
LLL12B, a Novel Small-Molecule STAT3 Inhibitor, Induces Apoptosis and Suppresses Cell Migration and Tumor Growth in Triple-Negative Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. SiRNA and Transfection
2.3. MTT Cell Viability Assay
2.4. Western Blot Analysis
2.5. Immunofluorescence Staining
2.6. Caspase-3/7 Activity Assay
2.7. Flow Cytometry Analysis
2.8. Colony Formation Assay
2.9. Wound Healing Assay
2.10. Orthotopic Mammary Fat Pad Tumor Model In Vivo
2.11. Statistical Analysis
3. Results
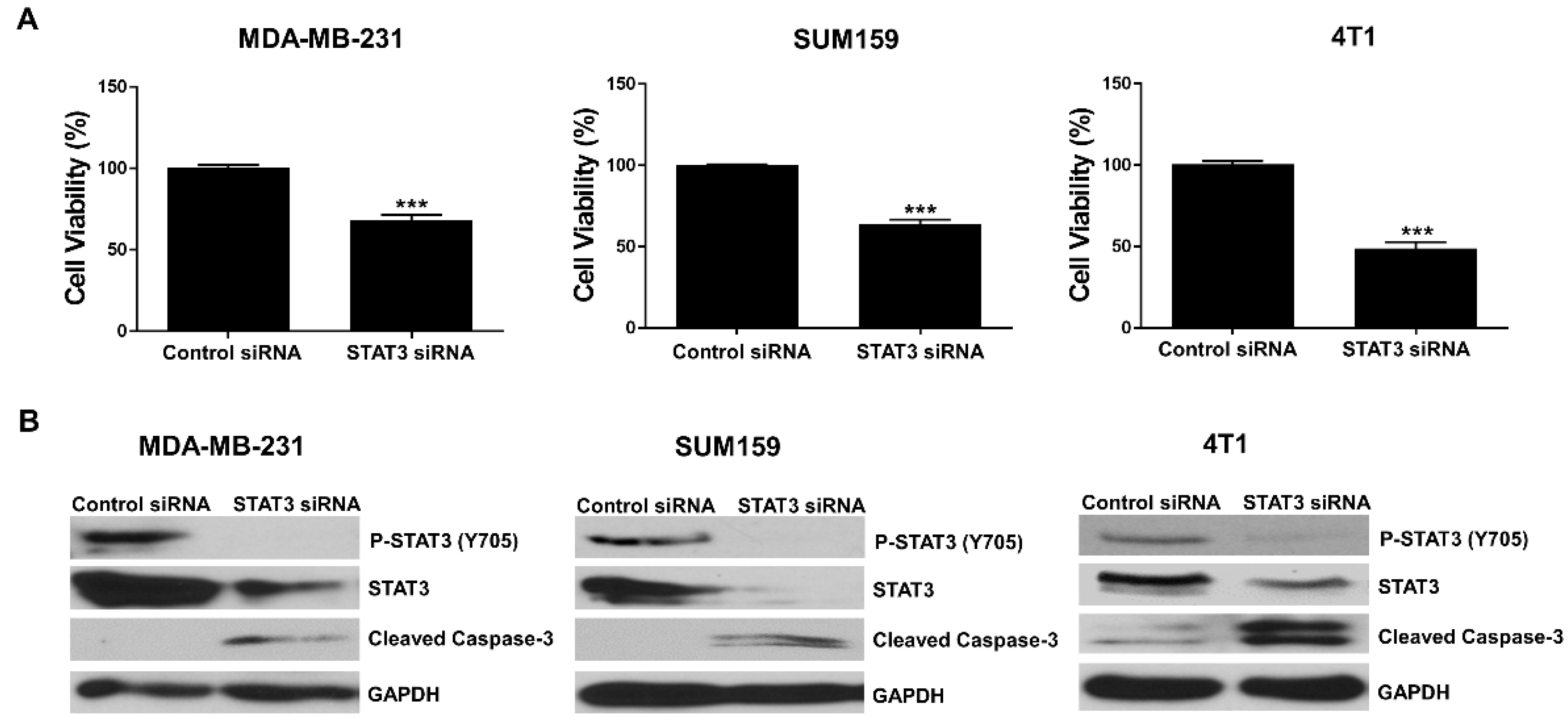
3.1. Knockdown of STAT3 Inhibits TNBC Cell Viability
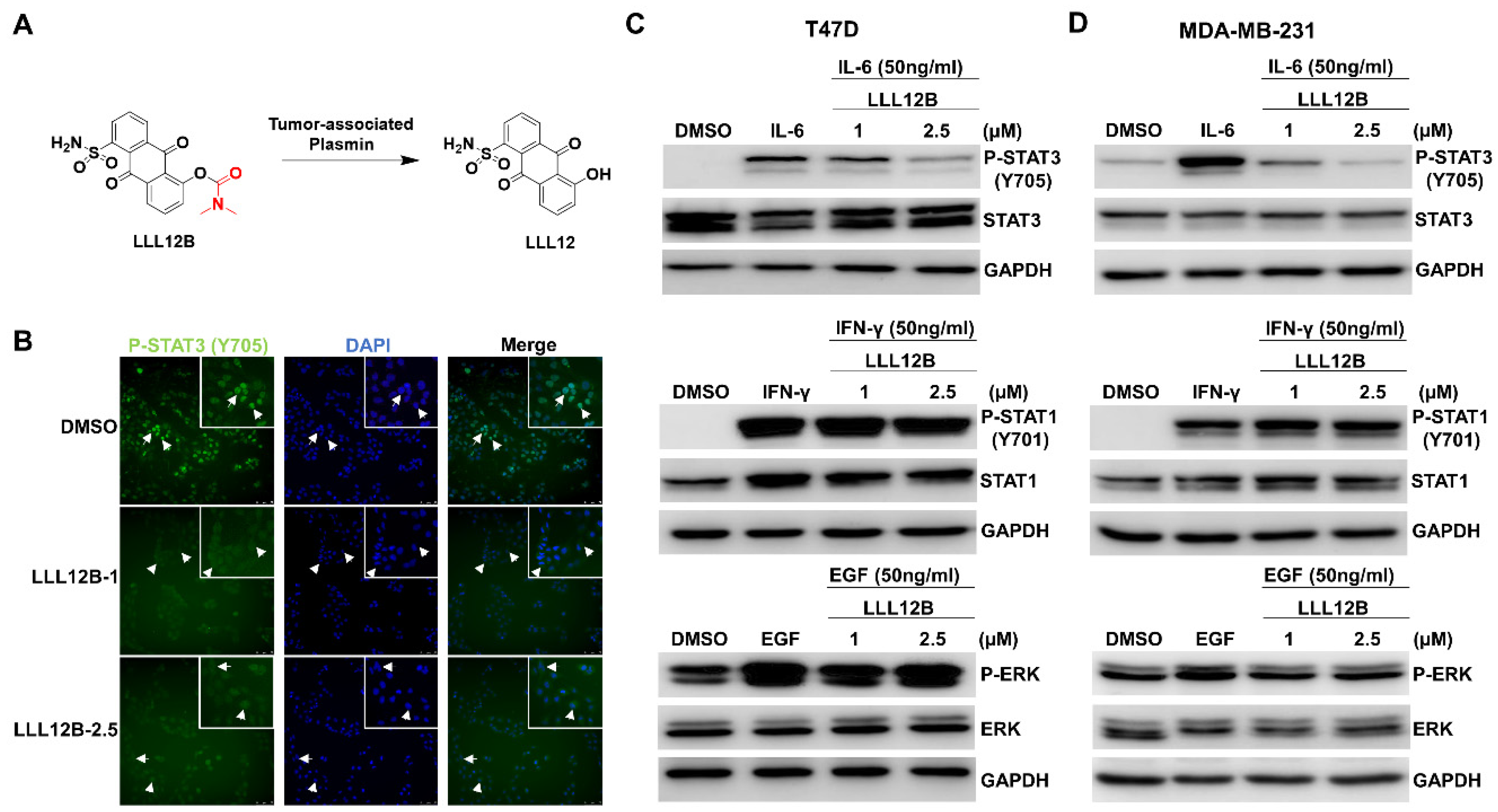
3.2. LLL12B Inhibits STAT3 Nuclear Translocation and Blocks IL-6-Induced STAT3 Phosphorylation
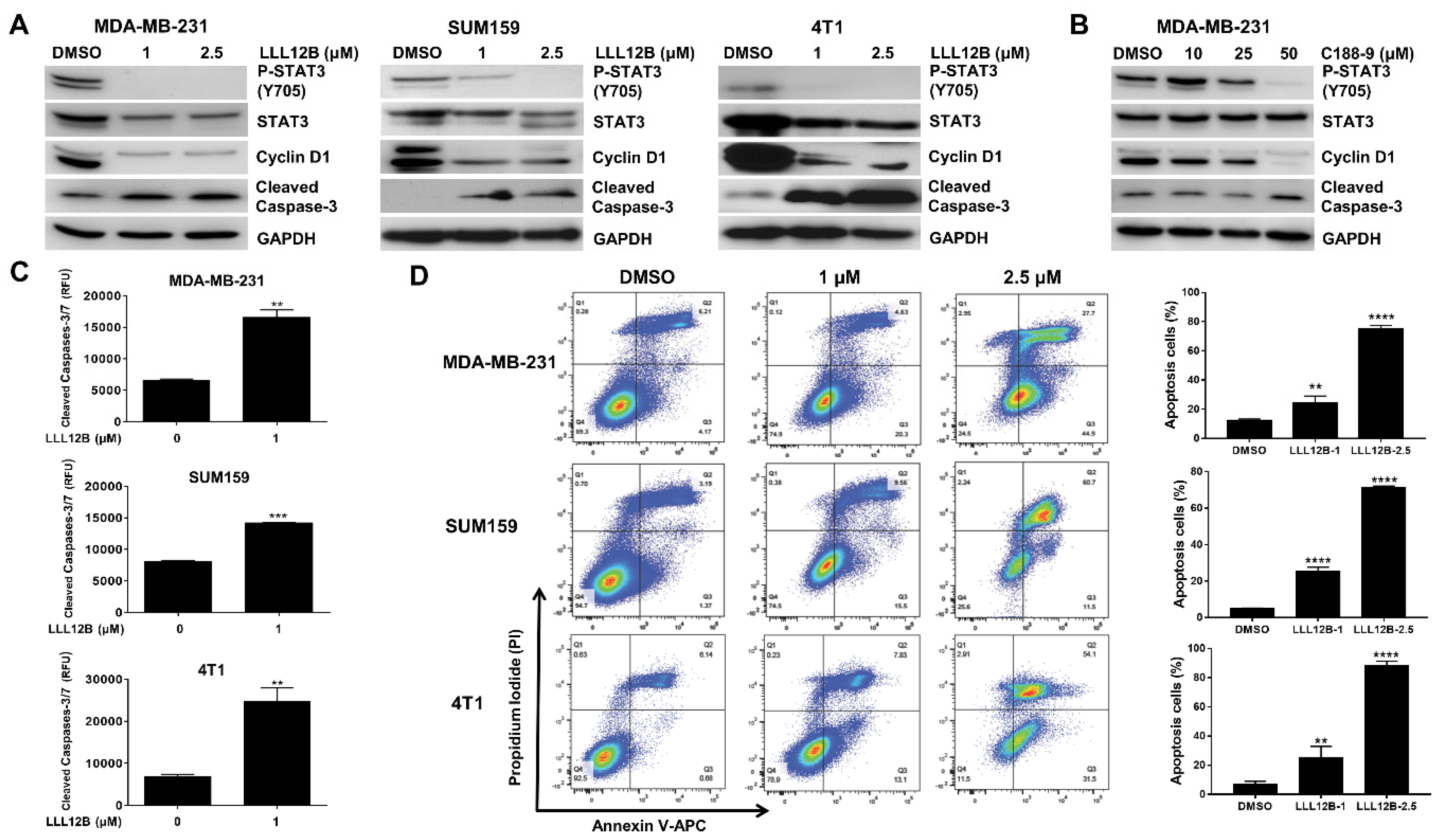
3.3. LLL12B Inhibits STAT3 Activation and Induces TNBC Cell Apoptosis
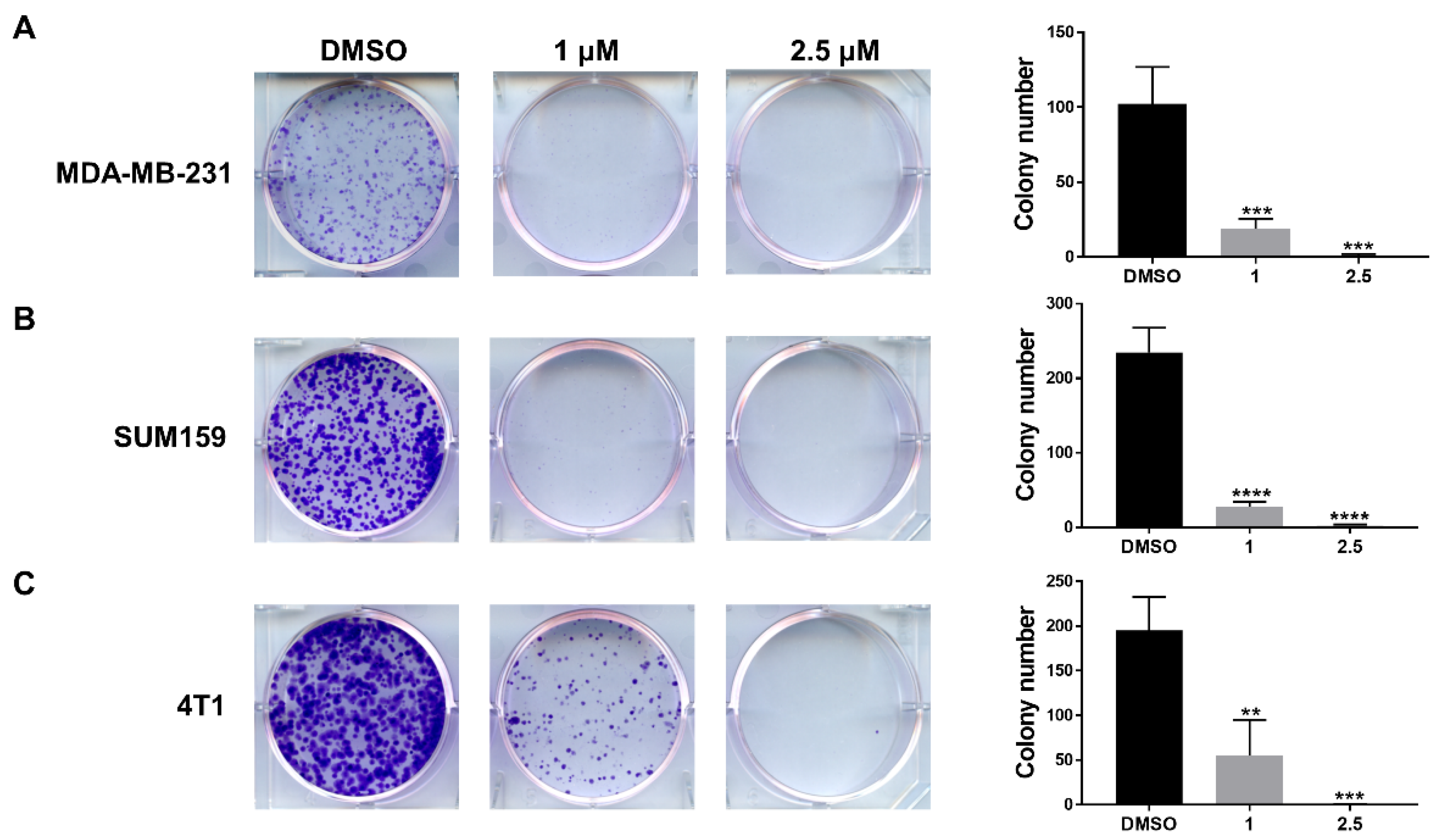
3.4. LLL12B Inhibits Colony Formation in the TNBC Cells
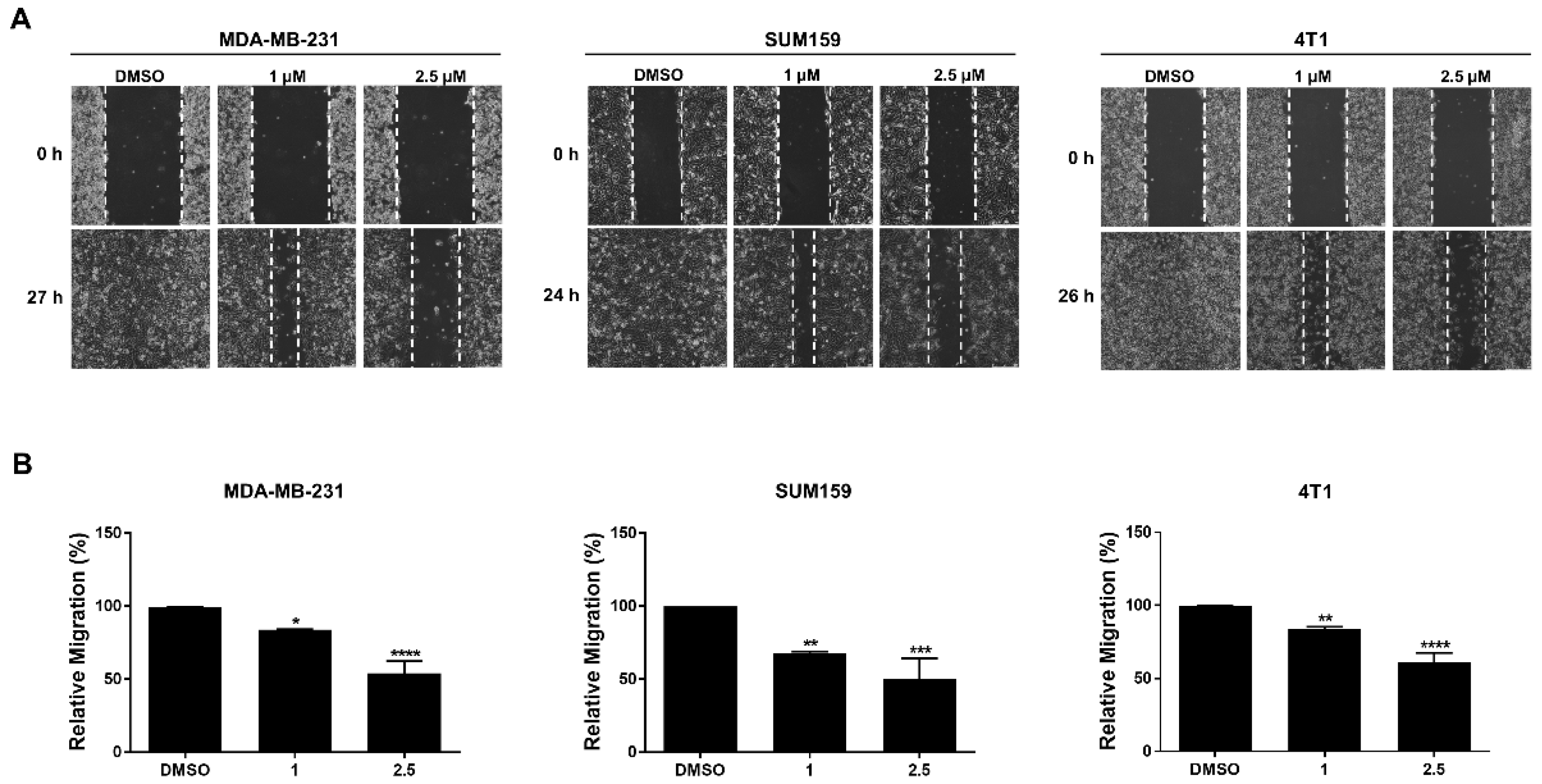
3.5. LLL12B Inhibits Cell Migration in the TNBC Cells
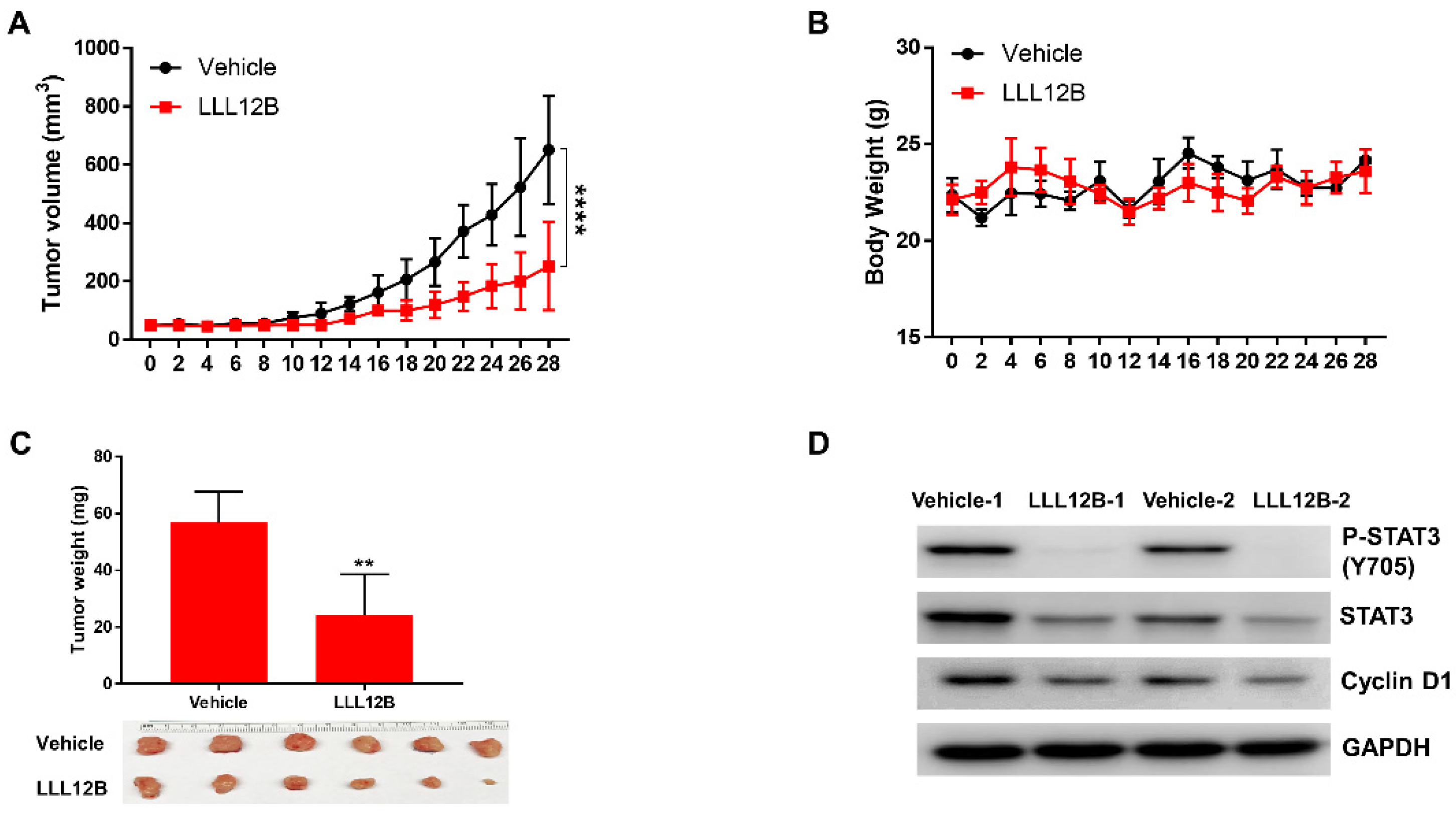
3.6. LLL12B Suppresses Tumor Growth in the MDA-MB-231 Orthotopic Tumor Model In Vivo
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.W.; Jimenez, C.R.; Boven, E. Breast cancer classification by proteomic technologies: Current state of knowledge. Cancer Treat. Rev. 2014, 40, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 212540. [Google Scholar] [CrossRef]
- Soare, G.R.; Soare, C.A. Immunotherapy for Breast Cancer: First FDA Approved Regimen. Discoveries 2019, 7, e91. [Google Scholar] [CrossRef]
- Bagegni, N.A.; Davis, A.A.; Clifton, K.K.; Ademuyiwa, F.O. Targeted Treatment for High-Risk Early-Stage Triple-Negative Breast Cancer: Spotlight on Pembrolizumab. Breast Cancer Targets Ther. (Dove Med. Press) 2022, 14, 113–123. [Google Scholar] [CrossRef]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef]
- Sporikova, Z.; Koudelakova, V.; Trojanec, R.; Hajduch, M. Genetic Markers in Triple-Negative Breast Cancer. Clin. Breast Cancer 2018, 18, e841–e850. [Google Scholar] [CrossRef]
- Won, K.A.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Qin, J.J.; Yan, L.; Zhang, J.; Zhang, W.D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, F.C.; Cheng, G.; Lin, J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem. Biophys. Res. Commun. 2005, 335, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Berishaj, M.; Gao, S.P.; Ahmed, S.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.L.; Bornmann, W.; Bromberg, J.F. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007, 9, R32. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Zhou, J.X.; Calvet, J.P.; Godwin, A.K.; Jensen, R.A.; Li, X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018, 9, 326. [Google Scholar] [CrossRef]
- McDaniel, J.M.; Varley, K.E.; Gertz, J.; Savic, D.S.; Roberts, B.S.; Bailey, S.K.; Shevde, L.A.; Ramaker, R.C.; Lasseigne, B.N.; Kirby, M.K.; et al. Genomic regulation of invasion by STAT3 in triple negative breast cancer. Oncotarget 2017, 8, 8226–8238. [Google Scholar] [CrossRef]
- Moreira, M.P.; da Conceição Braga, L.; Cassali, G.D.; Silva, L.M. STAT3 as a promising chemoresistance biomarker associated with the CD44(+/high)/CD24(−/low)/ALDH(+) BCSCs-like subset of the triple-negative breast cancer (TNBC) cell line. Exp. Cell Res. 2018, 363, 283–290. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef]
- Park, S.K.; Byun, W.S.; Lee, S.; Han, Y.T.; Jeong, Y.S.; Jang, K.; Chung, S.J.; Lee, J.; Suh, Y.G.; Lee, S.K. A novel small molecule STAT3 inhibitor SLSI-1216 suppresses proliferation and tumor growth of triple-negative breast cancer cells through apoptotic induction. Biochem. Pharm. 2020, 178, 114053. [Google Scholar] [CrossRef]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Beebe, J.D.; Liu, J.Y.; Zhang, J.T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharm. Ther. 2018, 191, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Li, C.; Zhang, W.; Xia, Y.; Li, S.; Lin, J.Y.; Yu, K.; Liu, M.; Yang, L.; Luo, J.; et al. Discovery of an Orally Selective Inhibitor of Signal Transducer and Activator of Transcription 3 Using Advanced Multiple Ligand Simultaneous Docking. J. Med. Chem. 2017, 60, 2718–2731. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Chen, X.; Fu, S.; Yu, W.; Li, C.; Wang, T.; Lo, H.W.; Lin, J. LLY17, a novel small molecule STAT3 inhibitor induces apoptosis and suppresses cell migration and tumor growth in triple-negative breast cancer. Breast Cancer Res. Treat. 2020, 181, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, L.; Wei, J.; Zhang, R.; Yang, X.; Song, J.; Bai, R.Y.; Fu, S.; Pierson, C.R.; Finlay, J.L.; et al. LLL12B, a small molecule STAT3 inhibitor, induces growth arrest, apoptosis, and enhances cisplatin-mediated cytotoxicity in medulloblastoma cells. Sci. Rep. 2021, 11, 6517. [Google Scholar] [CrossRef] [PubMed]
- De Groot, F.M.; van Berkom, L.W.; Scheeren, H.W. Synthesis and biological evaluation of 2′-carbamate-linked and 2′-carbonate-linked prodrugs of paclitaxel: Selective activation by the tumor-associated protease plasmin. J. Med. Chem. 2000, 43, 3093–3102. [Google Scholar] [CrossRef]
- Lin, L.; Hutzen, B.; Li, P.K.; Ball, S.; Zuo, M.; DeAngelis, S.; Foust, E.; Sobo, M.; Friedman, L.; Bhasin, D.; et al. A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia 2010, 12, 39–50. [Google Scholar] [CrossRef]
- Aqel, S.I.; Yang, X.; Kraus, E.E.; Song, J.; Farinas, M.F.; Zhao, E.Y.; Pei, W.; Lovett-Racke, A.E.; Racke, M.K.; Li, C.; et al. A STAT3 inhibitor ameliorates CNS autoimmunity by restoring Teff:Treg balance. JCI Insight 2021, 6, e142376. [Google Scholar] [CrossRef]
- Matošević, A.; Bosak, A. Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents. Arh. Hig. Rada Toksikol. 2020, 71, 285–299. [Google Scholar] [CrossRef]
- Igarashi, Y.; Yanagisawa, E.; Ohshima, T.; Takeda, S.; Aburada, M.; Miyamoto, K. Synthesis and evaluation of carbamate prodrugs of a phenolic compound. Chem. Pharm. Bull. 2007, 55, 328–333. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, R.; Ma, L.; Pierson, C.R.; Finlay, J.L.; Li, C.; Lin, J. STAT3 inhibitor in combination with irradiation significantly inhibits cell viability, cell migration, invasion and tumorsphere growth of human medulloblastoma cells. Cancer Biol. 2021, 22, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Yoo, W.; Stevenson, H.L.; Deshpande, D.; Shen, H.; Gagea, M.; Yoo, S.Y.; Wang, J.; Eckols, T.K.; Bharadwaj, U.; et al. Multifunctional Effects of a Small-Molecule STAT3 Inhibitor on NASH and Hepatocellular Carcinoma in Mice. Clin. Cancer Res. 2017, 23, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Carcereri de Prati, A.; Mariotto, S. Redox Regulation of STAT1 and STAT3 Signaling. Int. J. Mol. Sci. 2020, 21, 7034. [Google Scholar] [CrossRef]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. Jakstat 2012, 1, 65–72. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Lieblein, J.C.; Ball, S.; Hutzen, B.; Sasser, A.K.; Lin, H.J.; Huang, T.H.; Hall, B.M.; Lin, J. STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer 2008, 8, 302. [Google Scholar] [CrossRef]
- Wake, M.S.; Watson, C.J. STAT3 the oncogene—Still eluding therapy? FEBS J. 2015, 282, 2600–2611. [Google Scholar] [CrossRef]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, M.; Zhang, S.; Xue, N.; Xu, H.; Lin, S.; Chen, X. Bt354 as a new STAT3 signaling pathway inhibitor against triple negative breast cancer. J. Drug Target 2018, 26, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yang, X.; Roque, D.M.; Li, C.; Lin, J. A novel small molecule LLL12B inhibits STAT3 signaling and sensitizes ovariancancer cell to paclitaxel and cisplatin. PLoS ONE 2021, 16, e0240145. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, L.; Chen, X.; Rassool, F.V.; Li, C.; Lin, J. LLL12B, a Novel Small-Molecule STAT3 Inhibitor, Induces Apoptosis and Suppresses Cell Migration and Tumor Growth in Triple-Negative Breast Cancer Cells. Biomedicines 2022, 10, 2003. https://doi.org/10.3390/biomedicines10082003
Pan L, Chen X, Rassool FV, Li C, Lin J. LLL12B, a Novel Small-Molecule STAT3 Inhibitor, Induces Apoptosis and Suppresses Cell Migration and Tumor Growth in Triple-Negative Breast Cancer Cells. Biomedicines. 2022; 10(8):2003. https://doi.org/10.3390/biomedicines10082003
Chicago/Turabian StylePan, Li, Xiang Chen, Feyruz Virgilia Rassool, Chenglong Li, and Jiayuh Lin. 2022. "LLL12B, a Novel Small-Molecule STAT3 Inhibitor, Induces Apoptosis and Suppresses Cell Migration and Tumor Growth in Triple-Negative Breast Cancer Cells" Biomedicines 10, no. 8: 2003. https://doi.org/10.3390/biomedicines10082003