
Notch Missense Mutations in Drosophila Reveal Functions of Specific EGF-like Repeats in Notch Folding, Trafficking, and Signaling

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drosophila Stocks and Crosses
2.2. Immunostaining
3. Results
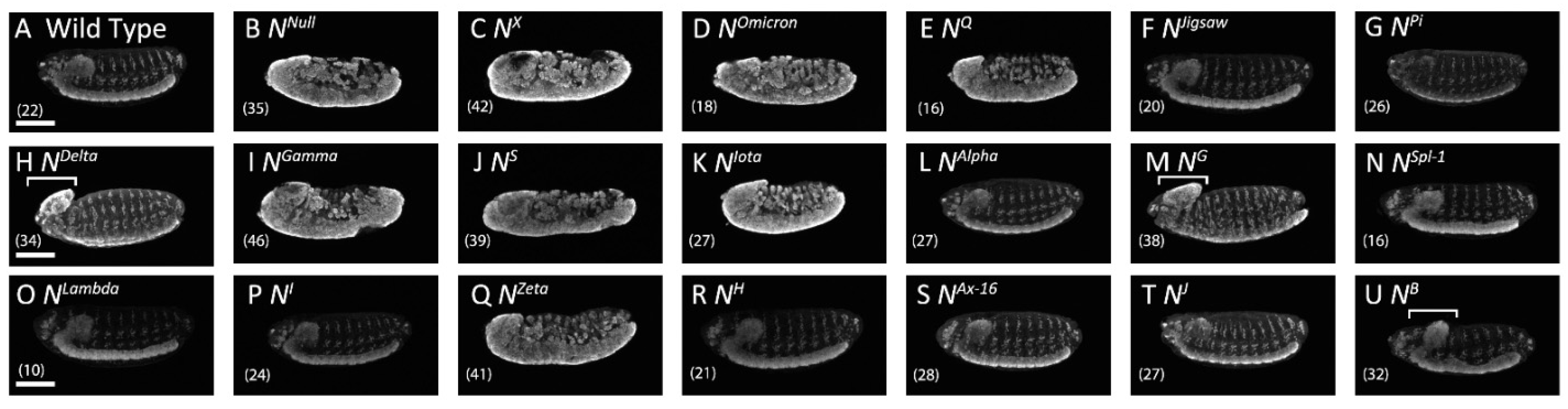
3.1. Notch Missense Mutations That Affected the Development of the Embryonic Nervous System
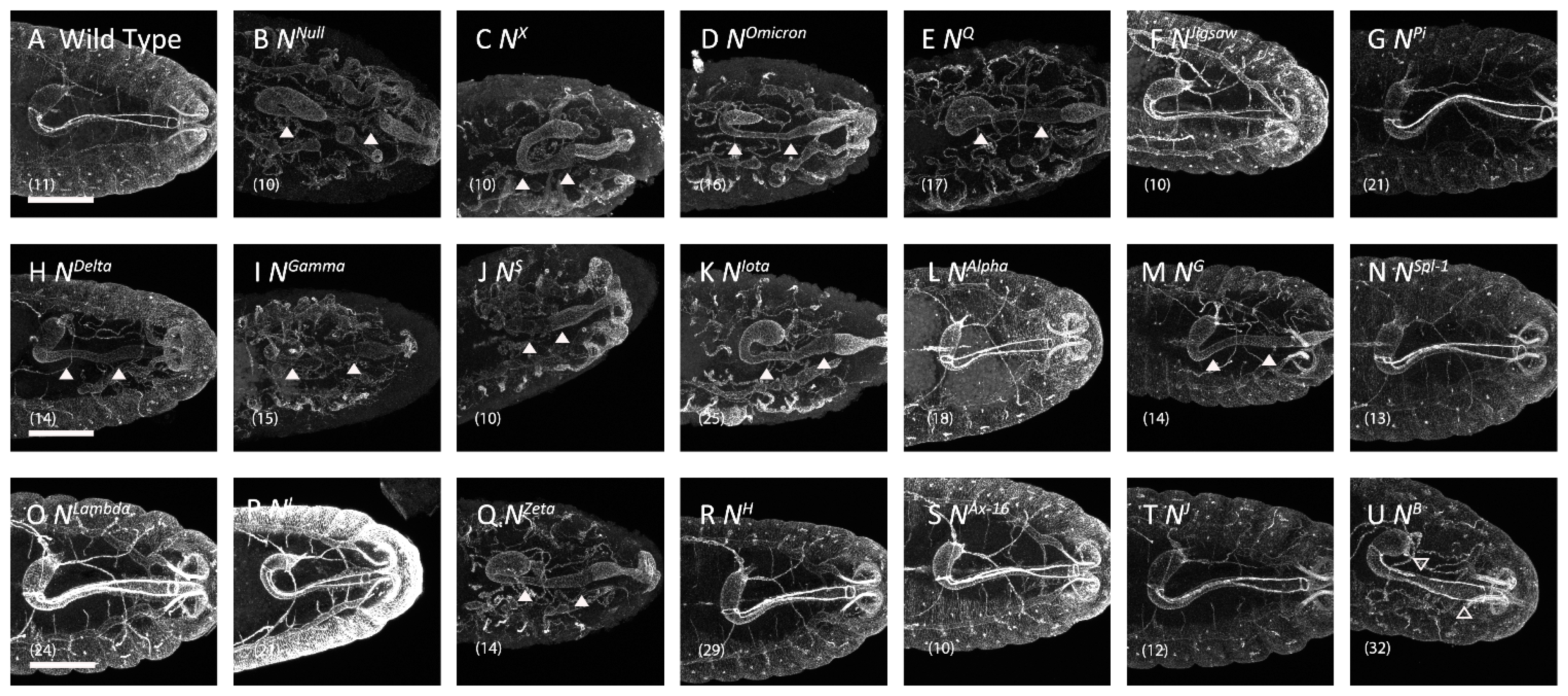
3.2. Missense Notch Mutations That Affected Boundary Cell Formation in the Hindgut
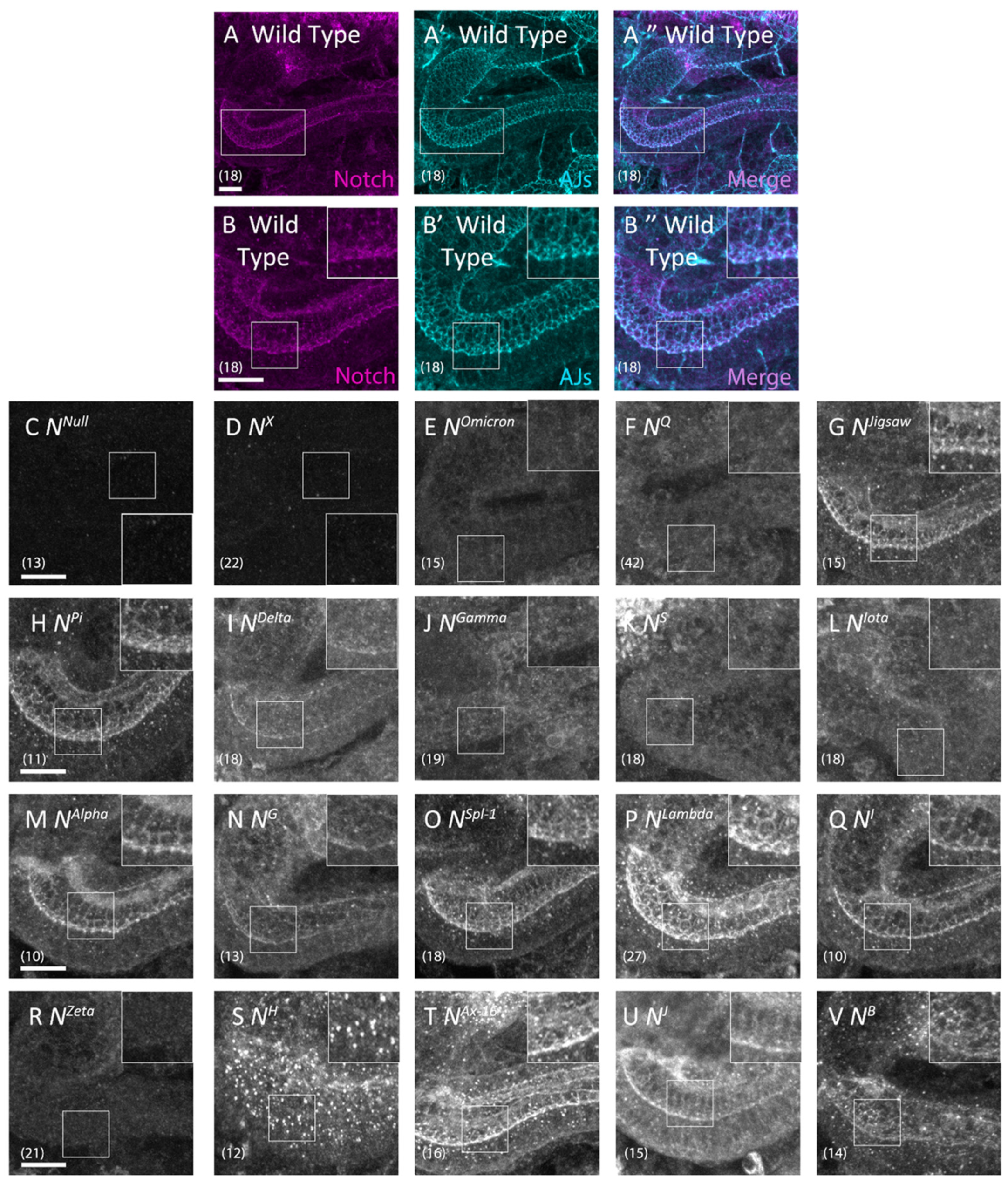
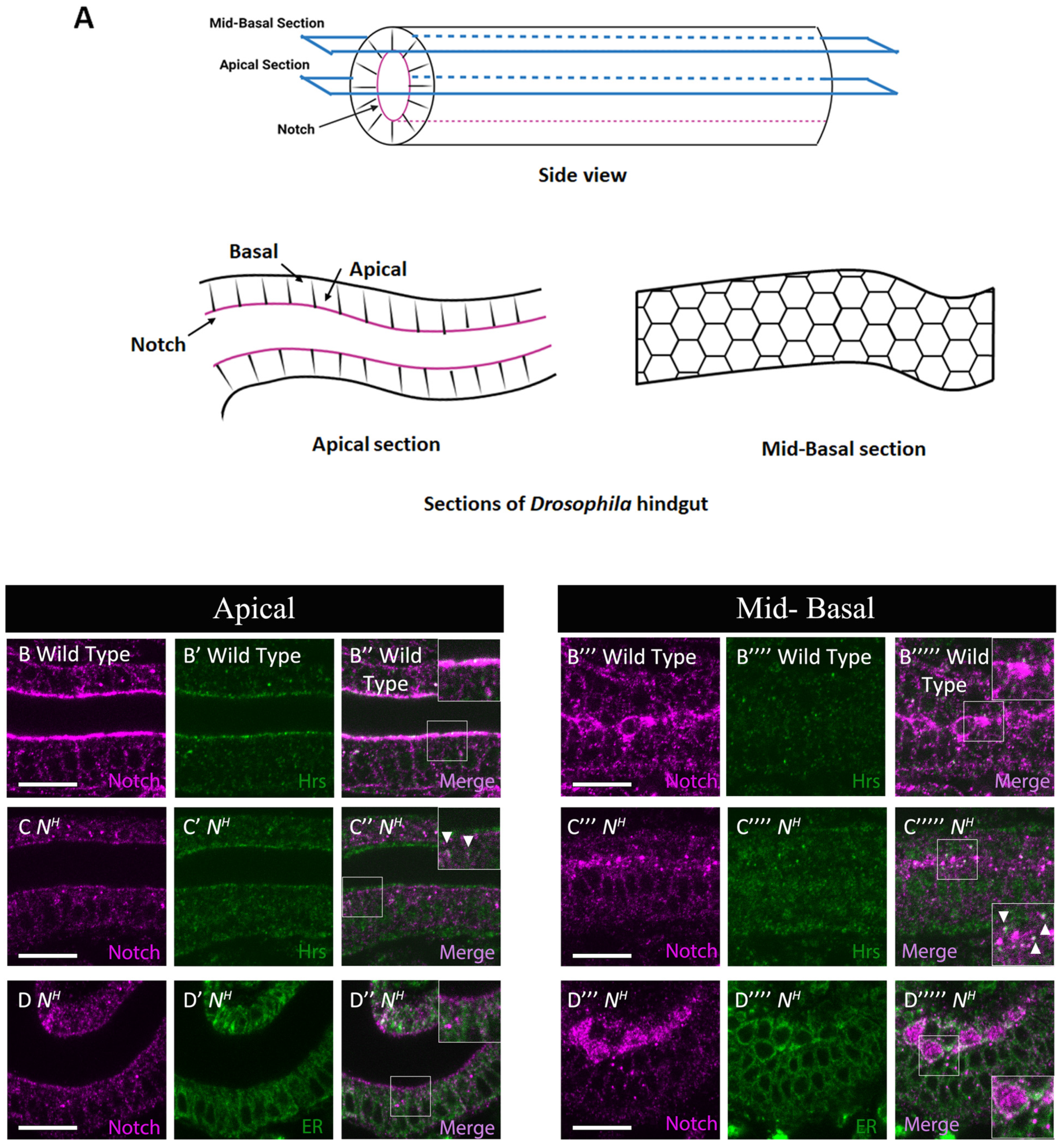
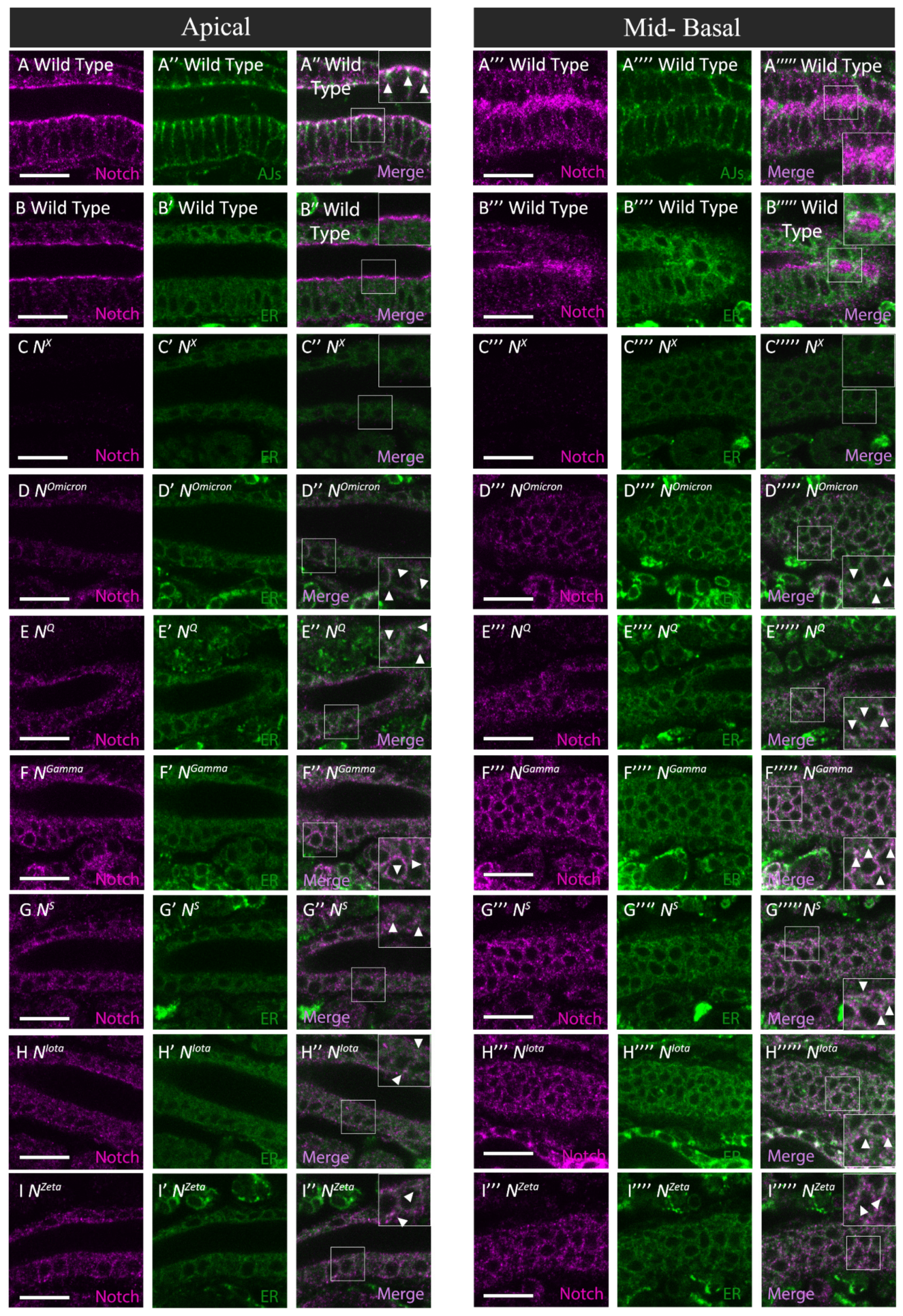
3.3. Notch Missense Mutations That Disrupted Intracellular Notch Trafficking
3.4. Defects in Notch Trafficking and Loss of Notch Activity Were Not Always Coupled
3.5. Notch Missense Mutations That Coupled Trafficking Defects with Loss of Notch Activity
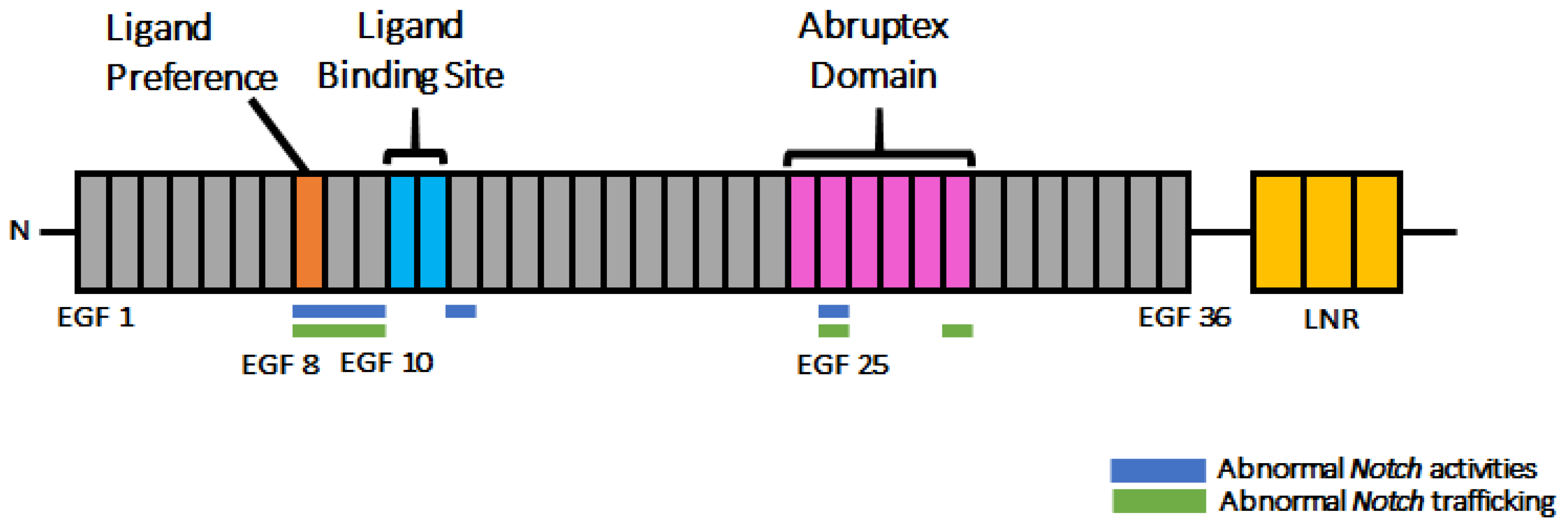
3.6. Disrupting Conserved Disulfide Bonds in Different EGF-like Repeats Induced Distinct Defects in Notch Activity and Trafficking
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazave, E.; Lapébie, P.; Richards, G.S.; Brunet, F.; Ereskovsky, A.; Degnan, B.M.; Borchiellini, C.; Vervoort, M.; Renard, E. Origin and evolution of the Notch signalling pathway: An overview from eukaryotic genomes. BMC Evol. Biol. 2009, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, M. An overview of the Notch signalling pathway. Semin. Cell Dev. Biol. 2003, 14, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Schweisguth, F. Regulation of Notch signaling activity. Curr. Biol. 2004, 14, R129–R138. [Google Scholar] [CrossRef]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2015, 52, 797–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chillakuri, C.R.; Sheppard, D.; Lea, S.M.; Handford, P.A. Notch receptor–ligand binding and activation: Insights from molecular studies. Semin. Cell Dev. Biol. 2012, 23, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Kidd, S.; Deutsch, W.A.; Young, M.W. Mutations altering the structure of epidermal growth factor-like coding sequences at the Drosophila Notch locus. Cell 1987, 51, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Haltom, A.R.; Jafar-Nejad, H. The multiple roles of epidermal growth factor repeat O-glycans in animal development. Glycobiology 2015, 25, 1027–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, M.A.; Rigoutsos, I.; Chu, C.K.; Feng, L.L.; Sparrow, D.B.; Dunwoodie, S.L. Evolution of distinct EGF domains with specific functions. Protein Sci. 2005, 14, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Rand, M.D.; Lindblom, A.; Carlson, J.; Villoutreix, B.O.; Stenflo, J. Calcium binding to tandem repeats of EGF-like modules. Expression and characterization of the EGF-like modules of human Notch-1 implicated in receptor-ligand interactions. Protein Sci. 2008, 6, 2059–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombling, B.J.; Wang, C.K.; Craik, D.J. EGF-like and other disulfide-rich microdomains as therapeutic scaffolds. Angew. Chem. Int. Ed. 2020, 59, 11218–11232. [Google Scholar] [CrossRef]
- Luca, V.C.; Jude, K.M.; Pierce, N.W.; Nachury M v Fischer, S.; Garcia, K.C. Structural basis for Notch1 engagement of Delta-like 4. Science 2015, 347, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luca, V.C.; Kim, B.C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger, R.S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [CrossRef] [Green Version]
- Stanley, P.; Okajima, T. Roles of Glycosylation in Notch Signaling. In Current Topics in Developmental Biology: Notch Signaling; Kopan, R., Ed.; Elsevier: Berkeley, CA, USA, 2010; pp. 131–164. [Google Scholar] [CrossRef]
- Pandey, A.; Harvey, B.M.; Lopez, M.F.; Ito, A.; Haltiwanger, R.S.; Jafar-Nejad, H. Glycosylation of specific Notch EGF repeats by O-Fut1 and fringe regulates notch signaling in drosophila. Cell Rep. 2019, 29, 2054–2066.e6. [Google Scholar] [CrossRef] [Green Version]
- Kakuda, S.; Haltiwanger, R.S. Deciphering the fringe-mediated Notch code: Identification of activating and inhibiting sites allowing discrimination between ligands. Dev. Cell 2017, 40, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Ayukawa, T.; Ishio, A.; Sasamura, T.; Yamakawa, T.; Matsuno, K. Dual roles of O-Glucose glycans redundant with monosaccharide O-Fucose on Notch in Notch trafficking. J. Biol. Chem. 2016, 291, 13743–13752. [Google Scholar] [CrossRef] [PubMed]
- De Celis, J.F.; Bray, S.J. The Abruptex domain of Notch regulates negative interactions between Notch, its ligands and Fringe. Development 2000, 127, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Baron, M. Combining genetic and biophysical approaches to probe the structure and function relationships of the Notch receptor. Mol. Membr. Biol. 2017, 34, 33–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S. Making sense out of missense mutations: Mechanistic dissection of Notch receptors through structure-function studies in Drosophila. Dev. Growth Differ. 2000, 62, 15–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Charng, W.-L.; Rana, N.A.; Kakuda, S.; Jaiswal, M.; Bayat, V.; Xiong, B.; Zhang, K.; Sandoval, H.; David, G.; et al. A mutation in EGF Repeat-8 of Notch discriminates between serrate/jagged and delta family ligands. Science 2012, 338, 1229–1232. [Google Scholar] [CrossRef] [Green Version]
- Johannes, B.; Preiss, A. Wing vein formation in Drosophila melanogaster: Hairless is involved in the cross-talk between Notch and EGF signaling pathways. Mech. Dev. 2002, 115, 3–14. [Google Scholar] [CrossRef] [PubMed]
- De Celis, J.F.; Garcia-Bellido, A.; Bray, S.J. Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development 1996, 122, 359–369. [Google Scholar] [CrossRef]
- Yamamoto, S.; Jaiswal, M.; Charng, W.-L.; Gambin, T.; Karaca, E.; Mirzaa, G.; Wiszniewski, W.; Sandoval, H.; Haelterman, N.A.; Xiong, B.; et al. A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 2014, 159, 200–214. [Google Scholar] [CrossRef] [Green Version]
- Haelterman, N.A.; Jiang, L.; Li, Y.; Bayat, V.; Sandoval, H.; Ugur, B.; Tan, K.L.; Zhang, K.; Bei, D.; Xiong, B.; et al. Large-scale identification of chemically induced mutations in Drosophila melanogaster. Genome Res. 2014, 24, 1707–1718. [Google Scholar] [CrossRef] [Green Version]
- Sjöqvist, M.; Andersson, E.R. Do as I say, Not(ch) as I do: Lateral control of cell fate. Dev. Biol. 2019, 447, 58–70. [Google Scholar] [CrossRef]
- Takashima, S.; Yoshimori, H.; Yamasaki, N.; Matsuno, K.; Murakami, R. Cell-fate choice and boundary formation by combined action of Notch and engrailed in the Drosophila hindgut. Dev. Genes Evol. 2002, 212, 534–541. [Google Scholar] [CrossRef]
- Fuß, B.; Hoch, M. Notch signaling controls cell fate specification along the dorsoventral axis of the drosophila gut. Curr. Biol. 2002, 12, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Sasamura, T.; Ishikawa, H.O.; Kanai, M.; Ueda, R.; Saigo, K.; Matsuno, K. Polarized exocytosis and transcytosis of Notch during its apical localization in Drosophila epithelial cells. Genes Cells 2007, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Okajima, T.; Xu, A.; Lei, L.; Irvine, K.D. Chaperone activity of protein O-Fucosyltransferase 1 promotes Notch receptor folding. Science 2005, 307, 1599–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Charng, W.-L.; Bellen, H.J. Endocytosis and Intracellular Trafficking of Notch and Its Ligands. In Current Topics in Developmental Biology: Notch Signaling; Kopan, R., Ed.; Elsevier: Berkeley, CA, USA, 2010; pp. 165–200. [Google Scholar] [CrossRef]
- Hounjet, J.; Vooijs, M. The role of intracellular trafficking of notch receptors in ligand-independent notch activation. Biomolecules 2021, 11, 1369. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.A.; Pierce, S.B. In vivo cross-linking of protein disulfide isomerase to immunoglobulins. Biochemistry 1987, 26, 4179–4182. [Google Scholar] [CrossRef] [PubMed]
- Rhyu, M.S.; Jan, L.Y.; Jan, Y.N. Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell 1994, 76, 477–491. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, E.M.; Rebay, I.; Tjian, R.; Rubin, G.M. The activities of two Ets-related transcription factors required for drosophila eye development are modulated by the Ras/MAPK pathway. Cell 1994, 78, 137–147. [Google Scholar] [CrossRef]
- Fehon, R.G.; Kooh, P.J.; Rebay, I.; Regan, C.L.; Xu, T.; Muskavitch MA, T.; Artavanis-Tsakonas, S. Molecular interactions between the protein products of the neurogenic loci Notch and Delta, two EGF-homologous genes in Drosophila. Cell 1990, 61, 523–534. [Google Scholar] [CrossRef]
- Tepass, U.; Knust, E. crumbs and stardust act in a genetic pathway that controls the organization of epithelia in drosophila melanogaster. Dev. Biol. 1993, 159, 311–326. [Google Scholar] [CrossRef]
- Oda, H.; Uemura, T.; Harada, Y.; Iwai, Y.; Takeichi, M. A drosophila homolog of cadherin associated with armadillo and essential for embryonic cell-cell adhesion. Dev. Biol. 1994, 165, 716–726. [Google Scholar] [CrossRef]
- Lloyd, T.E.; Atkinson, R.; Wu, M.N.; Zhou, Y.; Pennetta, G.; Bellen, H.J. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in drosophila. Cell 2002, 108, 261–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Nakamura, A. The endocytic pathway acts downstream of Oskar in Drosophila germ plasm assembly. Development 2008, 135, 1107–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, N.C.; Jahn, T.R.; Reid, E.; O’Kane, C.J. Reticulon-like-1, the Drosophila orthologue of the Hereditary Spastic Paraplegia gene reticulon 2, is required for organization of endoplasmic reticulum and of distal motor axons. Hum. Mol. Genet. 2012, 21, 3356–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Hara, Y.; Takagi, C.; Yamamoto, T.S.; Ueno, N. MID1 and MID2 are required for Xenopus neural tube closure through the regulation of microtubule organization. Development 2010, 137, 2329–2339. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, R.; Jimenez, F.; Dietrich, U.; Campos-Ortega, J.A. On the phenotype and development of mutants of early neurogenesis in Drosophila melanogaster. Wilhelm Roux’s Arch. Dev. Biol. 1983, 192, 62–74. [Google Scholar] [CrossRef]
- Berger, C.; Renner, S.; Lüer, K.; Technau, G.M. The commonly used marker ELAV is transiently expressed in neuroblasts and glial cells in the Drosophila embryonic CNS. Dev. Dyn. 2007, 236, 3562–3568. [Google Scholar] [CrossRef]
- Schweisguth, F. Asymmetric cell division in the Drosophila bristle lineage: From the polarization of sensory organ precursor cells to Notch-mediated binary fate decision. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 299–309. [Google Scholar] [CrossRef]
- Iwaki, D.D.; Lengyel, J.A. A Delta–Notch signaling border regulated by Engrailed/Invected repression specifies boundary cells in the Drosophila hindgut. Mech. Dev. 2002, 114, 71–84. [Google Scholar] [CrossRef]
- Kumichel, A.; Knust, E. Apical localisation of crumbs in the boundary cells of the drosophila hindgut is independent of its canonical interaction partner stardust. PLoS ONE 2014, 9, e94038. [Google Scholar] [CrossRef] [Green Version]
- Brennan, K.; Tateson, R.; Lewis, K.; Arias, A.M. A functional analysis of Notch mutations in Drosophila. Genetics 1997, 147, 177–188. [Google Scholar] [CrossRef]
- Pei, Z.; Baker, N.E. Competition between Delta and the Abruptex domain of Notch. BMC Dev. Biol. 2008, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Rebay, I.; Fleming, R.J.; Fehon, R.G.; Cherbas, L.; Cherbas, P.; Artavanis-Tsakonas, S. Specific EGF repeats of Notch mediate interactions with Delta and serrate: Implications for Notch as a multifunctional receptor. Cell 1991, 67, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Becam, I.; Fiuza, U.-M.; Arias, A.M.; Milán, M. A role of receptor Notch in ligand cis-inhibition in drosophila. Curr. Biol. 2010, 20, 554–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebel, C.; Lendahl, U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrawes, M.B.; Xu, X.; Liu, H.; Ficarro, S.B.; Marto, J.A.; Aster, J.C.; Blacklow, S.C. Intrinsic selectivity of Notch 1 for delta-like 4 over delta-like 1. J. Biol. Chem. 2013, 288, 25477–25489. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Name | EGF-like Repeat A | Mutation Position | Notch Activity | Notch Trafficking | Notch Localization B | ||
|---|---|---|---|---|---|---|---|---|
| Bristle Formation | Lateral Inhibition | Inductive Signaling | ||||||
| 1 | NX | EGF 8 | C343S (C2S) | Absent | Neurogenic | Depletion | Abnormal | Loss |
| 2 | NOmicron | EGF 8 | C343Y (C2Y) | Absent | Neurogenic | Depletion | Abnormal | ER |
| 3 | NQ | EGF 8 | D331N | Absent | Neurogenic | Depletion | Abnormal | ER |
| 4 | NJigsaw | EGF 8 | V361M | Normal | Normal | Normal | Normal | AJs |
| 5 | NPi | EGF 9 | D374G | Absent | Normal | Normal | Normal | AJs |
| 6 | NDelta | EGF 9 | D389N | Absent | Brain deformation | Depletion | Normal | AJs |
| 7 | NGamma | EGF 9 | C398Y (C5Y) | Absent | Neurogenic | Depletion | Abnormal | ER |
| 8 | NS | EGF 9 | C407S (C6S) | Absent | Neurogenic | Depletion | Abnormal | ER |
| 9 | NIota | EGF 10 | C413S (C1S) | Absent | Neurogenic | Depletion | Abnormal | ER |
| 10 | NAlpha | EGF 11 | E452K | Absent | Normal | Normal | Normal | AJs |
| 11 | NG | EGF 13 | C535S (C2S) | Absent | Brain deformation | Depletion | Normal | AJs |
| 12 | NSpl−1 | EGF 14 | I578T | Reduced | Normal | Normal | Normal | AJs |
| 13 | NLambda | EGF 16 | G668R | Absent | Normal | Normal | Normal | AJs |
| 14 | NI | EGF 16 | G671D | Absent | Normal | Normal | Normal | AJs |
| 15 | NZeta | EGF 25 | C993S (C2S) | Absent | Neurogenic | Depletion | Abnormal | ER |
| 16 | NH | EGF 29 | C1155S (C2S) | Absent | Normal | Normal | Abnormal | Early endosomes |
| 17 | NAx−16 | EGF 29 | G1174A | Reduced | Normal | Normal | Normal | AJs |
| 18 | NJ | EGF 34 | C1341Y(C1Y) | Absent | Normal | Normal | Normal | AJs |
| 19 | NB | TMD | I1751K | Normal | Brain deformation | Abnormal Gaps | Normal | AJs |
| Classes | Notch Activity in Neuron & Boundary Cell | Notch Trafficking | Notch Alleles |
|---|---|---|---|
| I | Normal | Normal | NJigsaw, NPi, NAlpha, NSpl−1, NLambda, NI, NAx−16, NJ |
| II | Normal | Abnormal | NH |
| III | Abnormal | Normal | NDelta, NG, NB |
| IV | Abnormal | Abnormal | NX, NOmicron, NQ, NS, NGamma, NIota, NZeta |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nurmahdi, H.; Hasegawa, M.; Mujizah, E.Y.; Sasamura, T.; Inaki, M.; Yamamoto, S.; Yamakawa, T.; Matsuno, K. Notch Missense Mutations in Drosophila Reveal Functions of Specific EGF-like Repeats in Notch Folding, Trafficking, and Signaling. Biomolecules 2022, 12, 1752. https://doi.org/10.3390/biom12121752
Nurmahdi H, Hasegawa M, Mujizah EY, Sasamura T, Inaki M, Yamamoto S, Yamakawa T, Matsuno K. Notch Missense Mutations in Drosophila Reveal Functions of Specific EGF-like Repeats in Notch Folding, Trafficking, and Signaling. Biomolecules. 2022; 12(12):1752. https://doi.org/10.3390/biom12121752
Chicago/Turabian StyleNurmahdi, Hilman, Mao Hasegawa, Elzava Yuslimatin Mujizah, Takeshi Sasamura, Mikiko Inaki, Shinya Yamamoto, Tomoko Yamakawa, and Kenji Matsuno. 2022. "Notch Missense Mutations in Drosophila Reveal Functions of Specific EGF-like Repeats in Notch Folding, Trafficking, and Signaling" Biomolecules 12, no. 12: 1752. https://doi.org/10.3390/biom12121752