
Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies
by
Andrew Manley
1,†, 
Bahar I. Meshkat
1,2,†,
Monica M. Jablonski
1,3,4,5 and
T.J. Hollingsworth
1,3,6,* 1
Department of Ophthalmology, Hamilton Eye Institute, College of Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA
2
Department of Physiology, University of Tennessee Health Science Center, Memphis, TN 38163, USA
3
Department of Anatomy and Neurobiology, University of Tennessee Health Science Center, Memphis, TN 38163, USA
4
Department of Pharmaceutical Sciences, University of Tennessee Health Science Center, Memphis, TN 38163, USA
5
Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, USA
6
Department of Microbiology, Immunology, and Biochemistry, University of Tennessee Health Science Center, Memphis, TN 38163, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomolecules 2023, 13(2), 271; https://doi.org/10.3390/biom13020271
Submission received: 21 December 2022
/
Revised: 23 January 2023
/
Accepted: 27 January 2023
/
Published: 1 February 2023
(This article belongs to the Collection Feature Papers in Section Molecular Medicine)
Abstract
:Inherited retinal dystrophies (IRDs) are congenital retinal degenerative diseases that have various inheritance patterns, including dominant, recessive, X-linked, and mitochondrial. These diseases are most often the result of defects in rod and/or cone photoreceptor and retinal pigment epithelium function, development, or both. The genes associated with these diseases, when mutated, produce altered protein products that have downstream effects in pathways critical to vision, including phototransduction, the visual cycle, photoreceptor development, cellular respiration, and retinal homeostasis. The aim of this manuscript is to provide a comprehensive review of the underlying molecular mechanisms of pathogenesis of IRDs by delving into many of the genes associated with IRD development, their protein products, and the pathways interrupted by genetic mutation.
1. Introduction
Inherited retinal dystrophies (IRDs) is an umbrella term for a multitude of retinal neurodegenerative diseases that are the result of genetic mutation derived from a myriad of genes whose protein products are inherently associated with a critical factor of retinal function and support [1,2,3,4,5,6,7,8]. Some examples of these protein products are shown in Figure 1, including their localization in the mouse retina using immunohistochemical labeling. Interestingly, the majority of proteins associated with IRDs are localized to the same cell types, organelles, and structures in mice as they are in humans, making them an important model for studying these diseases. The two main categories of IRDs are rod-cone dystrophies (RCDs) and cone-rod dystrophies (CRDs), with each group characterized by a primary loss of one of the specific photoreceptor cell types—rods or cones—and a secondary loss of the other cell type [1,6,9,10,11,12,13]. It is important to note that, while IRDs primarily affect the photoreceptors’ survival, the mutated genes causing these diseases are not necessarily expressed by the photoreceptors. In fact, congenital retinal pigment epithelium (RPE) dysfunction is a major cause of both RCDs and CRDs and is detailed in Section 4 below. Examples of RCDs include retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA) [6,10]. While both diseases cause primary rod cell death, they remain quite different from one another. LCA is often described as a more severe degeneration on the basis of age (early childhood) and rate of progression (blindness prior to or into early adulthood). Regarding genetic inheritance, LCA is usually recessively inherited. In contrast, RP maintains a more heterogenous group of diseases that includes dominant, recessive, mitochondrial, and X-linked inheritance, as well as a varying age of onset ranging from childhood to adulthood. Disease progression in RP is also varied from mutation to mutation and individual to individual [14,15]. LCA, a less common disease, affects 1:50,000 to 1:100,000 live births, while RP affects approximately 1:3000 to 1:5000 live births [6,10,14]. CRDs result in cone cell loss first, which, in humans, causes a rapid loss of visual acuity and central vision due to cone cell death in the foveal/macular region of the retina [1,5,11]. Diseases in this category include Stargardt’s disease and other macular dystrophies and are also able to be inherited heterogeneously, though less so compared to RP, and occur at a prevalence comparable to that of LCA [1,5,11,16]. Both RP and CRDs, unlike LCA, are capable of being inherited in a syndromic manner (i.e., the retinal disease maintains one phenotype in a primary, multisystemic disease such as Bardet–Biedl syndrome or Usher syndrome) or in a non-syndromic manner, with only the retina being affected [1,3,5,6,10,11,16,17,18,19]. Regardless of whether the IRD presents as a RCD or CRD, the ultimate outcome for the patient is blindness. The remainder of this review will focus on the underlying gene-dependent molecular mechanisms governing IRD pathogeneses, with our focus on the specific cellular organelles and molecular pathways affected by mutations in IRD-causing genes identified by perusing the literature, as well as RetNet (https://web.sph.uth.edu/RetNet/disease.htm, accessed on 28 August 2022). This review covers a majority of these genes, with an emphasis on those that are more commonly mutated.
2. Materials and Methods
2.1. Fluorescent Immunohistochemistry (fIHC)
Whole eyes from C57B/6J mice at 3 months of age were enucleated and fixed in 4% paraformaldehyde in PBS, pH 7.4 overnight at 4 °C. Fixation was quenched in 100 mM glycine in PBS, pH 7.4 for 10 min at room temperature and subsequently washed in PBS. Eyes were dehydrated with 30 min incubations in a graded ethanol series (50%, 70%, 85%, 95%, and 100%), then cleared via a 30 min incubation in graded xylenes (2:1, 1:1, and 1:2 ethanol:xylenes) and two 30 min incubations in 100% xylenes. Eyes were then infiltrated with paraffin using a graded paraffin series with 30 min incubations in 2:1, 1:1, and 1:2 xylenes:paraffin and two subsequent 1 h incubations in 100% paraffin. Paraffin-embedded tissue was then sectioned at 8 µm and sections deparaffinized and rehydrated, treated using heat-mediated antigen retrieval by heating slides at 95 °C in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0) for 1 h, washed in PBS twice, and subsequently blocked in 10% goat serum/5% BSA/0.5% TritonX-100 in PBS for 30 min at RT. Primary antibodies against markers for specific retinal cell types, cytoskeletal components, and IRD-associated genes were then applied at recommended dilutions and incubated overnight at 4 °C (Table 1). Slides were then washed in PBS, pH 7.4 three times for 10 min each. For standard fIHC, post-washing, slides were incubated in secondary antibodies conjugated to either AlexaFluor488l AlexaFluor568; or AlexaFluor647 (A21121, A21131, A21141, A11006, A21134, A11041, A11036, A21236, A21240, A21241, and A21449; ThermoFisher; 168 Third Avenue, Waltham, MA, USA) at 1:400 dilutions for 1 h and nuclei stained using a 1:10,000 dilution of 14.3 mM DAPI (D21490; ThermoFisher; 168 Third Avenue, Waltham, MA, USA) and mounted using Prolong Diamond Antifade mountant (P36961; ThermoFisher; 168 Third Avenue, Waltham, MA, USA). For multiplexed fIHC labeling, HRP-conjugated secondary antibodies were used (A16078, A10551, G21234, and A16005; ThermoFisher; 168 Third Avenue, Waltham, MA, USA), and, post-washing, tyramide signal amplification (TSA) was performed for 2 min with a 50 mM Tris-HCl, pH 7.4 reaction buffer containing 0.0015% H2O2 and tyramide reagent conjugated to either AZDye488; AZDye568; or AZDye647 (1538, 1541, and 1543; Click-ChemistryTools; 8341 E Gelding Drive, Scottsdale, AZ, USA 85260). After TSA, slides were washed in PBS three times and subsequently heated to 95 °C in PBS for 30 min to remove antibodies and subsequently probed using the second or third antibody in the multiplex label. After all TSA was performed, slides were stained with DAPI and washed in PBS, pH 7.4 four times and mounted using Prolong Diamond Antifade mountant (P36961; ThermoFisher; 168 Third Avenue, Waltham, MA, USA). After drying overnight, all sections were imaged using a Zeiss 710 laser scanning confocal microscope (LSM) using a 40× objective with 1.3 numerical aperture (NA) or 63× objective with 1.4 NA with or without an associated 1.7× zoom (~100×).
3. The Photoreceptor Outer Segment
The photoreceptor outer segment is a highly specialized primary cilium containing hundreds of membranous discs whose primary function is to house the components of the phototransduction cascade, which is responsible for converting light into a visual signal. A plethora of genes associated with IRDs affect some aspect of the outer segment, be it phototransduction, ciliary formation or trafficking, or disc morphogenesis, as seen detailed below.
3.1. Phototransduction
The phototransduction cascade is the pathway responsible for converting photons into an electrical signal that the brain is meant to interpret (Figure 2). Rods (and cones in bright light) are critical in this process, as they contain opsins, a group of specialized G-protein coupled receptors (GPCRs) that, when covalently bound to their ligand 11-cis retinal, are capable of absorbing visible light [20,21,22,23]. 11-cis retinal is one of the few GPCR ligands that covalently links to its receptor. In the case of retinal, the linkage is made through a protonated Schiff’s base [24,25,26]. When photons enter the eye and reach the retina, they are absorbed by the 11-cis retinal, converting it to all-trans retinal. Depending on the opsin to which it is bound, the wavelength of light the receptor absorbs changes, allowing for color discrimination [27,28]. Upon photoisomerization, activated opsins bind to and activate the heterotrimeric G-protein transducin (Gt). Opsin-initiated activation of Gt results in the exchange of a GDP (inactive Gt) for GTP (active Gt) on the α subunit of Gt (Gtα). Gtα dissociates from the remaining β and γ subunits and activates cyclic guanosine monophosphate (cGMP) phosphodiesterase 6 (PDE6). PDE6 is a highly efficient enzyme that hydrolyzes cGMP, a gate-keeping molecule that holds the cyclic nucleotide-gated (CNG) cation channel open into GMP. The decrease in intracellular cGMP concentration results in CNG channel closure and a subsequent hyperpolarization of the photoreceptor [20,21,22]. This causes glutamate release at the synapse to slow, resulting in signal propagation to the bipolar cells for eventual relay to the brain. Once turned on, the cascade must also be turned off. As described, phototransduction is a multistep process that includes numerous proteins (Figure 3). In the case of rod photoreceptors, the primary cells affected in RP, recoverin, an inhibitory protein of rhodopsin kinase (GRK1), releases its inhibition upon lower calcium levels subsequent to CNG channel closure. GRK1 then phosphorylates rhodopsin at its carboxyl terminus, a signal to the protein arrestin to cap the Gt activation site of rhodopsin [20,23,29]. With the site capped, Gt can no longer be activated. During rhodopsin shut off, Gt actively hydrolyzes GTP to GDP through an intrinsic GTPase activity, and this activity is further enhanced by a protein complex consisting of the regulator of G-protein signaling 9 (RGS9), RGS9 anchoring protein (R9AP), and guanine nucleotide-binding protein β5 (Gβ5) [30,31,32,33,34,35,36,37,38]. The decreased calcium levels serve another purpose of releasing the inhibition of GCAPs (guanylate cyclase activating proteins), which results in the activation of GCs (guanylate cyclases) of the retina that then restore the cGMP levels, effectively opening the CNG channels once again [9,11,12,21,39,40,41]. Ultimately, the all-trans retinal releases from the opsin molecule and is replaced with another molecule of the 11-cis retinal, which results in silencing the rhodopsin signaling and allows arrestin to disengage and the phosphates to be removed from the carboxyl terminus.
In the light activation pathway of phototransduction, there are a multitude of genes that, if mutated, have been shown to result in IRDs. RHO encodes one of the most critical proteins within the phototransduction cascade, rhodopsin (RHO), and has been identified as causing a considerable number of IRDs if mutated. Specifically, 30% of dominant RP cases have been linked to mutations in RHO [14,42,43]. RHO is a class A GPCR that, when activated by light, initiates the first step in vision. Failure of this protein to properly function causes RP by multiple mechanisms, including protein misfolding and aggregation (the most common mechanism of RHO-induced RP) and receptor mislocalization (the most severe forms of RHO-induced RP), as well as others such as constitutive activation and loss of Gt binding [14,43,44,45,46,47]. OPN1LW-OPN1MW, genes on the X chromosome, encode for red and green cone opsins, respectively. Mutations in these genes have been shown to present with X-linked cone dysfunction with and without degeneration [48]. GNAT1 and GNAT2, the genes encoding the α subunit of Gt in rods and cones, respectively, are necessary for propagation of the photon-induced signaling cascade, which continues with the activation of PDE6 and closure of the CNG channels with subsequent hyperpolarization. Therefore, GNAT1 and GNAT2 mutations are causative for IRDs through a loss of phototransduction or a gain-of-function resulting in constitutive Gt signaling [49,50,51,52]. The next protein involved in phototransduction progression is PDE6, a key regulator in phototransduction functioning to hydrolyze intracellular cGMP, resulting in the closing of CNG channels and subsequently causing a hyperpolarization in the membrane potential and propagation of the visual signal. PDE6 is a heterotetrameric enzyme composed of an α, β, and two γ subunits, in which the α and β subunits constitute the catalytic core while γ functions as the inhibitory subunit of PDE6 in both rod and cone cells. Mutations in genes encoding these subunits have been associated with IRDs. For instance, mutations in rod PDE6 subunit genes PDE6A, PDE6B, and PDE6G (encoding the α, β, and γ subunits, respectively) have been shown to cause autosomal recessive RP. Since PDE6 is critical for phototransduction, the discussed mutations not only impede the cascade but lead to an elevated level of cGMP, which rapidly becomes toxic to the cell [16,53,54]. PDE6C is a gene that encodes for the α subunit of the cone photoreceptor PDE6. The inhibitory γ subunit of cone PDE6 is encoded by PDE6H and is also implicated in IRDs [55]. In addition, AIPL1, a gene encoding chaperone aryl hydrocarbon receptor (AhR)-interacting protein-like 1, is critical to the functional integrity of PDE6 by promoting its correct folding and assembly. Mutations in AIPL1 have been associated with IRDs, including cone-rod dystrophies and LCA [56]. CNG channels, functioning directly downstream of PDE6, have also been linked to IRDs via mutations in genes encoding its α and β subunits that serve as the primary channel pore and regulatory calmodulin-binding sites, allowing for intracellular calcium level sensing and trafficking of the entire channel complex to the outer segment of the photoreceptor cell, respectively. Mutations in cone CNG channels, including CNGB3 (encoding the β subunit) and CNGA3 (encoding the α subunit), have been observed to cause autosomal recessive achromatopsia, a disorder characterized primarily by the absence of color vision [57,58]. On the other hand, mutations in the rod CNG channel’s α and β subunits, encoded by CNGA1 and CNGB1, respectively, have been associated with RCDs [5,59]. Mutated β subunits of the CNG channels can also affect channel regulation through calcium binding, as well as disc formation issues associated with the amino terminal GARPs (glutamic acid rich proteins), which have been shown to associate with both PDE6 and the disc proteins peripherin-2 (PRPH2) and rod outer segment membrane protein-1 (ROM-1) (discussed subsequently in Section 3.3) [60,61,62,63,64].
Additionally, IRD-associated gene mutations have been identified in components involved in phototransduction deactivation. Arrestin-1 is a pivotal protein involved in phototransduction, specifically for RHO deactivation. It does so via binding activated RHO after phosphorylation by GRK1, blocking Gt molecules from binding RHO and preventing the subsequent steps of phototransduction from taking place. The binding of arrestin-1 to RHO allows for the timely return of the photoreceptor cell to its inhibited dark state. Mutations in SAG, the gene coding for arrestin-1, have been shown to cause RP by causing the rod cell’s dysfunction in returning to its dark state and, therefore, preventing further phototransduction, which can cause both IRD and congenital stationary night blindness (CSNB) [52,65,66,67,68]. Interestingly, there are no known mutations in GRK1 that cause IRD but have been shown to cause CSNB [69]. Other gene mutations, such as in GNAT1 and GNAT2, have also been identified as factors affecting phototransduction deactivation. As previously discussed, GNAT1 and GNAT2 mutations have been shown to cause IRD through constitutive Gt signaling. However, Gt maintains intrinsic GTPase activity, which can also be hindered through mutations in GNAT1 and GNAT2. With that, these mutations have been shown to cause a loss or slowing of phototransduction deactivation [70].
As previously discussed, the closure of CNG channels via the hydrolysis of intracellular cGMP to GMP is important in the progression of phototransduction. In relation to this mechanism, the replenishment of intracellular cGMP is essential to the restoration of open CNG channels in order to deactivate phototransduction and return to the dark state. Photoreceptor guanylate cyclase, GC-D, is a calcium-sensitive protein that functions to synthesize intracellular cGMP and is further activated and regulated by the calcium-binding guanylate cyclase activating proteins or GCAPs. Loss-of-function mutations in GUCY2D, encoding GC-D, eventually lead to low cGMP production from GTP and therefore prolong the activity of the phototransduction cascade, inducing degeneration [41]. GUCA1A and GUCA1B encode GCAP1 and GCAP2 that sense changes in cytoplasmic-free calcium, communicate it to GC-D, and therein activate it. Abnormalities in these genes have been implicated in RP through dysregulation and dysfunction of the GC-D protein, causing low cGMP production and cation imbalances [9,11,12,13,40,71].
The phototransduction cascade maintains many important channels and transporters for both dark- and light-activated signal propagation, as multiple components are involved in the regulation of membrane potential via the transport of ions. Similar to CNG channels, Na+/Ca2+-K+ exchanger 1 (NCKX1), a potassium-dependent sodium/calcium antiporter, functions to balance the intracellular calcium levels in rod outer segments. Mutations in SLC24A1, the gene encoding this antiporter, have been linked to causing autosomal recessive congenital stationary night blindness (CSNB) due to the partial or complete loss of ion transfer, resulting in abnormal levels of intracellular calcium concentrations [72,73]. Additionally seen to cause CSNB, specifically CSNB2A, is a dysfunction in the L-type voltage-gated calcium channel (VGCC), caused by a mutation in CACNA1F, the gene encoding the pore-forming α1F subunit of this channel. These channels, localized to photoreceptor terminals, function in mediating the inward calcium current required for the subsequent release of glutamate. The absence of this function via genetic mutation causes impaired or absent synaptic transmission from both rod and cone photoreceptor cells [74,75]. Finally, the activation range of potassium channels within cone and rod photoreceptors plays an important role in setting the photoreceptor resting potential. Kv8.2, a voltage-gated potassium channel subunit, acts as a voltage response modulator via shifting the activation range of potassium channels. KCNV2-associated retinopathy, an autosomal recessive cone-rod dystrophy, has been associated with gene variants in KCNV2, which codes for Kv8.2 and is currently under investigation as a potential target for gene therapy [76]. Interestingly, purine nucleotide synthesis is also important in maintaining the phototransduction cascade. Insoine-5′-monophosphate dehydrogenase, IMPDH (coded by IMPDH1), is the first committed and rate-limiting step enzyme in de novo purine biosynthesis, functioning to catalyze the conversion of inosine monophosphate (IMP) into xanthine monophosphate (XMP) that subsequently produces downstream GMP and GTP. IMPDH has been identified as essential in sustaining the intracellular GTP pool. Mutations in IMPDH1, coding for isoform IMPDH1 in retina, have been linked to causing IRDs such as autosomal dominant RP and, rarely, LCA. This is due to photoreceptor sensitivity to a required balance of purine nucleotides, including cGMP, which is a key signaling molecule in phototransduction [77,78,79]. In addition to IMPDH1, PRPS1, coding for phosphoribosylpyrophosphate synthetase 1, the enzyme catalyzing the first step in general nucleotide synthesis causes IRD, similar to IMPDH1 [77,80,81,82].
3.2. Ciliary Formation/Trafficking
Cilia are small, projecting organelles that, until the last 20 to 30 years, were thought to be mostly vestigial. These organelles are now known to exist on almost all cell types and are essential to the cell’s survival, so that losing them can ultimately result in mortality [17,83,84]. Two types of cilia exist, motile and primary, also known as non-motile or sensory cilia. These ciliary types primarily differ in their structure and function. Motile cilia bear a 9 + 2 bundle of microtubules and, as their name suggests, are capable of movement and serve this function in regions such as the esophagus, lungs, and sperm (flagella), where they allow for cellular mobility (sperm) or the active movement of such things as mucus, food, etc. Primary cilia, however, have a 9 + 0 bundle of microtubules and serve a much different function dependent on cell type and tissue expression, from sensing urine flow in the kidneys to dictating the number of digits on a developing embryo’s hands [17]. In the retina, one of the primary roles of the cilium is established in the photoreceptor outer segment [85] (Figure 4). Within the photoreceptor outer segment, phototransduction proteins are integrated in, or associated with, the disc membranes (whose formation is discussed in Section 3.3) and the plasma membrane. The photoreceptor cilia, as sensory cilia, are arranged in a 9 + 0 bundle of microtubules coming from a single, tubulin-composed centrosomal basal body [86,87]. This cilium is termed the connecting cilium (CC), as it is a cellular and molecular highway for outer segment proteins (soluble proteins and integral membrane or membrane-associated proteins) and the vesicular transporters carrying them. One example of a major molecular transport function served by the CC is the flux of Gt into the inner segment and arrestin into the outer segment during phototransduction [88,89]. The movement between the inner segment and outer segment via the CC is driven by a process called intraflagellar transport (IFT) in which IFT proteins, kinesins, and dyneins transport a variety of products to their correct locations. Eventually, the tips of these cilia differentiate to form the outer segment, which contains RHO and other proteins necessary for phototransduction. Any abnormality in the generation of these cilia have been shown to lead to a lack of normal rod and/or cone function and diseases such as RP and LCA [90].
One of the major genes causing disease is RPGR. RPGR accounts for over 70% of X-linked RP and over 20% of non-syndromic RP [10,91,92]. RPGR codes for retinitis pigmentosa GTPase regulator (RPGR). This protein, when lost, results in the lack of any ciliary development and severe retinal diseases from birth. In its absence, the CC fails to form, and retinal cells begin to die from as early as 6 months of age in mice [92]. In a similar sense as RPGR, mutations in RPGRIP1 also cause retinal degenerations. RPGRIP1 encodes retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1), which plays an important role in the CC of photoreceptor cells. Similar to other ciliary proteins, losing functional RPGRIP1 results in the loss of proper vesicular protein trafficking and inner/outer segment flux, causing cell death [8,93]. Recent studies have identified biallelic missense mutations in RPGR-interacting domains of RPGRIP1 to cause less severe forms of cone-rod dystrophies, while biallelic null mutations in this domain cause more severe forms [93]. RP1L1 is a gene that encodes retinitis pigmentosa 1-like 1 (RP1L1), which encodes a component of the photoreceptor cilium and axoneme, the core structure of the cilium. Mutations in this gene cause photoreceptor disease and phenotypic retinal degeneration due to a loss of the synergistic interactions between it and RP1, causing a subsequent lack of ciliary formation [94,95]. In conjunction with RP1L1 is RP1. RP1, coding for retinitis pigmentosa 1 (previously known as oxygen-regulated protein 1), is a photoreceptor-specific protein that localizes to the ciliary axoneme of outer segments, where it is thought to assist in horizontal disc orientation from the CC and, thus, ciliary, disc, and outer segment formation [90,95,96]. Due to this, mutations in RP1 result in malformed cilia and outer segments. FAM161A is another IRD gene that encodes FAM161 centrosomal protein A, which is primarily localized to the CC, basal body, and adjacent centrioles. It has been shown as a part of microtubule organizing centers within the retina. The stabilization of the microtubules within the retina is critical to maintain the microtubule tracks in photoreceptor transport [97,98]. TOPORS, a gene encoding for a topoisomerase 1-binding arginine/serine rich (TOPORS), functions as a dual E3 ubiquitin and SUMO1 ligase and has been implicated in RP when mutated. This protein localizes to the basal bodies of the CC within photoreceptors and has been proposed to play a role in the regulation of primary cilia-dependent photoreceptor development and function. Within the cilium, TOPORS helps to localize and nucleate the microtubules from the centriole. Without this, cilia are incapable of properly developing, and retinal degeneration ensues [99,100,101,102].
The cilium also needs to be regulated in multiple aspects, including length, stability, and biomolecular composition. This is accomplished by multiple genes controlling protein trafficking, length, gating, etc. CEP290 is a gene that encodes for a critical transition zone protein needed within the ciliary base. The transition zone functions as a diffusion barrier that, if mutated, perturbs the proper flux of inner/outer segment proteins, resulting in degeneration. CEP290 recruits DAZ-interacting zinc finger protein 1, which coordinates early ciliary membrane formation and transition zone assembly. Mutations in this gene lead to a lack of proper transition zone assembly and consequential retinal degenerations such as RP and LCA [103]. Basal exon skipping has been proposed as an underlying mechanism resulting in disease-causing CEP290 mutations [104]. EYS is also a gene that has been investigated and shown to have mutations that cause IRD. Its protein product, eyes shut homolog (EYS), has been shown in photoreceptor cells to maintain the ciliary axoneme in a stable formation. It preferentially localizes to the cytoplasm of the cilium and is used as an organization tool. EYS is essential for the structural maintenance of the ciliary pocket and organization of the CC of vertebrate photoreceptors. Mutations in EYS can cause disruption of the photoreceptor structure and subsequent CRD [102,105,106]. One ciliary regulator is SPATA7, which encodes for spermatogenesis-associated protein 7 (SPATA7). SPATA7 is localized to the CC and the RPE. Loss of this protein is shown to alter protein trafficking, including RHO and RPGRIP1 in the CC. Therefore, abnormalities in this protein cause retinal degeneration [107]. IFT140 is a transport gene that is critical for proper retinal functioning. It encodes part of the IFT-A complex, which functions within IFT specifically by the retrograde transport of proteins from the ciliary tip. Biallelic variant mutations in this gene, including both missense and frameshift, have been seen to cause IRDs through abnormal protein localization and photoreceptor degeneration [19]. LCA5 is also involved in the IFT of photoreceptor cilia, as deletions show impaired IFT [108]. The MAK gene codes for male germ cell-associated kinase (MAK), which regulates the length of the primary cilium in a variety of cell types. Within the retina, MAK deficiency results in retinal degeneration characterized and induced by the elongation of photoreceptor connecting cilia [109]. Oral-facial-digital syndrome 1 (OFD1) is another protein critical in regulating the photoreceptor cilium length and number. OFD1 additionally functions to provide a neuroprotective effect from oxidative stress and apoptosis. Insertions between exons 9 and 10, causing a frameshift in OFD1 transcripts, the gene encoding OFD1, lead to the loss of primary cilia and therefore consequent photoreceptor degeneration [110,111]. RP2, encoded by RP2, is an outer segment trafficking protein and shares homology with tubulin-specific chaperone cofactor C. RP2 acts as a GTPase-activating protein (GAP) for ADP-ribosylation factor-like 3 (ARL3). ARL3, localized primarily to photoreceptor-connecting cilium, functions in trafficking lipidated proteins that are critical in phototransduction, such as Gt, GRK1, and PDE6, to the outer segment. Abnormalities in ARL3 activation caused by RP2 dysfunction prevent the transport of these proteins and consequentially result in an RP phenotype [112,113]. AHI1 is another gene which protein product is critical for ciliary trafficking mechanisms. It encodes for Abelson helper integration site-1 (AHI1), a signaling and scaffolding protein localized to photoreceptor cilia. Failure of this protein to function properly also leads to failure of the outer segment to properly assemble beyond the axoneme, leading to retinal dystrophy [114,115].
Though several genes associated with IRDs and the primary cilia are non-syndromic, a multitude of the genes in this category are syndromic, and several of the genes overlap between syndromic and non-syndromic (Figure 5). Interestingly, different types of mutations in these genes are a major contributing factor to the disease manifestations, be it non-syndromic or syndromic. As an example, a nonsense mutation in a gene associated with cilium development yielding no functional protein would be more likely to cause syndromic disease, while a nonsynonymous missense mutation in the same gene may cause a loss of a retina-specific interaction (i.e., LCA/RP proteins such as CEP290), causing non-syndromic IRD. Below details this subset of IRD genes.
The BBSome is a complex critical for ciliogenesis and ciliary protein trafficking through the recruitment of Rabin8 and Rab8 [116,117,118]. It is encoded by several genes that are implicated in a syndromic retinal dystrophy called Bardet–Biedl syndrome. These include BBS1-22 [119]. While 22 different genes are associated with Bardet–Biedl syndrome, many serve redundant functions and will not be discussed here. BBS1 has been shown to affect retrograde trafficking of GPCRs out of primary cilia, and mutations in the gene have been seen to cause retinal degeneration [120,121]. Mutations in BBS2, BBS4, BBS5, BBS7, BBS9, BBS17, and BBS18 all result in an RP phenotype due to a loss of ciliary formation and/or mislocalization of photoreceptor proteins, including RHO and arrestin-1 [116,119,120,121,122,123,124,125,126,127,128,129,130]. BBS8 (TTC8) is a gene that has been associated with Bardet–Biedl syndrome and non-syndromic RP that codes for a BBSome member and is necessary for the proper trafficking of ciliary/outer segment proteins. The functional loss of BBS8 results in the mislocalization of outer segment proteins to the inner segment and axons, as well as cytosolic and axonal protein mislocalization to the outer segment and other cellular domains. The functional loss of BBS8 also results in the loss of ciliary formation [18,131]. BBS10 is a part of the BBSome complex assembly process and functions to regulate the interaction of the BBS6–BBS12–BBS7 intermediate step of BBSome assembly [132]. Another gene with mutations exhibiting a Bardet–Biedl syndrome phenotype is MKKS/BBS6. The association of MKKS and CEP290 (also known as BBS6 and BBS14, respectively) at the ciliary transition zone and basal body affects the integrity of multiprotein complexes, including the BBSome. These are essential functions within ciliary biogenesis, and mutations in the gene show a lack of ciliary function and the associated IRD [133,134]. Another of the BBS proteins that is implicated in ciliary dysfunction is BBS3. BBS3 encodes for Arf-like 6 GTPase (ARL6), which recruits the BBSome to the ciliary membrane to interact with its trafficked proteins. Therefore, without proper function, the BBSome does not localize to the correct location, and the same mechanism of disease follows [135]. SDCCAG8, also known as BBS16, causes Bardet–Biedl syndrome through the loss of ciliary formation due to the carboxy terminus of the protein product, SDCCAG8, being directly involved in the process [136,137,138,139]. The final BBS-associated gene discussed that causes Bardet–Biedl syndrome is IFT27 (also known as BBS19). Intraflagellar transport 27, coded by IFT27, is crucial for BBSome trafficking and Sonic hedgehog signaling. Cells depleted of these proteins due to an IFT27 mutation accumulate abnormal collections of IFT complex B proteins and cause phenotypic Bardet–Biedl syndrome [140]. IFT172 and IFT74 also code for IFT proteins with similar effects, while BBS21 codes for cilia- and flagella-associated protein 418 (CFAP418), which is found at the base of the connecting cilium and associates with other proteins involved in the transition zone and disc formation (discussed in Section 3.3) [141,142,143,144].
Other genes causing IRD through a syndromic form have been linked to Usher syndrome, which results in both RP and typically deafness and presents as one of three types (USH1, 2, or 3). MYO7A (or USH1B) is a known Usher-associated gene that encodes for unconventional myosin VIIa. This protein is localized in the apical region of the RPE and the ciliary membrane of the photoreceptors. Its function lies in the apical localization of RPE melanosomes and in the removal of phagosomes from the apical RPE for their eventual delivery to the basal RPE lysosomes. When MYO7A is mutated, opsin accumulates abnormally in the transition zone of the cilium, which causes retinal dysfunction, as well as a deficiency in autophagy for RPE cells [145,146,147,148,149]. USH1C is a gene needed for proper photoreceptor ciliary and synaptic development that encodes for the PDZ-domain-containing protein harmonin. Harmonin functions as a key scaffolding protein that other USH proteins bind. This is essential for the synaptic and ciliary maturation and maintenance of Müller glial cells. Without proper cilia and synaptic development and glial maintenance, retinal cells degenerate [150]. Cadherin-related 23 (CDH23 or USH1D), encoded by CDH23, is a member of the cadherin family that is comprised of calcium dependent cell-cell adhesion glycoproteins and are commonly known for their role in maintaining and establishing the organizational integrity of stereocilia within the hair cells of cochlea [151]. The role of CDH23 is well characterized in the development of hair cells of the inner ear but has yet to be further elucidated in its retinal function. Mutations in the transmembrane cadherin-like domain region of CDH23 have been linked to phenotypes presenting in Usher syndrome [151]. Protocadherin-15 (PCDH15 or USH1F), encoded by PCDH15, is localizes to the photoreceptors where it participates in outer segment scaffolding and mutations cause loss of this structural component and degeneration [152,153,154,155]. Through the same mechanism, USH1G encodes scaffold protein containing ankyrin repeats and SAM domain (SANS). SANS is known to interact with myomegalin and other USH proteins and is needed for organizing the network of USH proteins necessary for the mechanical stabilization of the outer segment, as well as the intracellular transport processes. Therefore, this lack of USH proteins consequently leads to additional retinal degeneration [156,157,158]. USH2A, coding for USH2A or Usherin, is one of the most common genes associated with Usher syndrome. The function of USH2A is to interact with ADGRV1 and bridge the gap between the membranes of photoreceptor CC and the periciliary region. Lack of this gene leads to the clinical manifestation of Usher syndrome due to the lack of the described interaction [154,159]. ADGRV1, also known as USH2C, is a gene that encodes for adhesion G protein-coupled receptor V1 (ADGRV1). It is located in the periciliary region between the inner and outer segments and thought to function in stabilizing the CC. Defects in this gene have been shown to result in abnormalities in the fibrous links and adhesion between sensory cells that can lead to photoreceptor dysfunction [160,161]. USH2D codes from the protein whirlin, a gene named for the effect it has in mice bearing mutations in the gene where a loss of vestibular function causes the mice to circle. This gene also results in retinal degeneration as it interacts with SANS, USH2A, and ADGRV1 [157,162,163,164]. USH3A is a gene encoding clarin-1, a four transmembrane-domain protein whose role lies in the excitatory ribbon synapse junction in hair cells of the ear, as well as in analogous synapses within the retina. It has also been localized to Müller cells and photoreceptor inner segments where it associates with microtubules, the framework components of the primary cilium [165,166,167,168,169]. Clarin-1 may also have a role in cell-cell adhesion between Müller cells and photoreceptors. A pathophysiology shared between the vestibular and visual systems presents itself when clarin-1 is incapable of proper intracellular trafficking and plasma membrane insertion, causing deafness and RP in Usher syndrome patients and have been linked to deletions in both exon 2 and 3 of USH3A [166].
Aside from Bardet-Biedl Syndrome and Usher syndrome, other systemic disorders are associated with ciliary defects (called ciliopathies) and several of the genes overlap between disorders. Alström syndrome (ALMS) is a vision disorder primarily characterized by CRD and hearing loss and is attributed to a mutation in ALMS1 gene, encoding ALMS1 [170,171]. Although the protein’s direct function remains unclear, ALMS1 was shown to have a role in both endosomal and ciliary transport. It localizes to the centrosomes and basal bodies of ciliated cells where it helps with trafficking of transferrin, GLUT4, Notch1 and others [172]. Therefore, without proper functioning, protein trafficking becomes abnormal and initiates IRD [170,171]. NPHP1 is another ciliary transition zone gene that encodes nephrocystin-1 (NPHP1). Its function is required to prevent infiltration of inner segment plasma membrane proteins during the photoreceptors outer segment development. It also has been shown to have a function in protein trafficking within the ciliary compartment. Therefore, proteins become mislocalized with mutations and photoreceptor dysfunction occurs [173]. NPHP4 is a ciliary gene encoding for nephrocystin-4 (NPHP4) that when mutated, patient photoreceptor outer segments fail to form properly and certain proteins mislocalize to the inner segments and outer nuclear layer. This has suggested a function for NPHP4 in intraflagellar transport or sensory signal transduction [174]. NPHP5 mutations are also implicated in retinal dystrophies, as it encodes a protein that functions to help localize parts of the BBSome complex (BBS2 and BBS5). Patients deficient in this gene have been shown to have mislocalizations of BBSome components and are unable to form connecting cilia and proper outer segments [175]. This group of NPHP genes collectively cause a disease called Senior-Løken syndrome. CEP41, coding for centrosomal protein 41, is necessary for regulation of tubulin glutamylation which is subsequently required for cilia disassembly and endothelial cell dynamics such as migration and tubulogenesis. Therefore, without this protein, cilia do not function properly and in turn cause retinal degeneration [176]. INPP5E or pharbin is a ubiquitously expressed phosphatidylinositol polyphosphate 5-phosphatase that is located within the primary cilia and functions to regulate the membrane composition of phosphoinositide. Mutations in INPP5E cause ciliary dysfunction and loss of ciliary localization of signaling proteins [177]. Working along with INPP5E is another gene, MKS1 (also known as BBS13). MKS1 encodes for Meckel syndrome 1 (MKS1) which is localized to the transition zone at the base of the cilium and functions to regulate ciliary INPP5E content via ADP-ribosylation factor-like GTPase-13B (ARL13B). Complete loss of the protein product of MKS1 has been shown to negatively affect cilium formation through abnormal basal body docking [178]. Retinitis pigmentosa GTPase regulator interacting protein 1-like (RPGRIP1L) is a protein coded for by the RPGRIP1L gene which, by its name, is a protein with high homology to RPGRIP1 and interacts with similar protein partners. As one would expect, mutations in this gene result in a loss of proper ciliary formation in the retina and other organs causing the symptoms of a ciliopathy, including RP [179,180,181]. Intraflagellar transport complex A component IFT144, encoded by WDR19, is another ciliary protein functioning specifically in retrograde ciliary transport of cargo in the cilium. Abnormalities in WDR19 expression, specifically a missense mutation, have been shown to express abnormal ciliopathies, including Sensenbrenner, Jeune Syndrome, and some forms of nephronophthisis [182]. Moreover, another gene indicated in IRDs is KIF11, coding for kinesin family member 11 (KIF11), a molecular motor protein known for its role in bipolar spindle formation and protein trafficking and primarily localized to the ciliary regions of the retina [183,184,185]. Mutations in KIF11 have been shown to cause retinal hypovascularization due to stunted growth of retinal vasculature. The pathogenesis of mutant KIF11 is still under investigation [184,186]. Many of these genes are associated with other syndromic diseases such as Joubert syndrome and Meckel syndrome, both forms of ciliopathies. Mutations in the CHM gene, resulting in a loss of function for its product protein, Rab escort protein-1 (REP-1), has been linked to causing choroideremia (CHM) [187]. CHM is characterized as an X-linked disease stemming from a gradual degeneration of RPE, choroid, and photoreceptor cells resulting in peripheral field vision loss once reaching adulthood. REP-1 normally functions as a post-translational lipid modifier of vesicular trafficking and phagosome fusion protein, Rab GTPase [188]. Mutant CHM expression pathogenesis is currently under investigation in that researchers are currently attempting to create a stable knock-out mouse model [187,189]. Interestingly, some mutated genes, such as SCAPER and ABHD12, result in phenotypes associated with a ciliopathy, but no immediate biomolecular evidence has been presented as to how they cause these phenotypes. SCAPER, a gene identified in a group of patients observed to have multiple BBS phenotypes, have no directly observed function for SCAPER protein for the phenotype [190,191]. Another gene similar to SCAPER is ABHD12. This gene’s protein product is thought to be involved in cell signaling using the endocannabinoid system and lysophosphatidylserine [192]; however, its phenotype is a disease PHARC, which clinically appears comparable to Usher syndrome [192,193].
3.3. Disc Morphogenesis
Discs are a significantly critical component of the phototransduction cascade as they are the foundation and attachment point for the receptors, second messengers and enzymes necessary to run the process. These discs are located in the outer segments of photoreceptor cells and lack all other organelles. Instead, they contain a great number of discs that are large flattened membranous vesicles. Cone and rod photoreceptors differ in their disc morphologies. Rod photoreceptors possess “closed” discs separated from the plasma membrane while cones possess “open” discs formed by an invagination of the plasma membrane [194]. In addition, rod discs are held suspended separately from one another and held in place at the edges where they connect to the plasma membranes through protein-protein interactions [194]. As the formation of these discs within the rod photoreceptor is still a topic of ongoing research, they are believed to be formed by the evagination of the plasma membrane with the rods and eventually pinching off to form their own separate membrane bound structure [194,195]. Due to the requirement of these discs and their phototransduction contents, any dysmorphogenesis with these discs is implicative of forming retinal based diseases such as RP or LCA [196]. Several genes have been associated with disc formation, as well as their resultant diseases caused by mutation.
The primary gene associated with disc morphogenesis is PRPH2 which codes for the PRPH2, a photoreceptor specific peripherin generally responsible for the flattening of the vesicles forming the discs and their attachment to the plasma membrane [195,197,198,199,200]. PRPH2 is restricted to the disc lamellae rim where it co-localizes with ROM-1, rod outer segment membrane protein 1. If PRPH2 is mutated, discs fail to properly flatten and lose contact with the membrane [200]. Interestingly, ROM-1 allows for PRPH2 to be trafficked to the disc lamellae rim and their interaction is necessary for proper disc flattening. Without properly functioning ROM-1, PRPH2 would be trafficked elsewhere, and discs would fail to form. Thus, mutations in ROM-1 cause IRDs similarly to loss of PRPH2 [90]. Another of these critical disc genes is FSCN2. In mice, FCSN2 has been shown to code for the protein fascin actin-bundling protein 2 that functions to induce actin binding and bundling, a process critical in the morphogenesis of discs within the outer segment. Mutations in this gene in mice have showcased steadily worsening rod function over time in a manner that indicates FCSN2 as being critical for the elongation and maintenance of discs within the outer segment [201,202]. PRCD is also a gene necessary for disk formation. Mislocalization of the encoded protein leads to rapid degradation of the retina. In normal photoreceptor cells, discs are aligned in a precise way, but in PRCD mutated mice, these discs are misaligned and have bulges with an associated accumulation of vesicles in the outer segment. This leads to eventual toxicity and degeneration of the retinal photoreceptors [203]. PROM1 codes for prominin-1, a transmembrane protein localized to the photoreceptor outer segment that is critical for assembly of discs, specifically the evagination and remodeling processes. Although not observed often in the population, its mutation has been shown to cause less rapid retinal degeneration than other forms, similar to that of mutations in PRPH2 [204]. Mutations in PCDH21, which codes for protocadherin-21 (PCDH21), has been associated with IRDs. Its function is thought to be related to its location between the inner and outer segments. PCDH21 may be responsible for proper disc alignment, evagination, and tethering, making its loss through mutation detrimental to outer segment function and formation [90,205]. The final two genes to mention regarding disc morphogenesis are CFAP418 (mentioned previously) and PCARE. These two genes are hypothesized to be crucial for disc formation and protein trafficking to the outer segment and the actin dynamic organization in the outer segment base, respectively, and both result in RP when mutated [142,206]. It’s still not currently known whether these genes are more associated with disc formation or ciliary formation. Regardless, the loss of proper outer segments and protein mislocalization suggest it could be either or both.
4. The Retinal Pigment Epithelium (RPE)
The RPE is a monolayer of cells located at the back of the eye between the retinal photoreceptors and their primary blood supply, the choriocapillaris. The RPE serves a multitude of functions for the retina, including producing the opsin chromophore 11-cis retinaldehyde, phagocytosing overgrown outer segments, excretion of metabolic waste to the bloodstream, absorbing stray photons, and assisting with general homeostasis. Many genes in the RPE are associated with IRDs when mutated and these are outlined below.
4.1. The Visual Cycle
Phototransduction, as covered in Section 2.1, relies on the replenishment of the chromophore 11-cis retinal, the inverse agonist of opsins. The visual cycle, or retinol cycle, can be summarized by a series of enzymatic reactions that result in the re-isomerization of all-trans retinal to 11-cis retinal [207] (Figure 6). In general terms, during phototransduction, when the all-trans retinal is cleaved from rhodopsin (or cone opsins) via hydrolysis, it is released from the opsin apoprotein and diffuses laterally through the photoreceptor disc membrane and is trafficked to the RPE using a series of enzymes and transporters [208]. To do so, the all-trans retinal reacts with the primary amine of endogenous phosphatidylethanolamine (PE) to form N-retinylidene-phosphatidylethanolamine (N-Ret-PE), the primary substrate for retinal specific ATP binding cassette transporter 4 (ABCA4). ABCA4, located in the outer segments of photoreceptor cells on the rim of disc membranes, functions as a flippase. The binding of N-Ret-PE to ABCA4 results in a confirmational change in which N-Ret-PE is released from its binding site towards the cytoplasmic leaflet of the disc membrane and is released from the PE as the all-trans retinal form [209]. Once in the cytoplasmic leaflet of disc membranes, the aldehyde of the all-trans retinal is reduced by all-trans retinol dehydrogenase (atRDH) with use of the cofactor NADPH, resulting in the production of all-trans retinol. Upon this reduction, the retinol is trafficked out of the photoreceptor outer segments using interphotoreceptor retinol binding protein (IRBP) to the RPE [210]. IRBP is a soluble lipoglycoprotein that preferentially binds all-trans or 11-cis retinoids and functions to transport retinoids between photoreceptor outer segments and apical RPE [211]. Once in the RPE, lecithin retinol acyl transferase (LRAT) catalyzes the transfer of a fatty acyl group from endogenous phosphatidylcholine to the all-trans retinol forming all-trans retinyl esters, formulating a nontoxic storage form of all-trans retinol [212]. A majority of retinyl esters are stored in hepatic stellate cells via the use of retinal binding protein 4 (RBP4) [213]. Regarding the storage of retinoids, the hydrophobicity of retinoids limits their ability to diffuse in aqueous environments, creating a barrier for transport to storage sites. RBP4, a member of the lipocalin family of proteins, functions to reversibly bind and sequester retinoids in order to facilitate their diffusion, with its activity significantly reliant on retinoid homeostasis. This activity is highest for the mobilization of retinoids to the liver in which retinyl esters are hydrolyzed to retinol and bind RBP4 to be carried through the bloodstream [214].
As all-trans retinyl esters are being formed along the smooth endoplasmic reticulum of RPE cells by LRAT; the trans isoform is then isomerized via retinoid isomerohydrolase retinal pigment epithelium 65 kDa (RPE65). RPE65 is primarily expressed in RPE cells and isomerizes retinyl esters to result in production of 11-cis retinol via a trans-to-cis alkene bond isomerization followed by a unique O-alkyl bond cleavage resulting in the dissociation of the ester group. The production of 11-cis retinol is the rate limiting step of the visual cycle [215]. After isomerization, 11-cis retinol dehydrogenase (11-cRDH) then oxidizes the alcohol groups of 11-cis retinol to an aldehyde, resulting in the formation of 11-cis retinal [210]. 11-cis retinal is then chaperoned by cellular retinaldehyde binding protein (CRALBP), which functions to facilitate intracellular transport of retinoids in the RPE to protect them from further esterification or thermal-isomerization [210,216]. As its function has been previously described, IRBP preferentially binds 11-cis retinal and traffics it back to the outer segment of photoreceptor discs. Now, 11-cis retinal inside of the photoreceptor discs can once again bind opsin to regenerate functional rhodopsin (or cone opsins) for phototransduction to continue [208]. This series of enzymatic reactions and trafficking of retinoids describes the dark adaptation regeneration of 11-cis retinal and is thought to be the primary mechanism of chromophore regeneration [208]. A secondary pathway, commonly referred to as the RGR (retinal G-protein coupled receptor) pathway, provides a direct photoisomerization of retinoids back into the 11-cis isomer. This can occur either in the RPE or the Müller cells, as RGR is known to be expressed in both cell types [217]. Photoisomerization in the RPE begins with chaperoning all-trans retinol via CRALBP. Under light conditions, RGR in complex with 11-cRDH, functions to directly photoisomerize all-trans retinol, resulting in the production of 11-cis retinal. Similar to the RPE65-based 11-cis retinal regeneration, CRALBP protects 11-cis retinoids produced by RGR from further esterification or photoisomerization effects [218].
ABCA4, coding for a flippase and member of the ATP-binding cassette (ABC) superfamily, is localized to the edge of disc membranes in photoreceptor outer segments and primarily functions as a retinoid transporter, as well as the RPE [219,220]. It has been shown that ABCA4 is highly expressed in the retina and functions in the transport of N-Ret-PE from the lumen of disc membranes to the cytoplasmic leaflet powered via ATP hydrolysis and hypothesized in the RPE to recycle the all-trans retinal present in phagocytosed discs [220,221]. In general, this prevents the accumulation of retinals and their toxic byproducts in disc membranes and the RPE. As previously described N-Ret-PE, the primary substrate of ABCA4, is synthesized via the reaction between all-trans retinal and endogenous phosphatidylethanolamine (PE), which results in the formation of pyridinium bis-retinoid (A2E), a prevalent lipid precursor to lipofuscin [222,223]. It has been shown that failures in ABCA4 function exacerbate the formation of A2E. The accumulation of cytotoxic biproducts such as lipofuscin are phenotypic hallmarks of multiple IRDs, including autosomal recessive Stargardt’s disease [222,224].
RPE65 is critical to the visual cycle in which its protein product RPE65 functions to convert all-trans retinyl esters to 11-cis retinol. RPE65 catalyzes this the enzymatic isomerization of all-trans retinyl esters via a radical cation mechanism to yield 11-cis retinol that is subsequently oxidized to 11-cis retinal [225]. Without properly functioning RPE65, there is substantial reduction in the levels of 11-cis retinal in addition to the accumulation of retinyl esters in the RPE [226]. RPE65 is one of the first RPE-specific proteins that has been identified to be associated with human IRDs, most severely seen in LCA and mildly in autosomal recessive RP, diseases that can be generalized as blindness from birth or early childhood [227]. Interestingly, patients with RPE65 deficient LCA showcase preserved retinal structure prior to the decline of visual function, providing the opportunity for investigation of potential therapeutic intervention prior to permanent vision loss. There are currently several human clinical trials, including gene replacement and augmentation therapy options for RPE65-associated dystrophies underway, as well as one that has been FDA approved [228,229,230,231].
As previously described, the secondary pathway of the visual cycle that functions to provide direct stereospecific photoisomerization of retinoids into the 11-cis isomer, requires the activity of the gene product of RGR, a seven transmembrane domain receptor localized to the intracellular membranes of both RPE and Müller cells. RGR, a close relative to RHO, is coupled to all-trans retinal that is then, upon light exposure, isomerized to 11-cis retinal [232]. Through studies involving RGR knockout mice, it has been shown that RGR is necessary in maintaining a normal rate of 11-cis retinal production in both dark and light conditions [233]. However, specified ocular diseases have been rarely reported to be in direct association with an RGR mutation. Although, both heterozygous frameshift and missense human RGR mutations have been closely associated with autosomal recessive RP [234].
LRAT, coding for a member of the NlpC/P60 thiol peptidase protein family, plays a role controlling the concentration of retinoic acid in the RPE. LRAT functions in the visual cycle as the main enzyme responsible for the conversion of all-trans retinols into all-trans retinyl esters [235,236]. LRAT has additionally been identified in the regulation of intestinal retinoid biosynthesis via negative feedback regulation [237]. LRAT activity, which can be defined by the transfer of an acyl moiety from the -sn1 position of phosphatidylcholine to all-trans retinols, has been identified to play a key role in maintaining optimal retinoid levels throughout the body [238]. The first reported LRAT human mutation has been associated with an early onset, severe retinal dystrophies, including rod cone dystrophy and has been compared to phenotypes similar to that resulting from mutation in RPE65. Studies in Lrat −/− mice have showcased LRAT as a critical enzyme for photoreceptor health and ocular retinoid homeostasis due to its role in the uptake and retention of vitamin A in the RPE which greatly impacts the production of 11-cis retinal [239]. This phenomenon has been shown to attribute to pathology seen in LCA patients, which was observed to result in disordered vectorial transport of cone visual pigments lacking bound chromophore. Currently, LRAT gene replacement therapy is under investigation as a potential treatment for RP associated with LRAT loss of function mutation [240].
The RDH5 gene, encoding for 11-cis retinol dehydrogenase, is abundantly expressed in RPE cells and functions in the oxidation of 11-cis retinol to 11-cis retinal prior to its transport to photoreceptor outer segments [241]. RDH5, in association with RDH11, plays an important role in avoiding the accumulation of 11-cis retinol in RPE cells, as well as providing photoreceptors with 11-cis retinal for proper visual cycle processes [242]. Mutations in RDH5 have been found to result in an autosomal recessive pattern of fundus albipunctatus, also referred to as retinol dehydrogenase 5 retinopathy, which is a form of CSNB and complaints of delayed dark adaptation after bright light exposure attributed to a lack of 11-cis retinal. An oral 9-cis-retinoid therapy has been proposed to function in the recovery of retinoids via bypassing the function of RDH5 and is currently under clinical investigation [243]. RDH12, as mentioned, is a member of a dehydrogenase/reductase family and is highly expressed in photoreceptor cells outer segments. In association with RDH8, RDH12 converts all-trans retinal to all-trans retinol in order to prevent the accumulation of toxic aldehyde by-products during the visual cycle via the reduction of all-trans retinal [244,245]. The accumulation of aldehydes in photoreceptor outer segments is closely related to mutations in RDH12 seen in ocular diseases, including up to 4% of the recessive form of LCA [246], as well as autosomal recessive retinitis pigmentosa, CRDs, and even macular dystrophies. RDH12 mutations maintain a broad retinal phenotypic spectrum with significant age variability [247]. In studies involving mouse models with disrupted RDH12 gene expression, although there is not a significant lack of chromophore regeneration, the retina was observed to become more susceptible to the toxic effects of light. With that, it is important to note a fundamental difference in the nocturnal nature of rodents in that their visual cycle processes retinoids slower than humans [248]. While the human retina’s visual cycle is much faster and the retina is twice as rich in cone pigments, it is susceptible to a more sensitive effect of toxic light damage. It has been proposed that patients maintaining an RDH12 null allele may reduce the rate of retinal degeneration via avoidance and protection from intense illumination [248,249].
RLBP1 encodes the visual cycle protein CRALBP, which functions to enzymatically facilitate the transport of retinoids, specifically 11-cis retinal, and protect it from further esterification/photo-isomerization and is expressed in abundance in both RPE and Müller cells [250]. CRALBP functions in the visual cycle by the reduction of spontaneous isomerization which allows for the ensured efficient yielding of 11-cis retinol and accumulation of aldehyde toxicity [251]. Mutations in RLBP1 cause autosomal recessive retinal diseases such as retinitis punctata albescens, recessive RP, and Newfoundland RCD and is phenotypically characterized by the degree of delayed dark adaptation and macular atrophy [252,253,254]. As previously defined, RBP4 functions in the storage of retinyl esters in hepatic stellate cells via facilitated diffusion and is the only known specific binding protein in circulation, a process essential for retinoid homeostasis [214]. In previous studies, RBP4 knockout mice resulted in the decreased uptake of retinol into the retina which subsequently caused an expected reduction in levels of chromophore, as well as impaired vision [255]. Interestingly, recent studies have shown that overexpression of RBP4 in mice resulted in the early onset of retinal degeneration [256]. There have been studies identifying a homozygous splice variant in RBP4 that has been associated with severe autosomal recessive RP in a pedigree of European ancestry in addition to a few members suffering from chorioretinal and iris colobomas [257]. RBP4 mutations, however, are not limited to ocular diseases and contribute to a multitude of lipid homeostasis diseases, glucose and cardiovascular dysfunctions, and many other human conditions [258,259].
4.2. Phagocytosis
Another critical maintenance process within the retina is the turnover of the outer segment. This turnover of the outer segment region of photoreceptor cells involves the RPE, a region of single layered cells directly adjacent to rod outer segments [260]. As previously mentioned, these outer segments are turned over at rapid rates when exposed to light. Each day, around 10% of the outer segment is phagocytosed to allow for new disc formation at the outer segment base [260]. Specifically, respective light and dark associated molecules dopamine and melatonin, along with the circadian clock genes PER1 and PER2 and L-type Ca2+, K+ and Cl− channels, have been shown to have a role in the rhythmic shedding of outer segments [261,262]. This leads to the outer segment being turned over completely around every 10 days. If this turnover is not properly regulated, it can lead to photoreceptor damage and/or a variety of visual dysfunctions. Of the many key functions done by the RPE, turnover of photoreceptor outer segments is one of the most critical when related to retinal dystrophies. This process repeats daily, so any malfunction in the required proteins necessary lead to abnormal balances and apoptosis [263].
One of the most critical of phagocytosis genes is MERTK. MERTK encodes a receptor, c-mer tyrosine kinase (MERTK), on the apical surface of the RPE that functions to internalize the outer segment prior to phagocytosis. As it is necessary for both MERTK to be present for phagocytosis and the retina needing to consistently phagocytose older portions of the outer segment, the mutations in the coding gene can cause buildup of shed photoreceptor disc membranes and the biomolecules contained within which can be significantly toxic to the retinal environment. Mutations in MERTK have been shown to cause steady retinal degeneration, leading to early signs of vision loss and blindness [264,265]. In addition, the IRD-associated protein ceramide kinase-like protein (CERKL), has been shown to regulate MERTK and thus influence the natural phagocytic turnover of the outer segment [266,267]. Therefore, abnormalities in CERKL functionality can induce either too much or too little turnover of the outer segment which requires a very tightly regulated balance [267].
A multifunctional gene, TULP1, has also been seen to cause IRDs. This gene codes for a protein, tubby-like protein 1 (TULP1), that localizes to multiple regions of the photoreceptor, ranging from the ribbon synapse up to the outer segments in which it functions in vesicular trafficking, endocytosis, and molecular recognition. Studies in mice maintaining deleterious mutations within TULP1 have shown shortened outer segments and lower rod length early in development. This is quickly followed by apoptosis and complete loss of functional retinal tissue [268]. It has also been shown that TULP1 may also act as a recognition protein for MERTK to initiate outer segment phagocytosis by the RPE [269]. The gene RAB28, coding for a ubiquitously expressed, farnesylated small GTPase, has also been implicated in the proper phagocytosis of photoreceptor outer segments, specifically those of cone photoreceptors [270]. The protein-protein interactions between RAB28 and Kir7.1, an inwardly rectifying potassium channel, in the RPE microvilli is required for proper shedding of cone photoreceptor outer segments [270,271]. Nonsense mutations in RAB28 have been identified to cause recessive cone-rod dystrophy 18, as well as RPE atrophy and progressive loss of visual acuity [270,271].
4.3. Other RPE Functions: Pigment Formation, Ionic Regulation, and Lipid Homeostasis
Aside from retinal/retinol recycling and phagocytosis, the RPE also plays other important roles in maintaining a retinal environment conducive for visual processes. One of these roles includes the protection of the retina from excessive illumination and UV light through the production of pigment. As seen in ocular albinism, the lack of retinal pigment (melanin) can lead to serious retinal degeneration attributed to lacking protection from light damage [272,273]. While pigment is important for light protection, it also plays a role in promoting expression of neurotrophic factors from the RPE. The gene GPR143 plays a major role in this facet. It is hypothesized that the protein product G protein-coupled receptor 143 (GPR143) acts as a receptor for the reaction by-product of melanin synthesis, L-DOPA, and that by binding L-DOPA, GPR143 signaling causes release of pigment epithelium derived neurotrophic factor (PEDF), a pro-survival factor for the retinal neurons [274]. It has also been shown that concomitant mutations in both GPR143 and cone opsin genes can result in foveal hypoplasia with monochromatism, as well as albinism type I [275].
In addition to pigment signaling, the RPE also works to regulate ionic homeostasis by using ion channels. Bestrophin-1 (BEST-1, coded for by the BEST-1 gene) is a pentameric ion channel that localizes to the basolateral side of the RPE near Bruch’s membrane and at the endoplasmic reticulum [276]. Due to its localization and interactions with calcium channels, BEST-1 is thought to be necessary for proper maintenance of intracellular calcium stores and retaining proper RPE cell morphology which, if lost, can results in RPE cell death and IRD [262,276,277,278,279,280].
In terms of lipid regulation, the RPE expresses multiple genes that act to oxidize retinal lipids or otherwise alter their structures. CYP4V2, a major cytochrome in present in ARPE-19 cells, is a principal metabolizer of polyunsaturated fatty acids, such as docosahexaenoic acid and eicosapentaenoic acid, which are disc membrane lipids internalized by RPE cells during phagocytosis of the outer segments. The accumulation of these lipids is a primary characteristic of Bietti’s crystalline corneoretinal dystrophy (BCD) and has been linked to mutations in CYP4V2, the gene encoding CYP4V2 [281,282]. Loss of this gene causes accumulation of these lipids and subsequent RPE cell death and IRD. Regarding lipid-associated RPE genes, HADHA, the gene encoding for hydroxyacyl-CoA dehydrogenase, is the alpha subunit of a trifunctional lipid oxidation enzyme complex that functions to catalyze the final steps in fatty acid oxidation [283]. Loss of this protein’s function was shown to lead to an increase in cytosolic lipids in RPE cells and a subsequent loss of tight junction integrity between cells, resulting in RPE cell death and IRD similar to CYP4V2 dysfunction [283,284,285,286,287].
5. Retinal and Photoreceptor Development, Metabolism, and Homeostasis
The retina is a highly ordered laminar structure containing a multitude of different cell types: neuronal, glial, and immune. As the retina develops in stages (ganglion cells forming first, followed by photoreceptors, and Müller glia forming last), any alterations in the gene expressions and timing of the process can cause deleterious effects, including retinal degeneration [288,289,290,291,292]. These players include transcription factors, enzymes, receptors, and other important physiological proteins.
5.1. Transcription Factors and Nuclear Proteins
A myriad of transcription factors (TFs) and nuclear proteins are necessary for the proper development and function of the retina. Many of these TFs are responsible for determining the cell fate of retinal progenitor cells. There are four TFs with known IRD-associations: CRX, NR2E3, NRL, and OTX2. These code for cone rod homeobox (CRX), nuclear receptor subfamily 2 group E member 3 (NR2E3), neural retina leucine zipper (NRL), and orthodenticle homeobox 2 (OTX2), respectively. CRX, NR2E3, and NRL promote the expression of photoreceptor-specific genes such as RHO, GNAT2, and OPN1SW, without which, one or more photoreceptor types will not develop or develop abnormally and cause an IRD [293,294]. This is especially notable in the rod-dominated retinas of mammals where a loss of CRX or NRL leads to IRD due to the loss of all rod cells [293,294,295]. CRX is also responsible for expression of the RP gene FAM161A, mentioned previously [98]. OTX2 is responsible for expression of multiple neuronal and ocular developmental genes, including RAX, PAX6, HES1, and SIX3, making its loss through mutation detrimental to proper development of the eye as a whole but especially outer retinal components, including photoreceptors, RPE, and Müller cells [296,297,298]. Another transcription factor associated with IRD is ZNF408 which codes for zinc finger protein 408 (ZNF408). ZNF408 has been shown to be associated with exudative vitreoretinopathy due to its role in the developing vasculature of the retina [299,300]. Other genes coding for nuclear proteins associated with IRDs that are not TFs include SAMD11 coding for sterile alpha motif (SAM) domain protein 11 (SAMD11), ATXN7 coding for ataxin-7 (ATXN7), and CHD7 coding for chromatin helicase DNA-binding protein 7 (CHD7). SAMD11 is known to play a role in the polycomb repressive complex 1, a complex required for the proper development of rod cells [301]. ATXN7 which causes spinocerebellar ataxia type 7, a neurodegenerative disease, results in the death of rod cells as well, likely through the downregulation of rod cell proteins, including those for phototransduction and development [302,303,304]. Lastly, CHD7, a chromatin remodeling protein that exposes genes for transcription, has been shown to cause CHARGE syndrome, which has an associated retinopathy and is, in part, attributed to the loss of the genes downstream of CHD7′s function (i.e., genes located in the remodeled chromatin) [305,306,307].
5.2. Transcriptional and Translational Regulation
As one would expect, loss of proper regulation of transcriptional and translational activity can lead to highly deleterious results as these processes are of the utmost importance for cell survival. This is no different in the retina. Currently, eight genes associated with transcriptional regulation are known to be causal in IRDs. These genes, PRPF3, PRPF4, PRPF6, PRPF8, PRPF31, SNRNP200, RP9 (or PAP-1), and CWC27 code for the proteins pre-mRNA processing factors 3, 4, 6, 8, and 31, small nuclear ribonucleoprotein U5 subunit 200, PIM-1 associated protein 1, and CWC27 spliceosome- associated cyclophilin. These proteins are all essential for the correct post-transcriptional processing of messenger RNAs, specifically splicing and spliceosome formation [308,309,310,311,312,313,314,315,316,317,318,319,320,321,322]. Without proper functioning of these spliceosome components, many of the genes associated with photoreceptor function and health are not properly expressed, including RHO, resulting in IRD [308,309,310,311,312,313,314,315,316,317,318,319,320,321,322,323,324,325]. In terms of translation, the gene OAT is also associated with IRD. This gene codes for ornithine δ-aminotransferase, the enzyme responsible for conversion of the amino acid ornithine to the amino acid proline. Mutations in this gene result in two deleterious events where the cell loses its ability to generate proline while also accumulating ornithine, causing degeneration in the form of gyrate atrophy of the choroid and retina [326,327,328]. Retina and anterior neural folding homeobox 2 (RAX2) acts as a transcriptional coactivator in that it cooperatively interacts with CRX to trans activate photoreceptor-specific genes [329].Alternative splicing variant mutations in Rax2, the gene encoding RAX2, have been associated with cone-rod dystrophy type 11 and RP [330]. Interestingly, the gene CERKL, previously associated with outer segment phagocytosis, has also been associated with translational regulation through interactions with components eIF3B and PABP, indicating this role could also have an effect on photoreceptor function through polysome formation, a process necessary for production of many overly expressed proteins such as RHO [331].
5.3. Retinal Architecture: Cell Adhesion, Extracellular Matrix (ECM), and Basement Membranes (BMs)
CRB1, a major gene involved in retinal development and homeostasis, especially in photoreceptors and Müller glia, is crumbs homolog 1 (CRB1). CRB1 is mainly localized apically in the retina and is necessary to maintain retinal polarity and helps to sustain proper cell-to-cell contacts between different cell types, including Müller cells, photoreceptors, and RPE. Without these, the intercellular connections of photoreceptors and Müller glia begin to degrade, and the cells are no longer able to properly interact and function. The loss of these cell-to-cell junctions results in decreased retinal integrity and accumulation of toxic photoreceptor products in the outer retina and subsequent degeneration of photoreceptors leading to the clinical presentation of IRD [1,332,333,334,335]. While CRB1 is a major protein involved in retinal integrity, other genes are also involved in creating and maintaining cell-to-cell adhesions and when mutated cause IRDs. CDHR1 and CDH3 are all genes coding for cell adhesion proteins responsible for different cellular connections in the retina. Cadherin-related family member 1 (CDHR1), encoded by CDHR1, plays a key role in maintaining the junction of the inner and outer segments of photoreceptors, maintaining their structure and integrity [186,336,337,338]. Cadherin 3 (CDH3), encoded by CDH3, has been localized to the cell-to cell junctions between RPE monolayer cells and is likely part of the blood-retinal barrier created by the RPE [186,338,339]. The last gene thought to affect cell-to-cell adhesion is C1QTNF5, which codes for C1q-tumor necrosis factor 5 (C1QTNF5), a secretory and membrane-associated protein. Mutation in C1QTNF5 have been identified to cause late-onset retinal macular degeneration (L-ORMD), characterized by lipid-rich deposits between the RPE and Bruch’s membrane. A Ser-to-Arg mutation at amino acid residue 163 (S163R) within C1QTNF5 has been proposed as the underlying cause in the proteins lack of adhesive function in creating cell-to-cell adhesions, resulting in deficient retinal integrity and homeostasis [186,280,338,340,341]. Interestingly, C1QTNF5 also appears to be a component of basement membranes (BMs), another critical aspect of retinal integrity and homeostasis discussed hereafter.
Another important aspect of the development and homeostasis of the retina is production and maintenance of the extracellular matrix (ECM) and, as stated previously, BMs. IMPG1 and IMPG2 code for interphotoreceptor matrix proteoglycan 1 and 2 (IMPG1 and IMPG2, respectively). These proteins are contained within the retina and are necessary for the formation and maintenance of the ECM around the photoreceptors themselves and between the RPE and photoreceptors. IMPG2 is thought to be necessary for proper localization of IMPG1, meaning genetic mutations in either will result in loss of the proper ECM formation and retinal degeneration [342,343,344,345]. Abnormalities in this gene have also been shown to lead to toxic accumulations in the subretinal space that result in photoreceptor degeneration [342]. Mutations in genes responsible for maintaining and altering the ECM, TIMP3 and ADAM9, are both known as causal genes for IRD [346,347,348,349,350]. These genes code for tissue inhibitor of metalloproteinase 3 (TIMP3) and A disintegrin and metalloproteinase domain-containing protein 9 (ADAM9), respectively. ADAM9 is a matrix metalloproteinase that breaks down and remodels the ECM and may have a role in the outer segment phagocytosis of the RPE. Inversely, TIMP3 functions as an inhibitory protein acting to halt enzymes that function to degrade matrix components, such as metalloproteinases. Mutations in ADAM9 have been found to cause CRD, while mutations in TIMP3 have been shown to cause RP due to disrupted ECM remodeling [346,347,348,349,350]. In terms of components of BMs, certain collagen genes are known to associate with IRDs. COL2A1 coding for collagen type II α1 (COL2A1) is a component of BMs and the vitreous humor. Mutations in COL2A1 result in dysregulated fibrillar lamellar development of the vitreous humor, as well as abnormal collagen helices. Mutations in COL2A1 have been linked to causing Wagner syndrome, characterized as an autosomal-dominant vitreoretinopathy [282]. In addition, COL4A5, another collagen-coding gene, results in an X-linked maculopathy when mutated due to a similar loss of BM integrity [351]. Finally, EGF containing fibulin extracellular matrix protein 1 (EFEMP1) is a glycoprotein that plays a role in extracellular matrix organization, primarily in Bruch’s membrane [352,353]. An SSCP analysis study identified a potential disease-causing exon shift in EFEMP1, the gene encoding EFEMP1, in patients with both Doyne Honeycomb and Malaria Leventinese Retinal Dystrophy [354]. Mutation EFEMP1 have been associated with the development of drusen deposits and retinal neovascularization, two primary characteristics identified in both Doyne Honeycomb Retinal Dystrophy and Malaria Leventinese [352,353,354].
5.4. Retinal Energetic Metabolism
A cell’s ability to produce and use its own energy is a critical aspect of cellular biology. Of all the tissues in the body, the retina is one of, if not the highest consumers of oxygen and ATP [355]. As such, any moderate loss of energetic metabolism could result in loss-of-function of many of the processes occurring during phototransduction, the visual cycle, or any number of the pathways associated with retinal integrity and homeostasis. Thus, mutations in genes associated with glycolytic or mitochondrial function could result in an IRD. Alternatively, loss of proper energetic metabolism will typically also have a deleterious effect on other highly metabolically active tissues. This is the case of known IRD-associated gene mutations involved in energetic metabolism.
CERKL, a gene previously mentioned to influence photoreceptor outer segment turnover, has also been identified to impact energetic metabolism when mutated. The gene product, ceramide kinase-like protein (CERKL), shown to additionally localize to mitochondria of retinal ganglion cells, functions in the regulation of mitochondrial metabolism in the retina via dysfunctional mitochondrial bioenergetics and altered mitochondrial distribution, seen to cause RP [356,357,358]. Additionally, CERKL shows protective function for the photoreceptors against oxidative stress through the antioxidant pathway [357]. A second gene implicated as important in cellular respiration is NMNAT1, encoding for nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1). Proper NMNAT1 function is necessary for nuclear NAD+ biosynthesis within all nucleated cells, meaning loss of this enzyme would result in a depletion of primary electron carriers in glycolysis and oxidative phosphorylation [359,360]. Approximately 5% of LCA cases are attributed to mutations in NMNAT1 and are observed to cause elevated levels of oxidative stress and damaging effects leading to retinal degeneration [359,360,361]. In fact, in a recent study involving mutant NMNAT1 mice, the first cell type to show degeneration were photoreceptors, exemplifying how important proper functioning NAD+ biosynthesis is to cellular maintenance [361]. Another seemingly ubiquitously expressed gene implicated in IRDs is HK1. HK1 encodes for the human hexokinase 1 (HK1), which is critical in the metabolism pathway of glycolysis, where it phosphorylates glucose to prevent it from diffusing out of the cell [362]. As the retina requires a high energy cost, mutations in HK1 have been shown to decrease energy utilization and lead to photoreceptor degeneration, especially in rods that heavily use glycolysis for their energy needs [362,363,364]. PANK2, the last gene discussed in respiration associated IRD, encodes pantothenate kinase 2 (PANK2). PANK2 is trafficked to the mitochondria, which indicates it is likely necessary within energy metabolism [365]. In fact, it is a necessary regulatory enzyme with the synthesis of coenzyme A (CoA). As CoA, or its alkylated derivative acetyl-CoA, is necessary for all metabolic functions regardless of carbon source, the loss of its proper function in photoreceptor cells would result in dire consequences, including the loss of energy production needed to maintain the function and production of fatty acids [365,366].
5.5. Cell Signaling, Iron Regulation, Autophagy, and Peroxisome Activity
Several signaling receptors, iron regulatory proteins, and autophagy-related or peroxisome-related proteins are necessary for development and homeostasis and are associated with IRD. Jagged-1 (JAG1), known for its role as a ligand in NOTCH signaling pathways, including the proper development of multiple sensory cell types and angiogenesis via JAG1-NOTCH interactions causing targeted gene transcription. Mutations in JAG1 have been associated with familial exudative vitreoretinopathy (FEVR) and is characterized by incomplete vascularization of the peripheral retina [186,367,368,369,370]. Similarly, frizzled-4 (FZD4) acts as a receptor downstream of Norrin/Wnt signaling and is involved in angiogenesis and retinal development [371,372]. Comparable to mutations in JAG1, mutations in FZD4 (the gene encoding for FZD4) have also been linked to causing FEVR via its effects on dysregulated blood-retina-barrier integrity [371,372,373,374].
Iron regulation is a homoeostatic function of necessity as iron maintains a multitude of purposes, including heme production, enzyme cofactors, and transcriptional regulators. Unfortunately, iron can also act to a cell’s detriment by oxidizing cellular components and inducing iron overload. The gene FLVCR1, coding for feline leukemia virus subgroup C receptor 1 (FLVCR1), is a gene involved heavily in iron regulation and causes IRD when mutated [375,376,377]. The molecular mechanism underlying this is thought to be a loss of proper localization of the protein to the cell membrane where it works to transport iron in and out of the cell. Dysfunctions in the process have been seen to result in excessive iron within the cell and subsequent cell death [375,376,377].
In terms of autophagy and lysosomal deficiency, multiple mutated genes have been shown to cause IRDs. CLN8 codes for ceroid lipofuscinosis type 8 (CLN8) which functions to transfer lysosomal enzymes that are necessary for lysosome biogenesis from the endoplasmic reticulum to the Golgi complex [378,379]. Without proper enzyme trafficking attributed to mutations in CLN8, there is a consequential lysosomal accumulation due to lack of enzymes for breakdown of lysosomal cargo and resultant cell death, including photoreceptors and RPE [379,380]. CLN3, another gene related to CLN8 and lysosomes, codes for ceroid lipofuscinosis type 3 (CLN3). Although the function of CLN3 remains unclear, it has been suspected to play a role in clearance of glycerophosphodiesters from lysosomes. In recent studies generating a CLN3 gene deletion in a mouse model, researchers noted RPE atrophy and degeneration, followed by significant vision impairment. It is likely the loss of CLN3 induces lysosomal storage disease and subsequent cell death. It was also proposed that in the RPE, the lack of functioning CLN3 induces a decrease in mitochondrial function with an increase in autophagy, likely degrading mitochondria and causing cell death [381,382]. Another gene familiar to this review and associated with autophagy regulation is CERKL. In addition to its other functions mentioned, CERKL has also been shown to regulate autophagy via its regulation of sirtuin-1 (SIRT1), a deacetylase of components essential to proper autophagy. Dysregulations in autophagy caused by mutations in CERKL have been seen to negatively impact the photoreceptor outer segment phagocytosis identified in RP [357,383]. The two final genes known to cause IRD through lysosomal dysfunction are HGSNAT and VSP13B. HGSNAT encodes heparan-α-glucosaminide-N-acetyltransferase (HGSNAT), a lysosomal acetyl transferase that functions to catalyze the transmembrane acetylation of heparan sulfate in BMs and allows for the step-wise breakdown of glycosaminoglycans (GAGs). Mutations in HGSNAT induce lysosomal storage disorder as unacetylated heparan sulfate is toxic to cells and accumulates in lysosomes, resulting in RP. VPS13B encodes an autophagy-associated protein called vacuolar protein sorting 13 homolog B (VPS13B). Mutations in VPS13B appear to cause increased lysosomal function, resulting in excessive degradation of cellular components [384,385,386,387,388]. Mutations in VPS13B have also been identified in many people with Cohen’s syndrome which is a rare autosomal recessive disorder characterized by a multitude of neurodevelopmental disorders, including retinal dystrophy [387].
The final genes to mention in relation to homeostasis are genes associated with the peroxisome and its functions. Acyl-CoA binding-domain-containing-5 (ACBD5), coded for by the ACBD5 gene, is a gene critical for the peroxisomes long-chain fatty acid oxidation [389,390,391]. ACBD5 is proposed to be anchored in the peroxisomal membrane and a transporter of very long chain fatty acids (VLCFAs) into the peroxisome for oxidation [389,390,391]. Loss of this gene results in increased levels of VLCFA incorporation into phospholipids and a resultant cell death and retinal dystrophy [389,390,391]. Another IRD-associated gene involved in fatty acid metabolism and the peroxisome is FALDH. FALDH encodes for fatty aldehyde dehydrogenase (FALDH). The primary role of this protein within the retina is the oxidation of medium and long chain aldehyde derivatives preventing oxidative stress to the peroxisome and subsequent cellular damage [392,393]. When this gene and subsequent protein are dysfunctional, there is an accumulation of fatty alcohols and fatty aldehydes in the retina. When these accumulate, the cellular membrane integrity is compromised by lipid peroxidation leading to abnormal photoreceptor function [392,393,394,395].The last genes to mention in peroxisomal function are PEX1 and PEX6 which code for the proteins peroxisomal biogenesis factors 1 (PEX1) and 6 (PEX6), respectively. These proteins are localized to the cytoplasm but become anchored in the peroxisomal membrane during formation and are both required for the proper import of peroxisome-targeted biomolecules. The dysfunction of either protein results in a loss of peroxisome formation and function and ultimately cell death [396,397,398,399,400]. In recent studies, hypomorphic mutations in either PEX1 or PEX6 have been observed to cause Heimler syndrome, a recessively inherited disease causing multiple sensory and neurological defects, including visual [397,400,401].
6. Proinflammatory Signaling and Leukocyte Activation and Invasion
A final, and notably one of the most recent, mechanism of degeneration associated with IRDs is retinal inflammation, specifically the effects of damage-associated molecular patterns (DAMPs), proinflammatory cytokine release, and microglial/monocytic invasion of the retina [402,403,404,405,406,407,408,409,410,411]. While IRDs are initiated by a mutation in any one of the many genes listed above (as well as others not covered here), studies are now showing that the degenerative phenotype observed is progressed by the retina’s cellular and molecular responses to the mutation’s effects. Multiple aspects of retinal inflammation have been proposed to contribute to the photoreceptor loss and these pathways have been shown in animal models, humans, or both [4,15,403,404,405,408,409,412,413,414,415,416]. As the photoreceptors fall victim to the disrupted pathways induced by the IRD mutation, their stress signals and initiation of apoptosis induces responses in multiple cell types, including Müller glia and the resident macrophages of the retina, microglia. DAMPs sensed by these cell types induce cytokine release, especially by microglia, which become M1 activated and release multiple cytokines, including interleukins (IL-1β, IL-6, and IL-18) and tumor necrosis factor alpha (TNFα) [4,15,403,404,405,409,412,413,414,415,417]. These cytokines activate multiple pathways in retinal cells, including the Janus kinase/Signal transducer and activator of transcription (JAK/STAT) pathway; nuclear factor-kappa B (NF-κB) activation; and induction of the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome [15,412,414,418]. A particularly interesting aspect of these pathways is a possible positive feedback loop inducing further exacerbation of the M1 microglial activation, causing overly phagocytic microglia and, as shown recently, invading monocytes from circulation to perform a process called phagoptosis [15,408,410,411,414,419,420]. Phagoptosis, discovered in the last decade, is a form of cell death instigated by phagocytosis of non-apoptosing, still viable cells. This exacerbated M1 state also causes further release of cytokines that induce cell death of photoreceptors, including TNFα and IL-1β. These cytokines and others such as IL-6 can induce the activation of STAT3, NF-κB, and the NLRP3 inflammasome in other cell types, including RPE and downstream neurons of the retina [4,15,414,421]. What results is enhanced photoreceptor loss due to mutation-induced apoptosis, cytokine-induced apoptosis, and macrophage-induced phagoptosis advancing the degeneration and rate of vision loss. As a result, treatments specifically targeting these inflammatory pathways and cells have become a hot topic area of research in recent years [4,15,404,405,407,409,413,414,415,416,422].
7. Concluding Remarks
IRDs, while rarer than other more prominent blinding diseases such as glaucoma and age-related macular degeneration, exhibit much more severe and early-onset degenerative phenotypes progressing much more rapidly often with little to no window for treatment. In this review, we delved into myriad pathogenetic molecular mechanisms, which all result in a similar outcome—complete loss of photoreceptors and blindness. Gene therapies for these congenital disorders are slow to produce and lack expedience when presented for FDA approval, especially when these treatments must be applied to children in cases such as LCA, Stargardt’s disease, and severe RP, where the disease symptoms begin in childhood and leave them blind by adolescence. Due to this unfortunate situation, therapies aimed at slowing the rate of photoreceptor degeneration and preserving sight for as long as possible are coveted in the realm of IRDs. Interestingly, our lab and others have shown that chronic inflammation and overactivation of macrophages in the outer retina progresses the rate of degeneration in mouse models of IRDs [4,15,403,404,407,408,414,416,420,423]. It is to this end that studies aimed at targeting the macrophages and/or cytokines associated with the proinflammatory environment are underway to serve this unmet need in the vision community with the hope to preserve the retinas of these burdened patients while cures are being developed.
Author Contributions
Conceptualization, A.M. and T.J.H.; resources, M.M.J.; writing—original draft preparation, A.M., B.I.M., and T.J.H.; writing—review and editing, B.I.M., T.J.H., and M.M.J.; visualization, A.M., B.I.M., and T.J.H.; project administration, T.J.H. and M.M.J.; and funding acquisition, T.J.H. and M.M.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by a Research to Prevent Blindness (RPB) Challenge Grant to the Hamilton Eye Institute, as well as an RPB/International Retina Research Foundation Catalyst Award for Innovative Research Approaches to Age-related Macular Degeneration (M.M.J.) and a Knights Templar Eye Foundation Career-Starter Research Grant (T.J.H.).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of University of Tennessee Health Science Center (Protocol 20-0194.0 Approved: 24 November 2020).
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cremers, F.P.; Maugeri, A.; den Hollander, A.I.; Hoyng, C.B. The expanding roles of ABCA4 and CRB1 in inherited blindness. Novartis Found. Symp. 2004, 255, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Yang, H.; Vollrath, D.; Yasumura, D.; Matthes, M.T.; Trautmann, N.; Chappelow, A.V.; Feng, W.; Earp, H.S.; Matsushima, G.K.; et al. Inherited retinal dystrophy in Mer knockout mice. Adv. Exp. Med. Biol. 2003, 533, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Jordan, K.E.; Jablonski, M.M.; Hollingsworth, T.J. Inflammation in the Pathogenesis of Progressive Retinal Dystrophies. J. Ophthalmic. Res. Vis. Care 2021, 1, 54289. [Google Scholar] [CrossRef]
- Nassisi, M.; Smirnov, V.M.; Solis Hernandez, C.; Mohand-Said, S.; Condroyer, C.; Antonio, A.; Kuhlewein, L.; Kempf, M.; Kohl, S.; Wissinger, B.; et al. CNGB1-related rod-cone dystrophy: A mutation review and update. Hum. Mutat. 2021, 42, 641–666. [Google Scholar] [CrossRef]
- O’Neal, T.B.; Luther, E.E. Retinitis Pigmentosa; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2022, 89, 101029. [Google Scholar] [CrossRef]
- Zou, G.; Zhang, T.; Cheng, X.; Igelman, A.D.; Wang, J.; Qian, X.; Fu, S.; Wang, K.; Koenekoop, R.K.; Fishman, G.A.; et al. Noncoding mutation in RPGRIP1 contributes to inherited retinal degenerations. Mol. Vis. 2021, 27, 95–106. [Google Scholar]
- Dell’Orco, D.; Behnen, P.; Linse, S.; Koch, K.W. Calcium binding, structural stability and guanylate cyclase activation in GCAP1 variants associated with human cone dystrophy. Cell Mol. Life Sci. 2010, 67, 973–984. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Hunt, D.M.; Wilkie, S.E.; Newbold, R.; Deery, E.; Warren, M.J.; Bhattacharya, S.S.; Zhang, K. Dominant cone and cone-rod dystrophies: Functional analysis of mutations in retGC1 and GCAP1. Novartis Found. Symp. 2004, 255, 37–49. [Google Scholar] [CrossRef]
- Jiang, L.; Baehr, W. GCAP1 mutations associated with autosomal dominant cone dystrophy. Adv. Exp. Med. Biol. 2010, 664, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Kitiratschky, V.B.; Glockner, C.J.; Kohl, S. Mutation screening of the GUCA1B gene in patients with autosomal dominant cone and cone rod dystrophy. Ophthalmic. Genet. 2011, 32, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, T.J.; Gross, A.K. Defective trafficking of rhodopsin and its role in retinal degenerations. Int. Rev. Cell Mol. Biol. 2012, 293, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, T.J.; Wang, X.; White, W.A.; Simpson, R.N.; Jablonski, M.M. Chronic Proinflammatory Signaling Accelerates the Rate of Degeneration in a Spontaneous Polygenic Model of Inherited Retinal Dystrophy. Front. Pharmacol. 2022, 13, 839424. [Google Scholar] [CrossRef] [PubMed]
- Daich Varela, M.; Ullah, E.; Yousaf, S.; Brooks, B.P.; Hufnagel, R.B.; Huryn, L.A. PDE6C: Novel Mutations, Atypical Phenotype, and Differences Among Children and Adults. Investig. Ophthalmol. Vis. Sci. 2020, 61, 1. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Makelainen, S.; Hellsand, M.; van der Heiden, A.D.; Andersson, E.; Thorsson, E.; Holst, B.S.; Haggstrom, J.; Ljungvall, I.; Mellersh, C.; Hallbook, F.; et al. Deletion in the Bardet-Biedl Syndrome Gene TTC8 Results in a Syndromic Retinal Degeneration in Dogs. Genes 2020, 11, 1090. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yang, L.; Wang, F.; Li, H.; Wang, X.; Wang, W.; Ge, Z.; Wang, K.; Zhao, L.; Li, H.; et al. Mutations in human IFT140 cause non-syndromic retinal degeneration. Hum. Genet. 2015, 134, 1069–1078. [Google Scholar] [CrossRef]
- Chen, C.K. The vertebrate phototransduction cascade: Amplification and termination mechanisms. Rev. Physiol. Biochem. Pharmacol. 2005, 154, 101–121. [Google Scholar] [CrossRef]
- Lamb, T.D.; Hunt, D.M. Evolution of the vertebrate phototransduction cascade activation steps. Dev. Biol. 2017, 431, 77–92. [Google Scholar] [CrossRef]
- Lamb, T.D.; Patel, H.; Chuah, A.; Natoli, R.C.; Davies, W.I.; Hart, N.S.; Collin, S.P.; Hunt, D.M. Evolution of Vertebrate Phototransduction: Cascade Activation. Mol. Biol. Evol. 2016, 33, 2064–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.; Zuker, C. Lights out: Deactivation of the phototransduction cascade. Trends Biochem. Sci. 1997, 22, 350–354. [Google Scholar] [CrossRef]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef]
- Ritter, E.; Elgeti, M.; Bartl, F.J. Activity switches of rhodopsin. Photochem. Photobiol. 2008, 84, 911–920. [Google Scholar] [CrossRef]
- Singh, D.; Hudson, B.S.; Middleton, C.; Birge, R.R. Conformation and orientation of the retinyl chromophore in rhodopsin: A critical evaluation of recent NMR data on the basis of theoretical calculations results in a minimum energy structure consistent with all experimental data. Biochemistry 2001, 40, 4201–4204. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, G.H. Primate photopigments and primate color vision. Proc. Natl. Acad. Sci. USA 1996, 93, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T. Molecular basis for color vision. Biophys. Chem. 1994, 50, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K. Recoverin and rhodopsin kinase. Adv. Exp. Med. Biol. 2002, 514, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.W.; He, W.; Wensel, T.G. RGS proteins: Lessons from the RGS9 subfamily. Prog. Nucleic Acid Res. Mol. Biol. 2001, 65, 341–359. [Google Scholar] [CrossRef]
- He, W.; Cowan, C.W.; Wensel, T.G. RGS9, a GTPase accelerator for phototransduction. Neuron 1998, 20, 95–102. [Google Scholar] [CrossRef]
- Hu, G.; Wensel, T.G. R9AP, a membrane anchor for the photoreceptor GTPase accelerating protein, RGS9-1. Proc. Natl. Acad. Sci. USA 2002, 99, 9755–9760. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, B.G.; Morgans, C.W.; Puthussery, T.; Wensel, T.G.; Burke, N.S.; Brown, R.L.; Duvoisin, R.M. R9AP stabilizes RGS11-G beta5 and accelerates the early light response of ON-bipolar cells. Vis. Neurosci. 2010, 27, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Lyubarsky, A.L.; Naarendorp, F.; Zhang, X.; Wensel, T.; Simon, M.I.; Pugh, E.N., Jr. RGS9-1 is required for normal inactivation of mouse cone phototransduction. Mol. Vis. 2001, 7, 71–78. [Google Scholar] [PubMed]
- Masuho, I.; Celver, J.; Kovoor, A.; Martemyanov, K.A. Membrane anchor R9AP potentiates GTPase-accelerating protein activity of RGS11 x Gbeta5 complex and accelerates inactivation of the mGluR6-G(o) signaling. J. Biol. Chem. 2010, 285, 4781–4787. [Google Scholar] [CrossRef]
- Morhardt, D.R.; Guido, W.; Chen, C.K. The role of Gbeta5 in vision. Prog. Mol. Biol. Transl. Sci. 2009, 86, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Slepak, V.Z. Structure, function, and localization of Gbeta5-RGS complexes. Prog. Mol. Biol. Transl. Sci. 2009, 86, 157–203. [Google Scholar] [CrossRef] [PubMed]
- Wensel, T.G. RGS9-1 phosphorylation and Ca2+. Adv. Exp. Med. Biol. 2002, 514, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Avesani, A.; Bielefeld, L.; Weisschuh, N.; Marino, V.; Mazzola, P.; Stingl, K.; Haack, T.B.; Koch, K.W.; Dell’Orco, D. Molecular Properties of Human Guanylate Cyclase-Activating Protein 3 (GCAP3) and Its Possible Association with Retinitis Pigmentosa. Int. J. Mol. Sci. 2022, 23, 3240. [Google Scholar] [CrossRef] [PubMed]
- Makino, C.L.; Peshenko, I.V.; Wen, X.H.; Olshevskaya, E.V.; Barrett, R.; Dizhoor, A.M. A role for GCAP2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J. Biol. Chem. 2008, 283, 29135–29143. [Google Scholar] [CrossRef]
- Sharon, D.; Wimberg, H.; Kinarty, Y.; Koch, K.W. Genotype-functional-phenotype correlations in photoreceptor guanylate cyclase (GC-E) encoded by GUCY2D. Prog. Retin. Eye Res. 2018, 63, 69–91. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Hahn, L.B.; Cowley, G.S.; Olsson, J.E.; Reichel, E.; Sandberg, M.A.; Berson, E.L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990, 323, 1302–1307. [Google Scholar] [CrossRef]
- Hollingsworth, T.J.; Gross, A.K. The severe autosomal dominant retinitis pigmentosa rhodopsin mutant Ter349Glu mislocalizes and induces rapid rod cell death. J. Biol. Chem. 2013, 288, 29047–29055. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye. Res. 2018, 62, 1–23. [Google Scholar] [CrossRef]
- Araujo, N.A.; Sanz-Rodriguez, C.E.; Bubis, J. Binding of rhodopsin and rhodopsin analogues to transducin, rhodopsin kinase and arrestin-1. World J. Biol. Chem. 2014, 5, 254–268. [Google Scholar] [CrossRef]
- Concepcion, F.; Chen, J. Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: A mouse model of Q344ter-induced retinal degeneration. PLoS ONE 2010, 5, e10904. [Google Scholar] [CrossRef]
- Orlans, H.O.; Barnard, A.R.; Patricio, M.I.; McClements, M.E.; MacLaren, R.E. Effect of AAV-Mediated Rhodopsin Gene Augmentation on Retinal Degeneration Caused by the Dominant P23H Rhodopsin Mutation in a Knock-In Murine Model. Hum. Gene Ther. 2020, 31, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Stingl, K.; Baumann, B.; De Angeli, P.; Vincent, A.; Heon, E.; Cordonnier, M.; De Baere, E.; Raskin, S.; Sato, M.T.; Shiokawa, N.; et al. Novel OPN1LW/OPN1MW Exon 3 Haplotype-Associated Splicing Defect in Patients with X-Linked Cone Dysfunction. Int. J. Mol. Sci. 2022, 23, 6868. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Yang, V.; Park, H.N.; Landis, E.G.; Dhakal, S.; Motz, C.T.; Bergen, M.A.; Iuvone, P.M.; Pardue, M.T. Lack of cone mediated retinal function increases susceptibility to form-deprivation myopia in mice. Exp. Eye Res. 2019, 180, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Calvert, P.D.; Krasnoperova, N.V.; Lyubarsky, A.L.; Isayama, T.; Nicolo, M.; Kosaras, B.; Wong, G.; Gannon, K.S.; Margolskee, R.F.; Sidman, R.L.; et al. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha -subunit. Proc. Natl. Acad. Sci. USA 2000, 97, 13913–13918. [Google Scholar] [CrossRef]
- Mejecase, C.; Laurent-Coriat, C.; Mayer, C.; Poch, O.; Mohand-Said, S.; Prevot, C.; Antonio, A.; Boyard, F.; Condroyer, C.; Michiels, C.; et al. Identification of a Novel Homozygous Nonsense Mutation Confirms the Implication of GNAT1 in Rod-Cone Dystrophy. PLoS ONE 2016, 11, e0168271. [Google Scholar] [CrossRef]
- Ringens, P.J.; Fang, M.; Shinohara, T.; Bridges, C.D.; Lerea, C.L.; Berson, E.L.; Dryja, T.P. Analysis of genes coding for S-antigen, interstitial retinol binding protein, and the alpha-subunit of cone transducin in patients with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1990, 31, 1421–1426. [Google Scholar]
- Willems, S.; Shenasa, M.; Borggrefe, M.; Hindricks, G.; Chen, X.; Rotman, B.; Kottkamp, H.; Haverkamp, W.; Breithardt, G. Atrioventricular nodal reentry tachycardia: Electrophysiologic comparisons in patients with and without 2:1 infra-His block. Clin. Cardiol. 1993, 16, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Robson, A.G.; Singh, N.; Pontikos, N.; Kane, T.; Hirji, N.; Ripamonti, C.; Rotsos, T.; Dubra, A.; Kalitzeos, A.; et al. Deep Phenotyping of PDE6C-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5112–5123. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Coppieters, F.; Meire, F.; Schaich, S.; Roosing, S.; Brennenstuhl, C.; Bolz, S.; van Genderen, M.M.; Riemslag, F.C.; European Retinal Disease, C.; et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am. J. Hum. Genet. 2012, 91, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Artemyev, N.O. AIPL1: A specialized chaperone for the phototransduction effector. Cell Signal 2017, 40, 183–189. [Google Scholar] [CrossRef]
- Aweidah, H.; Salameh, M.; Yahalom, C.; Blumenfeld, A.; Macarov, M.; Weisschuh, N.; Kohl, S.; Banin, E.; Sharon, D. A deep intronic substitution in CNGB3 is one of the major causes of achromatopsia among Jewish patients. Mol. Vis. 2021, 27, 588–600. [Google Scholar]
- Kohl, S.; Varsanyi, B.; Antunes, G.A.; Baumann, B.; Hoyng, C.B.; Jagle, H.; Rosenberg, T.; Kellner, U.; Lorenz, B.; Salati, R.; et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur. J. Hum. Genet. 2005, 13, 302–308. [Google Scholar] [CrossRef]
- Saito, K.; Gotoh, N.; Kang, I.; Shimada, T.; Usui, T.; Terao, C. A case of retinitis pigmentosa homozygous for a rare CNGA1 causal variant. Sci. Rep. 2021, 11, 4681. [Google Scholar] [CrossRef]
- Haber-Pohlmeier, S.; Abarca-Heidemann, K.; Korschen, H.G.; Dhiman, H.K.; Heberle, J.; Schwalbe, H.; Klein-Seetharaman, J.; Kaupp, U.B.; Pohlmeier, A. Binding of Ca2+ to glutamic acid-rich polypeptides from the rod outer segment. Biophys. J. 2007, 92, 3207–3214. [Google Scholar] [CrossRef]
- DeRamus, M.L.; Stacks, D.A.; Zhang, Y.; Huisingh, C.E.; McGwin, G.; Pittler, S.J. GARP2 accelerates retinal degeneration in rod cGMP-gated cation channel beta-subunit knockout mice. Sci. Rep. 2017, 7, 42545. [Google Scholar] [CrossRef]
- Michalakis, S.; Zong, X.; Becirovic, E.; Hammelmann, V.; Wein, T.; Wanner, K.T.; Biel, M. The glutamic acid-rich protein is a gating inhibitor of cyclic nucleotide-gated channels. J. Neurosci. 2011, 31, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Batra-Safferling, R.; Abarca-Heidemann, K.; Korschen, H.G.; Tziatzios, C.; Stoldt, M.; Budyak, I.; Willbold, D.; Schwalbe, H.; Klein-Seetharaman, J.; Kaupp, U.B. Glutamic acid-rich proteins of rod photoreceptors are natively unfolded. J. Biol. Chem. 2006, 281, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Milstein, M.L.; Kimler, V.A.; Ghatak, C.; Ladokhin, A.S.; Goldberg, A.F.X. An inducible amphipathic helix within the intrinsically disordered C terminus can participate in membrane curvature generation by peripherin-2/rds. J. Biol. Chem. 2017, 292, 7850–7865. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.S.; Bowne, S.J.; Koboldt, D.C.; Cadena, E.L.; Heckenlively, J.R.; Branham, K.E.; Wheaton, D.H.; Jones, K.D.; Ruiz, R.S.; Pennesi, M.E.; et al. A Novel Dominant Mutation in SAG, the Arrestin-1 Gene, Is a Common Cause of Retinitis Pigmentosa in Hispanic Families in the Southwestern United States. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2774–2784. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Wada, Y.; Tamai, M. Arrestin gene mutations in autosomal recessive retinitis pigmentosa. Arch. Ophthalmol. 1998, 116, 498–501. [Google Scholar] [CrossRef]
- Sippel, K.C.; DeStefano, J.D.; Berson, E.L.; Dryja, T.P. Evaluation of the human arrestin gene in patients with retinitis pigmentosa and stationary night blindness. Investig. Ophthalmol. Vis. Sci. 1998, 39, 665–670. [Google Scholar]
- Nishiguchi, K.M.; Ikeda, Y.; Fujita, K.; Kunikata, H.; Akiho, M.; Hashimoto, K.; Hosono, K.; Kurata, K.; Koyanagi, Y.; Akiyama, M.; et al. Phenotypic Features of Oguchi Disease and Retinitis Pigmentosa in Patients with S-Antigen Mutations: A Long-Term Follow-up Study. Ophthalmology 2019, 126, 1557–1566. [Google Scholar] [CrossRef]
- Yamamoto, S.; Khani, S.C.; Berson, E.L.; Dryja, T.P. Evaluation of the rhodopsin kinase gene in patients with retinitis pigmentosa. Exp. Eye Res. 1997, 65, 249–253. [Google Scholar] [CrossRef]
- Barren, B.; Natochin, M.; Artemyev, N.O. Mutation R238E in transducin-alpha yields a GTPase and effector-deficient, but not dominant-negative, G-protein alpha-subunit. Mol. Vis. 2006, 12, 492–498. [Google Scholar]
- Kitiratschky, V.B.; Wilke, R.; Renner, A.B.; Kellner, U.; Vadala, M.; Birch, D.G.; Wissinger, B.; Zrenner, E.; Kohl, S. Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5015–5023. [Google Scholar] [CrossRef]
- Sharon, D.; Yamamoto, H.; McGee, T.L.; Rabe, V.; Szerencsei, R.T.; Winkfein, R.J.; Prinsen, C.F.; Barnes, C.S.; Andreasson, S.; Fishman, G.A.; et al. Mutated alleles of the rod and cone Na-Ca+K-exchanger genes in patients with retinal diseases. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1971–1979. [Google Scholar]
- Vinberg, F.; Wang, T.; Molday, R.S.; Chen, J.; Kefalov, V.J. A new mouse model for stationary night blindness with mutant Slc24a1 explains the pathophysiology of the associated human disease. Hum. Mol. Genet. 2015, 24, 5915–5929. [Google Scholar] [CrossRef]
- Waldner, D.M.; Ito, K.; Chen, L.L.; Nguyen, L.; Chow, R.L.; Lee, A.; Rancourt, D.E.; Tremblay, F.; Stell, W.K.; Bech-Hansen, N.T. Transgenic Expression of Cacna1f Rescues Vision and Retinal Morphology in a Mouse Model of Congenital Stationary Night Blindness 2A (CSNB2A). Transl. Vis. Sci. Technol. 2020, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, R.; Mantyjarvi, M.; Tobias, R.; Isosomppi, J.; Sankila, E.M.; Alitalo, T.; Bech-Hansen, N.T. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J. Med. Genet. 2006, 43, 699–704. [Google Scholar] [CrossRef]
- Guimaraes, T.A.C.; Georgiou, M.; Robson, A.G.; Michaelides, M. KCNV2 retinopathy: Clinical features, molecular genetics and directions for future therapy. Ophthalmic. Genet. 2020, 41, 208–215. [Google Scholar] [CrossRef]
- Al-Maawali, A.; Dupuis, L.; Blaser, S.; Heon, E.; Tarnopolsky, M.; Al-Murshedi, F.; Marshall, C.R.; Paton, T.; Scherer, S.W.; Consortium, F.C.; et al. Prenatal growth restriction, retinal dystrophy, diabetes insipidus and white matter disease: Expanding the spectrum of PRPS1-related disorders. Eur. J. Hum. Genet. 2015, 23, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, A.L.; Kollman, J.M. IMPDH dysregulation in disease: A mini review. Biochem. Soc. Trans. 2022, 50, 71–82. [Google Scholar] [CrossRef]
- Bowne, S.J.; Sullivan, L.S.; Mortimer, S.E.; Hedstrom, L.; Zhu, J.; Spellicy, C.J.; Gire, A.I.; Hughbanks-Wheaton, D.; Birch, D.G.; Lewis, R.A.; et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 34–42. [Google Scholar] [CrossRef]
- Plana-Bonamaiso, A.; Lopez-Begines, S.; Fernandez-Justel, D.; Junza, A.; Soler-Tapia, A.; Andilla, J.; Loza-Alvarez, P.; Rosa, J.L.; Miralles, E.; Casals, I.; et al. Post-translational regulation of retinal IMPDH1 in vivo to adjust GTP synthesis to illumination conditions. eLife 2020, 9, e56418. [Google Scholar] [CrossRef]
- Fiorentino, A.; Fujinami, K.; Arno, G.; Robson, A.G.; Pontikos, N.; Arasanz Armengol, M.; Plagnol, V.; Hayashi, T.; Iwata, T.; Parker, M.; et al. Missense variants in the X-linked gene PRPS1 cause retinal degeneration in females. Hum. Mutat. 2018, 39, 80–91. [Google Scholar] [CrossRef]
- Georgiou, M.; Grewal, P.S.; Narayan, A.; Alser, M.; Ali, N.; Fujinami, K.; Webster, A.R.; Michaelides, M. Sector Retinitis Pigmentosa: Extending the Molecular Genetics Basis and Elucidating the Natural History. Am. J. Ophthalmol. 2021, 221, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Christensen, S.T. Structure and function of mammalian cilia. Histochem. Cell Biol. 2008, 129, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Youn, Y.H.; Han, Y.G. Primary Cilia in Brain Development and Diseases. Am. J. Pathol. 2018, 188, 11–22. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Neuhauss, S.C. The photoreceptor cilium and its diseases. Curr. Opin. Genet. Dev. 2019, 56, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Barker, A.R.; McIntosh, K.V.; Dawe, H.R. Centrosome positioning in non-dividing cells. Protoplasma 2016, 253, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Tapia Contreras, C.; Hoyer-Fender, S. The Transformation of the Centrosome into the Basal Body: Similarities and Dissimilarities between Somatic and Male Germ Cells and Their Relevance for Male Fertility. Cells 2021, 10, 2266. [Google Scholar] [CrossRef]
- Giessl, A.; Trojan, P.; Rausch, S.; Pulvermuller, A.; Wolfrum, U. Centrins, gatekeepers for the light-dependent translocation of transducin through the photoreceptor cell connecting cilium. Vision Res. 2006, 46, 4502–4509. [Google Scholar] [CrossRef]
- Slepak, V.Z.; Hurley, J.B. Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: Interaction-restricted diffusion. IUBMB Life 2008, 60, 2–9. [Google Scholar] [CrossRef]
- Goldberg, A.F.; Moritz, O.L.; Williams, D.S. Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res. 2016, 55, 52–81. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Barbaro, V.; De Nadai, K.; Lavezzo, E.; Toppo, S.; Chizzolini, M.; Palu, G.; Parolin, C.; Di Iorio, E. Identification of novel X-linked gain-of-function RPGR-ORF15 mutation in Italian family with retinitis pigmentosa and pathologic myopia. Sci. Rep. 2016, 6, 39179. [Google Scholar] [CrossRef]
- Patnaik, S.R.; Raghupathy, R.K.; Zhang, X.; Mansfield, D.; Shu, X. The Role of RPGR and Its Interacting Proteins in Ciliopathies. J. Ophthalmol. 2015, 2015, 414781. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Aweidah, H.; Carrero Valenzuela, R.D.; Berman, M.; Iguzquiza, O.; Cremers, F.P.M.; Khan, M.I.; Swaroop, A.; Amer, R.; Khateb, S.; et al. Retinal Degeneration Associated With RPGRIP1: A Review of Natural History, Mutation Spectrum, and Genotype-Phenotype Correlation in 228 Patients. Front Cell Dev. Biol. 2021, 9, 746781. [Google Scholar] [CrossRef] [PubMed]
- Noel, N.C.L.; MacDonald, I.M. RP1L1 and inherited photoreceptor disease: A review. Surv. Ophthalmol. 2020, 65, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Liu, J.; Gao, J.; LeNoue, S.; Wang, C.; Kaminoh, J.; Bowne, S.J.; Sullivan, L.S.; Daiger, S.P.; Zhang, K.; et al. Essential and synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis pigmentosa. J. Neurosci. 2009, 29, 9748–9760. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, J.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; Smith, J.E.; Sullivan, L.S.; Zuo, J.; Milam, A.H.; Pierce, E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Investig. Ophthalmol. Vis. Sci. 2002, 43, 22–32. [Google Scholar]
- Zach, F.; Grassmann, F.; Langmann, T.; Sorusch, N.; Wolfrum, U.; Stohr, H. The retinitis pigmentosa 28 protein FAM161A is a novel ciliary protein involved in intermolecular protein interaction and microtubule association. Hum. Mol. Genet. 2012, 21, 4573–4586. [Google Scholar] [CrossRef]
- Zach, F.; Stohr, H. FAM161A, a novel centrosomal-ciliary protein implicated in autosomal recessive retinitis pigmentosa. Adv. Exp. Med. Biol. 2014, 801, 185–190. [Google Scholar] [CrossRef]
- Czub, B.; Shah, A.Z.; Alfano, G.; Kruczek, P.M.; Chakarova, C.F.; Bhattacharya, S.S. TOPORS, a Dual E3 Ubiquitin and Sumo1 Ligase, Interacts with 26 S Protease Regulatory Subunit 4, Encoded by the PSMC1 Gene. PLoS ONE 2016, 11, e0148678. [Google Scholar] [CrossRef]
- He, K.; Zhou, Y.; Li, N. Mutations of TOPORS identified in families with retinitis pigmentosa. Ophthalmic. Genet. 2022, 43, 371–377. [Google Scholar] [CrossRef]
- Chakarova, C.F.; Khanna, H.; Shah, A.Z.; Patil, S.B.; Sedmak, T.; Murga-Zamalloa, C.A.; Papaioannou, M.G.; Nagel-Wolfrum, K.; Lopez, I.; Munro, P.; et al. TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum. Mol. Genet. 2011, 20, 975–987. [Google Scholar] [CrossRef]
- Chakarova, C.F.; Papaioannou, M.G.; Khanna, H.; Lopez, I.; Waseem, N.; Shah, A.; Theis, T.; Friedman, J.; Maubaret, C.; Bujakowska, K.; et al. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am. J. Hum. Genet. 2007, 81, 1098–1103. [Google Scholar] [CrossRef]
- Wu, Z.; Pang, N.; Zhang, Y.; Chen, H.; Peng, Y.; Fu, J.; Wei, Q. CEP290 is essential for the initiation of ciliary transition zone assembly. PLoS BIOL 2020, 18, e3001034. [Google Scholar] [CrossRef] [PubMed]
- Drivas, T.G.; Wojno, A.P.; Tucker, B.A.; Stone, E.M.; Bennett, J. Basal exon skipping and genetic pleiotropy: A predictive model of disease pathogenesis. Sci. Transl. Med. 2015, 7, 291ra297. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Delgado, A.B.; Valdes-Sanchez, L.; Morillo-Sanchez, M.J.; Ponte-Zuniga, B.; Diaz-Corrales, F.J.; de la Cerda, B. Dissecting the role of EYS in retinal degeneration: Clinical and molecular aspects and its implications for future therapy. Orphanet. J. Rare Dis. 2021, 16, 222. [Google Scholar] [CrossRef] [PubMed]
- Alfano, G.; Kruczek, P.M.; Shah, A.Z.; Kramarz, B.; Jeffery, G.; Zelhof, A.C.; Bhattacharya, S.S. EYS Is a Protein Associated with the Ciliary Axoneme in Rods and Cones. PLoS ONE 2016, 11, e0166397. [Google Scholar] [CrossRef]
- Eblimit, A.; Agrawal, S.A.; Thomas, K.; Anastassov, I.A.; Abulikemu, T.; Moayedi, Y.; Mardon, G.; Chen, R. Conditional loss of Spata7 in photoreceptors causes progressive retinal degeneration in mice. Exp. Eye Res. 2018, 166, 120–130. [Google Scholar] [CrossRef]
- Qu, Z.; Yimer, T.A.; Xie, S.; Wong, F.; Yu, S.; Liu, X.; Han, S.; Ma, J.; Lu, Z.; Hu, X.; et al. Knocking out lca5 in zebrafish causes cone-rod dystrophy due to impaired outer segment protein trafficking. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2694–2705. [Google Scholar] [CrossRef]
- Tucker, B.A.; Burnight, E.R.; Cranston, C.M.; Ulferts, M.J.; Luse, M.A.; Westfall, T.; Scott, C.A.; Marsden, A.; Gibson-Corley, K.; Wiley, L.A.; et al. Development and biological characterization of a clinical gene transfer vector for the treatment of MAK-associated retinitis pigmentosa. Gene Ther. 2022, 29, 259–288. [Google Scholar] [CrossRef]
- Wang, J.; Chen, X.; Wang, F.; Zhang, J.; Li, P.; Li, Z.; Xu, J.; Gao, F.; Jin, C.; Tian, H.; et al. OFD1, as a Ciliary Protein, Exhibits Neuroprotective Function in Photoreceptor Degeneration Models. PLoS ONE 2016, 11, e0155860. [Google Scholar] [CrossRef]
- Webb, T.R.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E.; Zito, I.; Thiselton, D.L.; Ressa, J.H.; Apergi, M.; et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem. Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef]
- Patil, S.B.; Hurd, T.W.; Ghosh, A.K.; Murga-Zamalloa, C.A.; Khanna, H. Functional analysis of retinitis pigmentosa 2 (RP2) protein reveals variable pathogenic potential of disease-associated missense variants. PLoS ONE 2011, 6, e21379. [Google Scholar] [CrossRef]
- Louie, C.M.; Caridi, G.; Lopes, V.S.; Brancati, F.; Kispert, A.; Lancaster, M.A.; Schlossman, A.M.; Otto, E.A.; Leitges, M.; Grone, H.J.; et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 2010, 42, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Chen, L.; Yan, L.; Perkins, B.D.; Li, S.; Li, B.; Xu, H.A.; Li, X.J. Mutant Ahi1 Affects Retinal Axon Projection in Zebrafish via Toxic Gain of Function. Front. Cell Neurosci 2019, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Klink, B.U.; Gatsogiannis, C.; Hofnagel, O.; Wittinghofer, A.; Raunser, S. Structure of the human BBSome core complex. eLife 2020, 9, e53910. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peranen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef]
- Nachury, M.V. Tandem affinity purification of the BBSome, a critical regulator of Rab8 in ciliogenesis. Methods Enzymol. 2008, 439, 501–513. [Google Scholar] [CrossRef]
- Hsu, Y.; Seo, S.; Sheffield, V.C. Photoreceptor cilia, in contrast to primary cilia, grant entry to a partially assembled BBSome. Hum. Mol. Genet. 2021, 30, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, G.; Bin, J.; Fieggen, K.J.; Duncan, J.L.; Gerth, C.; Ogata, K.; Wodak, S.S.; Traboulsi, E.I.; Fishman, G.A.; Paterson, A.; et al. Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. J. Med. Genet. 2010, 47, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, S.; Katoh, Y.; Kobayashi, T.; Nakayama, K. BBS1 is involved in retrograde trafficking of ciliary GPCRs in the context of the BBSome complex. PLoS ONE 2018, 13, e0195005. [Google Scholar] [CrossRef]
- Nishimura, D.Y.; Fath, M.; Mullins, R.F.; Searby, C.; Andrews, M.; Davis, R.; Andorf, J.L.; Mykytyn, K.; Swiderski, R.E.; Yang, B.; et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 16588–16593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Nishimura, D.; Vogel, T.; Shao, J.; Swiderski, R.; Yin, T.; Searby, C.; Carter, C.S.; Kim, G.; Bugge, K.; et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J. Cell Sci. 2013, 126, 2372–2380. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.L.; Bentley, M.R.; Croyle, M.J.; Kesterson, R.A.; Yoder, B.K.; Gross, A.K. BBSome Component BBS5 Is Required for Cone Photoreceptor Protein Trafficking and Outer Segment Maintenance. Investig. Ophthalmol. Vis. Sci. 2020, 61, 17. [Google Scholar] [CrossRef]
- Scheidecker, S.; Etard, C.; Pierce, N.W.; Geoffroy, V.; Schaefer, E.; Muller, J.; Chennen, K.; Flori, E.; Pelletier, V.; Poch, O.; et al. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J. Med. Genet. 2014, 51, 132–136. [Google Scholar] [CrossRef]
- Abd-El-Barr, M.M.; Sykoudis, K.; Andrabi, S.; Eichers, E.R.; Pennesi, M.E.; Tan, P.L.; Wilson, J.H.; Katsanis, N.; Lupski, J.R.; Wu, S.M. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome. Vision Res. 2007, 47, 3394–3407. [Google Scholar] [CrossRef]
- Veleri, S.; Bishop, K.; Dalle Nogare, D.E.; English, M.A.; Foskett, T.J.; Chitnis, A.; Sood, R.; Liu, P.; Swaroop, A. Knockdown of Bardet-Biedl syndrome gene BBS9/PTHB1 leads to cilia defects. PLoS ONE 2012, 7, e34389. [Google Scholar] [CrossRef]
- Smith, T.S.; Spitzbarth, B.; Li, J.; Dugger, D.R.; Stern-Schneider, G.; Sehn, E.; Bolch, S.N.; McDowell, J.H.; Tipton, J.; Wolfrum, U.; et al. Light-dependent phosphorylation of Bardet-Biedl syndrome 5 in photoreceptor cells modulates its interaction with arrestin1. Cell Mol. Life Sci. 2013, 70, 4603–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azari, A.A.; Aleman, T.S.; Cideciyan, A.V.; Schwartz, S.B.; Windsor, E.A.; Sumaroka, A.; Cheung, A.Y.; Steinberg, J.D.; Roman, A.J.; Stone, E.M.; et al. Retinal disease expression in Bardet-Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina-wide degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5004–5010. [Google Scholar] [CrossRef]
- Jiang, J.; Promchan, K.; Jiang, H.; Awasthi, P.; Marshall, H.; Harned, A.; Natarajan, V. Depletion of BBS Protein LZTFL1 Affects Growth and Causes Retinal Degeneration in Mice. J. Genet. Genom. 2016, 43, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Dilan, T.L.; Singh, R.K.; Saravanan, T.; Moye, A.; Goldberg, A.F.X.; Stoilov, P.; Ramamurthy, V. Bardet-Biedl syndrome-8 (BBS8) protein is crucial for the development of outer segments in photoreceptor neurons. Hum. Mol. Genet. 2018, 27, 283–294. [Google Scholar] [CrossRef]
- Alvarez-Satta, M.; Castro-Sanchez, S.; Valverde, D. Bardet-Biedl Syndrome as a Chaperonopathy: Dissecting the Major Role of Chaperonin-Like BBS Proteins (BBS6-BBS10-BBS12). Front. Mol. Biosci. 2017, 4, 55. [Google Scholar] [CrossRef] [PubMed]
- Rachel, R.A.; May-Simera, H.L.; Veleri, S.; Gotoh, N.; Choi, B.Y.; Murga-Zamalloa, C.; McIntyre, J.C.; Marek, J.; Lopez, I.; Hackett, A.N.; et al. Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J. Clin. Investig. 2012, 122, 1233–1245. [Google Scholar] [CrossRef]
- Hulleman, J.D.; Nguyen, A.; Ramprasad, V.L.; Murugan, S.; Gupta, R.; Mahindrakar, A.; Angara, R.; Sankurathri, C.; Mootha, V.V. A novel H395R mutation in MKKS/BBS6 causes retinitis pigmentosa and polydactyly without other findings of Bardet-Biedl or McKusick-Kaufman syndrome. Mol. Vis. 2016, 22, 73–81. [Google Scholar]
- Xue, B.; Liu, Y.X.; Dong, B.; Wingfield, J.L.; Wu, M.; Sun, J.; Lechtreck, K.F.; Fan, Z.C. Intraflagellar transport protein RABL5/IFT22 recruits the BBSome to the basal body through the GTPase ARL6/BBS3. Proc. Natl. Acad. Sci. USA 2020, 117, 2496–2505. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Chaya, T.; Tsujii, T.; Furukawa, T. The carboxyl-terminal region of SDCCAG8 comprises a functional module essential for cilia formation as well as organ development and homeostasis. J. Biol. Chem. 2022, 298, 101686. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.L.; Zhang, H.B.; Li, L.; Yang, Z.L.; Jiang, L. Characterization of two novel knock-in mouse models of syndromic retinal ciliopathy carrying hypomorphic Sdccag8 mutations. Zool. Res. 2022, 43, 442–456. [Google Scholar] [CrossRef]
- Schaefer, E.; Zaloszyc, A.; Lauer, J.; Durand, M.; Stutzmann, F.; Perdomo-Trujillo, Y.; Redin, C.; Bennouna Greene, V.; Toutain, A.; Perrin, L.; et al. Mutations in SDCCAG8/NPHP10 Cause Bardet-Biedl Syndrome and Are Associated with Penetrant Renal Disease and Absent Polydactyly. Mol. Syndromol. 2011, 1, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Bahmanpour, Z.; Daneshmandpour, Y.; Kazeminasab, S.; Khalil Khalili, S.; Alehabib, E.; Chapi, M.; Soosanabadi, M.; Darvish, H.; Emamalizadeh, B. A novel splice site mutation in the SDCCAG8 gene in an Iranian family with Bardet-Biedl syndrome. Int. Ophthalmol. 2021, 41, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Eguether, T.; San Agustin, J.T.; Keady, B.T.; Jonassen, J.A.; Liang, Y.; Francis, R.; Tobita, K.; Johnson, C.A.; Abdelhamed, Z.A.; Lo, C.W.; et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell 2014, 31, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Heon, E.; Kim, G.; Qin, S.; Garrison, J.E.; Tavares, E.; Vincent, A.; Nuangchamnong, N.; Scott, C.A.; Slusarski, D.C.; Sheffield, V.C. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum. Mol. Genet. 2016, 25, 2283–2294. [Google Scholar] [CrossRef]
- Sharif, A.S.; Yu, D.; Loertscher, S.; Austin, R.; Nguyen, K.; Mathur, P.D.; Clark, A.M.; Zou, J.; Lobanova, E.S.; Arshavsky, V.Y.; et al. C8ORF37 Is Required for Photoreceptor Outer Segment Disc Morphogenesis by Maintaining Outer Segment Membrane Protein Homeostasis. J. Neurosci. 2018, 38, 3160–3176. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Neveling, K.; Kohl, S.; Banin, E.; Rotenstreich, Y.; Sharon, D.; Falik-Zaccai, T.C.; Hipp, S.; Roepman, R.; Wissinger, B.; et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am. J. Hum. Genet. 2012, 90, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Rahner, N.; Nuernberg, G.; Finis, D.; Nuernberg, P.; Royer-Pokora, B. A novel C8orf37 splice mutation and genotype-phenotype correlation for cone-rod dystrophy. Ophthalmic. Genet. 2016, 37, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.S.; Lopes, V.S. The many different cellular functions of MYO7A in the retina. Biochem. Soc. Trans. 2011, 39, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Hasson, T.; Heintzelman, M.B.; Santos-Sacchi, J.; Corey, D.P.; Mooseker, M.S. Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc. Natl. Acad. Sci. USA 1995, 92, 9815–9819. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.; Azarian, S.M.; Lillo, C.; Kitamoto, J.; Klomp, A.E.; Steel, K.P.; Libby, R.T.; Williams, D.S. Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J. Cell Sci. 2004, 117, 6473–6483. [Google Scholar] [CrossRef]
- Kussel-Andermann, P.; El-Amraoui, A.; Safieddine, S.; Hardelin, J.P.; Nouaille, S.; Camonis, J.; Petit, C. Unconventional myosin VIIA is a novel A-kinase-anchoring protein. J. Biol. Chem. 2000, 275, 29654–29659. [Google Scholar] [CrossRef]
- Soni, L.E.; Warren, C.M.; Bucci, C.; Orten, D.J.; Hasson, T. The unconventional myosin-VIIa associates with lysosomes. Cell Motil. Cytoskelet. 2005, 62, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.B.; Blanco-Sanchez, B.; Lentz, J.J.; Tallafuss, A.; Khanobdee, K.; Sampath, S.; Jacobs, Z.G.; Han, P.F.; Mishra, M.; Titus, T.A.; et al. Harmonin (Ush1c) is required in zebrafish Muller glial cells for photoreceptor synaptic development and function. Dis. Model Mech. 2011, 4, 786–800. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, J.; Zhou, Q.; Khan, M.A.; Fu, J.; Duan, C.; Sun, S.; Lv, H.; Fu, J. Targeted Next-Generation Sequencing Identified Novel Compound Heterozygous Variants in the CDH23 Gene Causing Usher Syndrome Type ID in a Chinese Patient. Front. Genet. 2020, 11, 422. [Google Scholar] [CrossRef]
- Alagramam, K.N.; Yuan, H.; Kuehn, M.H.; Murcia, C.L.; Wayne, S.; Srisailpathy, C.R.; Lowry, R.B.; Knaus, R.; Van Laer, L.; Bernier, F.P.; et al. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum. Mol. Genet. 2001, 10, 1709–1718. [Google Scholar] [CrossRef]
- Miles, A.; Blair, C.; Emili, A.; Tropepe, V. Usher syndrome type 1-associated gene, pcdh15b, is required for photoreceptor structural integrity in zebrafish. Dis. Model Mech. 2021, 14, dmm048965. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Sumaroka, A.; Roman, A.J.; Gardner, L.M.; Prosser, H.M.; Mishra, M.; Bech-Hansen, N.T.; Herrera, W.; et al. Usher syndromes due to MYO7A, PCDH15, USH2A or GPR98 mutations share retinal disease mechanism. Hum. Mol. Genet. 2008, 17, 2405–2415. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.M.; Riazuddin, S.; Ahmad, J.; Bernstein, S.L.; Guo, Y.; Sabar, M.F.; Sieving, P.; Riazuddin, S.; Griffith, A.J.; Friedman, T.B.; et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 2003, 12, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, A.; Mozaffari-Jovin, S.; Wallisch, A.K.; Schafer, J.; Ludwig, S.E.J.; Urlaub, H.; Luhrmann, R.; Wolfrum, U. SANS (USH1G) regulates pre-mRNA splicing by mediating the intra-nuclear transfer of tri-snRNP complexes. Nucleic Acids Res. 2021, 49, 5845–5866. [Google Scholar] [CrossRef]
- Sorusch, N.; Bauss, K.; Plutniok, J.; Samanta, A.; Knapp, B.; Nagel-Wolfrum, K.; Wolfrum, U. Characterization of the ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum. Mol. Genet. 2017, 26, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Overlack, N.; Kilic, D.; Bauss, K.; Marker, T.; Kremer, H.; van Wijk, E.; Wolfrum, U. Direct interaction of the Usher syndrome 1G protein SANS and myomegalin in the retina. Biochim. Biophys. Acta 2011, 1813, 1883–1892. [Google Scholar] [CrossRef] [PubMed]
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; Vorthoren, L.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021, 29, 2441–2455. [Google Scholar] [CrossRef]
- Knapp, B.; Roedig, J.; Roedig, H.; Krzysko, J.; Horn, N.; Guler, B.E.; Kusuluri, D.K.; Yildirim, A.; Boldt, K.; Ueffing, M.; et al. Affinity Proteomics Identifies Interaction Partners and Defines Novel Insights into the Function of the Adhesion GPCR VLGR1/ADGRV1. Molecules 2022, 27, 3108. [Google Scholar] [CrossRef]
- Yan, W.; Long, P.; Chen, T.; Liu, W.; Yao, L.; Ren, Z.; Li, X.; Wang, J.; Xue, J.; Tao, Y.; et al. A Natural Occurring Mouse Model with Adgrv1 Mutation of Usher Syndrome 2C and Characterization of its Recombinant Inbred Strains. Cell Physiol. Biochem. 2018, 47, 1883–1897. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Zhao, Y.; Adamian, M.; Pawlyk, B.; Sun, X.; McMillan, D.R.; Liberman, M.C.; Li, T. Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 2010, 6, e1000955. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, E.; van der Zwaag, B.; Peters, T.; Zimmermann, U.; Te Brinke, H.; Kersten, F.F.; Marker, T.; Aller, E.; Hoefsloot, L.H.; Cremers, C.W.; et al. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet. 2006, 15, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zou, J.; Shen, Z.; Song, E.; Yang, J. Whirlin interacts with espin and modulates its actin-regulatory function: An insight into the mechanism of Usher syndrome type II. Hum. Mol. Genet. 2012, 21, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Herrera, W.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Banin, E.; Ben-Yosef, T.; Gardner, L.M.; Sumaroka, A.; Windsor, E.A.; Schwartz, S.B.; et al. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Adato, A.; Vreugde, S.; Joensuu, T.; Avidan, N.; Hamalainen, R.; Belenkiy, O.; Olender, T.; Bonne-Tamir, B.; Ben-Asher, E.; Espinos, C.; et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 2002, 10, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Bolch, S.N.; Santiago, C.P.; Dyka, F.M.; Akil, O.; Lobanova, E.S.; Wang, Y.; Martemyanov, K.A.; Hauswirth, W.W.; Smith, W.C.; et al. Clarin-1 expression in adult mouse and human retina highlights a role of Muller glia in Usher syndrome. J. Pathol. 2020, 250, 195–204. [Google Scholar] [CrossRef]
- Cosgrove, D.; Zallocchi, M. Clarin-1 protein expression in photoreceptors. Hear. Res. 2010, 259, 117. [Google Scholar] [CrossRef]
- Zallocchi, M.; Meehan, D.T.; Delimont, D.; Askew, C.; Garige, S.; Gratton, M.A.; Rothermund-Franklin, C.A.; Cosgrove, D. Localization and expression of clarin-1, the Clrn1 gene product, in auditory hair cells and photoreceptors. Hear. Res. 2009, 255, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, J.D.; Muller, J.; Collin, G.B.; Milan, G.; Kingsmore, S.F.; Dinwiddie, D.; Farrow, E.G.; Miller, N.A.; Favaretto, F.; Maffei, P.; et al. Alstrom Syndrome: Mutation Spectrum of ALMS1. Hum. Mutat. 2015, 36, 660–668. [Google Scholar] [CrossRef]
- Srikrupa, N.N.; Sripriya, S.; Pavithra, S.; Sen, P.; Gupta, R.; Mathavan, S. Whole-exome sequencing identifies two novel ALMS1 mutations in Indian patients with Leber congenital amaurosis. Hum. Genome Var. 2021, 8, 12. [Google Scholar] [CrossRef]
- Collin, G.B.; Marshall, J.D.; King, B.L.; Milan, G.; Maffei, P.; Jagger, D.J.; Naggert, J.K. The Alstrom syndrome protein, ALMS1, interacts with alpha-actinin and components of the endosome recycling pathway. PLoS ONE 2012, 7, e37925. [Google Scholar] [CrossRef]
- Datta, P.; Cribbs, J.T.; Seo, S. Differential requirement of NPHP1 for compartmentalized protein localization during photoreceptor outer segment development and maintenance. PLoS ONE 2021, 16, e0246358. [Google Scholar] [CrossRef]
- Won, J.; Marin de Evsikova, C.; Smith, R.S.; Hicks, W.L.; Edwards, M.M.; Longo-Guess, C.; Li, T.; Naggert, J.K.; Nishina, P.M. NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum. Mol. Genet. 2011, 20, 482–496. [Google Scholar] [CrossRef]
- Hanke-Gogokhia, C.; Chiodo, V.A.; Hauswirth, W.W.; Frederick, J.M.; Baehr, W. Rescue of cone function in cone-only Nphp5 knockout mouse model with Leber congenital amaurosis phenotype. Mol. Vis. 2018, 24, 834–846. [Google Scholar]
- Ki, S.M.; Kim, J.H.; Won, S.Y.; Oh, S.J.; Lee, I.Y.; Bae, Y.K.; Chung, K.W.; Choi, B.O.; Park, B.; Choi, E.J.; et al. CEP41-mediated ciliary tubulin glutamylation drives angiogenesis through AURKA-dependent deciliation. EMBO Rep. 2020, 21, e48290. [Google Scholar] [CrossRef]
- Sharif, A.S.; Gerstner, C.D.; Cady, M.A.; Arshavsky, V.Y.; Mitchell, C.; Ying, G.; Frederick, J.M.; Baehr, W. Deletion of the phosphatase INPP5E in the murine retina impairs photoreceptor axoneme formation and prevents disc morphogenesis. J. Biol. Chem. 2021, 296, 100529. [Google Scholar] [CrossRef]
- Slaats, G.G.; Isabella, C.R.; Kroes, H.Y.; Dempsey, J.C.; Gremmels, H.; Monroe, G.R.; Phelps, I.G.; Duran, K.J.; Adkins, J.; Kumar, S.A.; et al. MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J. Med. Genet. 2016, 53, 62–72. [Google Scholar] [CrossRef]
- Reble, E.; Feng, Y.; Wigg, K.G.; Barr, C.L. DNA Variant in the RPGRIP1L Gene Influences Alternative Splicing. Mol. Neuropsychiatry 2020, 5, 97–106. [Google Scholar] [CrossRef]
- Vossing, C.; Atigbire, P.; Eilers, J.; Markus, F.; Stieger, K.; Song, F.; Neidhardt, J. The Major Ciliary Isoforms of RPGR Build Different Interaction Complexes with INPP5E and RPGRIP1L. Int. J. Mol. Sci. 2021, 22, 3583. [Google Scholar] [CrossRef]
- Wiegering, A.; Dildrop, R.; Vesque, C.; Khanna, H.; Schneider-Maunoury, S.; Gerhardt, C. Rpgrip1l controls ciliary gating by ensuring the proper amount of Cep290 at the vertebrate transition zone. Mol. Biol. Cell 2021, 32, 675–689. [Google Scholar] [CrossRef]
- Coussa, R.G.; Otto, E.A.; Gee, H.Y.; Arthurs, P.; Ren, H.; Lopez, I.; Keser, V.; Fu, Q.; Faingold, R.; Khan, A.; et al. WDR19: An ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin. Genet. 2013, 84, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Nadar, V.C.; Ketschek, A.; Myers, K.A.; Gallo, G.; Baas, P.W. Kinesin-5 is essential for growth-cone turning. Curr. Biol. 2008, 18, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; Gliem, M.; Mangold, E.; Tebbe, L.; Spier, I.; Muller, P.L.; Holz, F.G.; Neuhaus, C.; Wolfrum, U.; Bolz, H.J.; et al. Novel Insights Into the Phenotypical Spectrum of KIF11-Associated Retinopathy, Including a New Form of Retinal Ciliopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3950–3959. [Google Scholar] [CrossRef]
- Wang, Y.; Smallwood, P.M.; Williams, J.; Nathans, J. A mouse model for kinesin family member 11 (Kif11)-associated familial exudative vitreoretinopathy. Hum. Mol. Genet. 2020, 29, 1121–1131. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Muller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Sarkar, H.; Moosajee, M. Choroideremia: Molecular mechanisms and therapies. Trends Mol. Med. 2022, 28, 378–387. [Google Scholar] [CrossRef]
- Strunnikova, N.V.; Barb, J.; Sergeev, Y.V.; Thiagarajasubramanian, A.; Silvin, C.; Munson, P.J.; Macdonald, I.M. Loss-of-function mutations in Rab escort protein 1 (REP-1) affect intracellular transport in fibroblasts and monocytes of choroideremia patients. PLoS ONE 2009, 4, e8402. [Google Scholar] [CrossRef]
- Lazow, M.A.; Hood, D.C.; Ramachandran, R.; Burke, T.R.; Wang, Y.Z.; Greenstein, V.C.; Birch, D.G. Transition zones between healthy and diseased retina in choroideremia (CHM) and Stargardt disease (STGD) as compared to retinitis pigmentosa (RP). Investig. Ophthalmol. Vis. Sci. 2011, 52, 9581–9590. [Google Scholar] [CrossRef] [PubMed]
- Fasham, J.; Arno, G.; Lin, S.; Xu, M.; Carss, K.J.; Hull, S.; Lane, A.; Robson, A.G.; Wenger, O.; Self, J.E.; et al. Delineating the expanding phenotype associated with SCAPER gene mutation. Am. J. Med. Genet. A 2019, 179, 1665–1671. [Google Scholar] [CrossRef] [Green Version]
- Tatour, Y.; Sanchez-Navarro, I.; Chervinsky, E.; Hakonarson, H.; Gawi, H.; Tahsin-Swafiri, S.; Leibu, R.; Lopez-Molina, M.I.; Fernandez-Sanz, G.; Ayuso, C.; et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J. Med. Genet. 2017, 54, 698–704. [Google Scholar] [CrossRef]
- Tingaud-Sequeira, A.; Raldua, D.; Lavie, J.; Mathieu, G.; Bordier, M.; Knoll-Gellida, A.; Rambeau, P.; Coupry, I.; Andre, M.; Malm, E.; et al. Functional validation of ABHD12 mutations in the neurodegenerative disease PHARC. Neurobiol. Dis. 2017, 98, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Igelman, A.D.; Ku, C.; da Palma, M.M.; Georgiou, M.; Schiff, E.R.; Lam, B.L.; Sankila, E.M.; Ahn, J.; Pyers, L.; Vincent, A.; et al. Expanding the clinical phenotype in patients with disease causing variants associated with atypical Usher syndrome. Ophthalmic. Genet. 2021, 42, 664–673. [Google Scholar] [CrossRef]
- Spencer, W.J.; Lewis, T.R.; Pearring, J.N.; Arshavsky, V.Y. Photoreceptor Discs: Built Like Ectosomes. Trends Cell Biol. 2020, 30, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.R.; Makia, M.S.; Castillo, C.M.; Al-Ubaidi, M.R.; Naash, M.I.; Arshavsky, V.Y. Photoreceptor Disc Enclosure Is Tightly Controlled by Peripherin-2 Oligomerization. J. Neurosci. 2021, 41, 3588–3596. [Google Scholar] [CrossRef]
- Pugh, E.N., Jr. Photoreceptor disc morphogenesis: The classical evagination model prevails. J. Cell Biol. 2015, 211, 491–493. [Google Scholar] [CrossRef]
- Kajiwara, K.; Berson, E.L.; Dryja, T.P. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef]
- Milstein, M.L.; Cavanaugh, B.L.; Roussey, N.M.; Volland, S.; Williams, D.S.; Goldberg, A.F.X. Multistep peripherin-2/rds self-assembly drives membrane curvature for outer segment disk architecture and photoreceptor viability. Proc. Natl. Acad. Sci. USA 2020, 117, 4400–4410. [Google Scholar] [CrossRef]
- Molday, R.S.; Goldberg, A.F.X. Peripherin diverts ciliary ectosome release to photoreceptor disc morphogenesis. J. Cell Biol. 2017, 216, 1227–1229. [Google Scholar] [CrossRef]
- Conley, S.M.; Stuck, M.W.; Watson, J.N.; Zulliger, R.; Burnett, J.L.; Naash, M.I. Prph2 initiates outer segment morphogenesis but maturation requires Prph2/Rom1 oligomerization. Hum. Mol. Genet. 2019, 28, 459–475. [Google Scholar] [CrossRef]
- Yokokura, S.; Wada, Y.; Nakai, S.; Sato, H.; Yao, R.; Yamanaka, H.; Ito, S.; Sagara, Y.; Takahashi, M.; Nakamura, Y.; et al. Targeted disruption of FSCN2 gene induces retinopathy in mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2905–2915. [Google Scholar] [CrossRef]
- Wada, Y.; Abe, T.; Takeshita, T.; Sato, H.; Yanashima, K.; Tamai, M. Mutation of human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2395–2400. [Google Scholar]
- Spencer, W.J.; Ding, J.D.; Lewis, T.R.; Yu, C.; Phan, S.; Pearring, J.N.; Kim, K.Y.; Thor, A.; Mathew, R.; Kalnitsky, J.; et al. PRCD is essential for high-fidelity photoreceptor disc formation. Proc. Natl. Acad. Sci. USA 2019, 116, 13087–13096. [Google Scholar] [CrossRef] [PubMed]
- Maw, M.A.; Corbeil, D.; Koch, J.; Hellwig, A.; Wilson-Wheeler, J.C.; Bridges, R.J.; Kumaramanickavel, G.; John, S.; Nancarrow, D.; Roper, K.; et al. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum. Mol. Genet. 2000, 9, 27–34. [Google Scholar] [CrossRef]
- Ostergaard, E.; Batbayli, M.; Duno, M.; Vilhelmsen, K.; Rosenberg, T. Mutations in PCDH21 cause autosomal recessive cone-rod dystrophy. J. Med. Genet. 2010, 47, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D.Y.; Baye, L.M.; Perveen, R.; Searby, C.C.; Avila-Fernandez, A.; Pereiro, I.; Ayuso, C.; Valverde, D.; Bishop, P.N.; Manson, F.D.; et al. Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am. J. Hum. Genet. 2010, 86, 686–695. [Google Scholar] [CrossRef] [PubMed]
- von Lintig, J.; Kiser, P.D.; Golczak, M.; Palczewski, K. The biochemical and structural basis for trans-to-cis isomerization of retinoids in the chemistry of vision. Trends Biochem. Sci. 2010, 35, 400–410. [Google Scholar] [CrossRef]
- Lamb, T.D.; Pugh, E.N., Jr. Dark adaptation and the retinoid cycle of vision. Prog. Retin. Eye Res. 2004, 23, 307–380. [Google Scholar] [CrossRef]
- Tsybovsky, Y.; Molday, R.S.; Palczewski, K. The ATP-binding cassette transporter ABCA4: Structural and functional properties and role in retinal disease. Adv. Exp. Med. Biol. 2010, 703, 105–125. [Google Scholar] [CrossRef]
- Kiser, P.D.; Golczak, M.; Palczewski, K. Chemistry of the retinoid (visual) cycle. Chem. Rev. 2014, 114, 194–232. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, F.; Bevilacqua, T.; Lee, K.I.; Chandrashekar, R.; Hsu, L.; Garlipp, M.A.; Griswold, J.B.; Crouch, R.K.; Ghosh, D. Retinol-binding site in interphotoreceptor retinoid-binding protein (IRBP): A novel hydrophobic cavity. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5577–5586. [Google Scholar] [CrossRef]
- MacDonald, P.N.; Ong, D.E. A lecithin:retinol acyltransferase activity in human and rat liver. Biochem. Biophys. Res. Commun. 1988, 156, 157–163. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Blaner, W.S. Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Raz, A.; Goodman, D.S. Retinol-binding protein: The transport protein for vitamin A in human plasma. J. Clin. Investig. 1968, 47, 2025–2044. [Google Scholar] [CrossRef] [PubMed]
- McBee, J.K.; Van Hooser, J.P.; Jang, G.F.; Palczewski, K. Isomerization of 11-cis-retinoids to all-trans-retinoids in vitro and in vivo. J. Biol. Chem. 2001, 276, 48483–48493. [Google Scholar] [CrossRef]
- Saari, J.C.; Nawrot, M.; Kennedy, B.N.; Garwin, G.G.; Hurley, J.B.; Huang, J.; Possin, D.E.; Crabb, J.W. Visual cycle impairment in cellular retinaldehyde binding protein (CRALBP) knockout mice results in delayed dark adaptation. Neuron 2001, 29, 739–748. [Google Scholar] [CrossRef]
- Radu, R.A.; Hu, J.; Peng, J.; Bok, D.; Mata, N.L.; Travis, G.H. Retinal pigment epithelium-retinal G protein receptor-opsin mediates light-dependent translocation of all-trans-retinyl esters for synthesis of visual chromophore in retinal pigment epithelial cells. J. Biol. Chem. 2008, 283, 19730–19738. [Google Scholar] [CrossRef]
- Stecher, H.; Gelb, M.H.; Saari, J.C.; Palczewski, K. Preferential release of 11-cis-retinol from retinal pigment epithelial cells in the presence of cellular retinaldehyde-binding protein. J. Biol. Chem. 1999, 274, 8577–8585. [Google Scholar] [CrossRef]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Lenis, T.L.; Hu, J.; Ng, S.Y.; Jiang, Z.; Sarfare, S.; Lloyd, M.B.; Esposito, N.J.; Samuel, W.; Jaworski, C.; Bok, D.; et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E11120–E11127. [Google Scholar] [CrossRef]
- Sun, H.; Molday, R.S.; Nathans, J. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem. 1999, 274, 8269–8281. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Fishkin, N.; Zhou, J.; Cai, B.; Jang, Y.P.; Krane, S.; Itagaki, Y.; Nakanishi, K. A2E, a byproduct of the visual cycle. Vision Res. 2003, 43, 2983–2990. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cremers, F.P.M. ABCA4-Associated Stargardt Disease. Klin. Monbl. Augenheilkd. 2020, 237, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Conley, S.M.; Naash, M.I. RPE65: Role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic. Genet. 2009, 30, 57–62. [Google Scholar] [CrossRef]
- Redmond, T.M.; Yu, S.; Lee, E.; Bok, D.; Hamasaki, D.; Chen, N.; Goletz, P.; Ma, J.X.; Crouch, R.K.; Pfeifer, K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998, 20, 344–351. [Google Scholar] [CrossRef]
- Fan, J.; Rohrer, B.; Frederick, J.M.; Baehr, W.; Crouch, R.K. Rpe65-/- and Lrat-/- mice: Comparable models of leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2384–2389. [Google Scholar] [CrossRef]
- Cideciyan, A.V. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog. Retin. Eye Res. 2010, 29, 398–427. [Google Scholar] [CrossRef]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef]
- Narfstrom, K.; Katz, M.L.; Ford, M.; Redmond, T.M.; Rakoczy, E.; Bragadottir, R. In vivo gene therapy in young and adult RPE65-/- dogs produces long-term visual improvement. J. Hered. 2003, 94, 31–37. [Google Scholar] [CrossRef]
- Ku, C.A.; Pennesi, M.E. The new landscape of retinal gene therapy. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 846–859. [Google Scholar] [CrossRef]
- Hao, W.; Fong, H.K. The endogenous chromophore of retinal G protein-coupled receptor opsin from the pigment epithelium. J. Biol. Chem. 1999, 274, 6085–6090. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Van Hooser, J.P.; Driessen, C.A.; Filipek, S.; Janssen, J.J.; Palczewski, K. Evaluation of the role of the retinal G protein-coupled receptor (RGR) in the vertebrate retina in vivo. J. Neurochem. 2003, 85, 944–956. [Google Scholar] [CrossRef]
- Morimura, H.; Saindelle-Ribeaudeau, F.; Berson, E.L.; Dryja, T.P. Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat. Genet. 1999, 23, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Rando, R.R. Membrane-bound lecithin-retinol acyltransferase. Biochem. Biophys. Res. Commun. 2002, 292, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Saari, J.C.; Bredberg, D.L. Lecithin:retinol acyltransferase in retinal pigment epithelial microsomes. J. Biol. Chem. 1989, 264, 8636–8640. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, S.; Moon, J.; Golczak, M.; von Lintig, J. LRAT coordinates the negative-feedback regulation of intestinal retinoid biosynthesis from beta-carotene. J. Lipid Res. 2021, 62, 100055. [Google Scholar] [CrossRef]
- Ruiz, A.; Winston, A.; Lim, Y.H.; Gilbert, B.A.; Rando, R.R.; Bok, D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J. Biol. Chem. 1999, 274, 3834–3841. [Google Scholar] [CrossRef]
- Maeda, T.; Cideciyan, A.V.; Maeda, A.; Golczak, M.; Aleman, T.S.; Jacobson, S.G.; Palczewski, K. Loss of cone photoreceptors caused by chromophore depletion is partially prevented by the artificial chromophore pro-drug, 9-cis-retinyl acetate. Hum. Mol. Genet. 2009, 18, 2277–2287. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Sui, R.; Sallum, J.; van den Born, L.I.; Ajlan, R.; Khan, A.; den Hollander, A.I.; Cremers, F.P.; Mendola, J.D.; Bittner, A.K.; et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: An open-label phase 1b trial. Lancet 2014, 384, 1513–1520. [Google Scholar] [CrossRef]
- Simon, A.; Romert, A.; Gustafson, A.L.; McCaffery, J.M.; Eriksson, U. Intracellular localization and membrane topology of 11-cis retinol dehydrogenase in the retinal pigment epithelium suggest a compartmentalized synthesis of 11-cis retinaldehyde. J. Cell Sci. 1999, 112, 549–558. [Google Scholar] [CrossRef]
- Thompson, D.A.; Gal, A. Vitamin A metabolism in the retinal pigment epithelium: Genes, mutations, and diseases. Prog. Retin. Eye. Res. 2003, 22, 683–703. [Google Scholar] [CrossRef]
- Katagiri, S.; Hayashi, T.; Nakamura, M.; Mizobuchi, K.; Gekka, T.; Komori, S.; Ueno, S.; Terasaki, H.; Sakuramoto, H.; Kuniyoshi, K.; et al. RDH5-Related Fundus Albipunctatus in a Large Japanese Cohort. Investig. Ophthalmol. Vis. Sci. 2020, 61, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Thompson, D.A.; Koutalos, Y. Reduction of all-trans-retinal in vertebrate rod photoreceptors requires the combined action of RDH8 and RDH12. J. Biol. Chem. 2012, 287, 24662–24670. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Sun, W.; Zhang, H.; Baehr, W.; Palczewski, K. Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc. Natl. Acad. Sci. USA 2007, 104, 19565–19570. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Hanein, S.; Gerber, S.; Barbet, F.; Ducroq, D.; Dollfus, H.; Hamel, C.; Dufier, J.L.; Munnich, A.; Kaplan, J.; et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am. J. Hum. Genet. 2004, 75, 639–646. [Google Scholar] [CrossRef]
- Chacon-Camacho, O.F.; Jitskii, S.; Buentello-Volante, B.; Quevedo-Martinez, J.; Zenteno, J.C. Exome sequencing identifies RDH12 compound heterozygous mutations in a family with severe retinitis pigmentosa. Gene 2013, 528, 178–182. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Imanishi, Y.; Sun, W.; Jastrzebska, B.; Hatala, D.A.; Winkens, H.J.; Hofmann, K.P.; Janssen, J.J.; Baehr, W.; et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J. Biol. Chem. 2006, 281, 37697–37704. [Google Scholar] [CrossRef]
- Jeon, C.J.; Strettoi, E.; Masland, R.H. The major cell populations of the mouse retina. J. Neurosci. 1998, 18, 8936–8946. [Google Scholar] [CrossRef]
- Xue, Y.; Shen, S.Q.; Jui, J.; Rupp, A.C.; Byrne, L.C.; Hattar, S.; Flannery, J.G.; Corbo, J.C.; Kefalov, V.J. CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Investig. 2015, 125, 727–738. [Google Scholar] [CrossRef]
- Saari, J.C. Vitamin A and Vision. Subcell. Biochem. 2016, 81, 231–259. [Google Scholar] [CrossRef]
- Maw, M.A.; Kennedy, B.; Knight, A.; Bridges, R.; Roth, K.E.; Mani, E.J.; Mukkadan, J.K.; Nancarrow, D.; Crabb, J.W.; Denton, M.J. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat. Genet. 1997, 17, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Morimura, H.; Berson, E.L.; Dryja, T.P. Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1000–1004. [Google Scholar]
- Eichers, E.R.; Green, J.S.; Stockton, D.W.; Jackman, C.S.; Whelan, J.; McNamara, J.A.; Johnson, G.J.; Lupski, J.R.; Katsanis, N. Newfoundland rod-cone dystrophy, an early-onset retinal dystrophy, is caused by splice-junction mutations in RLBP1. Am. J. Hum. Genet. 2002, 70, 955–964. [Google Scholar] [CrossRef]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef]
- Du, M.; Phelps, E.; Balangue, M.J.; Dockins, A.; Moiseyev, G.; Shin, Y.; Kane, S.; Otalora, L.; Ma, J.X.; Farjo, R.; et al. Transgenic Mice Over-Expressing RBP4 Have RBP4-Dependent and Light-Independent Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4375–4383. [Google Scholar] [CrossRef] [PubMed]
- Cukras, C.; Gaasterland, T.; Lee, P.; Gudiseva, H.V.; Chavali, V.R.; Pullakhandam, R.; Maranhao, B.; Edsall, L.; Soares, S.; Reddy, G.B.; et al. Exome analysis identified a novel mutation in the RBP4 gene in a consanguineous pedigree with retinal dystrophy and developmental abnormalities. PLoS ONE 2012, 7, e50205. [Google Scholar] [CrossRef]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef]
- Sun, Q.; Kiernan, U.A.; Shi, L.; Phillips, D.A.; Kahn, B.B.; Hu, F.B.; Manson, J.E.; Albert, C.M.; Rexrode, K.M. Plasma retinol-binding protein 4 (RBP4) levels and risk of coronary heart disease: A prospective analysis among women in the nurses’ health study. Circulation 2013, 127, 1938–1947. [Google Scholar] [CrossRef]
- Matsumoto, B.; Defoe, D.M.; Besharse, J.C. Membrane turnover in rod photoreceptors: Ensheathment and phagocytosis of outer segment distal tips by pseudopodia of the retinal pigment epithelium. Proc. R Soc. Lond. B Biol. Sci. 1987, 230, 339–354. [Google Scholar] [CrossRef]
- Milicevic, N.; Ait-Hmyed Hakkari, O.; Bagchi, U.; Sandu, C.; Jongejan, A.; Moerland, P.D.; Ten Brink, J.B.; Hicks, D.; Bergen, A.A.; Felder-Schmittbuhl, M.P. Core circadian clock genes Per1 and Per2 regulate the rhythm in photoreceptor outer segment phagocytosis. FASEB J. 2021, 35, e21722. [Google Scholar] [CrossRef]
- Muller, C.; Mas Gomez, N.; Ruth, P.; Strauss, O. CaV1.3 L-type channels, maxiK Ca(2+)-dependent K(+) channels and bestrophin-1 regulate rhythmic photoreceptor outer segment phagocytosis by retinal pigment epithelial cells. Cell Signal. 2014, 26, 968–978. [Google Scholar] [CrossRef]
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Almedawar, S.; Vafia, K.; Schreiter, S.; Neumann, K.; Khattak, S.; Kurth, T.; Ader, M.; Karl, M.O.; Tsang, S.H.; Tanaka, E.M. MERTK-Dependent Ensheathment of Photoreceptor Outer Segments by Human Pluripotent Stem Cell-Derived Retinal Pigment Epithelium. Stem. Cell Rep. 2020, 14, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Mohand-Said, S.; Boulanger-Scemama, E.; Zanlonghi, X.; Condroyer, C.; Demontant, V.; Boyard, F.; Antonio, A.; Mejecase, C.; El Shamieh, S.; et al. MERTK mutation update in inherited retinal diseases. Hum. Mutat. 2018, 39, 887–913. [Google Scholar] [CrossRef] [PubMed]
- Domenech, E.B.; Andres, R.; Lopez-Iniesta, M.J.; Mirra, S.; Garcia-Arroyo, R.; Milla, S.; Sava, F.; Andilla, J.; Loza-Alvarez, P.; de la Villa, P.; et al. A New Cerkl Mouse Model Generated by CRISPR-Cas9 Shows Progressive Retinal Degeneration and Altered Morphological and Electrophysiological Phenotype. Investig. Ophthalmol. Vis. Sci. 2020, 61, 14. [Google Scholar] [CrossRef]
- Yu, S.; Li, C.; Biswas, L.; Hu, X.; Liu, F.; Reilly, J.; Liu, X.; Liu, Y.; Huang, Y.; Lu, Z.; et al. CERKL gene knockout disturbs photoreceptor outer segment phagocytosis and causes rod-cone dystrophy in zebrafish. Hum. Mol. Genet. 2017, 26, 2335–2345. [Google Scholar] [CrossRef]
- Palfi, A.; Yesmambetov, A.; Humphries, P.; Hokamp, K.; Farrar, G.J. Non-photoreceptor Expression of Tulp1 May Contribute to Extensive Retinal Degeneration in Tulp1-/- Mice. Front. Neurosci. 2020, 14, 656. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef]
- Ying, G.; Boldt, K.; Ueffing, M.; Gerstner, C.D.; Frederick, J.M.; Baehr, W. The small GTPase RAB28 is required for phagocytosis of cone outer segments by the murine retinal pigmented epithelium. J. Biol. Chem. 2018, 293, 17546–17558. [Google Scholar] [CrossRef]
- Kanzaki, Y.; Fujita, H.; Sato, K.; Hosokawa, M.; Matsumae, H.; Shiraga, F.; Morizane, Y.; Ohuchi, H. KCNJ13 Gene Deletion Impairs Cell Alignment and Phagocytosis in Retinal Pigment Epithelium Derived from Human-Induced Pluripotent Stem Cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 38. [Google Scholar] [CrossRef]
- Naash, M.I.; Ripps, H.; Li, S.; Goto, Y.; Peachey, N.S. Polygenic disease and retinitis pigmentosa: Albinism exacerbates photoreceptor degeneration induced by the expression of a mutant opsin in transgenic mice. J. Neurosci. 1996, 16, 7853–7858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalban-Soler, L.; Alarcon-Martinez, L.; Jimenez-Lopez, M.; Salinas-Navarro, M.; Galindo-Romero, C.; Bezerra de Sa, F.; Garcia-Ayuso, D.; Aviles-Trigueros, M.; Vidal-Sanz, M.; Agudo-Barriuso, M.; et al. Retinal compensatory changes after light damage in albino mice. Mol. Vis. 2012, 18, 675–693. [Google Scholar]
- McKay, B.S. Pigmentation and vision: Is GPR143 in control? J. Neurosci. Res. 2019, 97, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Iarossi, G.; Coppe, A.M.; Passarelli, C.; Maltese, P.E.; Sinibaldi, L.; Cappelli, A.; Cetola, S.; Novelli, A.; Buzzonetti, L. Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes. Int. J. Mol. Sci. 2021, 22, 8617. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.M.; Tamm, E.R.; Straubeta, O. Role of bestrophin-1 in store-operated calcium entry in retinal pigment epithelium. Pflugers. Arch. 2013, 465, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, A.; Brandl, C.; Milenkovic, V.M.; Jendryke, T.; Sirianant, L.; Wanitchakool, P.; Zimmermann, S.; Reiff, C.M.; Horling, F.; Schrewe, H.; et al. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc. Natl. Acad. Sci. USA 2015, 112, E2630–E2639. [Google Scholar] [CrossRef]
- Cordes, M.; Bucichowski, P.; Alfaar, A.S.; Tsang, S.H.; Almedawar, S.; Reichhart, N.; Strauss, O. Inhibition of Ca(2+) channel surface expression by mutant bestrophin-1 in RPE cells. FASEB J. 2020, 34, 4055–4071. [Google Scholar] [CrossRef]
- Singh, R.; Shen, W.; Kuai, D.; Martin, J.M.; Guo, X.; Smith, M.A.; Perez, E.T.; Phillips, M.J.; Simonett, J.M.; Wallace, K.A.; et al. iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum. Mol. Genet. 2013, 22, 593–607. [Google Scholar] [CrossRef]
- Dinculescu, A.; Min, S.H.; Dyka, F.M.; Deng, W.T.; Stupay, R.M.; Chiodo, V.; Smith, W.C.; Hauswirth, W.W. Pathological Effects of Mutant C1QTNF5 (S163R) Expression in Murine Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6971–6980. [Google Scholar] [CrossRef]
- Nakano, M.; Kelly, E.J.; Wiek, C.; Hanenberg, H.; Rettie, A.E. CYP4V2 in Bietti’s crystalline dystrophy: Ocular localization, metabolism of omega-3-polyunsaturated fatty acids, and functional deficit of the p.H331P variant. Mol. Pharmacol. 2012, 82, 679–686. [Google Scholar] [CrossRef]
- Chen, Z.J.; Lin, K.H.; Lee, S.H.; Shen, R.J.; Feng, Z.K.; Wang, X.F.; Huang, X.F.; Huang, Z.Q.; Jin, Z.B. Mutation spectrum and genotype-phenotype correlation of inherited retinal dystrophy in Taiwan. Clin. Exp. Ophthalmol. 2020, 48, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Pennesi, M.E.; Harding, C.O.; Weleber, R.G.; Gillingham, M.B. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Mol. Genet. Metab. 2012, 106, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessein, A.F.; Hebbar, E.; Vamecq, J.; Lebredonchel, E.; Devos, A.; Ghoumid, J.; Mention, K.; Dobbelaere, D.; Chevalier-Curt, M.J.; Fontaine, M.; et al. A novel HADHA variant associated with an atypical moderate and late-onset LCHAD deficiency. Mol. Genet. Metab. Rep. 2022, 31, 100860. [Google Scholar] [CrossRef] [PubMed]
- Polinati, P.P.; Ilmarinen, T.; Trokovic, R.; Hyotylainen, T.; Otonkoski, T.; Suomalainen, A.; Skottman, H.; Tyni, T. Patient-Specific Induced Pluripotent Stem Cell-Derived RPE Cells: Understanding the Pathogenesis of Retinopathy in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3371–3382. [Google Scholar] [CrossRef]
- Dulz, S.; Atiskova, Y.; Engel, P.; Wildner, J.; Tsiakas, K.; Santer, R. Retained visual function in a subset of patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). Ophthalmic. Genet. 2021, 42, 23–27. [Google Scholar] [CrossRef]
- Garcia Garcia, L.C.; Zamorano Martin, F.; Rocha de Lossada, C.; Garcia Lorente, M.; Luque Aranda, G.; Escudero Gomez, J. Retinitis pigmentosa as a clinical presentation of LCHAD deficiency: A clinical case and review of the literature. Arch. Soc. Esp. Oftalmol. 2021, 96, 496–499. [Google Scholar] [CrossRef]
- Streeten, B.W. Development of the human retinal pigment epithelium and the posterior segment. Arch. Ophthalmol. 1969, 81, 383–394. [Google Scholar] [CrossRef]
- Uga, S.; Smelser, G.K. Electron microscopic study of the development of retinal Mullerian cells. Investig. Ophthalmol. 1973, 12, 295–307. [Google Scholar]
- Jakel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef]
- Hollenberg, M.J.; Spira, A.W. Human retinal development: Ultrastructure of the outer retina. Am. J. Anat. 1973, 137, 357–385. [Google Scholar] [CrossRef]
- Bhattacharjee, J. Sequential differentiation of retinal cells in the mouse studied by diaphorase staining. J. Anat. 1977, 123, 273–282. [Google Scholar] [PubMed]
- Mansergh, F.C.; Carrigan, M.; Hokamp, K.; Farrar, G.J. Gene expression changes during retinal development and rod specification. Mol. Vis. 2015, 21, 61–87. [Google Scholar] [PubMed]
- Tran, N.M.; Zhang, A.; Zhang, X.; Huecker, J.B.; Hennig, A.K.; Chen, S. Mechanistically distinct mouse models for CRX-associated retinopathy. PLoS Genet. 2014, 10, e1004111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tan, H.; Zeng, J.; Tao, D.; Ma, Y.; Liu, Y. A novel CRX variant (p.R98X) is identified in a Chinese family of Retinitis pigmentosa with atypical and mild manifestations. Genes Genom. 2019, 41, 359–366. [Google Scholar] [CrossRef]
- Chan, C.S.Y.; Lonfat, N.; Zhao, R.; Davis, A.E.; Li, L.; Wu, M.R.; Lin, C.H.; Ji, Z.; Cepko, C.L.; Wang, S. Cell type- and stage-specific expression of Otx2 is regulated by multiple transcription factors and cis-regulatory modules in the retina. Development 2020, 147, dev187922. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kon, T.; Omori, Y.; Furukawa, T. Functional and Evolutionary Diversification of Otx2 and Crx in Vertebrate Retinal Photoreceptor and Bipolar Cell Development. Cell Rep. 2020, 30, 658–671.e655. [Google Scholar] [CrossRef]
- Beby, F.; Housset, M.; Fossat, N.; Le Greneur, C.; Flamant, F.; Godement, P.; Lamonerie, T. Otx2 gene deletion in adult mouse retina induces rapid RPE dystrophy and slow photoreceptor degeneration. PLoS ONE 2010, 5, e11673. [Google Scholar] [CrossRef]
- Karjosukarso, D.W.; Ali, Z.; Peters, T.A.; Zhang, J.Q.C.; Hoogendoorn, A.D.M.; Garanto, A.; van Wijk, E.; Jensen, L.D.; Collin, R.W.J. Modeling ZNF408-Associated FEVR in Zebrafish Results in Abnormal Retinal Vasculature. Investig. Ophthalmol. Vis. Sci. 2020, 61, 39. [Google Scholar] [CrossRef]
- Collin, R.W.; Nikopoulos, K.; Dona, M.; Gilissen, C.; Hoischen, A.; Boonstra, F.N.; Poulter, J.A.; Kondo, H.; Berger, W.; Toomes, C.; et al. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proc. Natl. Acad. Sci. USA 2013, 110, 9856–9861. [Google Scholar] [CrossRef]
- Kubo, S.; Yamamoto, H.; Kajimura, N.; Omori, Y.; Maeda, Y.; Chaya, T.; Furukawa, T. Functional analysis of Samd11, a retinal photoreceptor PRC1 component, in establishing rod photoreceptor identity. Sci. Rep. 2021, 11, 4180. [Google Scholar] [CrossRef]
- Yanicostas, C.; Barbieri, E.; Hibi, M.; Brice, A.; Stevanin, G.; Soussi-Yanicostas, N. Requirement for zebrafish ataxin-7 in differentiation of photoreceptors and cerebellar neurons. PLoS ONE 2012, 7, e50705. [Google Scholar] [CrossRef] [PubMed]
- Yvert, G.; Lindenberg, K.S.; Picaud, S.; Landwehrmeyer, G.B.; Sahel, J.A.; Mandel, J.L. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum. Mol. Genet. 2000, 9, 2491–2506. [Google Scholar] [CrossRef]
- Michalik, A.; Martin, J.J.; Van Broeckhoven, C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur. J. Hum. Genet. 2004, 12, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Holak, H.M.; Kohlhase, J.; Holak, S.A.; Holak, N.H. New recognized ophthalmic morphologic anomalies in CHARGE syndrome caused by the R2319C mutation in the CHD7 gene. Ophthalmic. Genet. 2008, 29, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, M.; Udaka, T.; Kosaki, R.; Makita, Y.; Okamoto, N.; Yoshihashi, H.; Oki, H.; Nanao, K.; Moriyama, N.; Oku, S.; et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J. Pediatr. 2006, 148, 410–414. [Google Scholar] [CrossRef]
- Patten, S.A.; Jacobs-McDaniels, N.L.; Zaouter, C.; Drapeau, P.; Albertson, R.C.; Moldovan, F. Role of Chd7 in zebrafish: A model for CHARGE syndrome. PLoS ONE 2012, 7, e31650. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.E.; Vaclavik, V.; Wu, H.; Bujakowska, K.; Chakarova, C.F.; Bhattacharya, S.S.; Warren, M.J.; Hunt, D.M. Disease mechanism for retinitis pigmentosa (RP11) caused by missense mutations in the splicing factor gene PRPF31. Mol. Vis. 2008, 14, 683–690. [Google Scholar]
- Tanackovic, G.; Ransijn, A.; Ayuso, C.; Harper, S.; Berson, E.L.; Rivolta, C. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 2011, 88, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Brocher, J.; Fischer, U.; Winkler, C. Mutant Prpf31 causes pre-mRNA splicing defects and rod photoreceptor cell degeneration in a zebrafish model for Retinitis pigmentosa. Mol. Neurodegener. 2011, 6, 56. [Google Scholar] [CrossRef]
- Yuan, L.; Kawada, M.; Havlioglu, N.; Tang, H.; Wu, J.Y. Mutations in PRPF31 inhibit pre-mRNA splicing of rhodopsin gene and cause apoptosis of retinal cells. J. Neurosci. 2005, 25, 748–757. [Google Scholar] [CrossRef]
- Comitato, A.; Spampanato, C.; Chakarova, C.; Sanges, D.; Bhattacharya, S.S.; Marigo, V. Mutations in splicing factor PRPF3, causing retinal degeneration, form detrimental aggregates in photoreceptor cells. Hum. Mol. Genet. 2007, 16, 1699–1707. [Google Scholar] [CrossRef]
- Martinez-Gimeno, M.; Gamundi, M.J.; Hernan, I.; Maseras, M.; Milla, E.; Ayuso, C.; Garcia-Sandoval, B.; Beneyto, M.; Vilela, C.; Baiget, M.; et al. Mutations in the pre-mRNA splicing-factor genes PRPF3, PRPF8, and PRPF31 in Spanish families with autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2171–2177. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zheng, D.; Pan, Q.; Liu, Z.; Xi, X.; Hu, Z.; Deng, H.; Liu, X.; Jiang, D.; Deng, H.; et al. A novel PRPF31 splice-site mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Mol. Vis. 2004, 10, 361–365. [Google Scholar] [PubMed]
- Walia, S.; Fishman, G.A.; Zernant-Rajang, J.; Raime, K.; Allikmets, R. Phenotypic expression of a PRPF8 gene mutation in a Large African American family. Arch. Ophthalmol. 2008, 126, 1127–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towns, K.V.; Kipioti, A.; Long, V.; McKibbin, M.; Maubaret, C.; Vaclavik, V.; Ehsani, P.; Springell, K.; Kamal, M.; Ramesar, R.S.; et al. Prognosis for splicing factor PRPF8 retinitis pigmentosa, novel mutations and correlation between human and yeast phenotypes. Hum. Mutat. 2010, 31, E1361–E1376. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Sheng, X.; Tam, P.O.; Zhao, K.; Chen, X.; Rong, W.; Liu, Y.; Liu, X.; Pan, X.; et al. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2014, 23, 2926–2939. [Google Scholar] [CrossRef]
- Azizzadeh Pormehr, L.; Ahmadian, S.; Daftarian, N.; Mousavi, S.A.; Shafiezadeh, M. PRPF31 reduction causes mis-splicing of the phototransduction genes in human organotypic retinal culture. Eur. J. Hum. Genet. 2020, 28, 491–498. [Google Scholar] [CrossRef]
- Valdes-Sanchez, L.; Calado, S.M.; de la Cerda, B.; Aramburu, A.; Garcia-Delgado, A.B.; Massalini, S.; Montero-Sanchez, A.; Bhatia, V.; Rodriguez-Bocanegra, E.; Diez-Lloret, A.; et al. Retinal pigment epithelium degeneration caused by aggregation of PRPF31 and the role of HSP70 family of proteins. Mol. Med. 2019, 26, 1. [Google Scholar] [CrossRef]
- Zhong, Z.; Yan, M.; Sun, W.; Wu, Z.; Han, L.; Zhou, Z.; Zheng, F.; Chen, J. Two novel mutations in PRPF3 causing autosomal dominant retinitis pigmentosa. Sci. Rep. 2016, 6, 37840. [Google Scholar] [CrossRef]
- Abovich, N.; Legrain, P.; Rosbash, M. The yeast PRP6 gene encodes a U4/U6 small nuclear ribonucleoprotein particle (snRNP) protein, and the PRP9 gene encodes a protein required for U2 snRNP binding. Mol. Cell Biol. 1990, 10, 6417–6425. [Google Scholar] [CrossRef]
- Maita, H.; Kitaura, H.; Keen, T.J.; Inglehearn, C.F.; Ariga, H.; Iguchi-Ariga, S.M. PAP-1, the mutated gene underlying the RP9 form of dominant retinitis pigmentosa, is a splicing factor. Exp. Cell Res. 2004, 300, 283–296. [Google Scholar] [CrossRef]
- Bertrand, R.E.; Wang, J.; Li, Y.; Cheng, X.; Wang, K.; Stoilov, P.; Chen, R. Cwc27, associated with retinal degeneration, functions as a splicing factor in vivo. Hum. Mol. Genet. 2022, 31, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Bowne, S.J.; Sullivan, L.S.; Avery, C.E.; Sasser, E.M.; Roorda, A.; Duncan, J.L.; Wheaton, D.H.; Birch, D.G.; Branham, K.E.; Heckenlively, J.R.; et al. Mutations in the small nuclear riboprotein 200 kDa gene (SNRNP200) cause 1.6% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2013, 19, 2407–2417. [Google Scholar] [PubMed]
- Busetto, V.; Barbosa, I.; Basquin, J.; Marquenet, E.; Hocq, R.; Hennion, M.; Paternina, J.A.; Namane, A.; Conti, E.; Bensaude, O.; et al. Structural and functional insights into CWC27/CWC22 heterodimer linking the exon junction complex to spliceosomes. Nucleic Acids Res. 2020, 48, 5670–5683. [Google Scholar] [CrossRef] [PubMed]
- Inana, G.; Hotta, Y.; Zintz, C.; Takki, K.; Weleber, R.G.; Kennaway, N.G.; Nakayasu, K.; Nakajima, A.; Shiono, T. Expression defect of ornithine aminotransferase gene in gyrate atrophy. Investig. Ophthalmol. Vis. Sci. 1988, 29, 1001–1005. [Google Scholar]
- Wang, T.; Milam, A.H.; Steel, G.; Valle, D. A mouse model of gyrate atrophy of the choroid and retina. Early retinal pigment epithelium damage and progressive retinal degeneration. J. Clin. Investig. 1996, 97, 2753–2762. [Google Scholar] [CrossRef]
- Sen, S.; Kannan, S.K.; Shanmugam, U.; Rajan, R.; Babu, N.; Vanniarajan, A. Variable phenotypes of gyrate atrophy in siblings with a nonsense mutation in OAT gene. Ophthalmic. Genet. 2021, 42, 300–303. [Google Scholar] [CrossRef]
- Irie, S.; Sanuki, R.; Muranishi, Y.; Kato, K.; Chaya, T.; Furukawa, T. Rax Homeoprotein Regulates Photoreceptor Cell Maturation and Survival in Association with Crx in the Postnatal Mouse Retina. Mol. Cell Biol. 2015, 35, 2583–2596. [Google Scholar] [CrossRef]
- Van de Sompele, S.; Smith, C.; Karali, M.; Corton, M.; Van Schil, K.; Peelman, F.; Cherry, T.; Rosseel, T.; Verdin, H.; Derolez, J.; et al. Biallelic sequence and structural variants in RAX2 are a novel cause for autosomal recessive inherited retinal disease. Genet. Med. 2019, 21, 1319–1329. [Google Scholar] [CrossRef]
- Fathinajafabadi, A.; Perez-Jimenez, E.; Riera, M.; Knecht, E.; Gonzalez-Duarte, R. CERKL, a retinal disease gene, encodes an mRNA-binding protein that localizes in compact and untranslated mRNPs associated with microtubules. PLoS ONE 2014, 9, e87898. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.S.; Tham, A.; Lopez, A.J.; Edwards, S.; Woods, S.; Chen, J.; Wong-Fortunato, J.; Quiroz Alonso, A.; Javier, S.; Au, I.; et al. Cytoglobin deficiency potentiates Crb1-mediated retinal degeneration in rd8 mice. Dev. Biol. 2020, 458, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.A.; Cochran, K.J.; Kay, J.N. The Enigma of CRB1 and CRB1 Retinopathies. Adv. Exp. Med. Biol. 2019, 1185, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.H.; Pellissier, L.P.; Wijnholds, J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog. Retin. Eye Res. 2014, 40, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Booij, J.C.; Florijn, R.J.; ten Brink, J.B.; Loves, W.; Meire, F.; van Schooneveld, M.J.; de Jong, P.T.; Bergen, A.A. Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J. Med. Genet. 2005, 42, e67. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, I.H.; McClements, M.E.; MacLaren, R.E.; Charbel Issa, P. Deep phenotyping of the Cdhr1(-/-) mouse validates its use in pre-clinical studies for human CDHR1-associated retinal degeneration. Exp. Eye Res. 2021, 208, 108603. [Google Scholar] [CrossRef]
- Dawood, M.; Lin, S.; Din, T.U.; Shah, I.U.; Khan, N.; Jan, A.; Marwan, M.; Sultan, K.; Nowshid, M.; Tahir, R.; et al. Novel mutations in PDE6A and CDHR1 cause retinitis pigmentosa in Pakistani families. Int. J. Ophthalmol. 2021, 14, 1843–1851. [Google Scholar] [CrossRef]
- Hayward, C.; Shu, X.; Cideciyan, A.V.; Lennon, A.; Barran, P.; Zareparsi, S.; Sawyer, L.; Hendry, G.; Dhillon, B.; Milam, A.H.; et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: A genetic model for age-related macular degeneration. Hum. Mol. Genet. 2003, 12, 2657–2667. [Google Scholar] [CrossRef]
- Yang, X.; Chung, J.Y.; Rai, U.; Esumi, N. Cadherins in the retinal pigment epithelium (RPE) revisited: P-cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PLoS ONE 2018, 13, e0191279. [Google Scholar] [CrossRef]
- Chavali, V.R.; Khan, N.W.; Cukras, C.A.; Bartsch, D.U.; Jablonski, M.M.; Ayyagari, R. A CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum. Mol. Genet. 2011, 20, 2000–2014. [Google Scholar] [CrossRef]
- Mandal, M.N.; Vasireddy, V.; Reddy, G.B.; Wang, X.; Moroi, S.E.; Pattnaik, B.R.; Hughes, B.A.; Heckenlively, J.R.; Hitchcock, P.F.; Jablonski, M.M.; et al. CTRP5 is a membrane-associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5505–5513. [Google Scholar] [CrossRef]
- Salido, E.M.; Ramamurthy, V. Proteoglycan IMPG2 Shapes the Interphotoreceptor Matrix and Modulates Vision. J. Neurosci. 2020, 40, 4059–4072. [Google Scholar] [CrossRef] [PubMed]
- Felemban, M.; Dorgau, B.; Hunt, N.C.; Hallam, D.; Zerti, D.; Bauer, R.; Ding, Y.; Collin, J.; Steel, D.; Krasnogor, N.; et al. Extracellular matrix component expression in human pluripotent stem cell-derived retinal organoids recapitulates retinogenesis in vivo and reveals an important role for IMPG1 and CD44 in the development of photoreceptors and interphotoreceptor matrix. Acta Biomater. 2018, 74, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.; Coulter, C.; Geldenhuys, W.J.; Rhodes, S.; Salido, E.M. Interphotoreceptor matrix proteoglycans IMPG1 and IMPG2 proteolyze in the SEA domain and reveal localization mutual dependency. Sci. Rep. 2022, 12, 15535. [Google Scholar] [CrossRef] [PubMed]
- Olivier, G.; Brabet, P.; Pirot, N.; Broyon, M.; Guillou, L.; Cazevieille, C.; Sar, C.; Quiles, M.; Sarzi, E.; Pequignot, M.; et al. SPACR Encoded by IMPG1 Is Essential for Photoreceptor Survival by Interplaying between the Interphotoreceptor Matrix and the Retinal Pigment Epithelium. Genes 2022, 13, 1508. [Google Scholar] [CrossRef] [PubMed]
- Jomary, C.; Neal, M.J.; Jones, S.E. Increased expression of retinal TIMP3 mRNA in simplex retinitis pigmentosa is localized to photoreceptor-retaining regions. J. Neurochem. 1995, 64, 2370–2373. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020, 104, 451–460. [Google Scholar] [CrossRef]
- Meunier, I.; Bocquet, B.; Labesse, G.; Zeitz, C.; Defoort-Dhellemmes, S.; Lacroux, A.; Mauget-Faysse, M.; Drumare, I.; Gamez, A.S.; Mathieu, C.; et al. A new autosomal dominant eye and lung syndrome linked to mutations in TIMP3 gene. Sci. Rep. 2016, 6, 32544. [Google Scholar] [CrossRef]
- Parry, D.A.; Toomes, C.; Bida, L.; Danciger, M.; Towns, K.V.; McKibbin, M.; Jacobson, S.G.; Logan, C.V.; Ali, M.; Bond, J.; et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am. J. Hum. Genet. 2009, 84, 683–691. [Google Scholar] [CrossRef]
- El-Haig, W.M.; Jakobsson, C.; Favez, T.; Schorderet, D.F.; Abouzeid, H. Novel ADAM9 homozygous mutation in a consanguineous Egyptian family with severe cone-rod dystrophy and cataract. Br. J. Ophthalmol. 2014, 98, 1718–1723. [Google Scholar] [CrossRef] [PubMed]
- Savige, J.; Wang, Y.; Crawford, A.; Smith, J.; Symons, A.; Mack, H.; Nicholls, K.; Wilson, D.; Colville, D. Bull’s eye and pigment maculopathy are further retinal manifestations of an abnormal Bruch’s membrane in Alport syndrome. Ophthalmic. Genet. 2017, 38, 238–244. [Google Scholar] [CrossRef]
- Fu, L.; Garland, D.; Yang, Z.; Shukla, D.; Rajendran, A.; Pearson, E.; Stone, E.M.; Zhang, K.; Pierce, E.A. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum. Mol. Genet. 2007, 16, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Chen, C.; Guo, W.; Liu, K.; Zhao, Q.; Lu, P.; Yu, F.; Xu, X. EFEMP1 Overexpression Contributes to Neovascularization in Age-Related Macular Degeneration. Front. Pharmacol. 2020, 11, 547436. [Google Scholar] [CrossRef]
- Stone, E.M.; Lotery, A.J.; Munier, F.L.; Heon, E.; Piguet, B.; Guymer, R.H.; Vandenburgh, K.; Cousin, P.; Nishimura, D.; Swiderski, R.E.; et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 1999, 22, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Giessl, A.; Laufs, T.; Hankeln, T.; Wolfrum, U.; Burmester, T. How does the eye breathe? Evidence for neuroglobin-mediated oxygen supply in the mammalian retina. J. Biol. Chem. 2003, 278, 1932–1935. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, L.; Zhang, J.; Huang, M.; Wong, F.; Liu, X.; Liu, F.; Cui, X.; Yang, G.; Chen, J.; et al. CERKL interacts with mitochondrial TRX2 and protects retinal cells from oxidative stress-induced apoptosis. Biochim. Biophys. Acta 2014, 1842, 1121–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirra, S.; Garcia-Arroyo, R.; Domènech, E.B.; Gavalda-Navarro, A.; Herrera-Ubeda, C.; Oliva, C.; Garcia-Fernandez, J.; Artuch, R.; Villarroya, F.; Marfany, G. CERKL, a retinal dystrophy gene, regulates mitochondrial function and dynamics in the mammalian retina. Neurobiol. Dis. 2021, 156, 105405. [Google Scholar] [CrossRef]
- Mandal, N.A.; Tran, J.T.; Saadi, A.; Rahman, A.K.; Huynh, T.P.; Klein, W.H.; Cho, J.H. Expression and localization of CERKL in the mammalian retina, its response to light-stress, and relationship with NeuroD1 gene. Exp. Eye Res. 2013, 106, 24–33. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Wang, H.; Majewski, J.; Wang, X.; Lopez, I.; Ren, H.; Chen, Y.; Li, Y.; Fishman, G.A.; Genead, M.; et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012, 44, 1035–1039. [Google Scholar] [CrossRef]
- Sokolov, D.; Sechrest, E.R.; Wang, Y.; Nevin, C.; Du, J.; Kolandaivelu, S. Nuclear NAD(+)-biosynthetic enzyme NMNAT1 facilitates development and early survival of retinal neurons. eLife 2021, 10, e71185. [Google Scholar] [CrossRef]
- Greenwald, S.H.; Brown, E.E.; Scandura, M.J.; Hennessey, E.; Farmer, R.; Du, J.; Wang, Y.; Pierce, E.A. Mutant Nmnat1 leads to a retina-specific decrease of NAD+ accompanied by increased poly(ADP-ribose) in a mouse model of NMNAT1-associated retinal degeneration. Hum. Mol. Genet. 2021, 30, 644–657. [Google Scholar] [CrossRef]
- De Jesus, A.; Keyhani-Nejad, F.; Pusec, C.M.; Goodman, L.; Geier, J.A.; Stoolman, J.S.; Stanczyk, P.J.; Nguyen, T.; Xu, K.; Suresh, K.V.; et al. Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol. Cell 2022, 82, 1261–1277 e1269. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, B.; Xu, M.; Chang, E.Y.; Li, H.; Yang, L.; Wu, S.; Soens, Z.T.; Li, Y.; Wong, L.C.; et al. The phenotypic variability of HK1-associated retinal dystrophy. Sci. Rep. 2017, 7, 7051. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.S.; Koboldt, D.C.; Bowne, S.J.; Lang, S.; Blanton, S.H.; Cadena, E.; Avery, C.E.; Lewis, R.A.; Webb-Jones, K.; Wheaton, D.H.; et al. A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7147–7158. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J. Defective pantothenate metabolism and neurodegeneration. Biochem. Soc. Trans. 2014, 42, 1063–1068. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Duncan, J.L.; Westaway, S.K.; Yang, H.; Nune, G.; Xu, E.Y.; Hayflick, S.J.; Gitschier, J. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum. Mol. Genet. 2005, 14, 49–57. [Google Scholar] [CrossRef]
- Cheema, M.R.; Stone, L.G.; Sellar, P.W.; Quinn, S.; Clark, S.C.; Martin, R.J.; O’Brien, J.M.; Warriner, C.; Browning, A.C. Long-term follow-up of a patient with JAG1-associated retinopathy. Doc. Ophthalmol. 2021, 143, 237–247. [Google Scholar] [CrossRef]
- Jahan, I.; Pan, N.; Fritzsch, B. Opportunities and limits of the one gene approach: The ability of Atoh1 to differentiate and maintain hair cells depends on the molecular context. Front. Cell Neurosci. 2015, 9, 26. [Google Scholar] [CrossRef]
- Macchiarulo, S.; Morrow, B.E. Tbx1 and Jag1 act in concert to modulate the fate of neurosensory cells of the mouse otic vesicle. Biol. Open 2017, 6, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Xu, H.; Huang, L.; Zhang, S.; Liu, W.; Yang, Y.; Fei, P.; Li, S.; Yang, M.; et al. Exome sequencing revealed Notch ligand JAG1 as a novel candidate gene for familial exudative vitreoretinopathy. Genet. Med. 2020, 22, 77–84. [Google Scholar] [CrossRef]
- Zhang, K.; Harada, Y.; Wei, X.; Shukla, D.; Rajendran, A.; Tawansy, K.; Bedell, M.; Lim, S.; Shaw, P.X.; He, X.; et al. An essential role of the cysteine-rich domain of FZD4 in Norrin/Wnt signaling and familial exudative vitreoretinopathy. J. Biol. Chem. 2011, 286, 10210–10215. [Google Scholar] [CrossRef]
- Chidiac, R.; Abedin, M.; Macleod, G.; Yang, A.; Thibeault, P.E.; Blazer, L.L.; Adams, J.J.; Zhang, L.; Roehrich, H.; Jo, H.N.; et al. A Norrin/Wnt surrogate antibody stimulates endothelial cell barrier function and rescues retinopathy. EMBO Mol. Med. 2021, 13, e13977. [Google Scholar] [CrossRef] [PubMed]
- Paes, K.T.; Wang, E.; Henze, K.; Vogel, P.; Read, R.; Suwanichkul, A.; Kirkpatrick, L.L.; Potter, D.; Newhouse, M.M.; Rice, D.S. Frizzled 4 is required for retinal angiogenesis and maintenance of the blood-retina barrier. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6452–6461. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lu, J.; Zhang, L.; Zhang, Z.; Sun, L.; Li, S.; Zhang, T.; Chen, L.; Cao, L.; Ding, X. Whole-Gene Deletions of FZD4 Cause Familial Exudative Vitreoretinopathy. Genes 2021, 12, 980. [Google Scholar] [CrossRef]
- Yanatori, I.; Yasui, Y.; Miura, K.; Kishi, F. Mutations of FLVCR1 in posterior column ataxia and retinitis pigmentosa result in the loss of heme export activity. Blood Cells Mol. Dis. 2012, 49, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Kuehlewein, L.; Schols, L.; Llavona, P.; Grimm, A.; Biskup, S.; Zrenner, E.; Kohl, S. Phenotypic spectrum of autosomal recessive retinitis pigmentosa without posterior column ataxia caused by mutations in the FLVCR1 gene. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, I.H.; Shanks, M.E.; Clouston, P.; MacLaren, R.E. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic. Genet. 2018, 39, 263–267. [Google Scholar] [CrossRef]
- di Ronza, A.; Bajaj, L.; Sharma, J.; Sanagasetti, D.; Lotfi, P.; Adamski, C.J.; Collette, J.; Palmieri, M.; Amawi, A.; Popp, L.; et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 2018, 20, 1370–1377. [Google Scholar] [CrossRef]
- Pesaola, F.; Quassollo, G.; Venier, A.C.; De Paul, A.L.; Noher, I.; Bisbal, M. The neuronal ceroid lipofuscinosis-related protein CLN8 regulates endo-lysosomal dynamics and dendritic morphology. Biol. Cell 2021, 113, 419–437. [Google Scholar] [CrossRef]
- Salpeter, E.M.; Leonard, B.C.; Lopez, A.J.; Murphy, C.J.; Thomasy, S.; Imai, D.M.; Grimsrud, K.; Lloyd, K.C.K.; Yan, J.; Sanchez Russo, R.; et al. Retinal degeneration in mice and humans with neuronal ceroid lipofuscinosis type 8. Ann. Transl. Med. 2021, 9, 1274. [Google Scholar] [CrossRef]
- Wright, G.A.; Georgiou, M.; Robson, A.G.; Ali, N.; Kalhoro, A.; Holthaus, S.K.; Pontikos, N.; Oluonye, N.; de Carvalho, E.R.; Neveu, M.M.; et al. Juvenile Batten Disease (CLN3): Detailed Ocular Phenotype, Novel Observations, Delayed Diagnosis, Masquerades, and Prospects for Therapy. Ophthalmol. Retina 2020, 4, 433–445. [Google Scholar] [CrossRef]
- Zhong, Y.; Mohan, K.; Liu, J.; Al-Attar, A.; Lin, P.; Flight, R.M.; Sun, Q.; Warmoes, M.O.; Deshpande, R.R.; Liu, H.; et al. Loss of CLN3, the gene mutated in juvenile neuronal ceroid lipofuscinosis, leads to metabolic impairment and autophagy induction in retinal pigment epithelium. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165883. [Google Scholar] [CrossRef]
- Hu, X.; Lu, Z.; Yu, S.; Reilly, J.; Liu, F.; Jia, D.; Qin, Y.; Han, S.; Liu, X.; Qu, Z.; et al. CERKL regulates autophagy via the NAD-dependent deacetylase SIRT1. Autophagy 2019, 15, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Durand, S.; Feldhammer, M.; Bonneil, E.; Thibault, P.; Pshezhetsky, A.V. Analysis of the biogenesis of heparan sulfate acetyl-CoA:alpha-glucosaminide N-acetyltransferase provides insights into the mechanism underlying its complete deficiency in mucopolysaccharidosis IIIC. J. Biol. Chem. 2010, 285, 31233–31242. [Google Scholar] [CrossRef]
- Nagel, L.; Oliveira, R.; Pshezhetsky, A.V.; Morales, C.R. HGSNAT enzyme deficiency results in accumulation of heparan sulfate in podocytes and basement membranes. Histol. Histopathol. 2019, 34, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Carrera, W.; Ng, C.; Burckhard, B.; Ng, J.; McDonald, H.R.; Agarwal, A. Non-syndromic retinitis pigmentosa with bilateral retinal neovascularization due to HGSNAT mutation. Retin. Cases Brief Rep. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lee, S.K.; Choi, S.; Huh, Y.H.; Kwak, J.H.; Lee, Y.S.; Jang, D.J.; Lee, J.H.; Lee, K.; Kaang, B.K.; et al. Autophagy pathway upregulation in a human iPSC-derived neuronal model of Cohen syndrome with VPS13B missense mutations. Mol. Brain 2020, 13, 69. [Google Scholar] [CrossRef]
- Gillentine, M.A.; Schaaf, C.P.; Patel, A. The importance of phase analysis in multiexon copy number variation detected by aCGH in autosomal recessive disorder loci. Am. J. Med. Genet. A 2017, 173, 2485–2488. [Google Scholar] [CrossRef]
- Yagita, Y.; Shinohara, K.; Abe, Y.; Nakagawa, K.; Al-Owain, M.; Alkuraya, F.S.; Fujiki, Y. Deficiency of a Retinal Dystrophy Protein, Acyl-CoA Binding Domain-containing 5 (ACBD5), Impairs Peroxisomal beta-Oxidation of Very-long-chain Fatty Acids. J. Biol. Chem. 2017, 292, 691–705. [Google Scholar] [CrossRef]
- Bartlett, M.; Nasiri, N.; Pressman, R.; Bademci, G.; Forghani, I. First reported adult patient with retinal dystrophy and leukodystrophy caused by a novel ACBD5 variant: A case report and review of literature. Am. J. Med. Genet. A 2021, 185, 1236–1241. [Google Scholar] [CrossRef]
- Gorukmez, O.; Havali, C.; Gorukmez, O.; Dorum, S. Newly defined peroxisomal disease with novel ACBD5 mutation. J. Pediatr. Endocrinol. Metab. 2022, 35, 11–18. [Google Scholar] [CrossRef]
- Ashibe, B.; Hirai, T.; Higashi, K.; Sekimizu, K.; Motojima, K. Dual subcellular localization in the endoplasmic reticulum and peroxisomes and a vital role in protecting against oxidative stress of fatty aldehyde dehydrogenase are achieved by alternative splicing. J. Biol. Chem. 2007, 282, 20763–20773. [Google Scholar] [CrossRef] [PubMed]
- Ashibe, B.; Motojima, K. Fatty aldehyde dehydrogenase is up-regulated by polyunsaturated fatty acid via peroxisome proliferator-activated receptor alpha and suppresses polyunsaturated fatty acid-induced endoplasmic reticulum stress. FEBS J. 2009, 276, 6956–6970. [Google Scholar] [CrossRef] [PubMed]
- Bindu, P.S. Sjogren-Larsson Syndrome: Mechanisms and Management. Appl. Clin. Genet. 2020, 13, 13–24. [Google Scholar] [CrossRef]
- van Domburg, P.H.; Willemsen, M.A.; Rotteveel, J.J.; de Jong, J.G.; Thijssen, H.O.; Heerschap, A.; Cruysberg, J.R.; Wanders, R.J.; Gabreels, F.J.; Steijlen, P.M. Sjogren-Larsson syndrome: Clinical and MRI/MRS findings in FALDH-deficient patients. Neurology 1999, 52, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, G.; Sanchez-Navarro, I.; Aller, E.; Jaijo, T.; Fuster-Garcia, C.; Rodriguez-Munoz, A.; Vallejo, E.; Telleria, J.J.; Vazquez, S.; Beltran, S.; et al. Exome sequencing identifies PEX6 mutations in three cases diagnosed with Retinitis Pigmentosa and hearing impairment. Mol. Vis. 2020, 26, 216–225. [Google Scholar] [PubMed]
- Mechaussier, S.; Marlin, S.; Kaplan, J.; Rozet, J.M.; Perrault, I. Genetic Deciphering of Early-Onset and Severe Retinal Dystrophy Associated with Sensorineural Hearing Loss. Adv. Exp. Med. Biol. 2019, 1185, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Papp, K.M.; Casey, G.A.; Radziwon, A.; St Laurent, C.D.; Doucette, L.P.; MacDonald, I.M. PEX6 Mutations in Peroxisomal Biogenesis Disorders: An Usher Syndrome Mimic. Ophthalmol. Sci. 2021, 1, 100028. [Google Scholar] [CrossRef] [PubMed]
- Raas-Rothschild, A.; Wanders, R.J.; Mooijer, P.A.; Gootjes, J.; Waterham, H.R.; Gutman, A.; Suzuki, Y.; Shimozawa, N.; Kondo, N.; Eshel, G.; et al. A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am. J. Hum. Genet. 2002, 70, 1062–1068. [Google Scholar] [CrossRef]
- Ratbi, I.; Jaouad, I.C.; Elorch, H.; Al-Sheqaih, N.; Elalloussi, M.; Lyahyai, J.; Berraho, A.; Newman, W.G.; Sefiani, A. Severe early onset retinitis pigmentosa in a Moroccan patient with Heimler syndrome due to novel homozygous mutation of PEX1 gene. Eur. J. Med. Genet. 2016, 59, 507–511. [Google Scholar] [CrossRef]
- Smith, C.E.; Poulter, J.A.; Levin, A.V.; Capasso, J.E.; Price, S.; Ben-Yosef, T.; Sharony, R.; Newman, W.G.; Shore, R.C.; Brookes, S.J.; et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 2016, 24, 1565–1571. [Google Scholar] [CrossRef]
- Pekny, M.; Wilhelmsson, U.; Pekna, M. The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 2014, 565, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Campos-Caro, A.; Aguilar-Diosdado, M.; Valverde, A.M. IGF-1, Inflammation and Retinal Degeneration: A Close Network. Front. Aging Neurosci. 2018, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Wooff, Y.; Man, S.M.; Aggio-Bruce, R.; Natoli, R.; Fernando, N. IL-1 Family Members Mediate Cell Death, Inflammation and Angiogenesis in Retinal Degenerative Diseases. Front. Immunol. 2019, 10, 1618. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2019, 74, 100778. [Google Scholar] [CrossRef]
- Li, L.; Eter, N.; Heiduschka, P. The microglia in healthy and diseased retina. Exp. Eye Res. 2015, 136, 116–130. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef]
- Ronning, K.E.; Karlen, S.J.; Miller, E.B.; Burns, M.E. Molecular profiling of resident and infiltrating mononuclear phagocytes during rapid adult retinal degeneration using single-cell RNA sequencing. Sci. Rep. 2019, 9, 4858. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.Y.; Wang, Y.Y.; Chen, L.S.; Li, W.D.; Shen, Y.; Jin, Y.; Yang, J.; Wang, Y.; Yuan, J.; Cheng, L. Implication of inflammatory cytokines in the aqueous humour for management of macular diseases. Acta Ophthalmol. 2019, 98, e309–e315. [Google Scholar] [CrossRef]
- Yu, C.; Roubeix, C.; Sennlaub, F.; Saban, D.R. Microglia versus Monocytes: Distinct Roles in Degenerative Diseases of the Retina. Trends Neurosci. 2020, 43, 433–449. [Google Scholar] [CrossRef]
- Ronning, K.E.; Karlen, S.J.; Burns, M.E. Structural and functional distinctions of co-resident microglia and monocyte-derived macrophages after retinal degeneration. J. Neuroinflammation 2022, 19, 299. [Google Scholar] [CrossRef]
- Ten Berge, J.C.; Fazil, Z.; van den Born, I.; Wolfs, R.C.W.; Schreurs, M.W.J.; Dik, W.A.; Rothova, A. Intraocular cytokine profile and autoimmune reactions in retinitis pigmentosa, age-related macular degeneration, glaucoma and cataract. Acta Ophthalmol. 2019, 97, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Hou, C.; Yan, N. Neuroinflammation in retinitis pigmentosa: Therapies targeting the innate immune system. Front. Immunol. 2022, 13, 1059947. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, T.J.; Hubbard, M.G.; Levi, H.J.; White, W.; Wang, X.; Simpson, R.; Jablonski, M.M.; Gross, A.K. Proinflammatory Pathways Are Activated in the Human Q344X Rhodopsin Knock-In Mouse Model of Retinitis Pigmentosa. Biomolecules 2021, 11, 1163. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Gonzalez, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef]
- Appelbaum, T.; Santana, E.; Aguirre, G.D. Strong upregulation of inflammatory genes accompanies photoreceptor demise in canine models of retinal degeneration. PLoS ONE 2017, 12, e0177224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Pandey, R.K.; Miller, L.J.; Singh, P.K.; Kanwar, M. Muller glia in retinal innate immunity: A perspective on their roles in endophthalmitis. Crit. Rev. Immunol. 2013, 33, 119–135. [Google Scholar] [CrossRef]
- Hohmann, H.P.; Remy, R.; Poschl, B.; van Loon, A.P. Tumor necrosis factors-alpha and -beta bind to the same two types of tumor necrosis factor receptors and maximally activate the transcription factor NF-kappa B at low receptor occupancy and within minutes after receptor binding. J. Biol. Chem. 1990, 265, 15183–15188. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem. Sci. 2012, 37, 325–332. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Hollingsworth, T.J.; Gross, A.K. Innate and Autoimmunity in the Pathogenesis of Inherited Retinal Dystrophy. Cells 2020, 9, 630. [Google Scholar] [CrossRef]
- Mirra, S.; Sanchez-Bellver, L.; Casale, C.; Pescatore, A.; Marfany, G. Ubiquitin Specific Protease USP48 Destabilizes NF-kappaB/p65 in Retinal Pigment Epithelium Cells. Int. J. Mol. Sci. 2022, 23, 9682. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Immunohistochemical localization of many common inherited retinal dystrophy (IRD)-associated proteins. (A,B) Outer segment (POS) localization of phototransduction proteins CNGB1 (A, red), OPN1MW (B, red), GUCY2E (B, green), and RHO (A,B, blue), as well as phagocytosis protein MERTK (A, green). (C) Connecting cilium (>) localization of proteins RPGR (red), CEP290 (green), and acetylated TUBA1, a marker of the axoneme (blue). (D) Outer limiting membrane (*) localization of proteins CRB1 (red), CTNNB1 (β-catenin, green), a marker of cadherin junctions, and GS (glutamine synthetase, blue), a marker of Müller glia. (E) Photoreceptor localization of ABCA4 (red) and LRAT (blue), and retinal pigment epithelial (RPE) localization of RPE65 (green) and LRAT (blue). Nuclei labeled with DAPI (grey). RPE, retinal pigment epithelium; POS/IS, photoreceptor outer segments/inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm (A,D) and 10 μm (B,C,E).
Figure 1.
Immunohistochemical localization of many common inherited retinal dystrophy (IRD)-associated proteins. (A,B) Outer segment (POS) localization of phototransduction proteins CNGB1 (A, red), OPN1MW (B, red), GUCY2E (B, green), and RHO (A,B, blue), as well as phagocytosis protein MERTK (A, green). (C) Connecting cilium (>) localization of proteins RPGR (red), CEP290 (green), and acetylated TUBA1, a marker of the axoneme (blue). (D) Outer limiting membrane (*) localization of proteins CRB1 (red), CTNNB1 (β-catenin, green), a marker of cadherin junctions, and GS (glutamine synthetase, blue), a marker of Müller glia. (E) Photoreceptor localization of ABCA4 (red) and LRAT (blue), and retinal pigment epithelial (RPE) localization of RPE65 (green) and LRAT (blue). Nuclei labeled with DAPI (grey). RPE, retinal pigment epithelium; POS/IS, photoreceptor outer segments/inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 20 μm (A,D) and 10 μm (B,C,E).

Figure 2.
Diagram of phototransduction activation pathway proteins. Upon activation of RHO by absorption of a photon of light by its chromophore 11-cis retinal, RHO shifts its helical structure to allow for activation of the G-protein transducin (GT). GT, composed of α, β, and γ subunits, exchanges a GTP for a GDP in its activation site, allowing for separation of the α subunit from the βγ subunit complex, which then goes to activate the PDE6 enzyme by releasing the inhibition of its γ subunit. PDE6 then hydrolyzes cGMP to GMP, causing closure of the CNG channels, composed of three α and one β subunit, and subsequent hyperpolarization of the photoreceptor. This slows glutamate release at the synapse and propagates the electrical signal to the second and third-order neurons of the retina, the bipolar cells, and ganglion cells, respectively.
Figure 2.
Diagram of phototransduction activation pathway proteins. Upon activation of RHO by absorption of a photon of light by its chromophore 11-cis retinal, RHO shifts its helical structure to allow for activation of the G-protein transducin (GT). GT, composed of α, β, and γ subunits, exchanges a GTP for a GDP in its activation site, allowing for separation of the α subunit from the βγ subunit complex, which then goes to activate the PDE6 enzyme by releasing the inhibition of its γ subunit. PDE6 then hydrolyzes cGMP to GMP, causing closure of the CNG channels, composed of three α and one β subunit, and subsequent hyperpolarization of the photoreceptor. This slows glutamate release at the synapse and propagates the electrical signal to the second and third-order neurons of the retina, the bipolar cells, and ganglion cells, respectively.

Figure 3.
Diagram of phototransduction inactivation pathway proteins. After RHO activation and the subsequent cascade, the resulting decrease in calcium due to CNG channel closure releases the inhibition of recoverin, allowing for it to interact with and activate GRK1. GRK1 then phosphorylates RHO on its carboxyl terminus up to 6 times on serine and threonine residues, inducing RHO capping by arrestin. The isomerized all-trans retinal chromophore (ATR) is then hydrolyzed from the opsin protein and sent to the RPE for cis isomerization. GT has an intrinsic GTPase activity that is further enhanced by the trio of RGS9, Gβ5, and R9AP causing hydrolysis of the γ phosphate of GTP forming GDP and inactivating GTα, allowing for the β and γ subunits to reassociate with the α subunit. Along with recoverin, decreased calcium levels from CNG channel closure also release inhibition on GCAPs, allowing for them to activate GCs to produce cGMP from GTP for reopening of the CNG channels and depolarization of the cell to the dark state.
Figure 3.
Diagram of phototransduction inactivation pathway proteins. After RHO activation and the subsequent cascade, the resulting decrease in calcium due to CNG channel closure releases the inhibition of recoverin, allowing for it to interact with and activate GRK1. GRK1 then phosphorylates RHO on its carboxyl terminus up to 6 times on serine and threonine residues, inducing RHO capping by arrestin. The isomerized all-trans retinal chromophore (ATR) is then hydrolyzed from the opsin protein and sent to the RPE for cis isomerization. GT has an intrinsic GTPase activity that is further enhanced by the trio of RGS9, Gβ5, and R9AP causing hydrolysis of the γ phosphate of GTP forming GDP and inactivating GTα, allowing for the β and γ subunits to reassociate with the α subunit. Along with recoverin, decreased calcium levels from CNG channel closure also release inhibition on GCAPs, allowing for them to activate GCs to produce cGMP from GTP for reopening of the CNG channels and depolarization of the cell to the dark state.

Figure 4.
Diagram of ciliary and disc formation proteins. The connecting cilium (CC) forms from the basal body (BB), where CEP290 assists in tubulin nucleation. Trafficking protein complexes MYO7A, IFT-A, and IFT-B work to traffic important ciliary proteins and cargo anterograde and retrograde. Usher syndrome proteins USH2A, ADGRV1, and WHRN synergistically work to tether the periciliary ridge to the base of the outer segment. Ciliary proteins RPGR and RPGRIP1 work in concert to stabilize the CC, while RP1 and RP1L1 work to initiate the process of disc formation in the outer segment. Actin bundling protein FSCN2 works at the base of the outer segment to form F-actin bundles for disc formation to ensue by PRPH2 and ROM1, two of the major disc formation proteins tasked with flattening the circular vesicles into discs, a less energetically favorable configuration.
Figure 4.
Diagram of ciliary and disc formation proteins. The connecting cilium (CC) forms from the basal body (BB), where CEP290 assists in tubulin nucleation. Trafficking protein complexes MYO7A, IFT-A, and IFT-B work to traffic important ciliary proteins and cargo anterograde and retrograde. Usher syndrome proteins USH2A, ADGRV1, and WHRN synergistically work to tether the periciliary ridge to the base of the outer segment. Ciliary proteins RPGR and RPGRIP1 work in concert to stabilize the CC, while RP1 and RP1L1 work to initiate the process of disc formation in the outer segment. Actin bundling protein FSCN2 works at the base of the outer segment to form F-actin bundles for disc formation to ensue by PRPH2 and ROM1, two of the major disc formation proteins tasked with flattening the circular vesicles into discs, a less energetically favorable configuration.

Figure 5.
Venn diagram representation of the overlap of genes between different forms of inherited retinal dystrophies (IRDs). While hundreds of genes may cause a form of IRD, including non-syndromic rod-cone dystrophies (RCDs, magenta), non-syndromic cone-rod dystrophies (CRDs, blue), or syndromic RCDs/CRDs (yellow), several genes can result in more than one form when mutated (non-syndromic RCDs and syndromic RCDs/CRDs, orange; non-syndromic CRDs and syndromic RCDs/CRDs, purple; non-syndromic RCDs, non-syndromic CRDs and syndromic RCDs/CRDs, light brown), all dependent on the mutation and its effects. Note the overlap between the different groups consists of primarily ciliopathy genes among non-syndromic RCDs/CRDs and syndromic RCDs/CRDs and retinal function and development genes overlapping between the non-syndromic RCDs and non-syndromic CRDs.
Figure 5.
Venn diagram representation of the overlap of genes between different forms of inherited retinal dystrophies (IRDs). While hundreds of genes may cause a form of IRD, including non-syndromic rod-cone dystrophies (RCDs, magenta), non-syndromic cone-rod dystrophies (CRDs, blue), or syndromic RCDs/CRDs (yellow), several genes can result in more than one form when mutated (non-syndromic RCDs and syndromic RCDs/CRDs, orange; non-syndromic CRDs and syndromic RCDs/CRDs, purple; non-syndromic RCDs, non-syndromic CRDs and syndromic RCDs/CRDs, light brown), all dependent on the mutation and its effects. Note the overlap between the different groups consists of primarily ciliopathy genes among non-syndromic RCDs/CRDs and syndromic RCDs/CRDs and retinal function and development genes overlapping between the non-syndromic RCDs and non-syndromic CRDs.

Figure 6.
Diagram of the visual cycle proteins. The primary role of the visual cycle is the replenishment of 11-cis retinal (11-cis RAL) in photoreceptor cells via the re-isomerization of all-trans retinal (all-trans RAL). Once the all-trans retinal is cleaved from rhodopsin during phototransduction, it reacts with phosphatidylethanolamine (PE) to form N-retinylidene-phosphatidylethanolamine (N-Ret-PE), which binds the flippase ABCA4, resulting in a confirmational change that releases all-trans retinal into the cytoplasmic leaflet of the disc membrane. All-trans retinal is then reduced by all-trans retinol dehydrogenase (atRET-DH), forming all-trans retinol (all-trans ROL). All-trans retinol is trafficked by the retinal-binding protein IRBP from the photoreceptor outer segment to the RPE. Once in the RPE, the acyl transferase LRAT reacts an acyl group with all-trans retinol to form an all-trans retinyl ester. All-trans retinyl esters are a nontoxic storage form of retinal, which may be trafficked for storage by another retinal-binding protein RBP4. The rate-limiting step of the visual cycle is the isomerization of the all-trans retinyl ester to form 11 cis-retinol (11-cis ROL), in which 11-cis retinol dehydrogenase (11-cRDH) then oxidizes 11-cis retinol to form 11-cis retinal. 11-cis retinal is then chaperoned by the retinal-binding protein CRALBP to protect it from further isomerization or esterification. Finally, 11-cis retinal is trafficked from the RPE to the outer segment of photoreceptor discs, where it may bind opsin to regenerate functional rhodopsin.
Figure 6.
Diagram of the visual cycle proteins. The primary role of the visual cycle is the replenishment of 11-cis retinal (11-cis RAL) in photoreceptor cells via the re-isomerization of all-trans retinal (all-trans RAL). Once the all-trans retinal is cleaved from rhodopsin during phototransduction, it reacts with phosphatidylethanolamine (PE) to form N-retinylidene-phosphatidylethanolamine (N-Ret-PE), which binds the flippase ABCA4, resulting in a confirmational change that releases all-trans retinal into the cytoplasmic leaflet of the disc membrane. All-trans retinal is then reduced by all-trans retinol dehydrogenase (atRET-DH), forming all-trans retinol (all-trans ROL). All-trans retinol is trafficked by the retinal-binding protein IRBP from the photoreceptor outer segment to the RPE. Once in the RPE, the acyl transferase LRAT reacts an acyl group with all-trans retinol to form an all-trans retinyl ester. All-trans retinyl esters are a nontoxic storage form of retinal, which may be trafficked for storage by another retinal-binding protein RBP4. The rate-limiting step of the visual cycle is the isomerization of the all-trans retinyl ester to form 11 cis-retinol (11-cis ROL), in which 11-cis retinol dehydrogenase (11-cRDH) then oxidizes 11-cis retinol to form 11-cis retinal. 11-cis retinal is then chaperoned by the retinal-binding protein CRALBP to protect it from further isomerization or esterification. Finally, 11-cis retinal is trafficked from the RPE to the outer segment of photoreceptor discs, where it may bind opsin to regenerate functional rhodopsin.


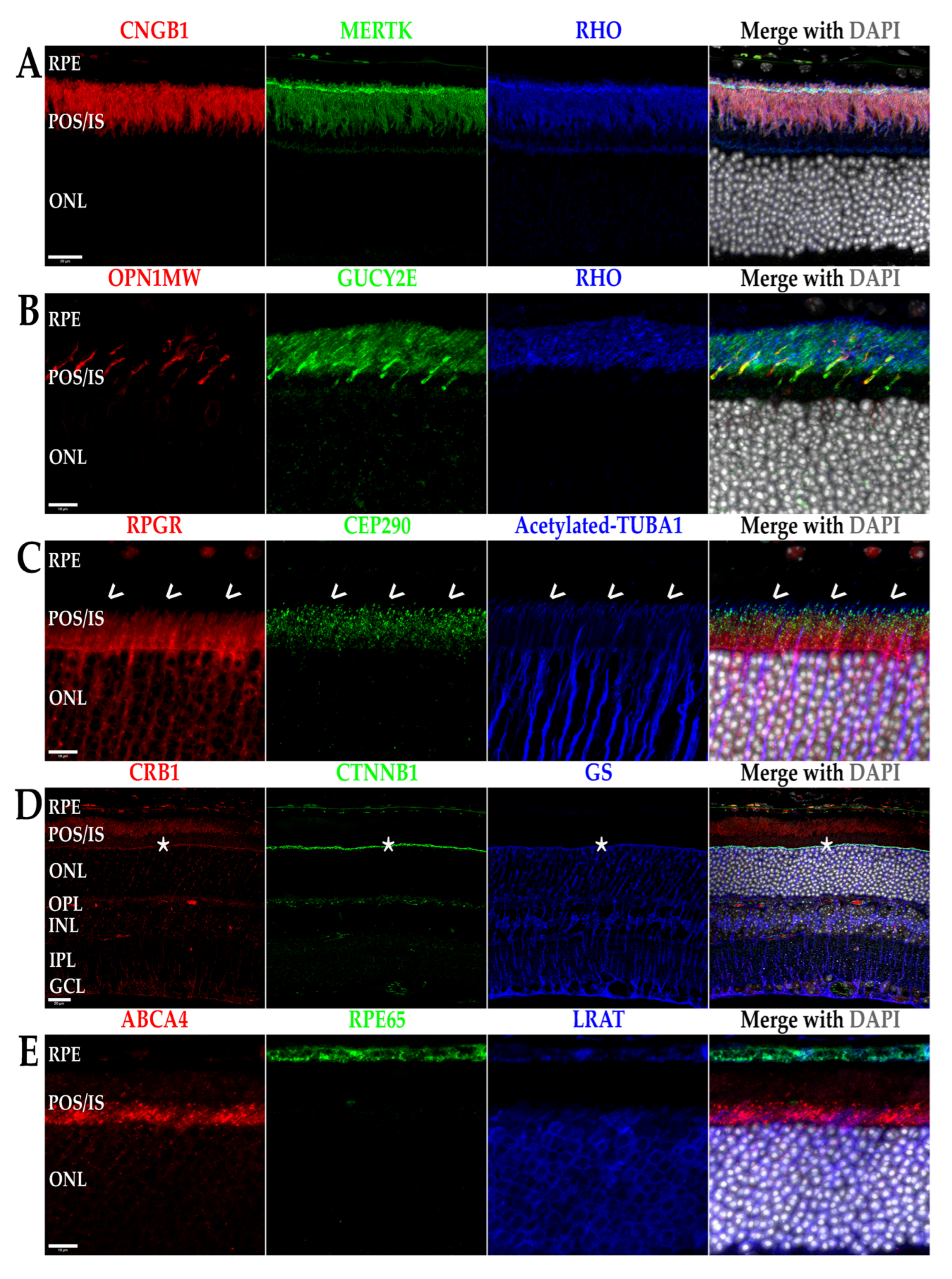{kind=link}
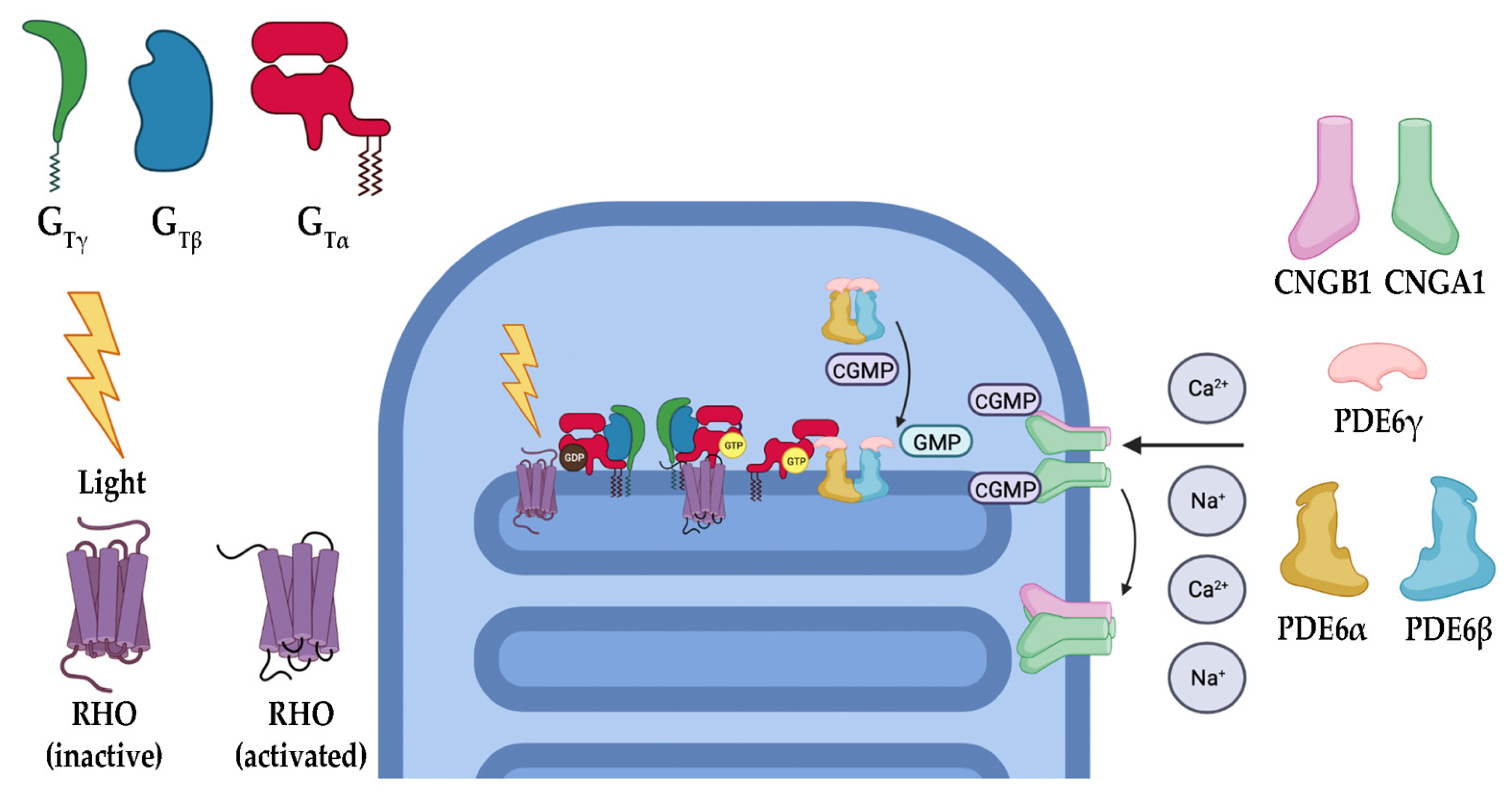{kind=link}
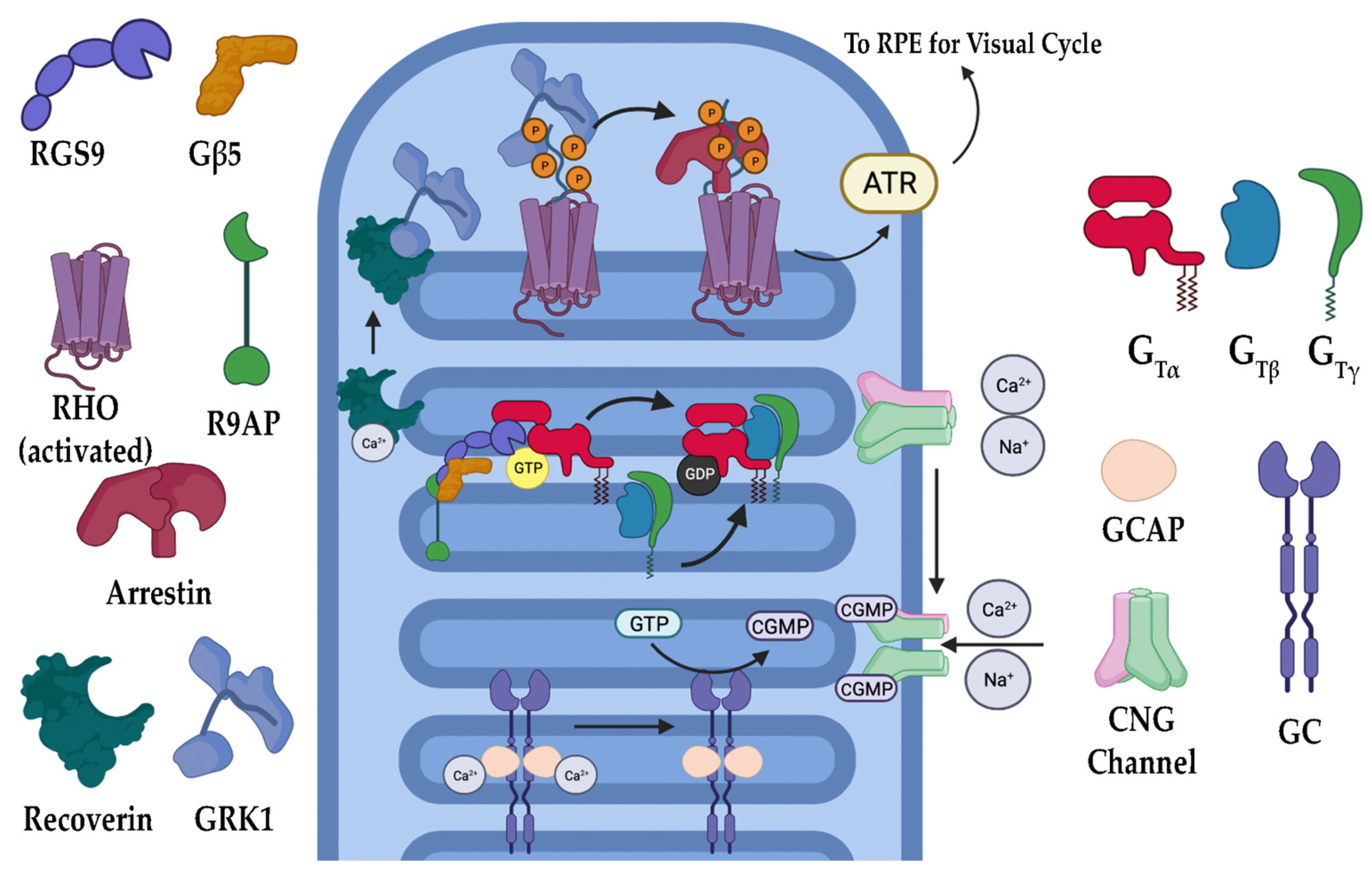{kind=link}
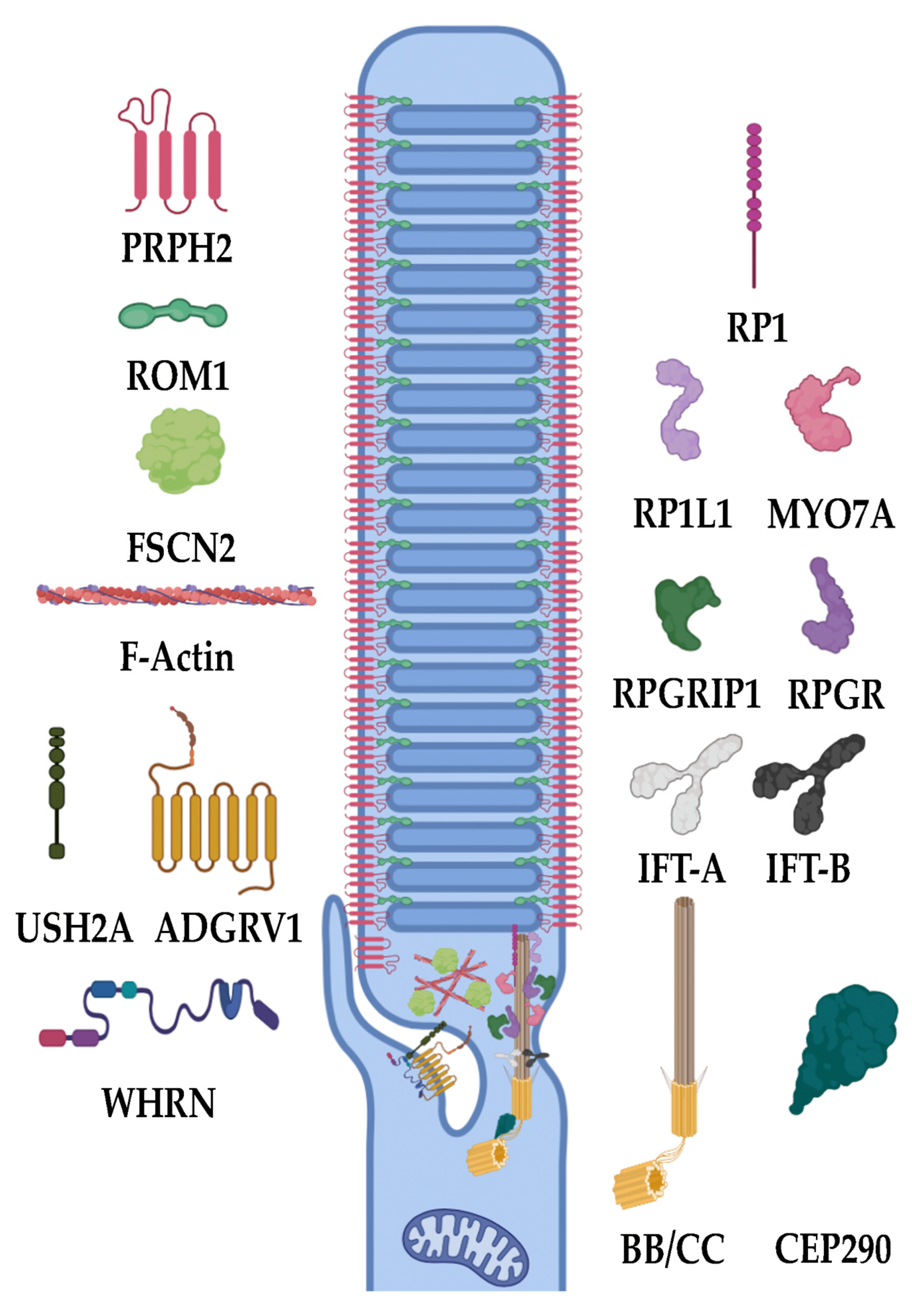{kind=link}
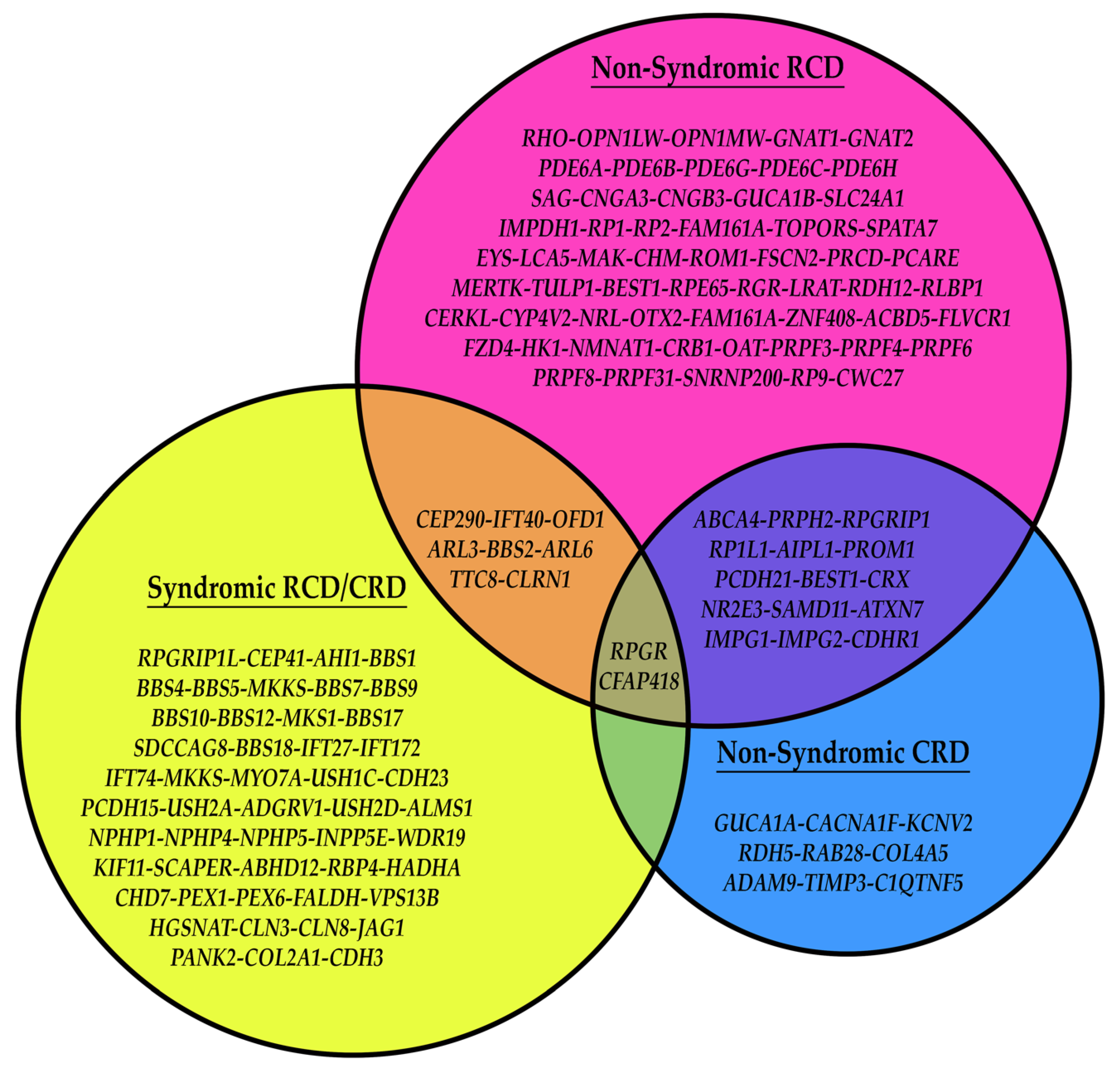{kind=link}
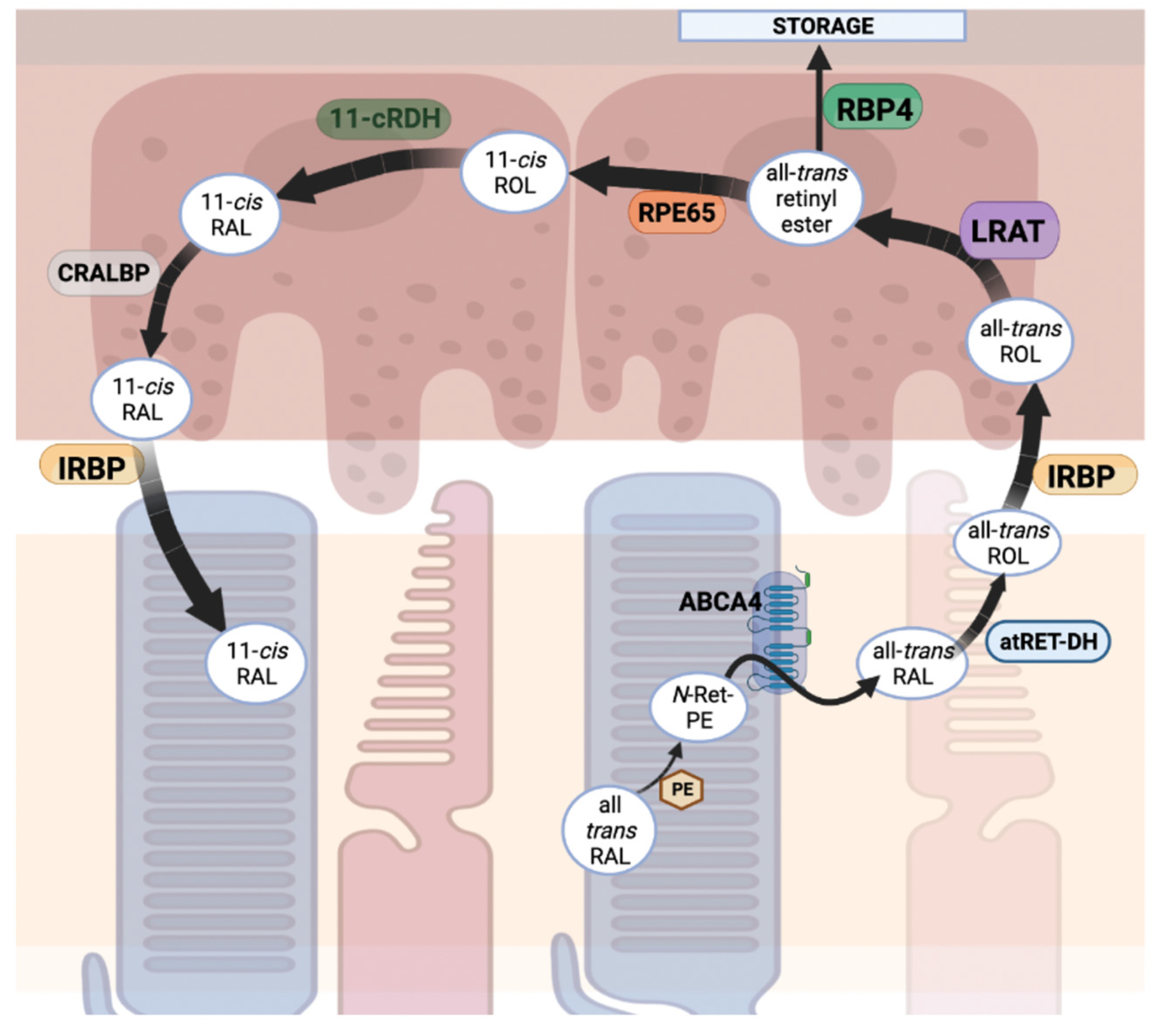{kind=link}
Table 1.
Antibodies used for Western blots and fIHC labeling of retinal sections.
| Antibody Target | Host Species & IgG Isoform | Dilution | Source |
|---|---|---|---|
| ABCA4 | Mouse IgG1 | 1:200 | EMDMillipore |
| CEP290 | Rabbit IgG | 1:200 | EMDMillipore |
| CNGB1 | Mouse IgG2a | 1:250 | EMDMillipore |
| CRB1 | Rabbit IgG | 1:100 | Boster Bio. Tech. |
| CTNNB1 | Mouse IgG2b | 1:200 | Origene |
| GS | Mouse IgG2a | 1:500 | BD Trans. Lab. |
| GUCY2E | Rabbit IgG | 1:100 | FabGennix |
| LRAT | Rabbit IgG | 1:100 | ThermoFisher |
| MERTK | Rat IgG2a | 1:100 | ThermoFisher |
| OPN1MW | Rabbit IgG | 1:100 | Novus Bio. |
| RHO | Mouse IgG1 | 1:500 | Santa Cruz Biotech. |
| RPE65 | Mouse IgG1 | 1:200 | ThermoFisher |
| RPGR | Rabbit IgG | 1:100 | abcam |
| TUBA1, Acetyl | Mouse IgG2b | 1:250 | EMDMillipore |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Manley, A.; Meshkat, B.I.; Jablonski, M.M.; Hollingsworth, T.J. Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies. Biomolecules 2023, 13, 271. https://doi.org/10.3390/biom13020271
AMA Style
Manley A, Meshkat BI, Jablonski MM, Hollingsworth TJ. Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies. Biomolecules. 2023; 13(2):271. https://doi.org/10.3390/biom13020271
Chicago/Turabian StyleManley, Andrew, Bahar I. Meshkat, Monica M. Jablonski, and T.J. Hollingsworth. 2023. "Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies" Biomolecules 13, no. 2: 271. https://doi.org/10.3390/biom13020271
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.