A.; Sala, C.; Rappuoli, R. Recent Progress in the Discovery and Development of Monoclonal Antibodies against Viral Infections. Biomedicines 2022, 10, 1861. [Google Scholar] [CrossRef]
- FDA. In Vitro Diagnostics EUAs-Molecular Diagnostic Tests for SARS-CoV-2. Available online: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-molecular-diagnostic-tests-sars-cov-2 (accessed on 15 February 2022).
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. Science Forum: SARS-CoV-2 (COVID-19) by the numbers. eLife 2020, 9, e57309. [Google Scholar] [CrossRef]
- Kingsmore, S.F. Multiplexed protein measurement: Technologies and applications of protein and antibody arrays. Nat. Rev. Drug Discov. 2006, 5, 310–321. [Google Scholar] [CrossRef] [Green Version]
- Karoonuthaisiri, N.; Charlermroj, R.; Uawisetwathana, U.; Luxananil, P.; Kirtikara, K.; Gajanandana, O. Development of antibody array for simultaneous detection of foodborne pathogens. Biosens. Bioelectron. 2009, 24, 1641–1648. [Google Scholar] [CrossRef]
- Lian, W.; Wu, D.; Lim, D.V.; Jin, S. Sensitive detection of multiplex toxins using antibody microarray. Anal. Biochem. 2010, 401, 271–279. [Google Scholar] [CrossRef]
- Rucker, V.C.; Havenstrite, K.L.; Herr, A.E. Antibody microarrays for native toxin detection. Anal. Biochem. 2005, 339, 262–270. [Google Scholar] [CrossRef]
- Cretich, M.; Damin, F.; Chiari, M. Protein microarray technology: How far off is routine diagnostics? Analyst 2014, 139, 528–542. [Google Scholar] [CrossRef]
- De Assis, R.R.; Jain, A.; Nakajima, R.; Jasinskas, A.; Felgner, J.; Obiero, J.M.; Norris, P.J.; Stone, M.; Simmons, G.; Bagri, A. Analysis of SARS-CoV-2 antibodies in COVID-19 convalescent blood using a coronavirus antigen microarray. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Assis, R.; Jain, A.; Nakajima, R.; Jasinskas, A.; Khan, S.; Palma, A.; Parker, D.M.; Chau, A.; Obiero, J.M.; Tifrea, D. Distinct SARS-CoV-2 antibody reactivity patterns elicited by natural infection and mRNA vaccination. NPJ Vaccines 2021, 6, 1–10. [Google Scholar] [CrossRef]
- Hedde, P.N.; Abram, T.J.; Jain, A.; Nakajima, R.; de Assis, R.R.; Pearce, T.; Jasinskas, A.; Toosky, M.N.; Khan, S.; Felgner, P.L. A modular microarray imaging system for highly specific COVID-19 antibody testing. Lab A Chip 2020, 20, 3302–3309. [Google Scholar] [CrossRef]
- Patterson, E.I.; Prince, T.; Anderson, E.R.; Casas-Sanchez, A.; Smith, S.L.; Cansado-Utrilla, C.; Solomon, T.; Griffiths, M.J.; Acosta-Serrano, Á.; Turtle, L. Methods of inactivation of SARS-CoV-2 for downstream biological assays. J. Infect. Dis. 2020, 222, 1462–1467. [Google Scholar] [CrossRef]
- Hernandez-Davies, J.E.; Felgner, J.; Strohmeier, S.; Pone, E.J.; Jain, A.; Jan, S.; Nakajima, R.; Jasinskas, A.; Strahsburger, E.; Krammer, F. Administration of Multivalent Influenza Virus Recombinant Hemagglutinin Vaccine in Combination-Adjuvant Elicits Broad Reactivity Beyond the Vaccine Components. Front. Immunol. 2021, 12, 2695. [Google Scholar] [CrossRef]
- Gross, F.L.; Bai, Y.; Jefferson, S.; Holiday, C.; Levine, M.Z. Measuring influenza neutralizing antibody responses to A (H3N2) viruses in human sera by microneutralization assays using MDCK-SIAT1 cells. JoVE (J. Vis. Exp.) 2017, 129, e56448. [Google Scholar] [CrossRef] [PubMed]
- Brauer, R.; Chen, P. Influenza virus propagation in embryonated chicken eggs. JoVE (J. Vis. Exp.) 2015, 97, e52421. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.H.; Liang, X.; Hernandez, J.E.; Randall, A.; Hirst, S.; Mu, Y.; Romero, K.M.; Nguyen, T.T.; Kalantari-Dehaghi, M.; Crotty, S. Profiling the humoral immune response to infection by using proteome microarrays: High-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. USA 2005, 102, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jain, A.; Taghavian, O.; Nakajima, R.; Jasinskas, A.; Supnet, M.; Felgner, J.; Davies, J.; de Assis, R.R.; Jan, S. Use of an influenza antigen microarray to measure the breadth of serum antibodies across virus subtypes. JoVE (J. Vis. Exp.) 2019, 149, e59973. [Google Scholar] [CrossRef]
- Haab, B.B.; Dunham, M.J.; Brown, P.O. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001, 2, 1–13. [Google Scholar] [CrossRef]
- Wisdom, G.B. Enzyme-immunoassay. Clin. Chem. 1976, 22, 1243–1255. [Google Scholar] [CrossRef]
- Wang, G.; Achim, C.L.; Hamilton, R.L.; Wiley, C.A.; Soontornniyomkij, V. Tyramide signal amplification method in multiple-label immunofluorescence confocal microscopy. Methods 1999, 18, 459–464. [Google Scholar] [CrossRef]
- Bobrow, M.N.; Harris, T.D.; Shaughnessy, K.J.; Litt, G.J. Catalyzed reporter deposition, a novel method of signal amplification application to immunoassays. J. Immunol. Methods 1989, 125, 279–285. [Google Scholar] [CrossRef]
- Hogan, C.A.; Sahoo, M.K.; Pinsky, B.A. Sample pooling as a strategy to detect community transmission of SARS-CoV-2. JAMA 2020, 323, 1967–1969. [Google Scholar] [CrossRef]
- Bourassa, L.; Perchetti, G.A.; Phung, Q.; Lin, M.J.; Mills, M.G.; Roychoudhury, P.; Harmon, K.G.; Reed, J.C.; Greninger, A.L. A SARS-CoV-2 nucleocapsid variant that affects antigen test performance. J. Clin. Virol. 2021, 141, 104900. [Google Scholar] [CrossRef]
- Shan, D.; Johnson, J.M.; Fernandes, S.C.; Suib, H.; Hwang, S.; Wuelfing, D.; Mendes, M.; Holdridge, M.; Burke, E.M.; Beauregard, K. N-protein presents early in blood, dried blood and saliva during asymptomatic and symptomatic SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wen, K.; Zhang, J.; Chen, J.; Han, C.; Chen, Y.; Wang, S.; Deng, G.; Zhou, H.; Wu, Y. Accuracy of a nucleocapsid protein antigen rapid test in the diagnosis of SARS-CoV-2 infection. Clin. Microbiol. Infect. 2021, 27, 289.e1–289.e4. [Google Scholar] [CrossRef] [PubMed]
- Wild, D. The Immunoassay Handbook: Theory and Applications of Ligand Binding, ELISA and Related Techniques; Newnes: Oxford, UK, 2013. [Google Scholar]
- Dudley, R.; Edwards, P.; Ekins, R.; Finney, D.; McKenzie, I.; Raab, G.; Rodbard, D.; Rodgers, R. Guidelines for immunoassay data processing. Clin. Chem. 1985, 31, 1264–1271. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2: Structure, biology, and structure-based therapeutics development. Front. Cell. Infect. Microbiol. 2020, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Dell’Angelica, E.C.; Springer, T.A. Immunoprecipitation. Curr. Protoc. Protein Sci. 1999, 18, 9.8.1–9.8.28. [Google Scholar] [CrossRef]
- Shidlovskaya, E.V.; Kuznetsova, N.A.; Divisenko, E.V.; Nikiforova, M.A.; Siniavin, A.E.; Ogarkova, D.A.; Shagaev, A.V.; Semashko, M.A.; Tkachuk, A.P.; Burgasova, O.A. The Value of Rapid Antigen Tests for Identifying Carriers of Viable SARS-CoV-2. Viruses 2021, 13, 2012. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Haage, V.C.; Bleicker, T.; Schmidt, M.L.; Mühlemann, B.; Zuchowski, M.; Jo, W.K.; Tscheak, P.; Möncke-Buchner, E.; Müller, M.A. Comparison of seven commercial SARS-CoV-2 rapid point-of-care antigen tests: A single-centre laboratory evaluation study. Lancet Microbe 2021, 2, e311–e319. [Google Scholar] [CrossRef]
- Lee, J.-H.; Jung, Y.; Lee, S.-K.; Kim, J.; Lee, C.-S.; Kim, S.; Lee, J.-S.; Kim, N.-H.; Kim, H.-G. Rapid Biosensor of SARS-CoV-2 Using Specific Monoclonal Antibodies Recognizing Conserved Nucleocapsid Protein Epitopes. Viruses 2022, 14, 255. [Google Scholar] [CrossRef]
- Pastorino, B.; Touret, F.; Gilles, M.; de Lamballerie, X.; Charrel, R.N. Heat inactivation of different types of SARS-CoV-2 samples: What protocols for biosafety, molecular detection and serological diagnostics? Viruses 2020, 12, 735. [Google Scholar] [CrossRef]
- WHO. Antigen-Detection in the Diagnosis of SARS-CoV-2 Infection. Available online: https://www.who.int/publications/i/item/antigen-detection-in-the-diagnosis-of-sars-cov-2infection-using-rapid-immunoassays (accessed on 15 February 2022).
- Pickering, S.; Batra, R.; Merrick, B.; Snell, L.B.; Nebbia, G.; Douthwaite, S.; Reid, F.; Patel, A.; Ik, M.T.K.; Patel, B. Comparative performance of SARS-CoV-2 lateral flow antigen tests and association with detection of infectious virus in clinical specimens: A single-centre laboratory evaluation study. Lancet Microbe 2021, 2, e461–e471. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A virus cell entry, replication, virion assembly and movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martínez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Velumani, S.; Du, Q.; Lim, C.W.; Ng, F.K.; Donis, R.; Kwang, J. Detection of H5 avian influenza viruses by antigen-capture enzyme-linked immunosorbent assay using H5-specific monoclonal antibody. Clin. Vaccine Immunol. 2007, 14, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Kampf, G.; Lemmen, S.; Suchomel, M. Ct values and infectivity of SARS-CoV-2 on surfaces. Lancet Infect. Dis. 2021, 21, e141. [Google Scholar] [CrossRef]
- Coyle, P.; Al Molawi, N.H.; Kacem, M.A.B.H.; El Kahlout, R.A.; Al Kuwari, E.; Al Khal, A.; Gilliani, I.; Jeremijenko, A.; Saeb, H.; Al Thani, S.M. Inclusion of cycle threshold (CT) values when reporting SARS-CoV-2 RT-PCR results improves clinical Interpretation in suspected and confirmed COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- CDC. Interim Guidance for Antigen Testing for SARS-CoV-2. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/resources/antigen-tests-guidelines.html (accessed on 15 February 2022).
- Plotnikova, M.A.; Klotchenko, S.A.; Lebedev, K.I.; Lozhkov, A.A.; Taraskin, A.S.; Gyulikhandanova, N.E.; Ramsay, E.S.; Vasin, A.V. Antibody microarray immunoassay for screening and differential diagnosis of upper respiratory tract viral pathogens. J. Immunol. Methods 2020, 478, 112712. [Google Scholar] [CrossRef] [PubMed]
- Boum, Y.; Fai, K.N.; Nikolay, B.; Mboringong, A.B.; Bebell, L.M.; Ndifon, M.; Abbah, A.; Essaka, R.; Eteki, L.; Luquero, F. Performance and operational feasibility of antigen and antibody rapid diagnostic tests for COVID-19 in symptomatic and asymptomatic patients in Cameroon: A clinical, prospective, diagnostic accuracy study. Lancet Infect. Dis. 2021, 21, 1089–1096. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beck, S.; Nakajima, R.; Jasinskas, A.; Abram, T.J.; Kim, S.J.; Bigdeli, N.; Tifrea, D.F.; Hernandez-Davies, J.; Huw Davies, D.; Hedde, P.N.; et al. A Protein Microarray-Based Respiratory Viral Antigen Testing Platform for COVID-19 Surveillance. Biomedicines 2022, 10, 2238. https://doi.org/10.3390/biomedicines10092238
Beck S, Nakajima R, Jasinskas A, Abram TJ, Kim SJ, Bigdeli N, Tifrea DF, Hernandez-Davies J, Huw Davies D, Hedde PN, et al. A Protein Microarray-Based Respiratory Viral Antigen Testing Platform for COVID-19 Surveillance. Biomedicines. 2022; 10(9):2238. https://doi.org/10.3390/biomedicines10092238
Chicago/Turabian StyleBeck, Sungjun, Rie Nakajima, Algis Jasinskas, Timothy J. Abram, Sun Jin Kim, Nader Bigdeli, Delia F. Tifrea, Jenny Hernandez-Davies, D. Huw Davies, Per Niklas Hedde, and et al. 2022. "A Protein Microarray-Based Respiratory Viral Antigen Testing Platform for COVID-19 Surveillance" Biomedicines 10, no. 9: 2238. https://doi.org/10.3390/biomedicines10092238