Linking Dysregulated AMPK Signaling and ER Stress in Ethanol-Induced Liver Injury in Hepatic Alcohol Dehydrogenase Deficient Deer Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Animal Experiments
2.3. Blood Alcohol and Acetaldehyde Levels, Plasma ALT and Hepatic CYP2E1 Activity
2.4. Morphological Studies and Immunohistochemical Staining
2.5. Hepatic Lipid Contents
2.6. Immunoblot Analysis
2.7. Statistical Analysis
3. Results
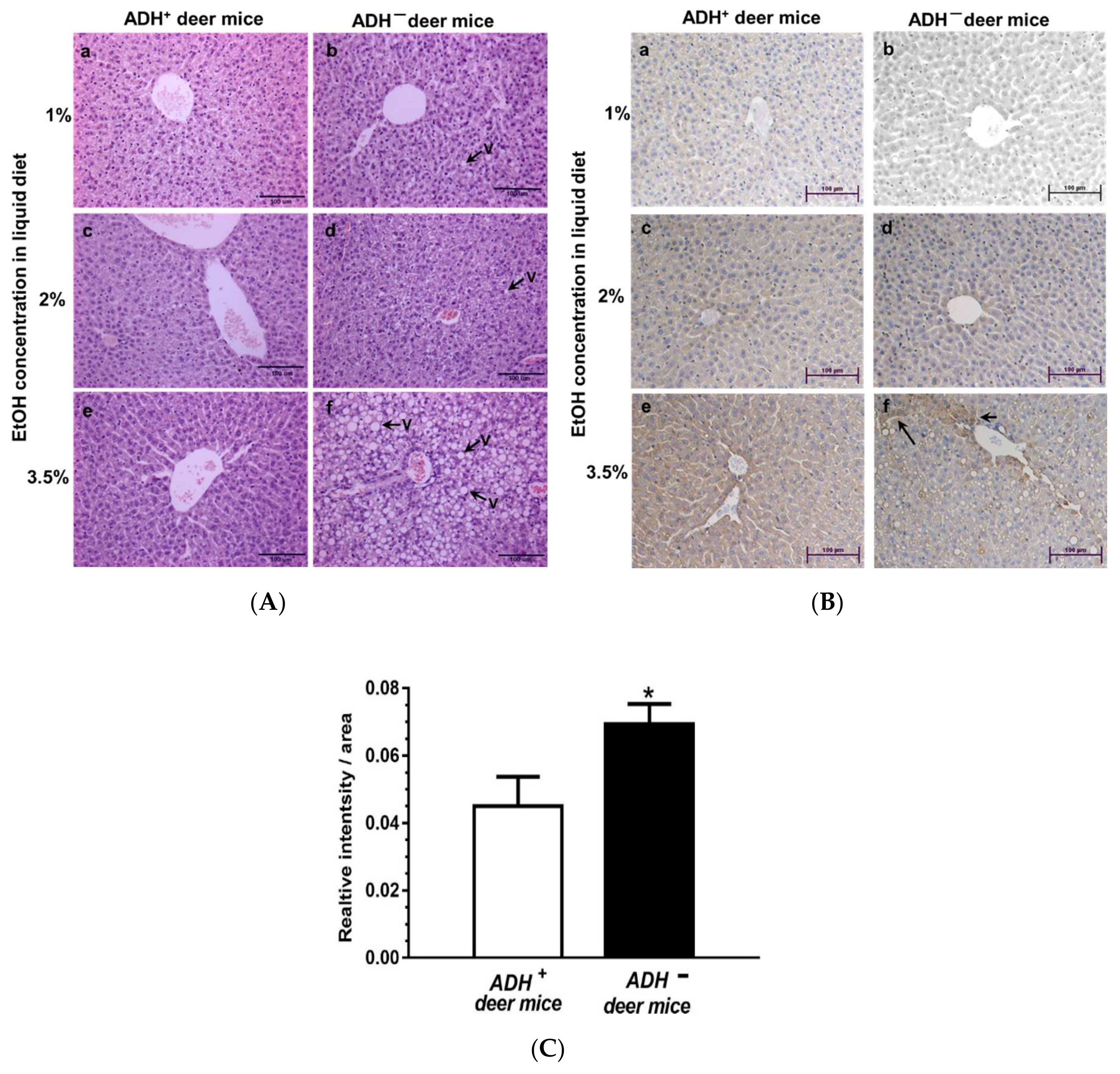
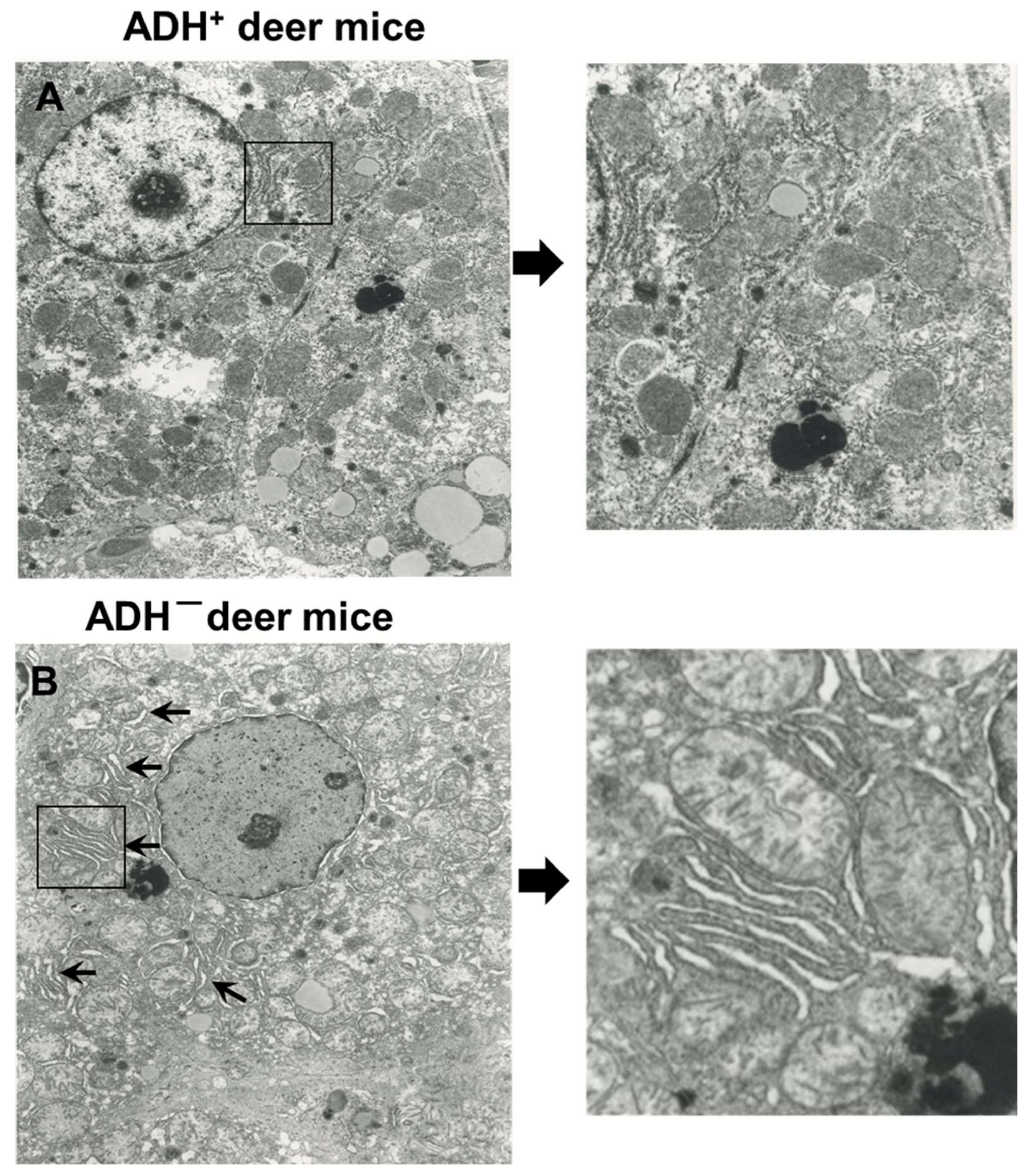
3.1. Morphological and Immunohistochemical Changes
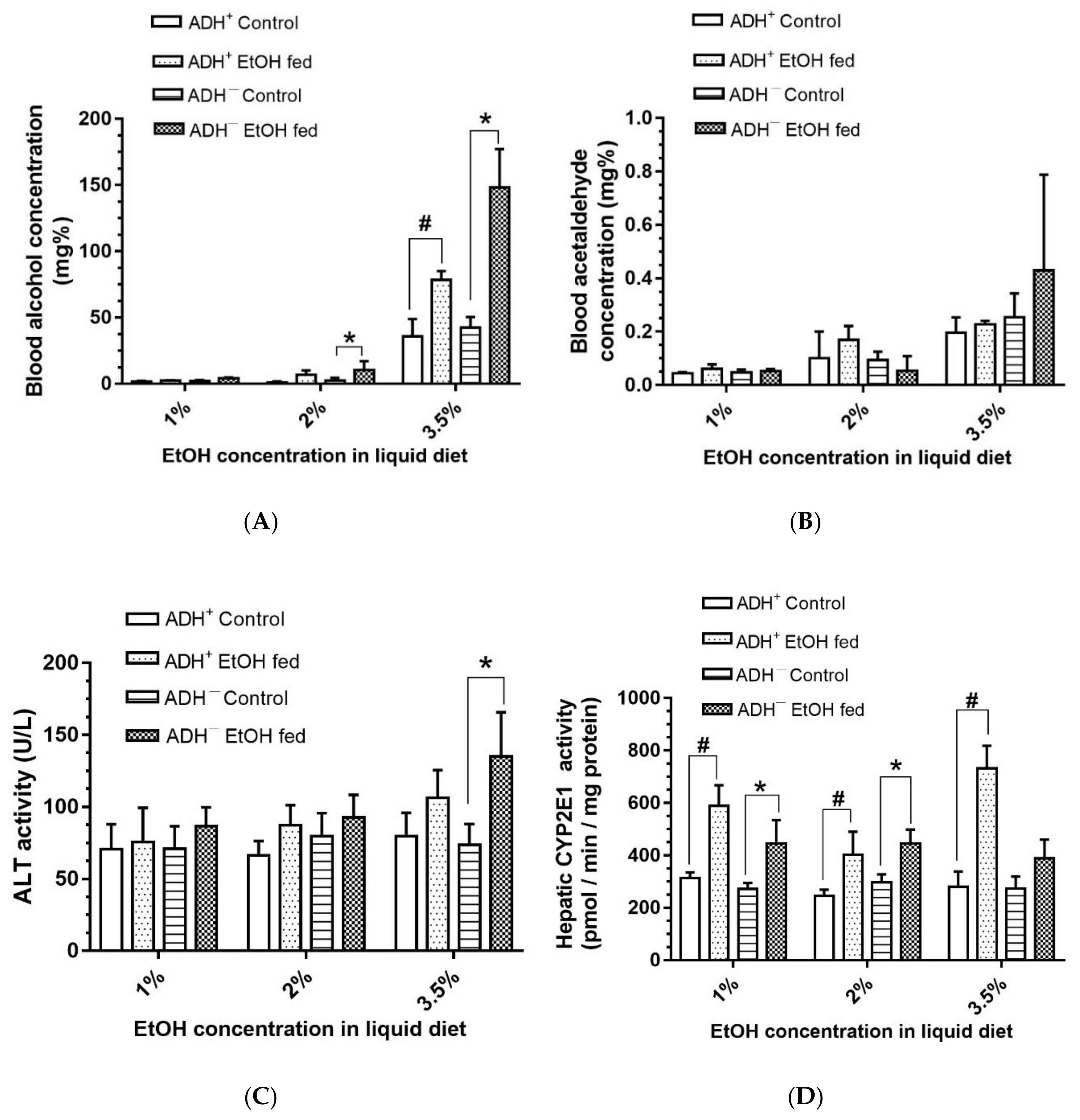
3.2. Blood Alcohol and Acetaldehyde Levels, and Levels of Hepatic Injury Marker and CYP2E1
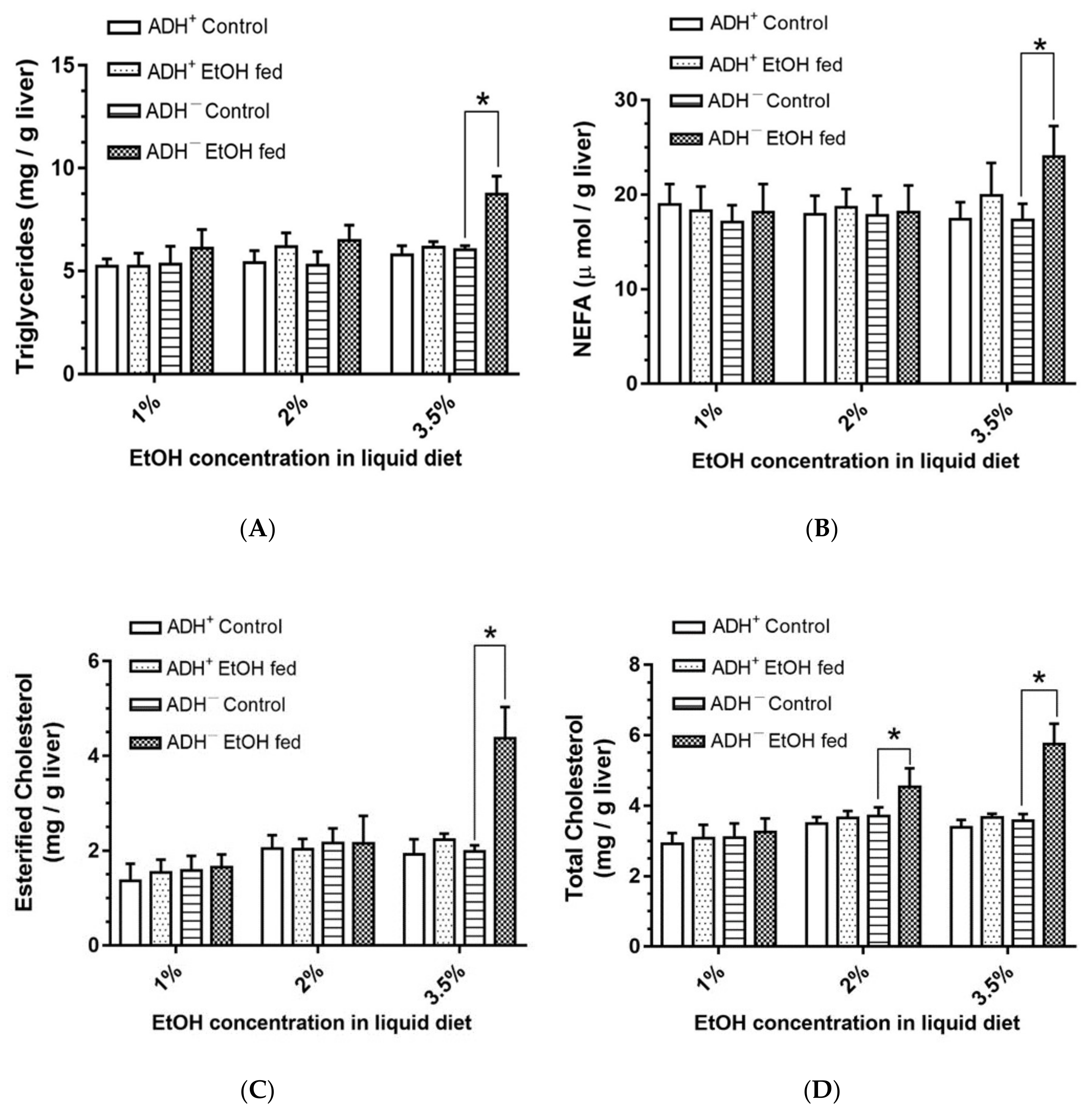
3.3. Hepatic Lipid Levels
3.4. ER Stress and Cell Death Pathways
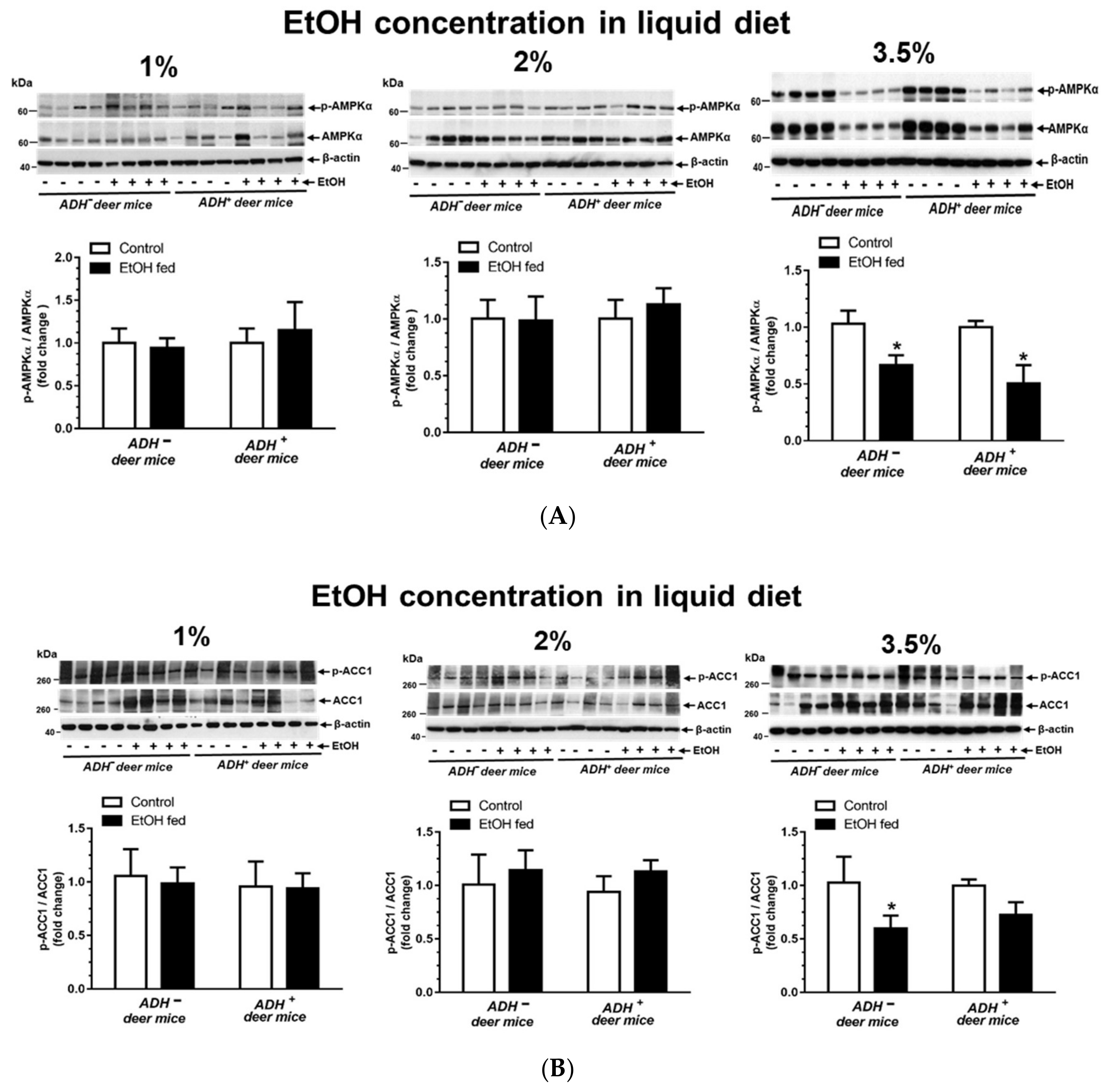
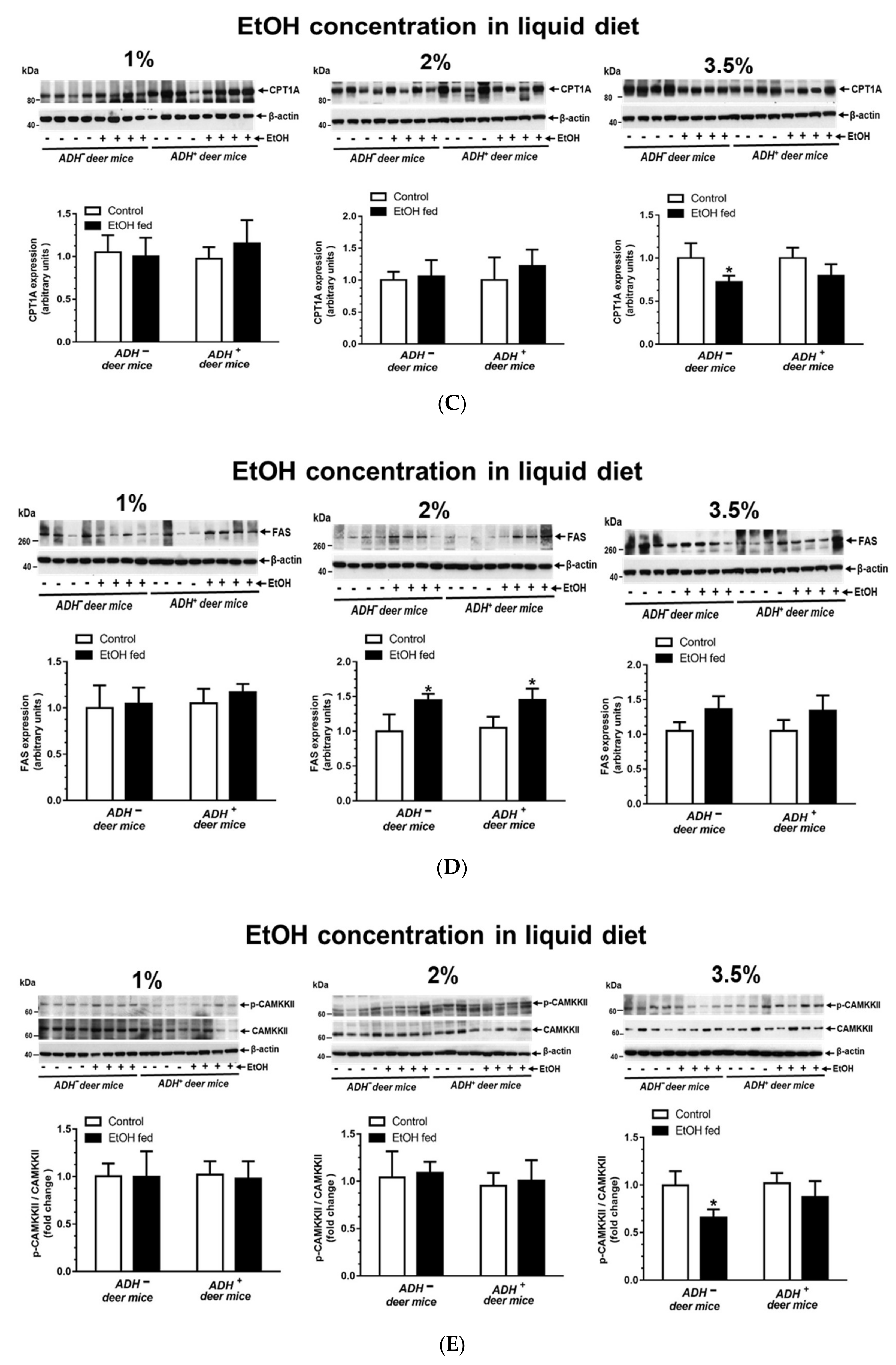
3.5. AMPKα Signaling
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Anand, B.S. Cirrhosis of liver. West. J. Med. 1999, 171, 110–115. [Google Scholar] [PubMed]
- Grant, B.F.; Goldstein, R.B.; Saha, T.D.; Chou, S.P.; Jung, J.; Zhang, H.; Pickering, R.P.; Ruan, W.J.; Smith, S.M.; Huang, B.; et al. Epidemiology of DSM-5 Alcohol Use Disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry 2015, 72, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Lucey, M.R.; Mathurin, P.; Morgan, T.R. Alcoholic hepatitis. N. Engl. J. Med. 2009, 360, 2758–2769. [Google Scholar] [CrossRef] [PubMed]
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 National and State Costs of Excessive Alcohol Consumption. Am. J. Prev. Med. 2015, 49, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Giacca, M.; Miglioli, L.; Monzoni, A.; Tiribelli, C. Risk factors for alcoholic liver disease. Addict. Biol. 2000, 5, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Clemens, M.; Diehl, A.M. Obesity increases sensitivity to endotoxin liver injury: Implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. USA 1997, 94, 2557–2562. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Lin, H.; Diehl, A.M. Fatty liver vulnerability to endotoxin-induced damage despite NF-kappaB induction and inhibited caspase 3 activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 281–392. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M. Nonalcoholic fatty liver disease: Implications for alcoholic liver disease pathogenesis. Alcohol. Clin. Exp. Res. 2001, 25, 8S–14S. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J.; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar]
- Anstee, Q.M.; Daly, A.K.; Day, C.P. Genetics of alcoholic and nonalcoholic fatty liver disease. Semin. Liver Dis 2011, 31, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Askgaard, G.; Gronbaek, M.; Kjaer, M.S.; Tjonneland, A.; Tolstrup, J.S. Alcohol drinking pattern and risk of alcoholic liver cirrhosis: A prospective cohort study. J. Hepatol. 2015, 62, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.E.; Smart, R.G.; Govoni, R. The epidemiology of alcoholic liver disease. Alcohol Res. Health 2003, 27, 209–219. [Google Scholar] [PubMed]
- Kaphalia, B.S.; Khan, M.F.; Carroll, R.M.; Aronson, J.; Ansari, G.A.S. Subchronic toxicity of 2-chloroethanol and 2-bromoethanol in rats. Res Commun Pharmacol Toxicol 1996, 1, 173–186. [Google Scholar]
- Nuutinen, H.; Lindros, K.O.; Salaspuro, M. Determinants of blood acetaldehyde level during ethanol oxidation in chronic alcoholics. Alcohol Clin. Exp. Res. 1983, 7, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Panes, J.; Caballeria, J.; Guitart, R.; Pares, A.; Soler, X.; Rodamilans, M.; Navasa, M.; Pares, X.; Bosch, J.; Rodes, J. Determinants of ethanol and acetaldehyde metabolism in chronic alcoholics. Alcohol Clin. Exp. Res. 1993, 17, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Sharkawi, M. In vivo inhibition of liver alcohol dehydrogenase by ethanol administration. Life Sci. 1984, 35, 2353–2357. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig. Dis. 2010, 28, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhai, Y.; Mu, Y.; Gong, H.; Uppal, H.; Toma, D.; Ren, S.; Evans, R.M.; Xie, W. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 2006, 281, 15013–15020. [Google Scholar] [CrossRef]
- Siler, S.Q.; Neese, R.A.; Hellerstein, M.K. De novo lipogenesis, lipid kinetics, and whole-body lipid balances in humans after acute alcohol consumption. Am. J. Clin. Nutr. 1999, 70, 928–936. [Google Scholar] [CrossRef]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Rasineni, K.; Donohue, T.M., Jr.; Thomes, P.G.; Yang, L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Ethanol-induced steatosis involves impairment of lipophagy, associated with reduced Dynamin2 activity. Hepatol. Commun. 2017, 1, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, R.J.; Rasineni, K.; Weller, S.G.; Schott, M.B.; Schroeder, B.; Casey, C.A.; McNiven, M.A. Ethanol exposure inhibits hepatocyte lipophagy by inactivating the small guanosine triphosphatase Rab7. Hepatol. Commun. 2017, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.J.; Smathers, R.L.; Shearn, C.T.; Fritz, K.S.; Backos, D.S.; Jiang, H.; Franklin, C.C.; Orlicky, D.J.; Maclean, K.N.; Petersen, D.R. Oxidative Stress and the ER Stress Response in a Murine Model for Early-Stage Alcoholic Liver Disease. J. Toxicol. 2012, 2012, 207594. [Google Scholar] [CrossRef]
- Zeng, H.; Guo, X.; Zhou, F.; Xiao, L.; Liu, J.; Jiang, C.; Xing, M.; Yao, P. Quercetin alleviates ethanol-induced liver steatosis associated with improvement of lipophagy. Food Chem. Toxicol. 2019, 125, 21–28. [Google Scholar] [CrossRef]
- Qiang, X.; Xu, L.; Zhang, M.; Zhang, P.; Wang, Y.; Wang, Y.; Zhao, Z.; Chen, H.; Liu, X.; Zhang, Y. Demethyleneberberine attenuates non-alcoholic fatty liver disease with activation of AMPK and inhibition of oxidative stress. Biochem. Biophys. Res. Commun. 2016, 472, 603–609. [Google Scholar] [CrossRef]
- Bhopale, K.K.; Wu, H.; Boor, P.J.; Popov, V.L.; Ansari, G.A.; Kaphalia, B.S. Metabolic basis of ethanol-induced hepatic and pancreatic injury in hepatic alcohol dehydrogenase deficient deer mice. Alcohol 2006, 39, 179–188. [Google Scholar] [CrossRef]
- Kaphalia, B.S.; Bhopale, K.K.; Kondraganti, S.; Wu, H.; Boor, P.J.; Ansari, G.A. Pancreatic injury in hepatic alcohol dehydrogenase-deficient deer mice after subchronic exposure to ethanol. Toxicol. Appl. Pharmacol. 2010, 246, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Cai, P.; Clemens, D.L.; Jerrells, T.R.; Ansari, G.A.; Kaphalia, B.S. Metabolic basis of ethanol-induced cytotoxicity in recombinant HepG2 cells: Role of nonoxidative metabolism. Toxicol. Appl. Pharmacol. 2006, 216, 238–247. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Adenovirus-mediated expression of CYP2E1 produces liver toxicity in mice. Toxicol. Sci. 2006, 91, 365–371. [Google Scholar] [CrossRef]
- Fernando, H.; Bhopale, K.K.; Boor, P.J.; Ansari, G.A.; Kaphalia, B.S. Hepatic lipid profiling of deer mice fed ethanol using (1)H and (3)(1)P NMR spectroscopy: A dose-dependent subchronic study. Toxicol. Appl. Pharmacol. 2012, 264, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Fernando, H.; Bhopale, K.K.; Kondraganti, S.; Kaphalia, B.S.; Shakeel Ansari, G.A. Lipidomic changes in rat liver after long-term exposure to ethanol. Toxicol. Appl. Pharmacol. 2011, 255, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG Clinical Guideline: Alcoholic Liver Disease. Am J Gastroenterol. 2018, 113, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly, Y.M.; Carling, D. Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, K.G.; Felder, M.R. Ethanol metabolism in Peromyscus genetically deficient in alcohol dehydrogenase. Biochem. Pharmacol. 1980, 29, 125–130. [Google Scholar] [CrossRef]
- Okuda, T.; Haseba, T.; Katsuyama, M.; Maruyama, M.; Akimoto, T.; Igarashi, T.; Ohno, Y. Metabolic pharmacokinetics of early chronic alcohol consumption mediated by liver alcohol dehydrogenases 1 and 3 in mice. J Gastroenterol. Hepatol. 2018, 33, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Deltour, L.; Foglio, M.H.; Duester, G. Metabolic deficiencies in alcohol dehydrogenase Adh1, Adh3, and Adh4 null mutant mice. Overlapping roles of Adh1 and Adh4 in ethanol clearance and metabolism of retinol to retinoic acid. J. Biol. Chem. 1999, 274, 16796–16801. [Google Scholar] [CrossRef]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J. Biol. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed]
- Sozio, M.S.; Liangpunsakul, S.; Crabb, D. The role of lipid metabolism in the pathogenesis of alcoholic and nonalcoholic hepatic steatosis. Semin. Liver Dis. 2010, 30, 378–390. [Google Scholar] [CrossRef]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef]
- DeZwaan-McCabe, D.; Sheldon, R.D.; Gorecki, M.C.; Guo, D.F.; Gansemer, E.R.; Kaufman, R.J.; Rahmouni, K.; Gillum, M.P.; Taylor, E.B.; Teesch, L.M.; et al. ER Stress Inhibits Liver Fatty Acid Oxidation while Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep. 2017, 19, 1794–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C. New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. Int. J. Hepatol. 2014, 2014, 513787. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Li, Z. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Front Genet. 2014, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, 48–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Shu, M.S.; Kim, J.Y.; Kim, Y.H.; Sim, K.H.; Sung, W.J.; Eun, J.R. Cilostazol protects hepatocytes against alcohol-induced apoptosis via activation of AMPK pathway. PLoS ONE 2019, 14, 14–e0211415. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, S.; Yang, J.H.; Kim, S.; Sim, J.; Kim, M.G.; Jeong, T.C.; Ku, S.K.; Cho, I.J.; Ki, S.H. Korean Red Ginseng attenuates ethanol-induced steatosis and oxidative stress via AMPK/Sirt1 activation. J. Ginseng Res. 2015, 39, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yang, C.; Thomes, P.G.; Kharbanda, K.K.; Casey, C.A.; McNiven, M.A.; Donohue, T.M., Jr. Lipophagy and Alcohol-Induced Fatty Liver. Front Pharmacol. 2019, 10, 495. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Changes in Protein Levels/Expression |
|---|---|
| p-IRE1α/IRE1α | No Change |
| Unspliced XBP1 * | Increased * |
| ATF-6 | No Change |
| PERK | No Change |
| p-EIF2α/EIF2α | No Change |
| CHOP | No Change |
| Caspase-1 | No Change |
| Caspase-3 | No Change |
| Caspase-8 | No Change |
| Protein | Changes in Protein Levels/Expression |
|---|---|
| p-AMPKα/AMPKα * | Decreased * |
| p-ACC/ACC * | Decreased * |
| FAS | No Change |
| CPT1A * | Decreased * |
| SREBP1 | No Change |
| p-CaMKKβ/CaMKKβ * | Decreased * |
| p-LKB1/LKB1 | No Change |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srinivasan, M.P.; Bhopale, K.K.; Amer, S.M.; Wan, J.; Kaphalia, L.; Ansari, G.S.; Kaphalia, B.S. Linking Dysregulated AMPK Signaling and ER Stress in Ethanol-Induced Liver Injury in Hepatic Alcohol Dehydrogenase Deficient Deer Mice. Biomolecules 2019, 9, 560. https://doi.org/10.3390/biom9100560
Srinivasan MP, Bhopale KK, Amer SM, Wan J, Kaphalia L, Ansari GS, Kaphalia BS. Linking Dysregulated AMPK Signaling and ER Stress in Ethanol-Induced Liver Injury in Hepatic Alcohol Dehydrogenase Deficient Deer Mice. Biomolecules. 2019; 9(10):560. https://doi.org/10.3390/biom9100560
Chicago/Turabian StyleSrinivasan, Mukund P., Kamlesh K. Bhopale, Samir M. Amer, Jie Wan, Lata Kaphalia, Ghulam S. Ansari, and Bhupendra S. Kaphalia. 2019. "Linking Dysregulated AMPK Signaling and ER Stress in Ethanol-Induced Liver Injury in Hepatic Alcohol Dehydrogenase Deficient Deer Mice" Biomolecules 9, no. 10: 560. https://doi.org/10.3390/biom9100560