Structural Characterization of Full-Length Human Dehydrodolichyl Diphosphate Synthase Using an Integrative Computational and Experimental Approach

 ,
,
Abstract
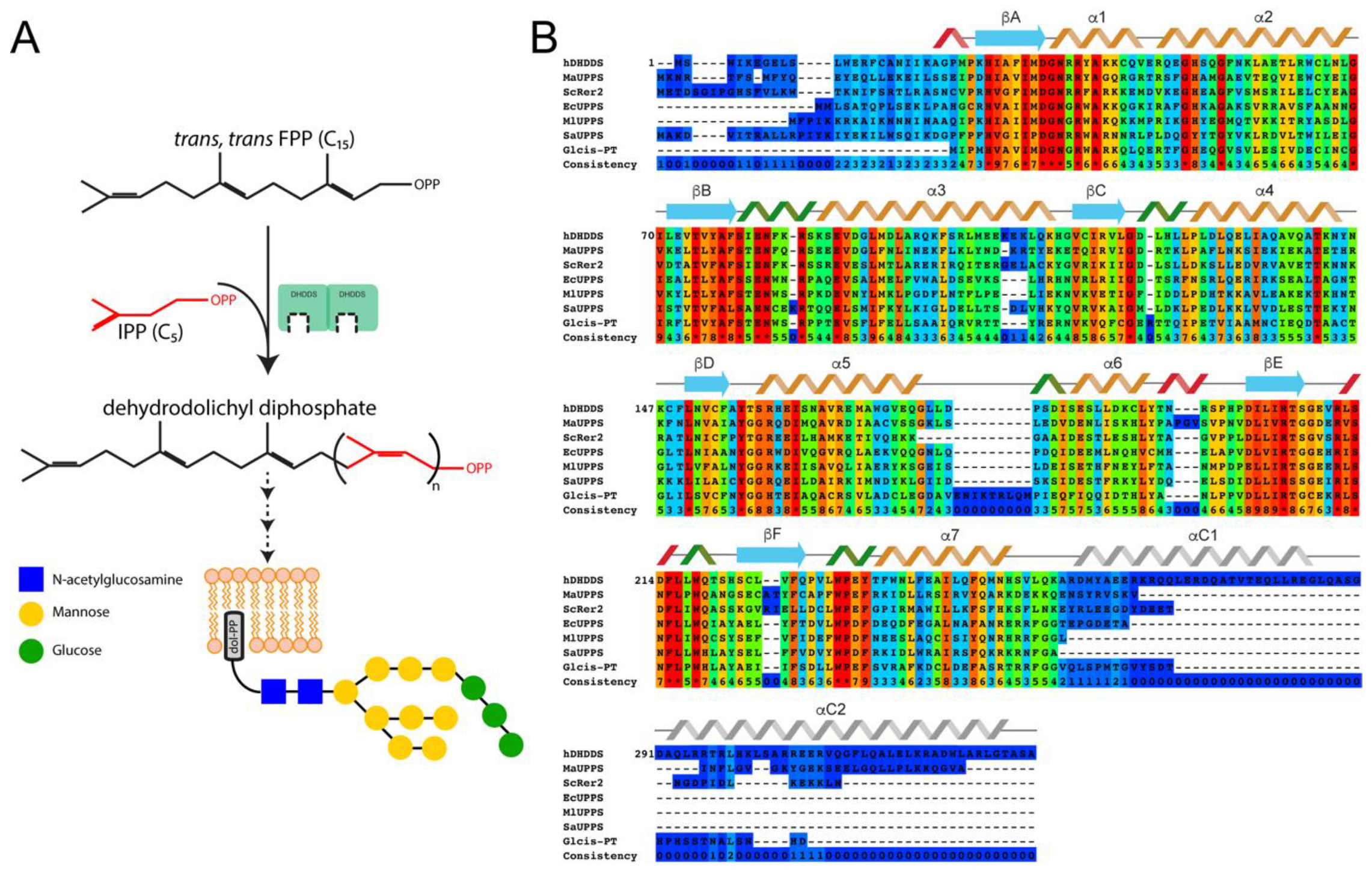
:1. Introduction
2. Materials and Methods
2.1. Cloning
2.2. Protein Expression and Purification
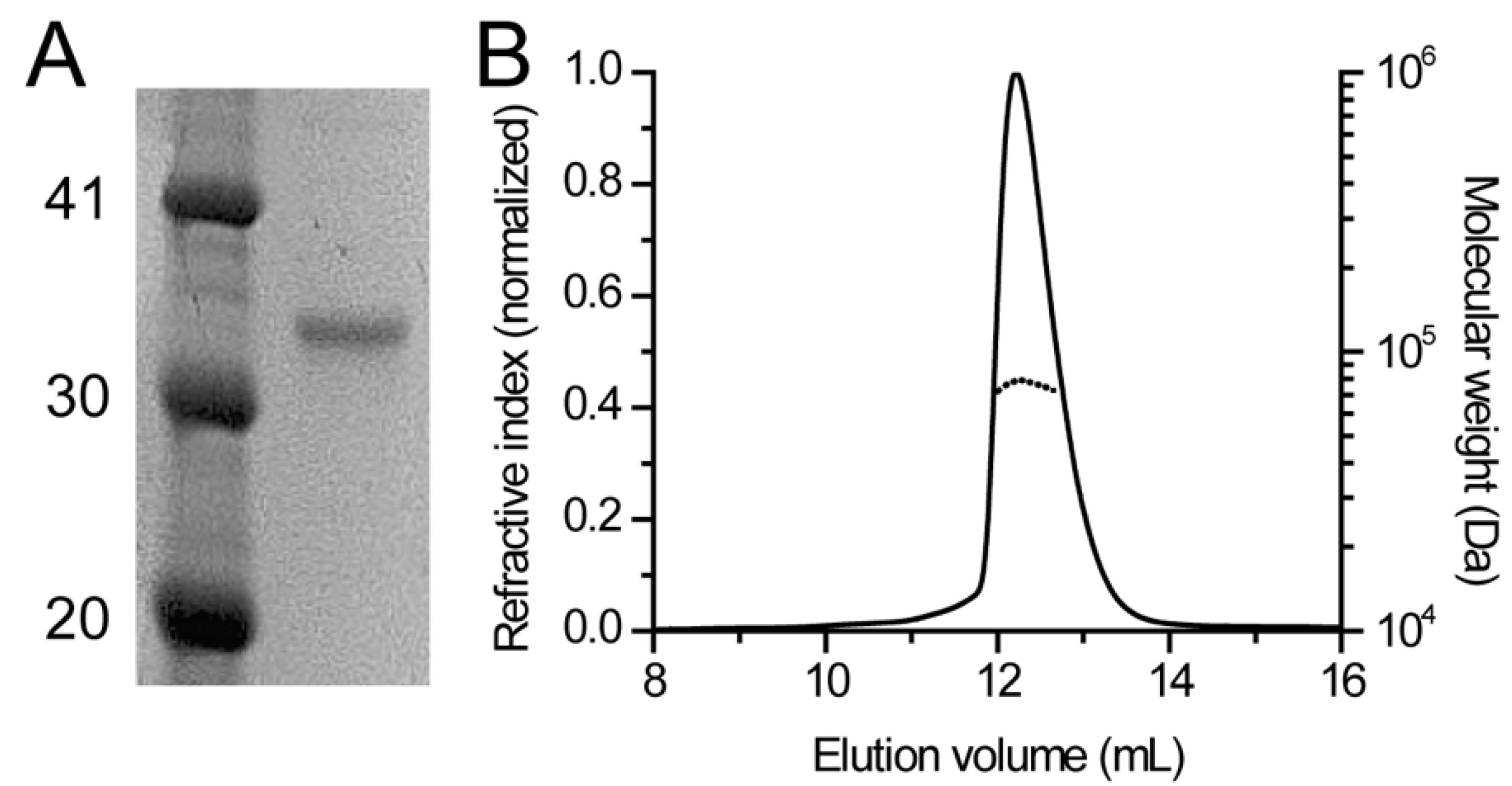
2.3. SEC-MALS
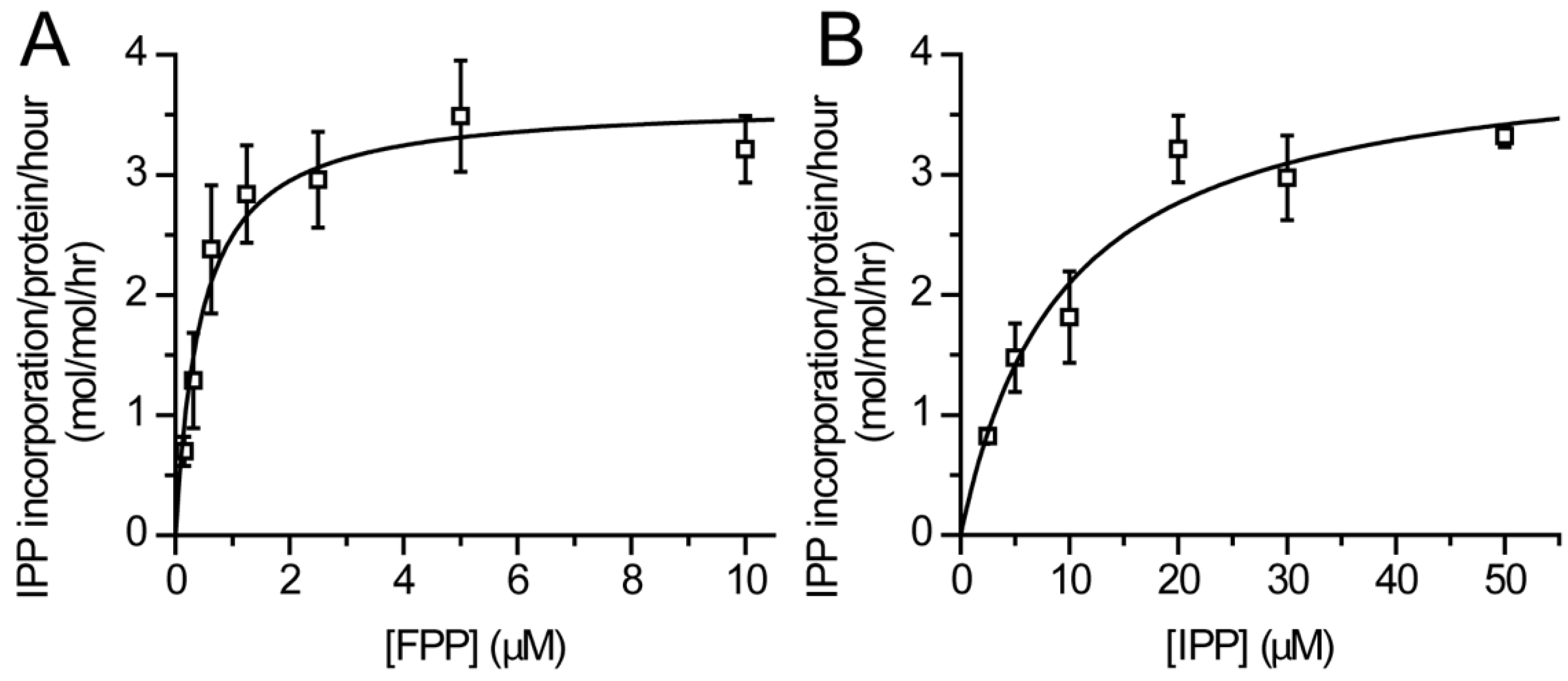
2.4. Enzyme Kinetics
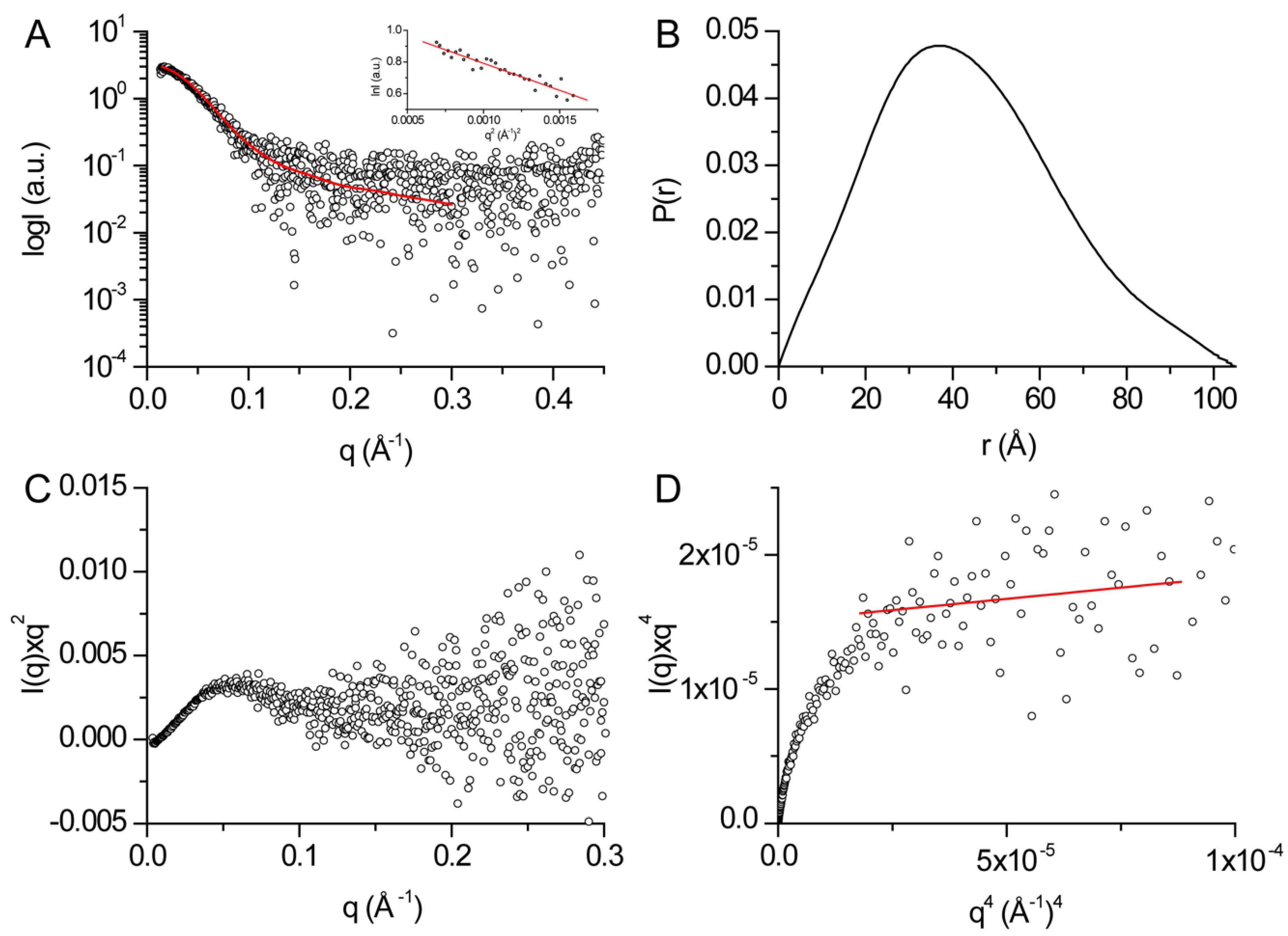
2.5. SAXS
2.6. HDX-MS
2.7. Structural Modeling
3. Results
3.1. Human DHDDS Forms a Monodisperse Homodimer
3.2. Catalytic Activity of Purified DHDDS
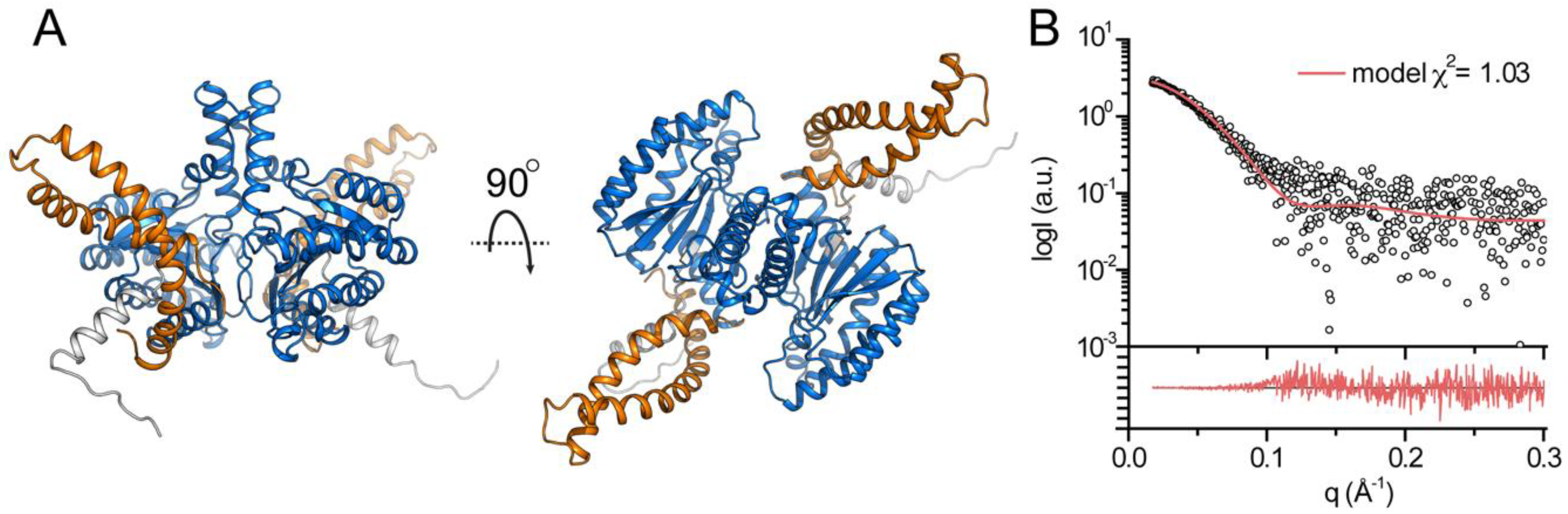
3.3. Structural Features of DHDDS in Solution
3.4. Structural Modeling of Full-Length DHDDS
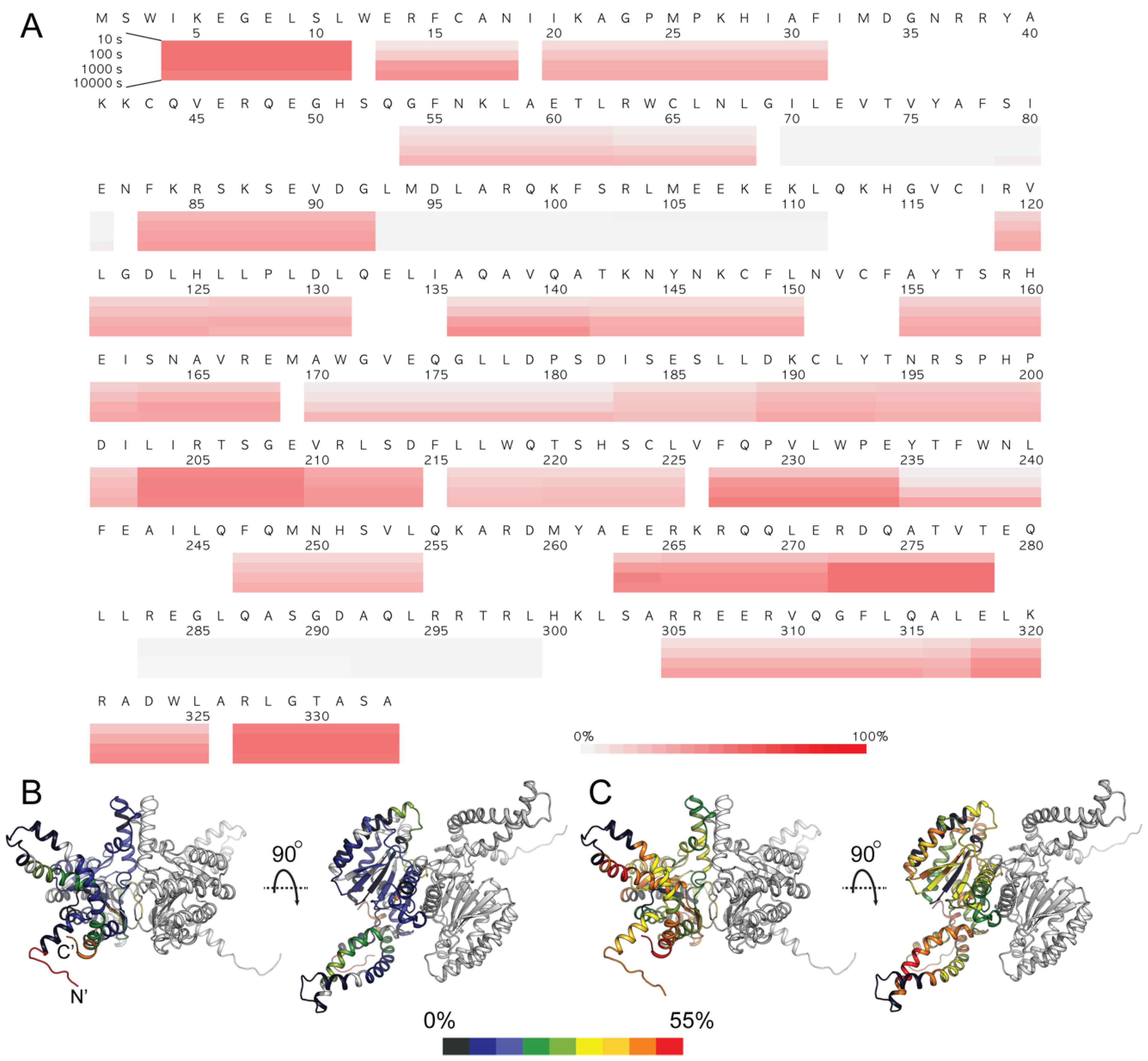
3.5. HDX-MS Analysis of Human DHDDS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Freeze, H.H. Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet. 2018, 34, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H. Understanding human glycosylation disorders: Biochemistry leads the charge. J. Biol. Chem. 2013, 288, 6936–6945. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.D.; Park, E.J.; Gao, N.; Kuo, A.; Rush, J.S.; Waechter, C.J.; Lehrman, M.A.; Sessa, W.C. Nogo-B receptor is necessary for cellular dolichol biosynthesis and protein N-glycosylation. EMBO J. 2011, 30, 2490–2500. [Google Scholar] [CrossRef]
- Grabińska, K.A.; Park, E.J.; Sessa, W.C. CIS-Prenyltransferase: New insights into protein glycosylation, rubber synthesis, and human diseases. J. Biol. Chem. 2016, 291, 18582–18590. [Google Scholar] [CrossRef]
- Ma, J.; Ko, T.P.; Yu, X.; Zhang, L.; Ma, L.; Zhai, C.; Guo, R.T.; Liu, W.; Li, H.Z.; Chen, C.C. Structural insights to heterodimeric cis-prenyltransferases through yeast dehydrodolichyl diphosphate synthase subunit Nus1. Biochem. Biophys. Res. Commun. 2019, 515, 621–626. [Google Scholar] [CrossRef]
- Zelinger, L.; Banin, E.; Obolensky, A.; Mizrahi-Meissonnier, L.; Beryozkin, A.; Bandah-Rozenfeld, D.; Frenkel, S.; Ben-Yosef, T.; Merin, S.; Schwartz, S.B.; et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in ashkenazi jews. Am. J. Hum. Genet. 2011, 88, 207–215. [Google Scholar] [CrossRef]
- Züchner, S.; Dallman, J.; Wen, R.; Beecham, G.; Naj, A.; Farooq, A.; Kohli, M.A.; Whitehead, P.L.; Hulme, W.; Konidari, I.; et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 2011, 88, 201–206. [Google Scholar] [CrossRef]
- Sabry, S.; Vuillaumier-Barrot, S.; Mintet, E.; Fasseu, M.; Valayannopoulos, V.; Héron, D.; Dorison, N.; Mignot, C.; Seta, N.; Chantret, I.; et al. A case of fatal Type i congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J. Rare Dis. 2016, 11. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Koyama, T. Structure and function of cis-prenyl chain elongating enzymes. Chem. Rec. 2006, 6, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.H.; Ko, T.P.; Wang, A.H.J. Structure, mechanism and function of prenyltransferases. Eur. J. Biochem. 2002, 269, 3339–3354. [Google Scholar] [CrossRef]
- Edri, I.; Goldenberg, M.; Lisnyansky, M.; Strulovich, R.; Newman, H.; Loewenstein, A.; Khananshvili, D.; Giladi, M.; Haitin, Y. Overexpression and Purification of Human Cis-prenyltransferase in Escherichia coli. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Giladi, M.; Edri, I.; Goldenberg, M.; Newman, H.; Strulovich, R.; Khananshvili, D.; Haitin, Y.; Loewenstein, A. Purification and characterization of human dehydrodolychil diphosphate synthase (DHDDS) overexpressed in E. coli. Protein Expr. Purif. 2017, 132, 138–142. [Google Scholar] [CrossRef]
- Rout, M.P.; Sali, A. Principles for Integrative Structural Biology Studies. Cell 2019, 177, 1384–1403. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J.A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 2011, 95, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinforma. 2016. [Google Scholar] [CrossRef]
- Chan, Y.T.; Ko, T.P.; Yao, S.H.; Chen, Y.W.; Lee, C.C.; Wang, A.H.J. Crystal Structure and Potential Head-to-Middle Condensation Function of a Z,Z-Farnesyl Diphosphate Synthase. ACS Omega 2017, 2, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.P.; Huang, C.H.; Lai, S.J.; Chen, Y. Structure of undecaprenyl pyrophosphate synthase from Acinetobacter baumannii. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 765–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.T.; Ko, T.P.; Chen, A.P.C.; Kuo, C.J.; Wang, A.H.J.; Liang, P.H. Crystal structures of undecaprenyl pyrophosphate synthase in complex with magnesium, isopentenyl pyrophosphate, and farnesyl thiopyrophosphate: Roles of the metal ion and conserved residues in catalysis. J. Biol. Chem. 2005, 280, 20762–20774. [Google Scholar] [CrossRef] [PubMed]
- Grabińska, K.A.; Edani, B.H.; Park, E.J.; Kraehling, J.R.; Sessa, W.C. A conserved C-terminal RXG motif in the NgBR subunit of cis-prenyltransferase is critical for prenyltransferase activity. J. Biol. Chem. 2017, 292, 17351–17361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M. Size Exclusion Chromatography (SEC). In Biopharmaceutical Processing: Development, Design, and Implementation of Manufacturing Processes; Elsevier: Amsterdam, The Netherlands, 2018; pp. 421–432. ISBN 9780128125526. [Google Scholar]
- Andersson, M.; Wittgren, B.; Wahlund, K.G. Accuracy in multiangle light scattering measurements for molar mass and radius estimations. Model calculations and experiments. Anal. Chem. 2003, 75, 4279–4291. [Google Scholar] [CrossRef] [PubMed]
- Folta-Stogniew, E. Oligomeric states of proteins determined by size-exclusion chromatography coupled with light scattering, absorbance, and refractive index detectors. Methods Mol. Biol. 2006, 328, 97–112. [Google Scholar] [PubMed]
- Dyer, K.N.; Hammel, M.; Rambo, R.P.; Tsutakawa, S.E.; Rodic, I.; Classen, S.; Tainer, J.A.; Hura, G.L. High-throughput SAXS for the characterization of biomolecules in solution: A practical approach. Methods Mol. Biol. 2014, 1091, 245–258. [Google Scholar]
- Koch, M.H.J.; Vachette, P.; Svergun, D.I. Small-angle scattering: A view on the properties, structures and structural changes of biological macromolecules in solution. Q. Rev. Biophys. 2003, 36, 147–227. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013, 105, 962–974. [Google Scholar] [CrossRef]
- Masson, G.R.; Jenkins, M.L.; Burke, J.E. An overview of hydrogen deuterium exchange mass spectrometry (HDX-MS) in drug discovery. Expert Opin. Drug Discov. 2017, 12, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Grabińska, K.A.; Guan, Z.; Stránecký, V.; Hartmannová, H.; Hodaňová, K.; Barešová, V.; Sovová, J.; Jozsef, L.; Ondrušková, N.; et al. Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab. 2014, 20, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.F.; Zhang, L.; Li, K.; Mei, J.P.; Xue, J.; Chen, J.; Tang, X.; Shen, L.; Jiang, H.; Chen, C.; et al. Coding mutations in NUS1 contribute to Parkinson’s disease. Proc. Natl. Acad. Sci. USA. 2018, 115, 11567–11572. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Yamaguchi, H.; Waki, T.; Aoki, Y.; Mizuno, M.; Yanbe, F.; Ishii, T.; Funaki, A.; Tozawa, Y.; Miyagi-Inoue, Y.; et al. Identification and reconstitution of the rubber biosynthetic machinery on rubber particles from Hevea brasiliensis. Elife 2016, 5, e19022. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection Parameters | |
|---|---|
| Beamline | ESRF BM29 |
| Beam geometry (mm2) | 0.7 × 0.7 |
| Wavelength (Å) | 1.0 |
| Q range (Å−1) | 0.0025–0.5 |
| Exposure per frame (seconds) | 1 |
| Temperature (°C) | 20 |
| Structural parameters | |
| Rg (Å)1 | 31.8 ± 1.4 |
| Dmax (Å)2 | 105 ± 11 |
| Porod volume [from P(r)] (103 Å3) | 132.8 |
| Estimated mass (kDa)2 | 78.1 ± 7.8 |
| Porod exponent | 3.6 |
| χ2 model | 1.03 |
| Software employed | |
| Primary data reduction | AUTOMAR |
| Data processing | PRIMUS, GNOM, ScÅtter |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisnyansky Bar-El, M.; Lee, S.Y.; Ki, A.Y.; Kapelushnik, N.; Loewenstein, A.; Chung, K.Y.; Schneidman-Duhovny, D.; Giladi, M.; Newman, H.; Haitin, Y. Structural Characterization of Full-Length Human Dehydrodolichyl Diphosphate Synthase Using an Integrative Computational and Experimental Approach. Biomolecules 2019, 9, 660. https://doi.org/10.3390/biom9110660
Lisnyansky Bar-El M, Lee SY, Ki AY, Kapelushnik N, Loewenstein A, Chung KY, Schneidman-Duhovny D, Giladi M, Newman H, Haitin Y. Structural Characterization of Full-Length Human Dehydrodolichyl Diphosphate Synthase Using an Integrative Computational and Experimental Approach. Biomolecules. 2019; 9(11):660. https://doi.org/10.3390/biom9110660
Chicago/Turabian StyleLisnyansky Bar-El, Michal, Su Youn Lee, Ah Young Ki, Noa Kapelushnik, Anat Loewenstein, Ka Young Chung, Dina Schneidman-Duhovny, Moshe Giladi, Hadas Newman, and Yoni Haitin. 2019. "Structural Characterization of Full-Length Human Dehydrodolichyl Diphosphate Synthase Using an Integrative Computational and Experimental Approach" Biomolecules 9, no. 11: 660. https://doi.org/10.3390/biom9110660