
Gut Microbiome in Non-Alcoholic Fatty Liver Disease: From Mechanisms to Therapeutic Role

 ,
,  , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
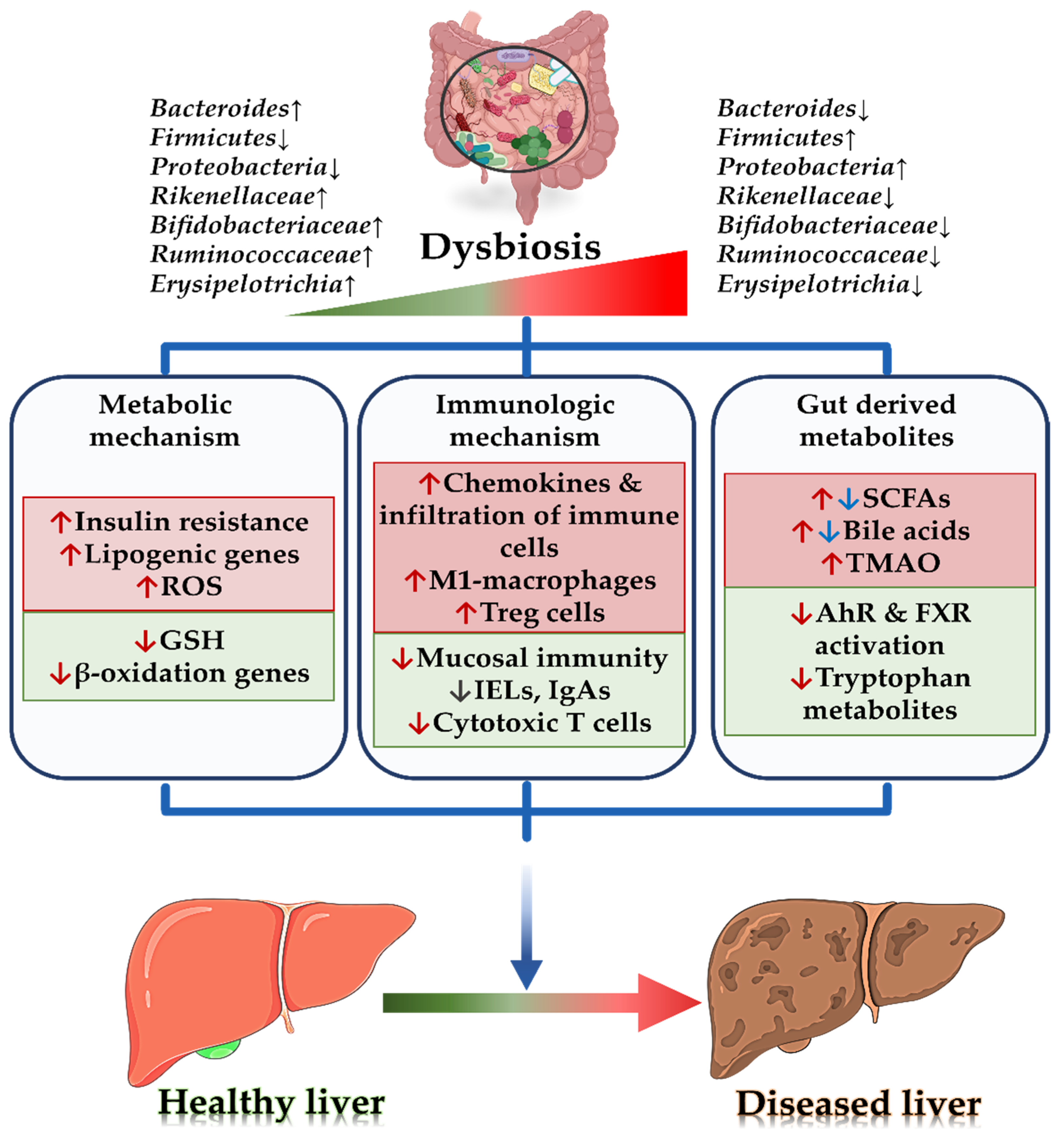
2. Microbial Pathophysiology Associated with Non-Alcoholic Liver Disease
2.1. Gut Microbiota and Metabolic Mechanism
2.1.1. Metaflammation and Lipotoxicity
2.1.2. Insulin Resistance, Oxidative Stress and Mitochondrial Dysfunction
2.1.3. De Novo Lipogenesis (DNL) Pathway
2.2. Gut Microbiota and Immunologic Mechanism
2.2.1. Lymphocytes and Macrophages
2.2.2. Chemokines
2.3. Gut Microbiota and Its Metabolites
2.3.1. Fermentable Dietary Nutrients
2.3.2. Amino Acid Metabolism and Its Byproducts
2.3.3. Bile Acids as Gut Microbial Messengers
2.3.4. Other Bacterial Metabolites
3. Therapeutic Approaches
3.1. Pharmacological Intervention
3.2. Microbiota-Based Intervention

{kind=link}
| Animal Study Model | Intervention | Co-Intervention | Effect on Gut | Effect on Liver Function | Metabolites | Ref. |
|---|---|---|---|---|---|---|
| HFD-NAFLD | Astragalus polysaccharides (prebiotic) | - | Desulfovibrio↑, Parabacteroides↑, Acetatifactor↑, Alistipes↑, F/B ratio↓ | TG↓, ALT↓, hepatic steatosis↓, hepatic inflammation↓, fatty acid oxidation↑ | Acetate↑ | [163] |
| HFD-NAFLD | Desulfovibrio vulgaris | - | Desulfovibrio vulgaris↑, | TG↓, ALT↓, hepatic steatosis↓, fatty acid oxidation↑ | [163] | |
| HFrD-NAFLD | Lactobacillus fermentum CECT5716 | FOS | F/B ratio↓, intestinal barrier integrity↑, Lactobacilli↑ | insulin resistance↓, hepatic steatosis↓, hepatic inflammation↓ | SCAs↓ | [164] |
| HFD-NAFLD | Lactobacillus acidophilus, Lactobacillus fermentum, Lactobacillus plantarum | - | F/B ratio↓, Lactobacillus↑ | TC↓, TG↓ | - | [151] |
| HFD-NAFLD | Bifidobacterium bifidum V Lactobacillus plantarum X | Salvia miltiorrhiza polysaccharide | Fecal TC↑, Cyanobacteria↓, F/B ratio↓ | TC↓, TG↓, LPS↓, hepatic steatosis↓, insulin resistance↓, hepatic inflammation↓ | SCAs↑ | [165] |
| HFD-NAFLD | Kefir (probiotic beverage) | - | Lactobacillus/Lactococcus↑, Bacteroides fragilis↓, Clostridiaceae↓, F/B ratio↓ | TC↓, fatty acid oxidation↑, hepatic inflammation and oxidative stress↓ | - | [166] |
| HFD-NAFLD | Bacillus mixture VSL#3 | - | Intestinal barrier integrity↑, Bacteroidetes↑ | Hepatic steatosis↓, insulin resistance↓, hepatic inflammation↓, fatty acid oxidation↑ | Acetate↓ | [167] |
| HFD-NAFLD | Lactobacillus rhamnosus GG | - | Desulfovibrionaceae↑, Lactobacillaceae↑ | LPS↓, hepatic steatosis↓ | FAs↓ | [168] |
| HFD-NAFLD | Lactobacillus lactis, Pediococcus pentosaceus | - | Intestinal barrier integrity↑, F/B ratio↓, Clostridium_g21↓ | TG↓, AST↓, TBil↓, TC↓, LPS↓, hepatic steatosis↓, hepatic inflammation↓, fatty acid oxidation↑ | Indole compounds↑ Acetate↑ Butyrate↑ Propionate↑ Primary BAs↑ | [110] |
| HFD/HFrD-NAFLD | Lactobacillus plantarum K2 and K6 | - | Bacteroides↑ | ALT↓, AST↓, ALP↓, TC↓, TG↓, MDA↓, hepatic steatosis↓, fatty acid oxidation↑, oxidative stress↓ | - | [169] |
| HSuD/HFD-NASH | Lactobacilli (9 species), Bifidobacteria (4 species), Streptococcus salivarius subsp (Thermophilus) | Inulin | Bacteroides↑ | ALT↓, AST↓, GGT↓, ALP↓, TBil↓, TC↓, TG↓, hepatic steatosis↓, fibrosis↓, hepatic inflammation↓ | - | [170] |
| HFD-NASH | Lactobacillus reuteri | Metformin | Bacteroidetes↓, Firmicutes↑ | ALT↓, AST↓, TC↓, TG↓, LPS↓, insulin resistance↓, oxidative stress↓, hepatic steatosis↓, fibrosis↓, hepatic inflammation↓ | Acetate↓ Butyrate↑ Propionate↑ | [171] |
| HFD-NAFLD | Polylactose | - | Bacteroides↑, Lactobacillus↑, Akkermansia muciniphila↑ F/B ratio↓, | TC↓, TG↓, insulin resistance↓, hepatic steatosis↓, hepatic inflammation↓ | Acetate↑ Propionate↑ | [172] |
| HFD-DSS-NAFLD | Schizophyllum commune-derived β-glucan | Probiotic mix (8 species) | Lactobacillus↑, Bifidobacterium↑, Akkermansia↑ | ALP↓, hepatic steatosis↓, hepatic inflammation↓ | Butyrate↑ | [173] |
| CDAA-NASH | MIYAIRI 588 | Losartan | Intestinal barrier integrity↑ | Hepatic steatosis↓, hepatic inflammation↓, fibrosis↓, early HCC↓ | - | [174] |
| HCholD | Lactobacillus paracasei, Lactobacillus rhamnosus, Lactobacillus acidophilus, Bifidobacterium lactis | FOS | - | TC↓, hepatic steatosis↓, hepatic inflammation↓ | - | [175] |
| MSG one dose (s.c.) | FOS | - | Clostridium cluster XI↑ Prevotella↓ | TC↓, ALT↓, LPS↓, insulin resistance↓, hepatic steatosis↓, hepatic inflammation↓ | Acetate↑ Butyrate↑ Propionate↑ | [176] |
| Human Study | Intervention | Outcomes | Ref. |
|---|---|---|---|
| 48 patients, type-2 diabetic with NAFLD | Multi-strain probiotic mixture (Bifidobacterium, Lactobacillus, Lactococcus, Propionibacterium) with omega-3 fatty acids once daily for 8 weeks | Fatty liver index↓, GGT↓, TG↓, TC↓, hepatic steatosis↓, inflammatory markers↓ | [177] |
| 58 patients, type-2 diabetic with NAFLD | Multi-strain probiotic mixture (Bifidobacterium, Lactobacillus, Lactococcus, Propionibacterium) once daily for 8 weeks | Fatty liver index↓, GGT↓, AST↓, hepatic steatosis↓, inflammatory markers↓ | [178] |
| 64 obese children with sonographic NAFLD | Probiotic mixture (Lactobacillus acidophilus, Bifidobacterium lactis, Bifidobacterium bifidum, Lactobacillus rhamnosus) | ALT↓, AST, TG↓, TC↓, hepatic steatosis↓ | [179] |
| 39 patients with NAFLD | Multi-strain probiotic mixture (Bifidobacterium, Lactobacillus, Streptococcus) for 1 year | ALT↓, LPS, hepatic steatosis↓, inflammatory markers↓ | [180] |
| 102 patients with NAFLD | Synbiotic yogurt (Bifidobacterium animalis and Inulin) for 24 weeks | ALT↓, AST, GGT↓, ALP↓, TG↓, TC↓, fatty liver index↓, insulin resistance↓ | [181] |
| 68 obese patients with NAFLD | Probiotic mixture (Lactobacillus, Pediococcus, Bifidobacterium) | TC↓, hepatic steatosis↓, inflammatory markers↓ | [182] |
| 75 patients with NASH | Probiotic cocktail (Lactobacillus, Streptococcus, Bifidobacterium) with FOS once daily for 12 weeks and on low-fat/low-calorie diet | TC↓, ALT↓, AST, liver stiffness↓ | [183] |
4. Limitations and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, X.; Liu, S.; Zhang, J.; Dong, M.; Wang, Y.; Wang, M.; Xin, Y. Proportion of NAFLD patients with normal ALT value in overall NAFLD patients: A systematic review and meta-analysis. BMC Gastroenterol. 2020, 20, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Sharma, S.P.; Suk, K.T.; Kim, D.J. Significance of gut microbiota in alcoholic and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2021, 27, 6161–6179. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Feldstein, A.E. Nonalcoholic steatohepatitis: Risk factors and diagnosis. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 623–635. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Van Natta, M.L.; Clark, J.; Neuschwander-Tetri, B.A.; Diehl, A.; Dasarathy, S.; Loomba, R.; Chalasani, N.; Kowdley, K.; Hameed, B.; et al. Prospective Study of Outcomes in Adults with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2021, 385, 1559–1569. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Ramos, M.J.; Bandiera, L.; Menolascina, F.; Fallowfield, J.A. In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications. iScience 2022, 25, 103549. [Google Scholar] [CrossRef]
- Pydyn, N.; Miękus, K.; Jura, J.; Kotlinowski, J. New therapeutic strategies in nonalcoholic fatty liver disease: A focus on promising drugs for nonalcoholic steatohepatitis. Pharmacol. Rep. 2020, 72, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Khoruts, A. Microbiota changes and intestinal microbiota transplantation in liver diseases and cirrhosis. J. Hepatol. 2020, 72, 1003–1027. [Google Scholar] [CrossRef] [Green Version]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Bergé, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef]
- Velazquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Miyajima, A. Liver regeneration and fibrosis after inflammation. Inflamm. Regen. 2016, 36, 19. [Google Scholar] [CrossRef] [Green Version]
- So, J.; Kim, A.; Lee, S.-H.; Shin, D. Liver progenitor cell-driven liver regeneration. Exp. Mol. Med. 2020, 52, 1230–1238. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Martin-Mateos, R.; Albillos, A. The Role of the Gut-Liver Axis in Metabolic Dysfunction-Associated Fatty Liver Disease. Front. Immunol. 2021, 12, 660179. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibbò, S.; Ianiro, G.; Dore, M.P.; Simonelli, C.; Newton, E.E.; Cammarota, G. Gut Microbiota as a Driver of Inflammation in Nonalcoholic Fatty Liver Disease. Mediat. Inflamm. 2018, 2018, 9321643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef]
- Crispe, I.N. Hepatocytes as Immunological Agents. J. Immunol. 2016, 196, 17–21. [Google Scholar] [CrossRef] [PubMed]
- du Plessis, J.; van Pelt, J.; Korf, H.; Mathieu, C.; van der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of Adipose Tissue Inflammation With Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 635–648.e614. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.M.; Go, Y.-M.; Jones, D.P. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 215–234. [Google Scholar] [CrossRef]
- de Groot, P.; Scheithauer, T.; Bakker, G.J.; Prodan, A.; Levin, E.; Khan, M.T.; Herrema, H.; Ackermans, M.; Serlie, M.J.M.; de Brauw, M.; et al. Donor metabolic characteristics drive effects of faecal microbiota transplantation on recipient insulin sensitivity, energy expenditure and intestinal transit time. Gut 2020, 69, 502. [Google Scholar] [CrossRef] [Green Version]
- Angelini, G.; Salinari, S.; Castagneto-Gissey, L.; Bertuzzi, A.; Casella-Mariolo, J.; Ahlin, S.; Boskoski, I.; Gaggini, M.; Raffaelli, M.; Costamagna, G.; et al. Small intestinal metabolism is central to whole-body insulin resistance. Gut 2021, 70, 1098. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Neish, A.S. Redox signaling mediated by the gut microbiota. Free Radic. Biol. Med. 2017, 105, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, G.L. Lipopolysaccharides in liver injury: Molecular mechanisms of Kupffer cell activation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2002, 283, G256–G265. [Google Scholar] [CrossRef] [Green Version]
- Tawadros, P.S.; Powers, K.A.; Ailenberg, M.; Birch, S.E.; Marshall, J.C.; Szaszi, K.; Kapus, A.; Rotstein, O.D. Oxidative Stress Increases Surface Toll-Like Receptor 4 Expression in Murine Macrophages Via Ceramide Generation. Shock 2015, 44, 157–165. [Google Scholar] [CrossRef]
- Akhter, N.; Madhoun, A.; Arefanian, H.; Wilson, A.; Kochumon, S.; Thomas, R.; Shenouda, S.; Al-Mulla, F.; Ahmad, R.; Sindhu, S. Oxidative Stress Induces Expression of the Toll-Like Receptors (TLRs) 2 and 4 in the Human Peripheral Blood Mononuclear Cells: Implications for Metabolic Inflammation. Cell Physiol. Biochem. 2019, 53, 1–18. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef]
- Borrelli, A.; Bonelli, P.; Tuccillo, F.M.; Goldfine, I.D.; Evans, J.L.; Buonaguro, F.M.; Mancini, A. Role of gut microbiota and oxidative stress in the progression of non-alcoholic fatty liver disease to hepatocarcinoma: Current and innovative therapeutic approaches. Redox Biol. 2018, 15, 467–479. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Qiao, Y.; Sun, J.; Ding, Y.; Le, G.; Shi, Y. Alterations of the gut microbiota in high-fat diet mice is strongly linked to oxidative stress. Appl. Microbiol. Biotechnol. 2013, 97, 1689–1697. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Shoaie, S.; Bergentall, M.; Ghaffari, P.; Zhang, C.; Larsson, E.; Bäckhed, F.; Nielsen, J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Rom, O.; Liu, Y.; Liu, Z.; Zhao, Y.; Wu, J.; Ghrayeb, A.; Villacorta, L.; Fan, Y.; Chang, L.; Wang, L.; et al. Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci. Transl. Med. 2020, 12, eaaz2841. [Google Scholar] [CrossRef] [PubMed]
- Davila, A.-M.; Blachier, F.; Gotteland, M.; Andriamihaja, M.; Benetti, P.-H.; Sanz, Y.; Tomé, D. Intestinal luminal nitrogen metabolism: Role of the gut microbiota and consequences for the host. Pharmacol. Res. 2013, 68, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Desai, C.; Darby, T.M.; Luo, L.; Wolfarth, A.A.; Scharer, C.D.; Ardita, C.S.; Reedy, A.R.; Keebaugh, E.S.; Neish, A.S. Lactobacilli Modulate Epithelial Cytoprotection through the Nrf2 Pathway. Cell Rep. 2015, 12, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, B.J.; Liu, K.H.; Owens, J.A.; Hunter-Chang, S.; Camacho, M.C.; Eboka, R.U.; Chandrasekharan, B.; Baker, N.F.; Darby, T.M.; Robinson, B.S.; et al. Gut-Resident Lactobacilli Activate Hepatic Nrf2 and Protect Against Oxidative Liver Injury. Cell Metab. 2020, 31, 956–968.e5. [Google Scholar] [CrossRef]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Marques-Lopes, I.; Ansorena, D.; Astiasaran, I.; Forga, L.; Martinez, J.A. Postprandial de novo lipogenesis and metabolic changes induced by a high-carbohydrate, low-fat meal in lean and overweight men. Am. J. Clin. Nutr. 2001, 73, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed]
- de Luis, D.A.; Izaola, O.; Aller, R.; de la Fuente, B.; Bachiller, R.; Romero, E. Effects of a high-protein/low carbohydrate versus a standard hypocaloric diet on adipocytokine levels and insulin resistance in obese patients along 9 months. J. Diabetes Complicat. 2015, 29, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Krebs, N.F.; Gao, D.; Gralla, J.; Collins, J.S.; Johnson, S.L. Efficacy and safety of a high protein, low carbohydrate diet for weight loss in severely obese adolescents. J. Pediatr. 2010, 157, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurtz, P.; Makinen, V.P.; Soininen, P.; Kangas, A.J.; Tukiainen, T.; Kettunen, J.; Savolainen, M.J.; Tammelin, T.; Viikari, J.S.; Ronnemaa, T.; et al. Metabolic signatures of insulin resistance in 7098 young adults. Diabetes 2012, 61, 1372–1380. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Yu, J.; Guo, Y.; Deng, J.; Li, K.; Du, Y.; Chen, S.; Zhu, J.; Sheng, H.; Guo, F. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism 2014, 63, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Charidemou, E.; Ashmore, T.; Li, X.; McNally, B.D.; West, J.A.; Liggi, S.; Harvey, M.; Orford, E.; Griffin, J.L. A randomized 3-way crossover study indicates that high-protein feeding induces de novo lipogenesis in healthy humans. JCI Insight 2019, 4, e124819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef]
- Kabat, A.M.; Srinivasan, N.; Maloy, K.J. Modulation of immune development and function by intestinal microbiota. Trends Immunol. 2014, 35, 507–517. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Olivares-Villagómez, D.; Van Kaer, L. Intestinal Intraepithelial Lymphocytes: Sentinels of the Mucosal Barrier. Trends Immunol. 2018, 39, 264–275. [Google Scholar] [CrossRef]
- McDonald, B.D.; Jabri, B.; Bendelac, A. Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2018, 18, 514–525. [Google Scholar] [CrossRef]
- Di Marco Barros, R.; Roberts, N.A.; Dart, R.J.; Vantourout, P.; Jandke, A.; Nussbaumer, O.; Deban, L.; Cipolat, S.; Hart, R.; Iannitto, M.L.; et al. Epithelia Use Butyrophilin-like Molecules to Shape Organ-Specific γδ T Cell Compartments. Cell 2016, 167, 203–218.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J. Pathol. 2006, 208, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4+CD8αα+ T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lu, D.; Zhuo, J.; Lin, Z.; Yang, M.; Xu, X. The Gut-liver Axis in Immune Remodeling: New insight into Liver Diseases. Int. J. Biol. Sci. 2020, 16, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Balmer, M.L.; Slack, E.; de Gottardi, A.; Lawson, M.A.; Hapfelmeier, S.; Miele, L.; Grieco, A.; Van Vlierberghe, H.; Fahrner, R.; Patuto, N.; et al. The Liver May Act as a Firewall Mediating Mutualism Between the Host and Its Gut Commensal Microbiota. Sci. Transl. Med. 2014, 6, 237ra266. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Kawaratani, H.; Tsujimoto, T.; Kitazawa, T.; Kitade, M.; Yoshiji, H.; Uemura, M.; Fukui, H. Innate immune reactivity of the liver in rats fed a choline-deficient L-amino-acid-defined diet. World J. Gastroenterol. 2008, 14, 6655–6661. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.L.; Gaudin, F.; Naveau, S.; Prevot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Ganini, D.; Tokar, E.J.; Kumar, A.; Das, S.; Corbett, J.; Kadiiska, M.B.; Waalkes, M.P.; Diehl, A.M.; Mason, R.P. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. J. Hepatol. 2013, 58, 778–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashimada, M.; Honda, M. Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics. Int. J. Mol. Sci. 2021, 22, 8008. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Ghazarian, M.; Revelo, X.S.; Nohr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci. Immunol. 2017, 2, eaai7616. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.A.; Bouladoux, N.; Sun, C.M.; Wohlfert, E.A.; Blank, R.B.; Zhu, Q.; Grigg, M.E.; Berzofsky, J.A.; Belkaid, Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 2008, 29, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.-H.; Liu, Z.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011, 140, 697–708.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behary, J.; Amorim, N.; Jiang, X.-T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 2020, 11, 4982. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- McMahan, R.H.; Porsche, C.E.; Edwards, M.G.; Rosen, H.R. Free Fatty Acids Differentially Downregulate Chemokines in Liver Sinusoidal Endothelial Cells: Insights into Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0159217. [Google Scholar] [CrossRef] [Green Version]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.-M.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. C-C Chemokine Receptor 2 (CCR2) Regulates the Hepatic Recruitment of Myeloid Cells That Promote Obesity-Induced Hepatic Steatosis. Diabetes 2010, 59, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.-I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of Distinct Subpopulations of Hepatic Macrophages in HFD/Obese Mice. Diabetes 2014, 64, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Li, L.; Xu, C.; Wang, Y.; Wang, Z.; Chen, M.; Jiang, Z.; Pan, J.; Yang, C.; Li, X.; et al. Cross-talk between the gut microbiota and monocyte-like macrophages mediates an inflammatory response to promote colitis-associated tumourigenesis. Gut 2021, 70, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.-S.; Seki, E. Chemokines and chemokine receptors in the development of NAFLD. Adv. Exp. Med. Biol. 2018, 1061, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chiwanda Kaminga, A.; Liu, A.; Wen, S.W.; Chen, J.; Luo, J. Chemokines in Non-alcoholic Fatty Liver Disease: A Systematic Review and Network Meta-Analysis. Front. Immunol. 2020, 11, 1802. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [Green Version]
- Bach Knudsen, K.E. Microbial Degradation of Whole-Grain Complex Carbohydrates and Impact on Short-Chain Fatty Acids and Health. Adv. Nutr. 2015, 6, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; Van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Sahuri-Arisoylu, M.; Brody, L.P.; Parkinson, J.R.; Parkes, H.; Navaratnam, N.; Miller, A.D.; Thomas, E.L.; Frost, G.; Bell, J.D. Reprogramming of hepatic fat accumulation and ‘browning’ of adipose tissue by the short-chain fatty acid acetate. Int. J. Obes. 2016, 40, 955–963. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- El Hage, R.; Hernandez-Sanabria, E.; Calatayud Arroyo, M.; Props, R.; Van de Wiele, T. Propionate-Producing Consortium Restores Antibiotic-Induced Dysbiosis in a Dynamic in vitro Model of the Human Intestinal Microbial Ecosystem. Front. Microbiol. 2019, 10, 1206. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Eur. Gastroenterol. J. 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-alcoholic Fatty Liver Disease. Sci. Rep. 2016, 6, 32002. [Google Scholar] [CrossRef]
- Yu, J.S.; Youn, G.S.; Choi, J.; Kim, C.-H.; Kim, B.Y.; Yang, S.-J.; Lee, J.H.; Park, T.-S.; Kim, B.K.; Kim, Y.B.; et al. Lactobacillus lactis and Pediococcus pentosaceus-driven reprogramming of gut microbiome and metabolome ameliorates the progression of non-alcoholic fatty liver disease. Clin. Transl. Med. 2021, 11, e634. [Google Scholar] [CrossRef]
- Dangana, E.O.; Omolekulo, T.E.; Areola, E.D.; Olaniyi, K.S.; Soladoye, A.O.; Olatunji, L.A. Sodium acetate protects against nicotine-induced excess hepatic lipid in male rats by suppressing xanthine oxidase activity. Chem.-Biol. Interact. 2020, 316, 108929. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.K.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744. [Google Scholar] [CrossRef] [Green Version]
- Endo, H.; Niioka, M.; Kobayashi, N.; Tanaka, M.; Watanabe, T. Butyrate-Producing Probiotics Reduce Nonalcoholic Fatty Liver Disease Progression in Rats: New Insight into the Probiotics for the Gut-Liver Axis. PLoS ONE 2013, 8, e63388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.H.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.C.; Feng, D.D.; Chen, C.; Lee, H.G.; et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099. [Google Scholar] [CrossRef] [Green Version]
- Aoki, R.; Onuki, M.; Hattori, K.; Ito, M.; Yamada, T.; Kamikado, K.; Kim, Y.-G.; Nakamoto, N.; Kimura, I.; Clarke, J.M.; et al. Commensal microbe-derived acetate suppresses NAFLD/NASH development via hepatic FFAR2 signalling in mice. Microbiome 2021, 9, 188. [Google Scholar] [CrossRef]
- Marinelli, L.; Martin-Gallausiaux, C.; Bourhis, J.-M.; Béguet-Crespel, F.; Blottière, H.M.; Lapaque, N. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci. Rep. 2019, 9, 643. [Google Scholar] [CrossRef]
- Ratzke, C.; Gore, J. Modifying and reacting to the environmental pH can drive bacterial interactions. PLoS Biol. 2018, 16, e2004248. [Google Scholar] [CrossRef] [Green Version]
- Mendes, B.G.; Schnabl, B. From intestinal dysbiosis to alcohol-associated liver disease. Clin. Mol. Hepatol. 2020, 26, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.; Suk, K.T.; Kim, D.J. Gut Microbiota at the Intersection of Alcohol, Brain, and the Liver. J. Clin. Med. 2021, 10, 541. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Wrzosek, L.; Ciocan, D.; Hugot, C.; Spatz, M.; Dupeux, M.; Houron, C.; Lievin-Le Moal, V.; Puchois, V.; Ferrere, G.; Trainel, N.; et al. Microbiota tryptophan metabolism induces aryl hydrocarbon receptor activation and improves alcohol-induced liver injury. Gut 2021, 70, 1299–1308. [Google Scholar] [CrossRef]
- Jin, U.H.; Lee, S.O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol. 2014, 85, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, R.; Ilha, M.; Vaittinen, M.; Kaminska, D.; Mannisto, V.; Karja, V.; Tuomainen, M.; Hanhineva, K.; Romeo, S.; Pajukanta, P.; et al. Indole-3-Propionic Acid, a Gut-Derived Tryptophan Metabolite, Associates with Hepatic Fibrosis. Nutrients 2021, 13, 3509. [Google Scholar] [CrossRef]
- Wlodarska, M.; Luo, C.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e6. [Google Scholar] [CrossRef] [Green Version]
- Masarone, M.; Troisi, J.; Aglitti, A.; Torre, P.; Colucci, A.; Dallio, M.; Federico, A.; Balsano, C.; Persico, M. Untargeted metabolomics as a diagnostic tool in NAFLD: Discrimination of steatosis, steatohepatitis and cirrhosis. Metabolomics 2021, 17, 12. [Google Scholar] [CrossRef]
- Staels, B.; Fonseca, V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes Care 2009, 32 (Suppl. S2), S237–S245. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Garcia-Irigoyen, O.; Cariello, M.; Scialpi, N.; Peres, C.; Vetrano, S.; Fiorino, G.; Danese, S.; Ko, B.; Luo, J.; et al. Fibroblast Growth Factor 19 modulates intestinal microbiota and inflammation in presence of Farnesoid X Receptor. EBioMedicine 2020, 54, 102719. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.-K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef]
- Stellwag, E.J.; Hylemon, P.B. 7alpha-Dehydroxylation of cholic acid and chenodeoxycholic acid by Clostridium leptum. J. Lipid Res. 1979, 20, 325–333. [Google Scholar] [CrossRef]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E.; Blaser, M.J. Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. mBio 2015, 6, e02481-14. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Flores-Guerrero, J.L.; Post, A.; van Dijk, P.R.; Connelly, M.A.; Garcia, E.; Navis, G.; Bakker, S.J.L.; Dullaart, R.P.F. Circulating trimethylamine-N-oxide is associated with all-cause mortality in subjects with nonalcoholic fatty liver disease. Liver Int. 2021, 41, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Aragones, G.; Colom-Pellicer, M.; Aguilar, C.; Guiu-Jurado, E.; Martinez, S.; Sabench, F.; Antonio Porras, J.; Riesco, D.; Del Castillo, D.; Richart, C.; et al. Circulating microbiota-derived metabolites: A "liquid biopsy? Int. J. Obes. (Lond.) 2020, 44, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Liu, X.; Xu, J.; Xue, C.; Xue, Y.; Wang, Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 2014, 118, 476–481. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Xin, F.Z.; Zhou, D.; Xue, Y.Q.; Liu, X.L.; Yang, R.X.; Pan, Q.; Fan, J.G. Trimethylamine N-oxide attenuates high-fat high-cholesterol diet-induced steatohepatitis by reducing hepatic cholesterol overload in rats. World J. Gastroenterol. 2019, 25, 2450–2462. [Google Scholar] [CrossRef]
- Dai, X.; Hou, H.; Zhang, W.; Liu, T.; Li, Y.; Wang, S.; Wang, B.; Cao, H. Microbial Metabolites: Critical Regulators in NAFLD. Front. Microbiol. 2020, 11, 2373. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, H.W.; Yoo, J.J.; Cho, Y.; Kim, S.U.; Lee, T.H.; Jang, B.K.; Kim, S.G.; Ahn, S.B.; Kim, H.; et al. KASL clinical practice guidelines: Management of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2021, 27, 363–401. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Friedman, S.L.; McCullough, A.J.; Dimick-Santos, L. Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: Findings and recommendations from an American Association for the Study of Liver Diseases-U.S. Food and Drug Administration Joint Workshop. Hepatology 2015, 61, 1392–1405. [Google Scholar] [CrossRef]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of Gut Microbiota in Hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.Y.; Suk, K.T. The Role of the Gut Microbiome in Liver Cirrhosis Treatment. Int. J. Mol. Sci. 2020, 22, 199. [Google Scholar] [CrossRef]
- Lee, N.Y.; Shin, M.J.; Youn, G.S.; Yoon, S.J.; Choi, Y.R.; Kim, H.S.; Gupta, H.; Han, S.H.; Kim, B.K.; Lee, D.Y.; et al. Lactobacillus attenuates progression of nonalcoholic fatty liver disease by lowering cholesterol and steatosis. Clin. Mol. Hepatol. 2021, 27, 110–124. [Google Scholar] [CrossRef]
- Machado, A.S.; Oliveira, J.R.; Lelis, D.d.F.; de Paula, A.M.B.; Guimarães, A.L.S.; Andrade, J.M.O.; Brandi, I.V.; Santos, S.H.S. Oral Probiotic Bifidobacterium Longum Supplementation Improves Metabolic Parameters and Alters the Expression of the Renin-Angiotensin System in Obese Mice Liver. Biol. Res. Nurs. 2020, 23, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, S.R.; Maraj, B.; Harding-Theobald, E.; Vittinghoff, E.; Terrault, N.A. Gut microbiome-targeted therapies in nonalcoholic fatty liver disease: A systematic review, meta-analysis, and meta-regression. Am. J.Clin. Nutr. 2019, 110, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Gómez, M.X.; Martínez, I.; Bottacini, F.; O’Callaghan, A.; Ventura, M.; van Sinderen, D.; Hillmann, B.; Vangay, P.; Knights, D.; Hutkins, R.W.; et al. Stable Engraftment of Bifidobacterium longum AH1206 in the Human Gut Depends on Individualized Features of the Resident Microbiome. Cell Host Microbe 2016, 20, 515–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, M.; Schuchmann, M. Long-term management of hepatic encephalopathy with lactulose and/or rifaximin: A review of the evidence. Eur. J. Gastroenterol. Hepatol. 2019, 31, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, A.N.; Goel, A.; Singh, A.; Sasi, A.; Aggarwal, R. Faecal bacterial microbiota in patients with cirrhosis and the effect of lactulose administration. BMC Gastroenterol. 2017, 17, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Heuman, D.M.; Sanyal, A.J.; Hylemon, P.B.; Sterling, R.K.; Stravitz, R.T.; Fuchs, M.; Ridlon, J.M.; Daita, K.; Monteith, P.; et al. Modulation of the Metabiome by Rifaximin in Patients with Cirrhosis and Minimal Hepatic Encephalopathy. PLoS ONE 2013, 8, e60042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadi, A.; Mohammadi, H.; Miraghajani, M.; Ghaedi, E. Efficacy of synbiotic supplementation in patients with nonalcoholic fatty liver disease: A systematic review and meta-analysis of clinical trials: Synbiotic supplementation and NAFLD. Crit. Rev. Food Sci. Nutr. 2019, 59, 2494–2505. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Afolabi, P.R.; Miles, E.A.; Smith, D.E.; Almehmadi, A.; Alshathry, A.; Childs, C.E.; Del Fabbro, S.; Bilson, J.; Moyses, H.E.; et al. Synbiotics Alter Fecal Microbiomes, But Not Liver Fat or Fibrosis, in a Randomized Trial of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1597–1610.e7. [Google Scholar] [CrossRef] [PubMed]
- Mofidi, F.; Poustchi, H.; Yari, Z.; Nourinayyer, B.; Merat, S.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic supplementation in lean patients with non-alcoholic fatty liver disease: A pilot, randomised, double-blind, placebo-controlled, clinical trial. Br. J. Nutr. 2017, 117, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga–Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.J.; Smits, L.P.; Pekmez, C.T.; Prodan, A.; Meijnikman, A.S.; Troelstra, M.A.; Bouter, K.E.C.; Herrema, H.; Levin, E.; Holleboom, A.G.; et al. Donor Fecal Microbiota Transplantation Alters Gut Microbiota and Metabolites in Obese Individuals With Steatohepatitis. Hepatol. Commun. 2020, 4, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Sheng, L.; Zhong, J.; Tao, X.; Zhu, W.; Ma, J.; Yan, J.; Zhao, A.; Zheng, X.; Wu, G.; et al. Desulfovibrio vulgaris, a potent acetic acid-producing bacterium, attenuates nonalcoholic fatty liver disease in mice. Gut Microbes 2021, 13, 1930874. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Gutiérrez, B.; Gámez-Belmonte, R.; Suárez, M.D.; Lavín, J.L.; Aransay, A.M.; Olivares, M.; Martínez-Augustin, O.; de Medina, F.S.; Zarzuelo, A. A synbiotic composed of Lactobacillus fermentum CECT5716 and FOS prevents the development of fatty acid liver and glycemic alterations in rats fed a high fructose diet associated with changes in the microbiota. Mol. Nutr. Food Res. 2017, 61, 1600622. [Google Scholar] [CrossRef]
- Wang, W.; Xu, A.L.; Li, Z.C.; Li, Y.; Xu, S.F.; Sang, H.C.; Zhi, F. Combination of Probiotics and Salvia miltiorrhiza Polysaccharide Alleviates Hepatic Steatosis via Gut Microbiota Modulation and Insulin Resistance Improvement in High Fat-Induced NAFLD Mice. Diabetes Metab. J. 2020, 44, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Kim, H.; Jeong, D.; Kang, I.-B.; Chon, J.-W.; Kim, H.-S.; Song, K.-Y.; Seo, K.-H. Kefir alleviates obesity and hepatic steatosis in high-fat diet-fed mice by modulation of gut microbiota and mycobiota: Targeted and untargeted community analysis with correlation of biomarkers. J. Nutr. Biochem. 2017, 44, 35–43. [Google Scholar] [CrossRef]
- Kim, B.; Kwon, J.; Kim, M.S.; Park, H.; Ji, Y.; Holzapfel, W.; Hyun, C.K. Protective effects of Bacillus probiotics against high-fat diet-induced metabolic disorders in mice. PLoS ONE 2018, 13, e0210120. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.R.; Park, H.-J.; Kang, D.; Chung, H.; Nam, M.H.; Lee, Y.; Park, J.-H.; Lee, H.-Y. A protective mechanism of probiotic Lactobacillus against hepatic steatosis via reducing host intestinal fatty acid absorption. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Park, E.-J.; Lee, Y.-S.; Kim, S.M.; Park, G.-S.; Lee, Y.H.; Jeong, D.Y.; Kang, J.; Lee, H.-J. Beneficial Effects of Lactobacillus plantarum Strains on Non-Alcoholic Fatty Liver Disease in High Fat/High Fructose Diet-Fed Rats. Nutrients 2020, 12, 542. [Google Scholar] [CrossRef] [Green Version]
- Gadallah, S.H.; Eissa, S.; Ghanem, H.M.; Ahmed, E.K.; Hasanin, A.H.; El Mahdy, M.M.; Matboli, M. Probiotic-prebiotic-synbiotic modulation of (YAP1, LATS1 and NF2 mRNAs/miR-1205/lncRNA SRD5A3-AS1) panel in NASH animal model. Biomed. Pharmacother. 2021, 140, 111781. [Google Scholar] [CrossRef]
- Ahmed, L.A.; Salem, M.B.; Seif el-Din, S.H.; El-Lakkany, N.M.; Ahmed, H.O.; Nasr, S.M.; Hammam, O.A.; Botros, S.S.; Saleh, S. Gut microbiota modulation as a promising therapy with metformin in rats with non-alcoholic steatohepatitis: Role of LPS/TLR4 and autophagy pathways. Eur. J. Pharmacol. 2020, 887, 173461. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, B.E.; Schoenfuss, T.C.; Bailey, A.S.; Gallaher, D.D. Polylactose Exhibits Prebiotic Activity and Reduces Adiposity and Nonalcoholic Fatty Liver Disease in Rats Fed a High-Fat Diet. J. Nutr. 2021, 151, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Vu, V.; Muthuramalingam, K.; Singh, V.; Hyun, C.; Kim, Y.M.; Unno, T.; Cho, M. Effects of β-glucan, probiotics, and synbiotics on obesity-associated colitis and hepatic manifestations in C57BL/6J mice. Eur. J. Nutr. 2022, 61, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Kawaratani, H.; Kubo, T.; Fujinaga, Y.; Furukawa, M.; Saikawa, S.; Sato, S.; Seki, K.; Takaya, H.; Okura, Y.; et al. Combining probiotics and an angiotensin-II type 1 receptor blocker has beneficial effects on hepatic fibrogenesis in a rat model of non-alcoholic steatohepatitis. Hepatol. Res. 2019, 49, 284–295. [Google Scholar] [CrossRef]
- Alves, C.C.; Waitzberg, D.L.; de Andrade, L.S.; Dos Santos Aguiar, L.; Reis, M.B.; Guanabara, C.C.; Junior, O.A.; Ribeiro, D.A.; Sala, P. Prebiotic and Synbiotic Modifications of Beta Oxidation and Lipogenic Gene Expression after Experimental Hypercholesterolemia in Rat Liver. Front. Microbiol. 2017, 8, 2010. [Google Scholar] [CrossRef] [Green Version]
- Takai, A.; Kikuchi, K.; Ichimura, M.; Tsuneyama, K.; Moritoki, Y.; Matsumoto, K.; Tsunashima, H.; Onda, T.; Kuniyoshi, N.; Nariyama, T.; et al. Fructo-oligosaccharides ameliorate steatohepatitis, visceral adiposity, and associated chronic inflammation via increased production of short-chain fatty acids in a mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 2020, 20, 46. [Google Scholar] [CrossRef]
- Kobyliak, N.; Abenavoli, L.; Falalyeyeva, T.; Mykhalchyshyn, G.; Boccuto, L.; Kononenko, L.; Kyriienko, D.; Komisarenko, I.; Dynnyk, O. Beneficial effects of probiotic combination with omega-3 fatty acids in NAFLD: A randomized clinical study. Minerva Med. 2018, 109, 418–428. [Google Scholar] [CrossRef]
- Kobyliak, N.; Abenavoli, L.; Mykhalchyshyn, G.; Kononenko, L.; Boccuto, L.; Kyriienko, D.; Dynnyk, O. A Multi-strain Probiotic Reduces the Fatty Liver Index, Cytokines and Aminotransferase levels in NAFLD Patients: Evidence from a Randomized Clinical Trial. J. Gastrointestin Liver Dis. 2018, 27, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Famouri, F.; Shariat, Z.; Hashemipour, M.; Keikha, M.; Kelishadi, R. Effects of Probiotics on Nonalcoholic Fatty Liver Disease in Obese Children and Adolescents. J. Pediatric Gastroenterol. Nutr. 2017, 64, 413–417. [Google Scholar] [CrossRef]
- Duseja, A.; Acharya, S.K.; Mehta, M.; Chhabra, S.; Shalimar; Rana, S.; Das, A.; Dattagupta, S.; Dhiman, R.K.; Chawla, Y.K. High potency multistrain probiotic improves liver histology in non-alcoholic fatty liver disease (NAFLD): A randomised, double-blind, proof of concept study. BMJ Open Gastroenterol. 2019, 6, e000315. [Google Scholar] [CrossRef] [Green Version]
- Bakhshimoghaddam, F.; Shateri, K.; Sina, M.; Hashemian, M.; Alizadeh, M. Daily Consumption of Synbiotic Yogurt Decreases Liver Steatosis in Patients with Nonalcoholic Fatty Liver Disease: A Randomized Controlled Clinical Trial. J. Nutr. 2018, 148, 1276–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.B.; Jun, D.W.; Kang, B.K.; Lim, J.H.; Lim, S.; Chung, M.J. Randomized, Double-blind, Placebo-controlled Study of a Multispecies Probiotic Mixture in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 5688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzhalii, E.; Virchenko, O.; Falalyeyeva, T.; Beregova, T.; Stremmel, W. Treatment efficacy of a probiotic preparation for non-alcoholic steatohepatitis: A pilot trial. J. Dig. Dis. 2017, 18, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Armet, A.M.; Finlay, B.B.; Shanahan, F. Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell 2020, 180, 221–232. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, H.; Min, B.-H.; Ganesan, R.; Gebru, Y.A.; Sharma, S.P.; Park, E.; Won, S.-M.; Jeong, J.-J.; Lee, S.-B.; Cha, M.-G.; et al. Gut Microbiome in Non-Alcoholic Fatty Liver Disease: From Mechanisms to Therapeutic Role. Biomedicines 2022, 10, 550. https://doi.org/10.3390/biomedicines10030550
Gupta H, Min B-H, Ganesan R, Gebru YA, Sharma SP, Park E, Won S-M, Jeong J-J, Lee S-B, Cha M-G, et al. Gut Microbiome in Non-Alcoholic Fatty Liver Disease: From Mechanisms to Therapeutic Role. Biomedicines. 2022; 10(3):550. https://doi.org/10.3390/biomedicines10030550
Chicago/Turabian StyleGupta, Haripriya, Byeong-Hyun Min, Raja Ganesan, Yoseph Asmelash Gebru, Satya Priya Sharma, Eunju Park, Sung-Min Won, Jin-Ju Jeong, Su-Been Lee, Min-Gi Cha, and et al. 2022. "Gut Microbiome in Non-Alcoholic Fatty Liver Disease: From Mechanisms to Therapeutic Role" Biomedicines 10, no. 3: 550. https://doi.org/10.3390/biomedicines10030550