
Downregulating Mitochondrial DNA Polymerase γ in the Muscle Stimulated Autophagy, Apoptosis, and Muscle Aging-Related Phenotypes in Drosophila Adults


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fly Stocks and Culture
2.2. Immunofluorescence
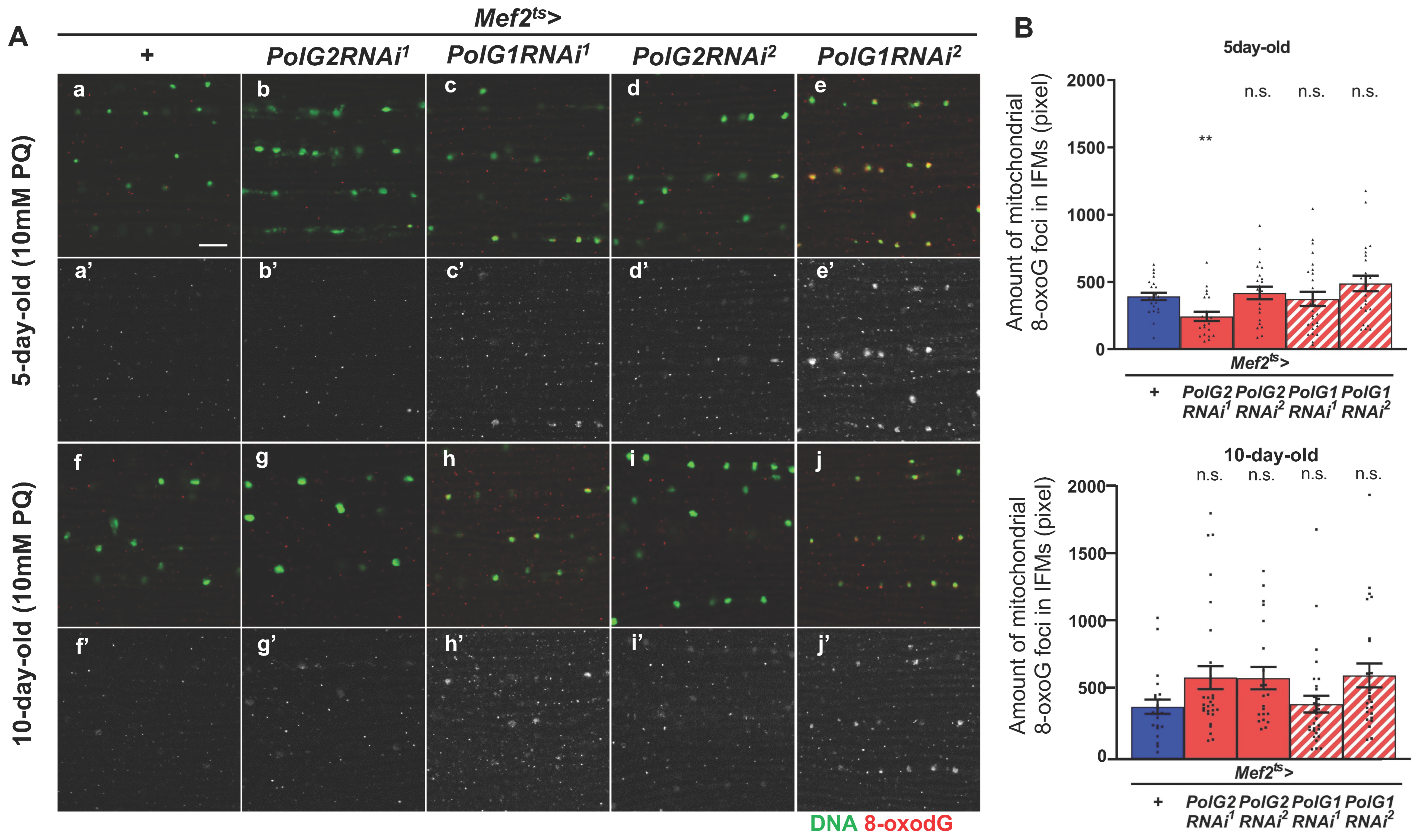
2.3. Quantification of 8-oxo-dG in Mitochondrial DNA
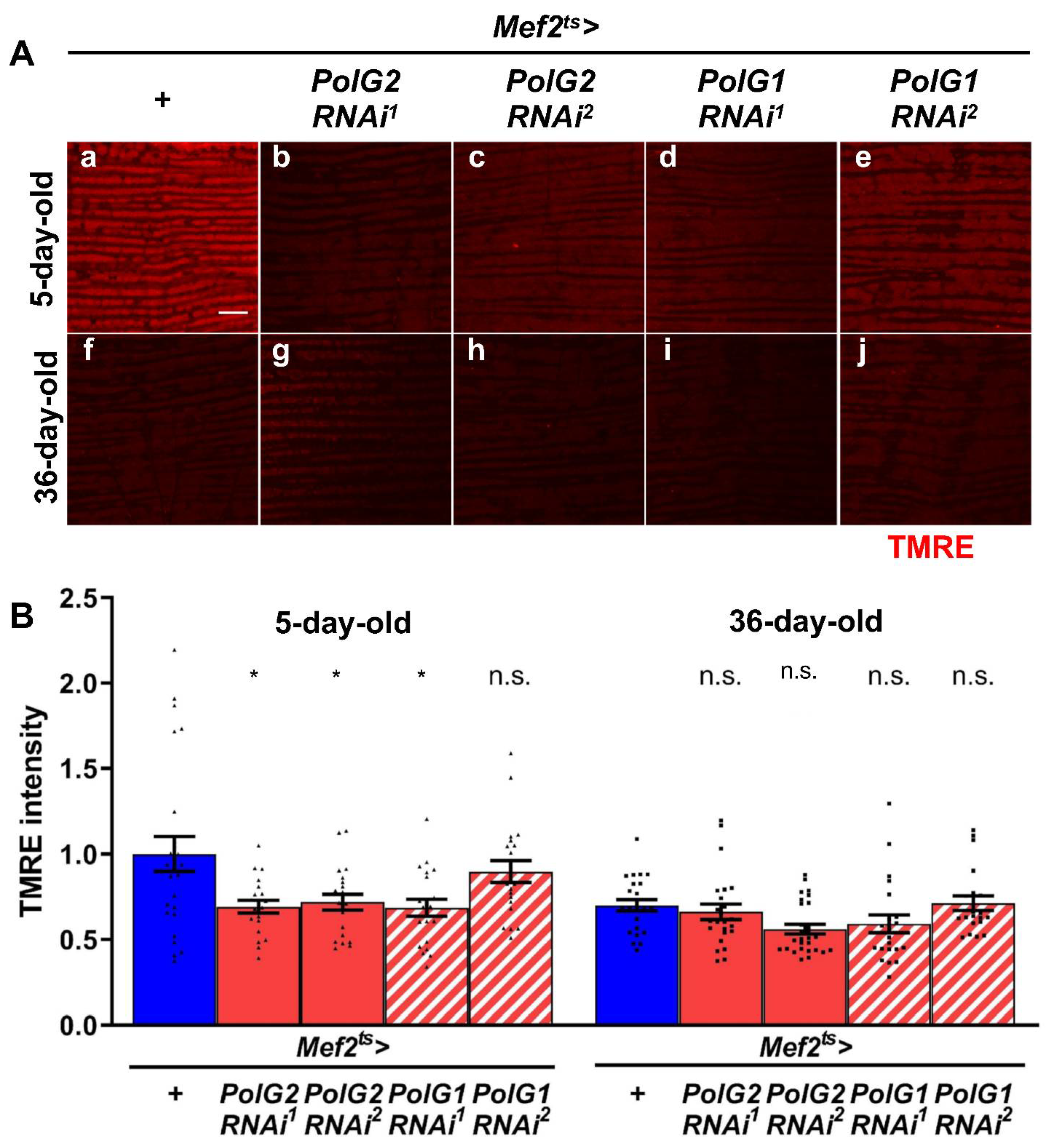
2.4. Tetramethylrhodamine Ethyl Ester (TMRE) Staining
2.5. Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
| RP49 | F: TTCCTGGTGCACAACGTG |
| R: TCTCCTTGCGCTTCTTGG | |
| PolG2 | F: CTTCTACAACATGCAGCGTGAG |
| R: TAGCTCGTGCGGATATCGATG | |
| PolG1 | F: ACAATGTCGCTGCACATGTG |
| R: CCTTCTTGGATTTGAGCATGGC | |
| COX III | F: TGACCATTAACAGGAGCTATCGG |
| R: CCTTCTCGTGATACATCTCGTCA |
2.6. Nucleic Acid Preparation for Quantitative PCR to Estimate mtDNA Copy Number
2.7. ATP Assay
2.8. Transmission Electron Microscope Observation of IFMs
2.9. Survival Assay
2.10. Climbing Assay
2.11. Locomotor Assay
2.12. Statistical Analysis
3. Results
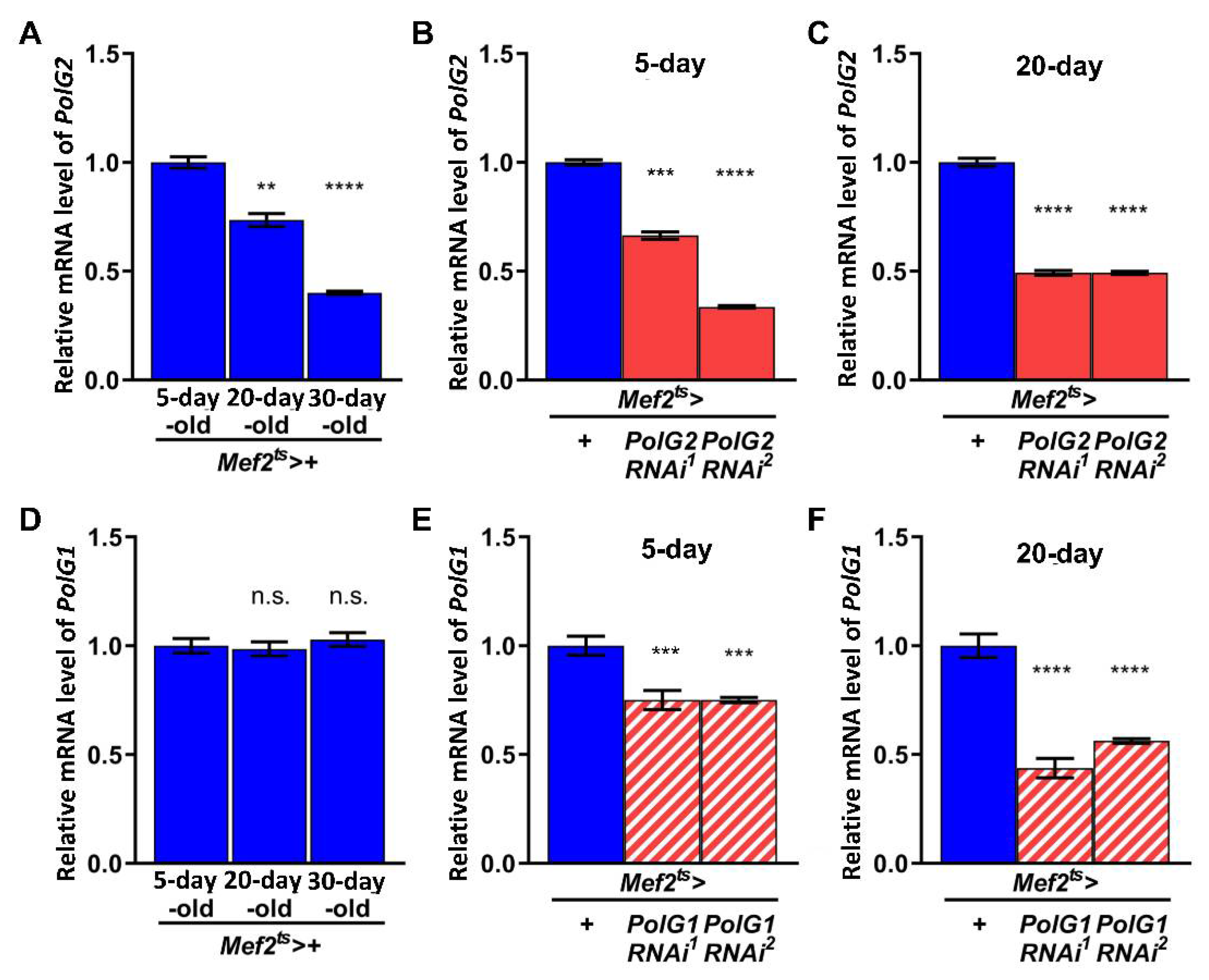
3.1. Adult Muscle-Specific Silencing of Polγ Subunit Genes Partially Inhibited Mitochondrial DNA Replication and Repair of Oxidatively Damaged DNA in Drosophila Adult Muscle
3.2. Reduced mRNA Levels of Polγ Genes Resulted in Partially Reduced Mitochondrial Activity in Adult Thoraxes Containing Flight Muscle as a Major Component
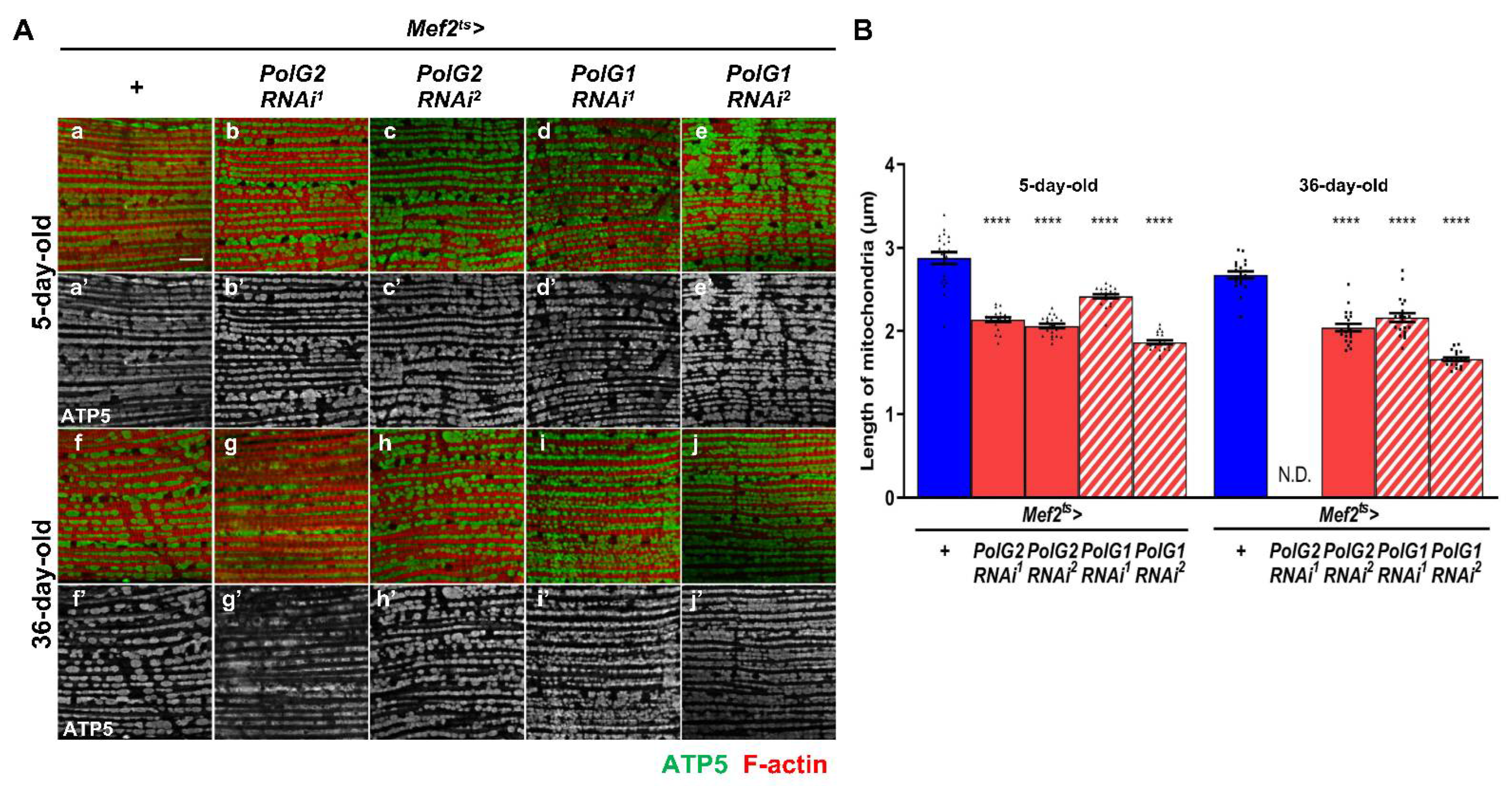
3.3. Enhancement of Mitochondrial Fission in the IFMs Harboring PolγRNAi
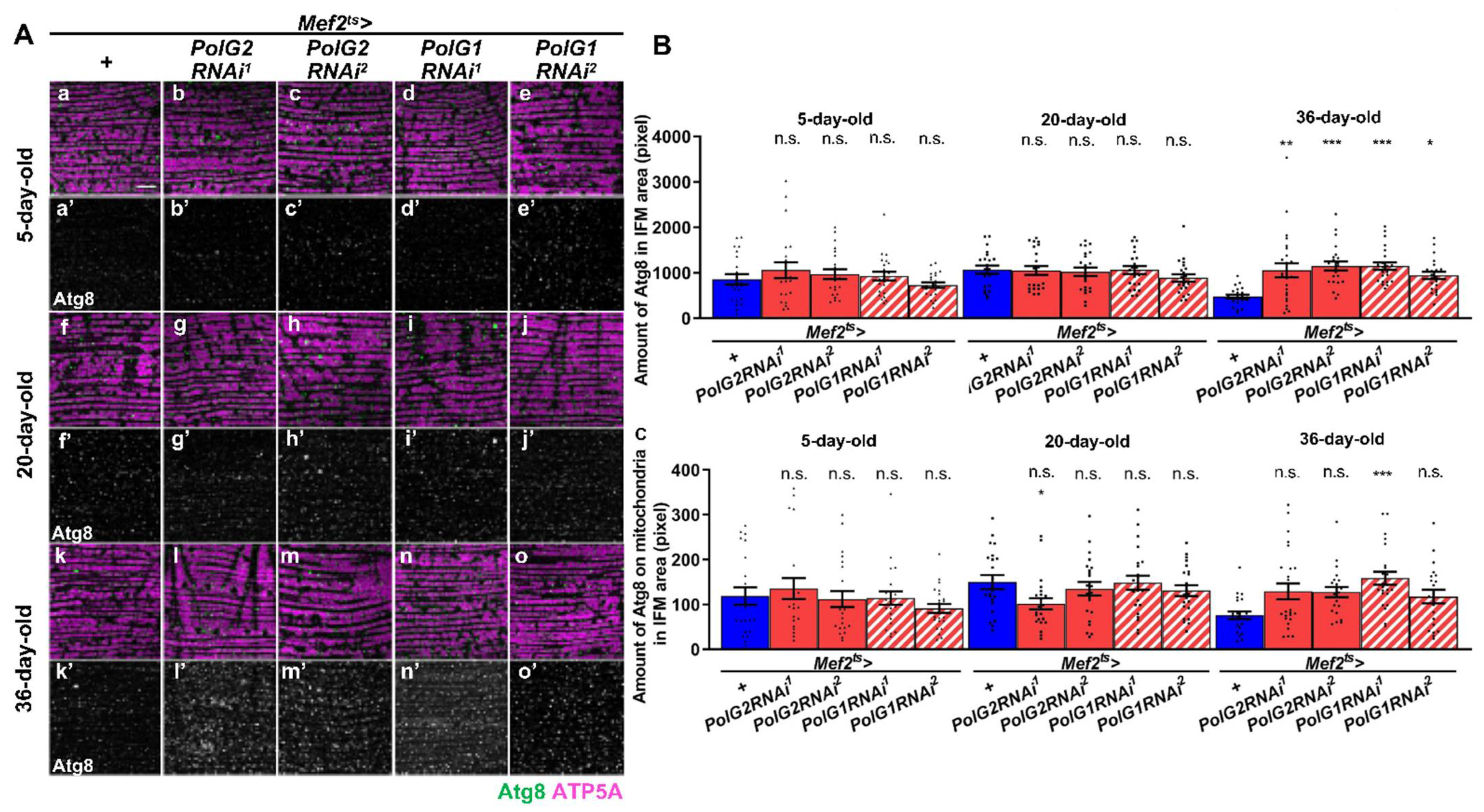
3.4. Stimulation of Autophagy in the IFMs Harboring PolγRNAi as the Flies Aged
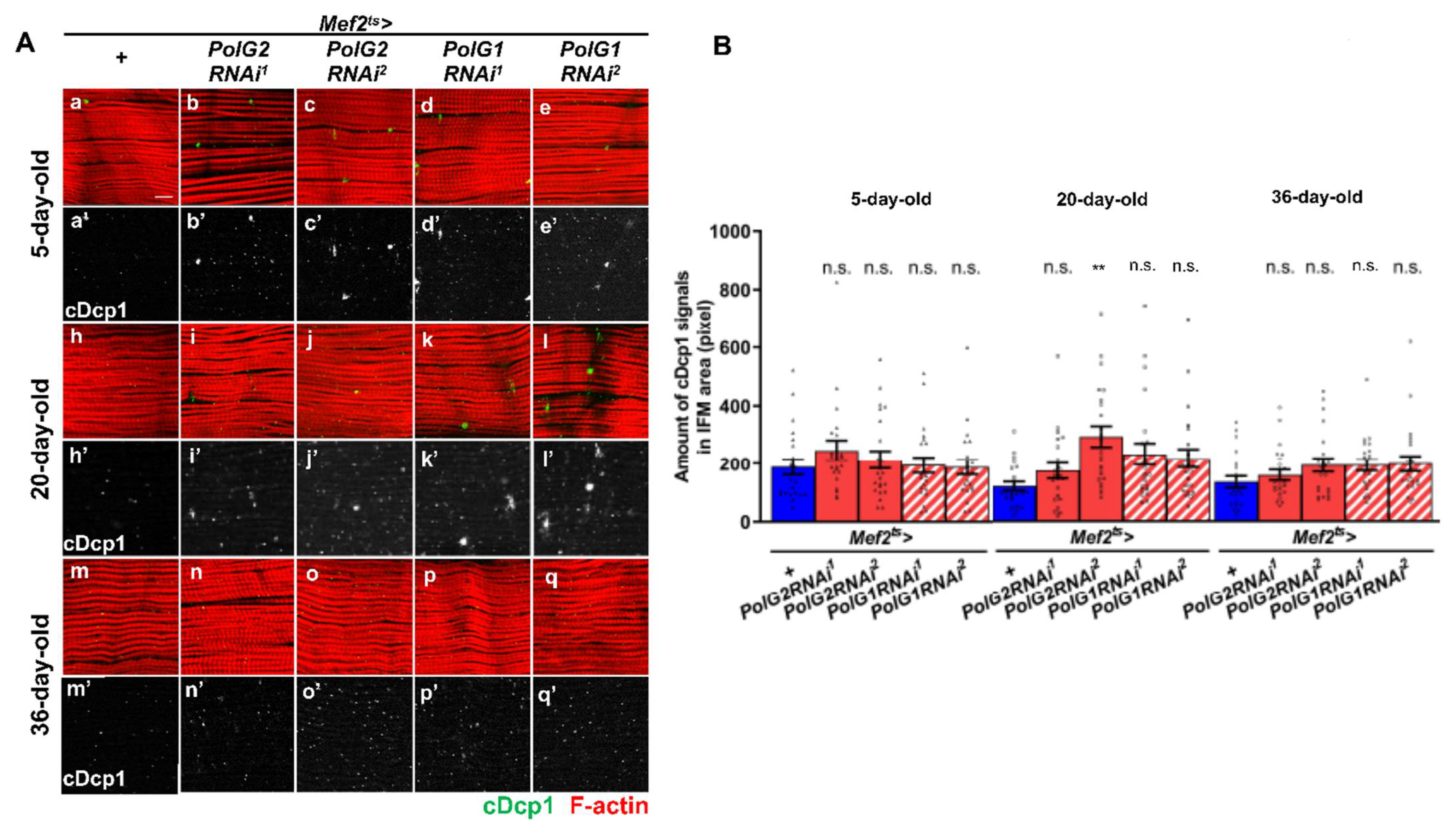
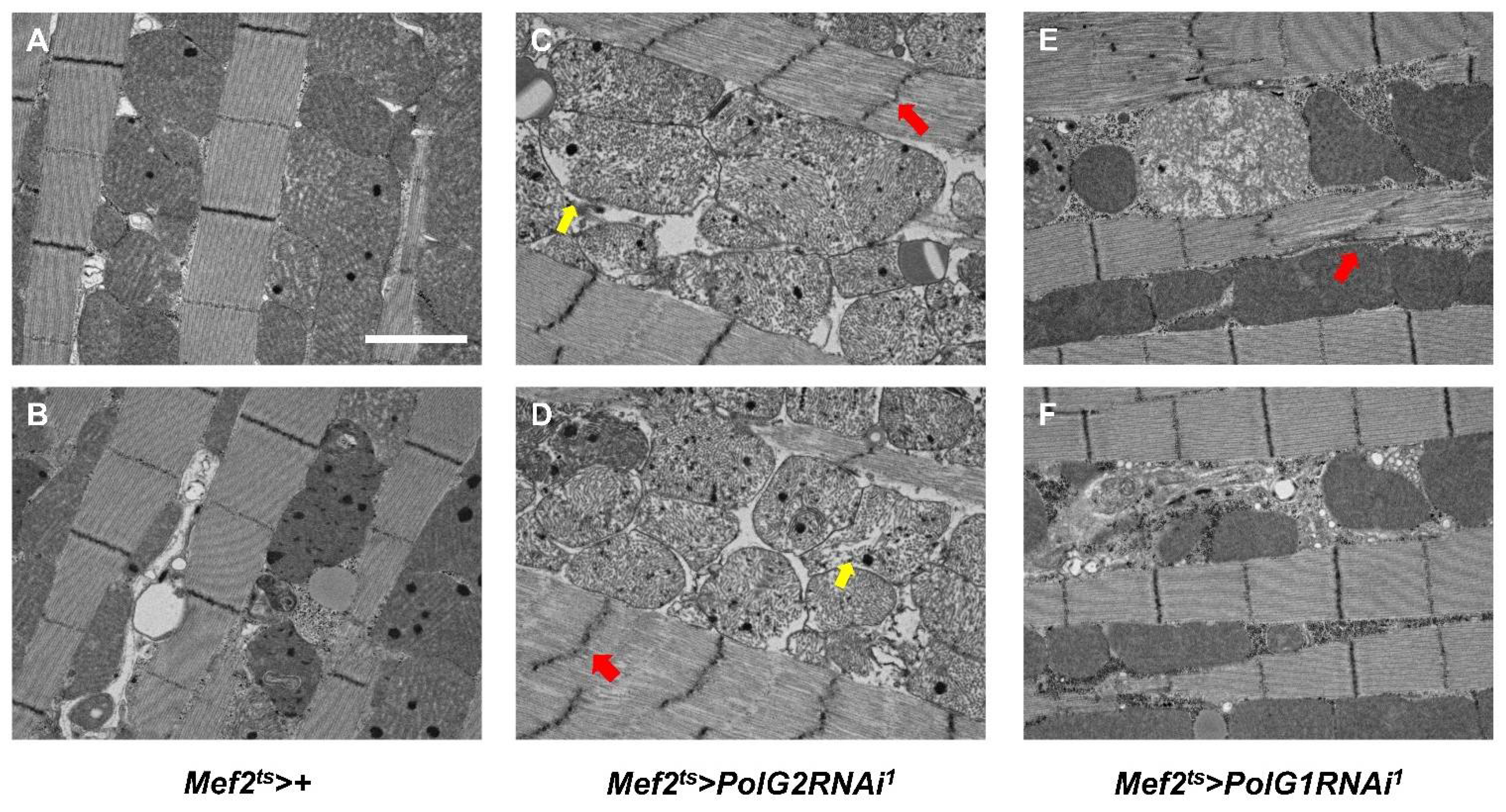
3.5. Continuous Silencing of the Polγ Genes in the Adult Muscle Resulted in Enhanced Apoptosis and Malformation of Muscle Mitochondria and Myofibrils
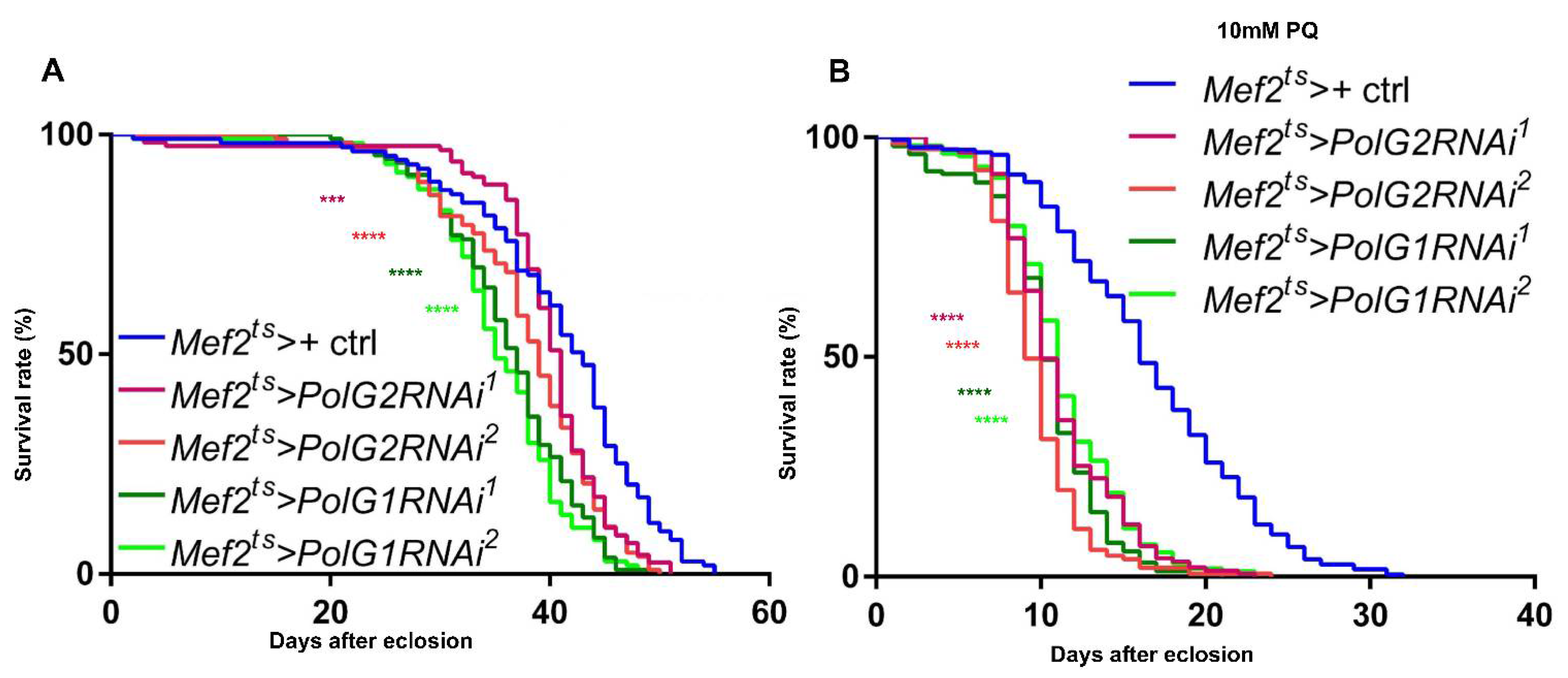
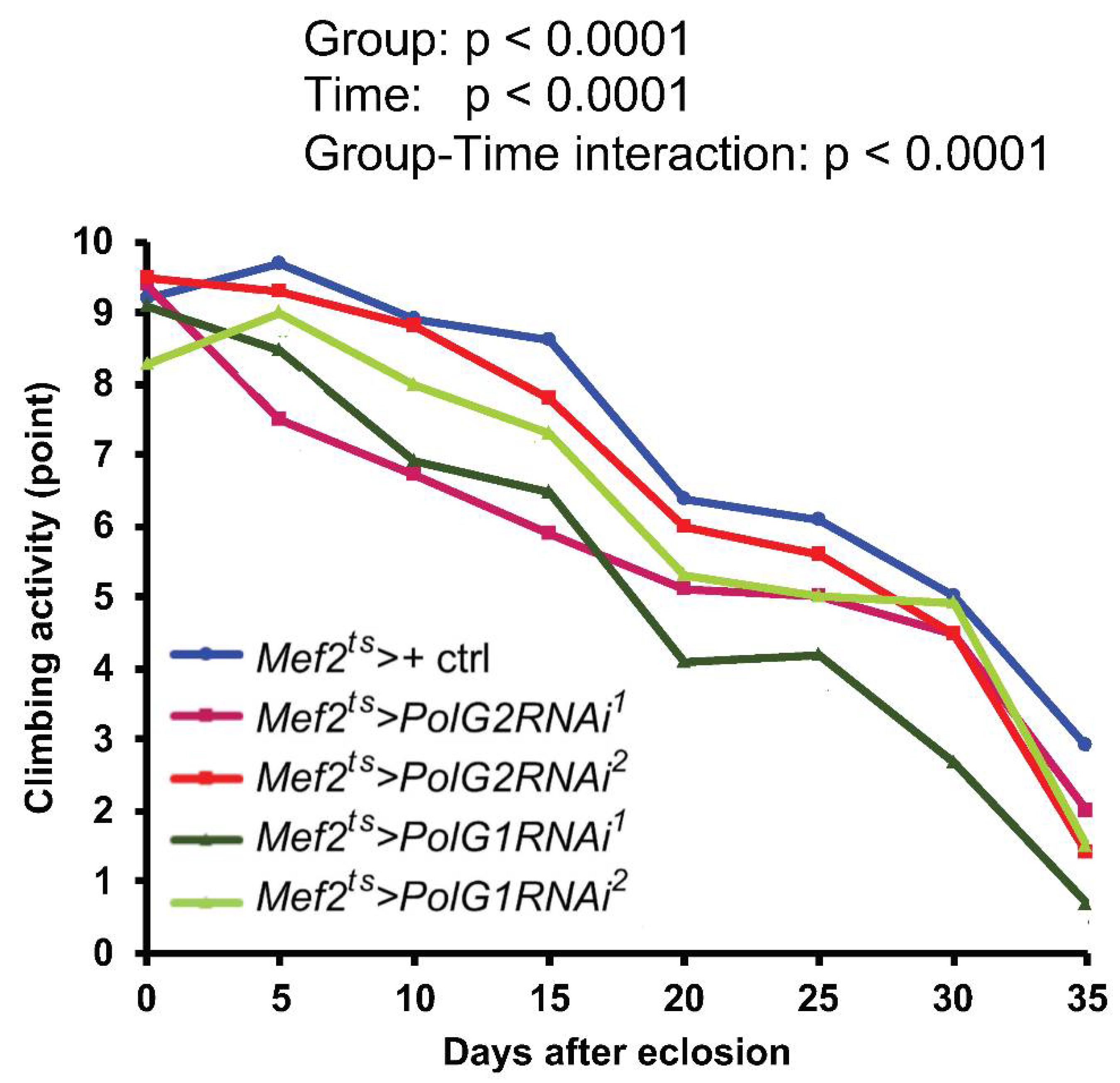
3.6. Downregulation of the Polγ Genes in the Muscle Shortened Adult Lifespan and Accelerated Age-Dependent Impairment of Locomotion
4. Discussion
4.1. Accumulation of Mitochondrial DNA Damage and Promotion of Mitochondrial Dysfunction in Polγ-Silenced Adult Muscle
4.2. Acceleration of Mitochondrial Fission, Followed by Mitophagy in Adult Muscle Carrying Abnormal Mitochondria with Accumulating DNA Damage
4.3. Age-Dependent Impairment of Mitochondrial Activity, and Aging-Related Phenotypes and Behavior in the Adult Muscle
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Cochemé, H.M.; Quin, C.; McQuaker, S.J.; Cabreiro, F.; Logan, A.; Prime, T.A.; Abakumova, I.; Patel, J.V.; Fearnley, I.M.; James, A.M.; et al. Measurement of H2O2 within Living Drosophila during Aging Using a Ratiometric Mass Spectrometry Probe Targeted to the Mitochondrial Matrix. Cell Metab. 2011, 13, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Gómez, L.A.; Hagen, T.M. Age-Related Decline in Mitochondrial Bioenergetics: Does Supercomplex Destabilization Determine Lower Oxidative Capacity and Higher Superoxide Production? Semin. Cell Dev. Biol. 2012, 23, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.L.; Van Remmen, H.V.; Drake, J.A.; Yang, H.; Guo, Z.M.; Kewitt, K.; Walter, C.A.; Richardson, A. Does Oxidative Damage to DNA Increase with Age? Proc. Natl. Acad. Sci. USA 2001, 98, 10469–10474. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Wallace, D.C.; Martin, G.M. The Mitochondrial Theory of Aging and Its Relationship to Reactive Oxygen Species Damage and Somatic mtDNA Mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18769–18770. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. BioMed Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The Role of Mitochondria in Aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Garesse, R. Drosophila melanogaster Mitochondrial DNA: Gene Organization and Evolutionary Considerations. Genetics 1988, 118, 649–663. [Google Scholar] [CrossRef]
- Perez-Campo, R.; López-Torres, M.; Cadenas, S.; Rojas, C.; Barja, G. The Rate of Free Radical Production as a Determinant of the Rate of Aging: Evidence from the Comparative Approach. J. Comp. Physiol. B 1998, 168, 149–158. [Google Scholar] [CrossRef]
- Martin, J.A.; Martini, A.; Molinari, A.; Morgan, W.; Ramalingam, W.; Buckwalter, J.A.; McKinley, T.O. Mitochondrial Electron Transport and Glycolysis Are Coupled in Articular Cartilage. Osteoarthr. Cartil. 2012, 20, 323–329. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive Sequencing Reveals an Age-Related Increase in Somatic Mitochondrial Mutations That Are Inconsistent with Oxidative Damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA Damage and Dysfunction Associated with Oxidative Stress in Failing Hearts after Myocardial Infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Moraes, C.T. Mechanisms Linking mtDNA Damage and Aging. Free Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef]
- Garesse, R.; Kaguni, L.S. A Drosophila Model of Mitochondrial DNA Replication: Proteins, Genes and Regulation. IUBMB Life 2005, 57, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Maryold, S.J.; Attrill, H.; Speretta, E.; Warner, K.; Magrane, M.; Berloco, M.; Cotterill, S.; McVey, M.; Rong, Y.; Yamaguchi, M. The DNA Polymerases of Drosophila melanogaster. Fly 2020, 14, 49–61. [Google Scholar] [CrossRef]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A.; et al. DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol. Cell Biol. 2017, 37, e00237-17. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature Ageing in Mice Expressing Defective Mitochondrial DNA Polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Khrapko, K.; Kraytsberg, Y.; de Grey, A.D.; Vijg, J.; Schon, E.A. Does Premature Aging of the mtDNA Mutator Mouse Prove that mtDNA Mutations Are Involved in Natural Aging? Aging Cell 2006, 5, 279–282. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 10, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Samstag, C.L.; Hoekstra, J.G.; Huang, C.H.; Chaisson, M.J.; Youle, R.J.; Kennedy, S.R.; Pallanck, L.J. Deleterious Mitochondrial DNA Point Mutations Are Overrepresented in Drosophila Expressing a Proofreading-defective DNA Polymerase γ. PLoS Genet. 2018, 14, e1007805. [Google Scholar] [CrossRef]
- Shirihai, O.S.; Song, M.; Dorn, G.W., 2nd. How Mitochondrial Dynamism Orchestrates Mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial Dynamics—Mitochondrial Fission and Fusion in Human Diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Filipovska, A. Organization and Expression of the Mammalian Mitochondrial Genome. Nat. Rev. Genet. 2022. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The Interplay between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Nezis, I.P.; Simonsen, A.; Sagona, A.P.; Finley, K.; Gaumer, S.; Contamine, D.; Rusten, T.E.; Stenmark, H.; Brech, A. Ref(2)P, the Drosophila melanogaster Homologue of Mammalian p62, Is Required for the Formation of Protein Aggregates in Adult Brain. J. Cell Biol. 2008, 180, 1065–1071. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular Machinery for Self-Eating. Cell Death Differ. 2005, 12 (Suppl. S2), 1542–1552. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-Mediated Mitochondrial Fission in Midlife Prolongs Healthy Lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Suster, M.L.; Seugnet, L.; Bate, M.; Sokolowski, M.B. Refining GAL4-Driven Transgene Expression in Drosophila with a GAL80 Enhancer-Trap. Genesis 2004, 39, 240–245. [Google Scholar] [CrossRef]
- Oka, S.; Hirai, J.; Yasukawa, T.; Nakahara, Y.; Inoue, Y.H. A Correlation of Reactive Oxygen Species Accumulation by Depletion of Superoxide Dismutases with Age-Dependent Impairment in the Nervous System and Muscles of Drosophila Adults. Biogerontology 2015, 16, 485–501. [Google Scholar] [CrossRef]
- Okumura, K.; Nishihara, S.; Inoue, Y.H. Genetic Identification and Characterization of Three Genes That Prevent Accumulation of Oxidative DNA Damage in Drosophila Adult Tissues. DNA Repair 2019, 78, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Le, T.D.; Nakahara, Y.; Ueda, M.; Okumura, K.; Hirai, J.; Sato, Y.; Takemoto, D.; Tomimori, N.; Ono, Y.; Nakai, M.; et al. Sesamin Suppresses Aging Phenotypes in Adult Muscular and Nervous Systems and Intestines in a Drosophila Senescence-Accelerated Model. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1826–1839. [Google Scholar] [CrossRef]
- Takeda, K.; Okumura, T.; Terahata, M.; Yamaguchi, M.; Taniguchi, K.; Adachi-Yamada, T. Drosophila Peptide Hormones Allatostatin A and Diuretic Hormone 31 Exhibiting Complementary Gradient Distribution in Posterior Midgut Antagonistically Regulate Midgut Senescence and Adult Lifespan. Zool. Sci. 2018, 35, 75–85. [Google Scholar] [CrossRef]
- Martinez, V.G.; Javadi, C.S.; Ngo, E.; Ngo, L.; Lagow, R.D.; Zhang, B. Age-related Changes in Climbing Behavior and Neural Circuit Physiology in Drosophila. Dev. Neurobiol. 2007, 67, 778–791. [Google Scholar] [CrossRef]
- Kim, T.; Shin, H.; Song, B.; Won, C.; Yoshida, H.; Yamaguchi, M.; Cho, K.S.; Lee, I.S. Overexpression of H3K36 Methyltransferase NSD in Glial Cells Affects Brain Development in Drosophila. Glia 2020, 68, 2503–2516. [Google Scholar] [CrossRef]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA Polymerase γ in Mitochondrial DNA Replication and Repair. Chem. Rev. 2006, 106, 383–405. [Google Scholar] [CrossRef]
- Cohen, B.H.; Naviaux, R.K. The Clinical Diagnosis of POLG Disease and Other Mitochondrial DNA Depletion Disorders. Methods 2010, 51, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., 2nd. Mitochondrial Fission and Fusion Factors Reciprocally Orchestrate Mitophagic Culling in Mouse Hearts and Cultured Fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef]
- Gomes, L.C.; Scorrano, L. High Levels of Fis1, a Pro-fission Mitochondrial Protein, Trigger Autophagy. Biochim. Biophys. Acta 2008, 1777, 860–866. [Google Scholar] [CrossRef]
- Iqbal, S.; Hood, D.A. Oxidative Stress-induced Mitochondrial Fragmentation and Movement in Skeletal Muscle Myoblasts. Am. J. Physiol. Cell Physiol. 2014, 306, C1176–C1183. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pinheiro, P.; Pardo-Hernández, C.; Reyes, A.; Tilokani, L.; Mishra, A.; Cerutti, R.; Li, S.; Rozsivalova, D.H.; Valenzuela, S.; Dogan, S.A.; et al. DNA Polymerase Gamma Mutations That Impair Holoenzyme Stability Cause Catalytic Subunit Depletion. Nucleic Acids Res. 2021, 49, 5230–5248. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA Damage and Reactive Oxygen Species in Neurodegenerative Disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Fayet, G.; Jansson, M.; Sternberg, D.; Moslemi, A.R.; Blondy, P.; Lombès, A.; Fardeau, M.; Oldfors, A. Ageing Muscle: Clonal Expansions of Mitochondrial DNA Point Mutations and Deletions Cause Focal Impairment of Mitochondrial Function. Neuromuscul. Disord. 2002, 12, 484–493. [Google Scholar] [CrossRef]
- Larsson, N.G. Somatic Mitochondrial DNA Mutations in Mammalian Aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and Disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Ameur, A.; Stewart, J.B.; Freyer, C.; Hagström, E.; Ingman, M.; Larsson, N.G.; Gyllensten, U. Ultra-Deep Sequencing of Mouse Mitochondrial DNA: Mutational Patterns and Their Origins. PLoS Genet. 2011, 7, e1002028. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive Oxygen Species Are Essential for Autophagy and Specifically Regulate the Activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Shirane, M.; Nakayama, K.I. Selective Escape of Proteins from the Mitochondria during Mitophagy. Nat. Commun. 2013, 4, 1410. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Hussien, R.; Brooks, G.A. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med. 2010, 49, 1646–1654. [Google Scholar] [CrossRef]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A Role for Autophagy in the Extension of Lifespan by Dietary Restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the Beclin 1-BCL2 Autophagy Regulatory Complex Promotes Longevity in Mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Bjedov, I.; Rallis, C. The Target of Rapamycin Signalling Pathway in Ageing and Lifespan Regulation. Genes 2020, 11, 1043. [Google Scholar] [CrossRef]
- Denton, D.; Nicolson, S.; Kumar, S. Cell Death by Autophagy: Facts and Apparent Artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef]
- Yuan, J.; Kroemer, G. Alternative Cell Death Mechanisms in Development and Beyond. Genes Dev. 2010, 24, 2592–2602. [Google Scholar] [CrossRef] [PubMed]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial Function and Apoptotic Susceptibility in Aging Skeletal Muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.E.; Van Zeeland, N.L.; Dahl, D.B.; Weindruch, R.; Aiken, J.M. Cellular Phenotypes of Age-Associated Skeletal Muscle Mitochondrial Abnormalities in Rhesus Monkeys. Mutat. Res. 2000, 452, 123–138. [Google Scholar] [CrossRef]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA Deletion Mutations colocalize with Segmental Electron Transport System Abnormalities, Muscle Fiber Atrophy, Fiber Splitting, and Oxidative Damage in Sarcopenia. FASEB J. 2001, 15, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, L.N.; Yee, Z.; Ng, L.F.; Gunawan, R.; Halliwell, B.; Gruber, J. Clonal Expansion of Mitochondrial DNA Deletions Is a Private Mechanism of Aging in Long-Lived Animals. Aging Cell 2018, 17, e12814. [Google Scholar] [CrossRef]
- Demontis, F.; Piccirillo, R.; Goldberg, A.L.; Perrimon, N. Mechanisms of Skeletal Muscle Aging: Insights from Drosophila and Mammalian Models. Dis. Model. Mech. 2013, 6, 1339–1352. [Google Scholar] [CrossRef]
- Ueyama, M.; Akimoto, Y.; Ichimiya, T.; Ueda, R.; Kawakami, H.; Aigaki, T.; Nishihara, S. Increased Apoptosis of Myoblasts in Drosophila Model for the Walker-Warburg Syndrome. PLoS ONE 2010, 5, e11557. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Piplani, H.; Sin, J.; Sawaged, S.; Hamid, S.M.; Taylor, D.J.; de Freitas Germano, J. At the Heart of Mitochondrial Quality Control: Many Roads to the Top. Cell. Mol. Life Sci. 2021, 78, 3791–3801. [Google Scholar] [CrossRef]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The Role of Dynamin-Related Protein 1, a Mediator of Mitochondrial Fission, in Apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.D.; Foretz, M.; et al. Mitochondrial Fission and Remodelling Contributes to Muscle Atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Fleming, J.E. A Two-Step Hypothesis on the Mechanisms of In Vitro Cell Aging: Cell Differentiation Followed by Intrinsic Mitochondrial Mutagenesis. Exp. Gerontol. 1984, 19, 31–36. [Google Scholar] [CrossRef]
- Choi, Y.I.; Park, D.K.; Chung, J.W.; Kim, K.O.; Kwon, K.A.; Kim, Y.J. Circadian Rhythm Disruption Is Associated with an Increased Risk of Sarcopenia: A Nationwide Population-Based Study in Korea. Sci. Rep. 2019, 9, 12015. [Google Scholar] [CrossRef] [PubMed]
- Mattis, J.; Sehgal, A. Circadian Rhythms, Sleep, and Disorders of Aging. Trends Endocrinol. Metab. 2016, 27, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Checcaglini, F.; Coscia, F.; Gigliotti, P.; Fulle, S.; Fanò-Illic, G. Biological Aspects of Selected Myokines in Skeletal Muscle: Focus on Aging. Int. J. Mol. Sci. 2021, 22, 8520. [Google Scholar] [CrossRef]
- Silva, B.S.A.; Uzeloto, J.S.; Lira, F.S.; Pereira, T.; Coelho-E-Silva, M.J.; Caseiro, A. Exercise as a Peripheral Circadian Clock Resynchronizer in Vascular and Skeletal Muscle Aging. Int. J. Environ. Res. Public Health 2021, 18, 12949. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozaki, M.; Le, T.D.; Inoue, Y.H. Downregulating Mitochondrial DNA Polymerase γ in the Muscle Stimulated Autophagy, Apoptosis, and Muscle Aging-Related Phenotypes in Drosophila Adults. Biomolecules 2022, 12, 1105. https://doi.org/10.3390/biom12081105
Ozaki M, Le TD, Inoue YH. Downregulating Mitochondrial DNA Polymerase γ in the Muscle Stimulated Autophagy, Apoptosis, and Muscle Aging-Related Phenotypes in Drosophila Adults. Biomolecules. 2022; 12(8):1105. https://doi.org/10.3390/biom12081105
Chicago/Turabian StyleOzaki, Mika, Tuan Dat Le, and Yoshihiro H. Inoue. 2022. "Downregulating Mitochondrial DNA Polymerase γ in the Muscle Stimulated Autophagy, Apoptosis, and Muscle Aging-Related Phenotypes in Drosophila Adults" Biomolecules 12, no. 8: 1105. https://doi.org/10.3390/biom12081105