
The Impact of Pancreatic Exocrine Diseases on the β-Cell and Glucose Metabolism—A Review with Currently Available Evidence


 ,
, 
Abstract
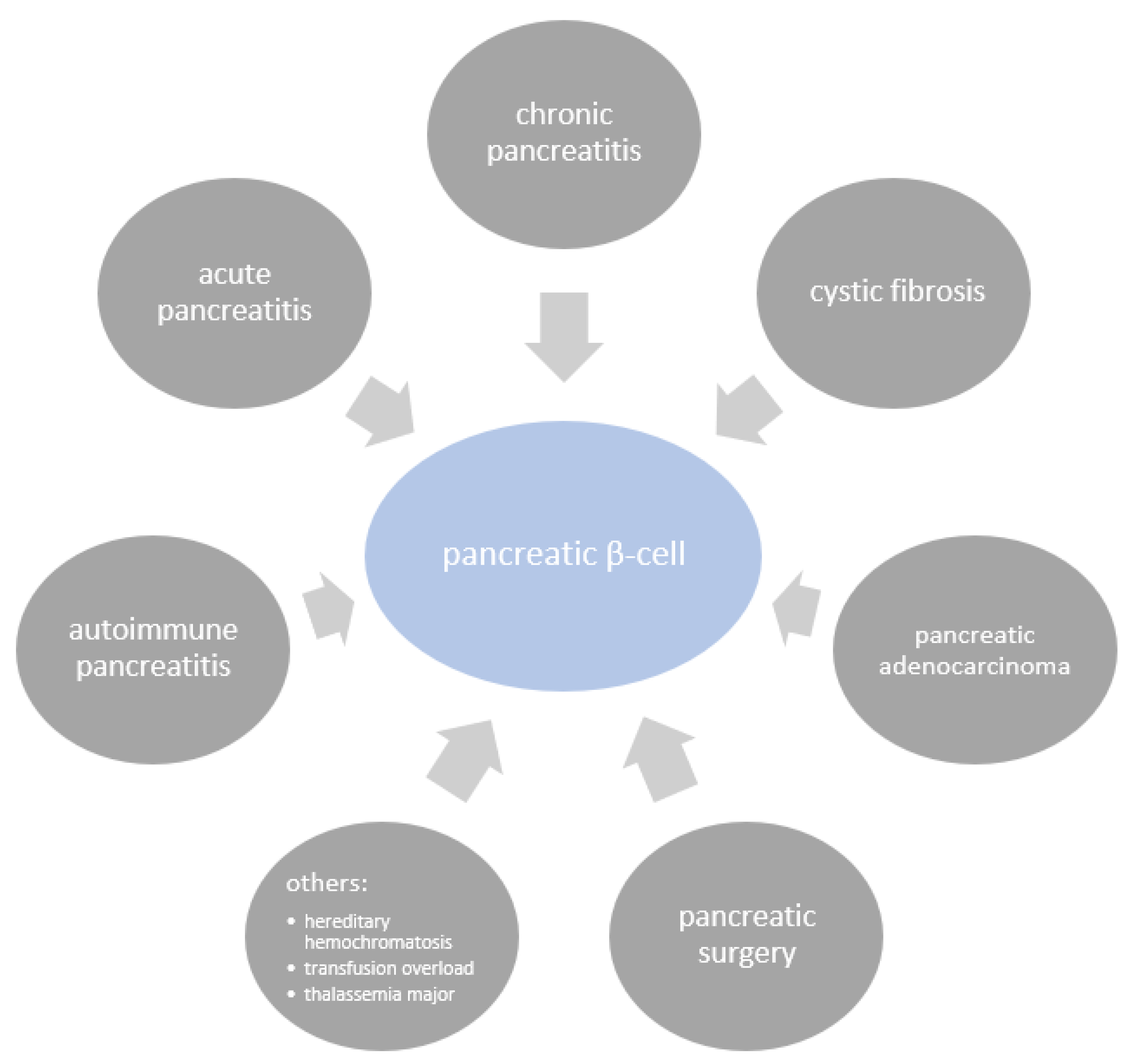
:1. Introduction
2. Search Strategy
3. Chronic Pancreatitis
3.1. Risk Factors
3.2. Timing
3.3. Mechanism
4. Acute Pancreatitis
4.1. Risk Factors
4.2. Timing
4.3. Mechanism
5. Cystic Fibrosis
5.1. Risk Factors and Timing
5.2. Mechanism
5.2.1. β-Cell Loss
5.2.2. Immunological Response
5.2.3. CFTR Expression
5.2.4. Impaired Insulin Secretion
5.2.5. The Genotype
6. Pancreatic Surgery
6.1. The Surgical Procedure
6.2. The Volume of Resection
6.3. Associated Risk Factors
7. Pancreatic Adenocarcinoma
7.1. Risk Factors
7.2. Timing
7.3. Mechanism

8. Autoimmune Pancreatitis
8.1. Risk Factors—Steroid Treatment
8.2. Timing
8.3. Mechanism
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ewald, N.; Kaufmann, C.; Raspe, A.; Kloer, H.U.; Bretzel, R.G.; Hardt, P.D. Prevalence of diabetes mellitus secondary to pancreatic diseases (type 3c): Prevalence of Type 3c Diabetes. Diabetes Metab. Res. Rev. 2012, 28, 338–342. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43 (Suppl. S1), S14–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendharkar, S.A.; Mathew, J.; Petrov, M.S. Age- and sex-specific prevalence of diabetes associated with diseases of the exocrine pancreas: A population-based study. Dig. Liver Dis. 2017, 49, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Woodmansey, C.; McGovern, A.P.; McCullough, K.A.; Whyte, M.B.; Munro, N.M.; Correa, A.C.; Gatenby, P.A.; Jones, S.A.; de Lusignan, S. Incidence, Demographics, and Clinical Characteristics of Diabetes of the Exocrine Pancreas (Type 3c): A Retrospective Cohort Study. Diabetes Care 2017, 40, 1486–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynne, K.; Devereaux, B.; Dornhorst, A. Diabetes of the exocrine pancreas: Diabetes of the exocrine pancreas. J. Gastroenterol. Hepatol. 2019, 34, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Stanojevic, V.; Habener, J.F. Evolving function and potential of pancreatic alpha cells. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Olesen, S.S.; Poulsen, J.L.; Novovic, S.; Nøjgaard, C.; Kalaitzakis, E.; Jensen, N.M.; Engjom, T.; Tjora, E.; Waage, A.; Hauge, T.; et al. Multiple risk factors for diabetes mellitus in patients with chronic pancreatitis: A multicentre study of 1117 cases. United Eur. Gastroenterol. J. 2020, 8, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Malka, D.; Hammel, P.; Sauvanet, A.; Rufat, P.; O’Toole, D.; Bardet, P.; Belghiti, J.; Bernades, P.; Ruszniewski, P.; Lévy, P. Risk factors for diabetes mellitus in chronic pancreatitis. Gastroenterology 2000, 119, 1324–1332. [Google Scholar] [CrossRef]
- Wang, W.; Guo, Y.; Liao, Z.; Zou, D.-W.; Jin, Z.-D.; Zou, D.-J.; Jin, G.; Hu, X.-G.; Li, Z.-S. Occurrence of and Risk Factors for Diabetes Mellitus in Chinese Patients With Chronic Pancreatitis. Pancreas 2011, 40, 206–212. [Google Scholar] [CrossRef]
- Pan, J.; Xin, L.; Wang, D.; Liao, Z.; Lin, J.-H.; Li, B.-R.; Du, T.-T.; Ye, B.; Zou, W.-B.; Chen, H.; et al. Risk Factors for Diabetes Mellitus in Chronic Pancreatitis: A Cohort of 2011 Patients. Medicine 2016, 95, e3251. [Google Scholar] [CrossRef]
- Sun, J.; Ni, Q.; Xie, J.; Xu, M.; Zhang, J.; Kuang, J.; Wang, Y.; Ning, G.; Wang, Q. β-Cell Dedifferentiation in Patients With T2D With Adequate Glucose Control and Nondiabetic Chronic Pancreatitis. J. Clin. Endocrinol. Metab. 2019, 104, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensellam, M.; Jonas, J.-C.; Laybutt, D.R. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in β-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef] [PubMed]
- Mitnala, S.; Pondugala, P.K.; Guduru, V.R.; Rabella, P.; Thiyyari, J.; Chivukula, S.; Boddupalli, S.; Hardikar, A.A.; Reddy, D.N. Reduced Expression of PDX-1 Is Associated With Decreased Beta Cell Function in Chronic Pancreatitis. Pancreas 2010, 39, 856–862. [Google Scholar] [CrossRef]
- Hart, P.A.; Bellin, M.D.; Andersen, D.K.; Bradley, D.; Cruz-Monserrate, Z.; Forsmark, C.E.; Goodarzi, M.O.; Habtezion, A.; Korc, M.; Kudva, Y.C.; et al. Type 3c (pancreatogenic) diabetes mellitus secondary to chronic pancreatitis and pancreatic cancer. Lancet Gastroenterol. Hepatol. 2016, 1, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, R.; Sasikala, M.; Pavan Kumar, P.; Rao, G.V.; Pradeep, R.; Reddy, D.N. T-Helper Cell–Mediated Islet Inflammation Contributes to β-Cell Dysfunction in Chronic Pancreatitis. Pancreas 2016, 45, 434–442. [Google Scholar] [CrossRef]
- Kumar, P.P.; Radhika, G.; Rao, G.; Pradeep, R.; Subramanyam, C.; Talukdar, R.; Reddy, D.; Sasikala, M. Interferon γ and glycemic status in diabetes associated with chronic pancreatitis. Pancreatology 2012, 12, 65–70. [Google Scholar] [CrossRef]
- Sasikala, M.; Talukdar, R.; Kumar, P.P.; Radhika, G.; Rao, G.V.; Pradeep, R.; Subramanyam, C.; Reddy, D.N. β-Cell Dysfunction in Chronic Pancreatitis. Dig. Dis. Sci. 2012, 57, 1764–1772. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-Cell Deficit and Increased β-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Mandrup-Poulsen, T. A choice of death—The signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Darville, M.I. beta-cell apoptosis and defense mechanisms: Lessons from type 1 diabetes. Diabetes 2001, 50 (Suppl. S1), S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-K.; Huang, M.-Y.; Hsu, C.-Y.; Su, Y.-C. Bidirectional Relationship Between Diabetes and Acute Pancreatitis: A Population-Based Cohort Study in Taiwan. Medicine 2016, 95, e2448. [Google Scholar] [CrossRef] [PubMed]
- Das, S.L.M.; Singh, P.P.; Phillips, A.R.J.; Murphy, R.; Windsor, J.A.; Petrov, M.S. Newly diagnosed diabetes mellitus after acute pancreatitis: A systematic review and meta-analysis. Gut 2014, 63, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Yang, Y.; Zhang, J.; Yang, Q.; Lu, G.; Li, B.; Tong, Z.; Ke, L.; Li, W.; Li, J. Effect of the disease severity on the risk of developing new-onset diabetes after acute pancreatitis. Medicine 2018, 97, e10713. [Google Scholar] [CrossRef] [PubMed]
- Vujasinovic, M. Pancreatic exocrine insufficiency, diabetes mellitus and serum nutritional markers after acute pancreatitis. World J. Gastroenterol. 2014, 20, 18432. [Google Scholar] [CrossRef] [PubMed]
- Soo, D.H.E.; Pendharkar, S.A.; Jivanji, C.J.; Gillies, N.A.; Windsor, J.A.; Petrov, M.S. Derivation and validation of the prediabetes self-assessment screening score after acute pancreatitis (PERSEUS). Dig. Liver Dis. 2017, 49, 1146–1154. [Google Scholar] [CrossRef]
- Petrov, M.S. Panorama of mediators in postpancreatitis diabetes mellitus. Curr. Opin. Gastroenterol. 2020, 36, 443–451. [Google Scholar] [CrossRef]
- Pendharkar, S.A.; Singh, R.G.; Bharmal, S.H.; Drury, M.; Petrov, M.S. Pancreatic Hormone Responses to Mixed Meal Test in New-onset Prediabetes/Diabetes After Non-necrotizing Acute Pancreatitis. J. Clin. Gastroenterol. 2020, 54, e11–e20. [Google Scholar] [CrossRef]
- Halangk, W.; Lerch, M.M. Early events in acute pancreatitis. Gastroenterol. Clin. North Am. 2004, 33, 717–731. [Google Scholar] [CrossRef]
- Kamboj, S.S.; Sandhir, R. Protective effect of N-acetylcysteine supplementation on mitochondrial oxidative stress and mitochondrial enzymes in cerebral cortex of streptozotocin-treated diabetic rats. Mitochondrion 2011, 11, 214–222. [Google Scholar] [CrossRef]
- Husain, S.Z.; Orabi, A.I.; Muili, K.A.; Luo, Y.; Sarwar, S.; Mahmood, S.M.; Wang, D.; Choo-Wing, R.; Singh, V.P.; Parness, J.; et al. Ryanodine receptors contribute to bile acid-induced pathological calcium signaling and pancreatitis in mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 302, G1423–G1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, N.; Aramandla, R.; Poffenberger, G.; Fayolle, C.; Thames, A.H.; Bautista, A.; Spigelman, A.F.; Babon, J.A.B.; DeNicola, M.E.; Dadi, P.K.; et al. Cystic fibrosis–related diabetes is caused by islet loss and inflammation. JCI Insight. 2018, 3, e98240. [Google Scholar] [CrossRef] [Green Version]
- Colomba, J.; Rabasa-Lhoret, R.; Bonhoure, A.; Bergeron, C.; Boudreau, V.; Tremblay, F.; Senior, P.; Potter, K. Dyslipidemia is not associated with the development of glucose intolerance or diabetes in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Konrad, K.; Scheuing, N.; Badenhoop, K.; Borkenstein, M.H.; Gohlke, B.; Schöfl, C. Cystic fibrosis-related diabetes compared to type 1 and type 2 diabetes in adults: CFRD in adulthood. Diabetes Metab. Res. Rev. 2013, 29, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Tierney, S.; Webb, K.; Jones, A.; Dodd, M.; Mckenna, D.; Rowe, R.; Whitehouse, J.; Deaton, C. Living with cystic fibrosis-related diabetes or type 1 diabetes mellitus: A comparative study exploring health-related quality of life and patients’ reported experiences of hypoglycaemia. Chronic. Illn. 2008, 4, 278–288. [Google Scholar] [CrossRef]
- Hull, R.L.; Gibson, R.L.; McNamara, S.; Deutsch, G.H.; Fligner, C.L.; Frevert, C.W.; Ramsey, B.W.; Sanda, S. Islet Interleukin-1β Immunoreactivity Is an Early Feature of Cystic Fibrosis That May Contribute to β-Cell Failure. Diabetes Care 2018, 41, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Barrio, R. Management of Endocrine Disease: Cystic fibrosis-related diabetes: Novel pathogenic insights opening new therapeutic avenues. Eur. J. Endocrinol. 2015, 172, R131–R141. [Google Scholar] [CrossRef] [Green Version]
- Hameed, S.; Jaffé, A.; Verge, C.F. Cystic Fibrosis Related Diabetes (CFRD)—The End Stage of Progressive Insulin Deficiency. Pediatr. Pulmonol. 2011, 46, 747–760. [Google Scholar] [CrossRef]
- Ali, B.R. Is cystic fibrosis-related diabetes an apoptotic consequence of ER stress in pancreatic cells? Med. Hypotheses 2009, 72, 55–57. [Google Scholar] [CrossRef]
- Bellin, M.D.; Laguna, T.; Leschyshyn, J.; Regelmann, W.; Dunitz, J.; Billings, J. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: A small pilot study: Insulin secretion following CFTR correction. Pediatr. Diabetes 2013, 14, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Cano Megías, M.; González Albarrán, O.; Guisado Vasco, P.; Lamas Ferreiro, A.; Máiz Carro, L. Resistencia insulínica, disfunción de la célula β pancreática y diferencias en los puntos intermedios de las curvas de glucemia e insulina tras una sobrecarga oral estándar de glucosa en adultos con fibrosis quística. Endocrinol. Nutr. 2015, 62, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Colomba, J.; Boudreau, V.; Lehoux-Dubois, C.; Desjardins, K.; Coriati, A.; Tremblay, F.; Rabasa-Lhoret, R. The main mechanism associated with progression of glucose intolerance in older patients with cystic fibrosis is insulin resistance and not reduced insulin secretion capacity. J. Cyst. Fibros. 2019, 18, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, V.; Coriati, A.; Hammana, I.; Ziai, S.; Desjardins, K.; Berthiaume, Y. Variation of glucose tolerance in adult patients with cystic fibrosis: What is the potential contribution of insulin sensitivity? J. Cyst. Fibros. 2016, 15, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Street, M.E.; Spaggiari, C.; Ziveri, M.A.; Rossi, M.; Volta, C.; Viani, I. Insulin production and resistance in cystic fibrosis: Effect of age, disease activity, and genotype. J. Endocrinol. Investig. 2012, 35, 246–253. [Google Scholar] [CrossRef]
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slezak, L.A.; Andersen, D.K. Pancreatic Resection: Effects on Glucose Metabolism. World J. Surg. 2001, 25, 452–460. [Google Scholar] [CrossRef]
- Ishida, J.; Toyama, H.; Matsumoto, I.; Shirakawa, S.; Terai, S.; Yamashita, H.; Yanagimoto, H.; Asari, S.; Kido, M.; Fukumoto, T. Glucose Tolerance after Pancreatectomy: A Prospective Observational Follow-Up Study of Pancreaticoduodenectomy and Distal Pancreatectomy. J. Am. Coll. Surg. 2021, 233, 753–762. [Google Scholar] [CrossRef]
- Balduzzi, A.; Marchegiani, G.; Andrianello, S.; Romeo, F.; Amodio, A.; De Pretis, N.; Zamboni, G.; Malleo, G.; Frulloni, L.; Salvia, R.; et al. Pancreaticoduodenectomy for paraduodenal pancreatitis is associated with a higher incidence of diabetes but a similar quality of life and pain control when compared to medical treatment. Pancreatology 2020, 20, 193–198. [Google Scholar] [CrossRef]
- King, J.; Kazanjian, K.; Matsumoto, J.; Reber, H.A.; Yeh, M.W.; Hines, O.J.; Eibl, G. Distal Pancreatectomy: Incidence of Postoperative Diabetes. J. Gastrointest. Surg. 2008, 12, 1548–1553. [Google Scholar] [CrossRef]
- Beger, H.G.; Mayer, B.; Poch, B. Resection of the duodenum causes long-term endocrine and exocrine dysfunction after Whipple procedure for benign tumors—Results of a systematic review and meta-analysis. HPB 2020, 22, 809–820. [Google Scholar] [CrossRef]
- Kumar, R.; Sahoo, J.P.; Pottakat, B.; Kamalanathan, S.; Mohan, P.; Kate, V.; Kar, S.S.; Selviambigapathy, J. Effect of Frey’s procedure on islet cell function in patients with chronic calcific pancreatitis. Hepatobiliary Pancreat Dis. Int. 2018, 17, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Keck, T.; Wellner, U.F.; Riediger, H.; Adam, U.; Sick, O.; Hopt, U.T.; Makowiec, F. Long-Term Outcome after 92 Duodenum-Preserving Pancreatic Head Resections for Chronic Pancreatitis: Comparison of Beger and Frey Procedures. J. Gastrointest. Surg. 2010, 14, 549–556. [Google Scholar] [CrossRef]
- Singh, A.N.; Pal, S.; Kilambi, R.; Madhusudhan, K.S.; Dash, N.R.; Tandon, N. Diabetes after pancreaticoduodenectomy: Can we predict it? J. Surg. Res. 2018, 227, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Sato, T.; Andoh, H.; Yasui, O.; Yoshioka, M.; Kurokawa, T.; Watanabe, G.; Ise, N.; Kotanagi, H.; Asanuma, Y.; et al. Outcomes and Indications of Segmental Pancreatectomy. Dig. Surg. 2004, 21, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.N.; Kingham, T.P.; D’Angelica, M.I.; DeMatteo, R.P.; Jarnagin, W.R.; Kalin, M.F.; Allen, P.J. Intraductal Papillary Mucinous Neoplasms and the Risk of Diabetes Mellitus in Patients Undergoing Resection Versus Observation. J. Gastrointest. Surg. 2015, 19, 1974–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, H.; Menge, B.A.; Zeidler, C.; Ritter, P.R.; Tannapfel, A.; Uhl, W.; Schmidt, W.E.; Meier, J.J. Determinants of glucose control in patients with chronic pancreatitis. Diabetologia 2010, 53, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Menge, B.A.; Breuer, T.G.K.; Ritter, P.R.; Uhl, W.; Schmidt, W.E.; Meier, J.J. Long-term recovery of β-cell function after partial pancreatectomy in humans. Metabolism 2012, 61, 620–624. [Google Scholar] [CrossRef]
- Sohn, S.Y.; Lee, E.K.; Han, S.-S.; Lee, Y.J.; Hwangbo, Y.; Kang, Y.H.; Lee, S.D.; Kim, S.H.; Woo, S.M.; Lee, W.J.; et al. Favorable glycemic response after pancreatoduodenectomy in both patients with pancreatic cancer and patients with non-pancreatic cancer. Medicine 2018, 97, e0590. [Google Scholar] [CrossRef]
- Pannala, R.; Leirness, J.B.; Bamlet, W.R.; Basu, A.; Petersen, G.M.; Chari, S.T. Prevalence and Clinical Profile of Pancreatic Cancer–Associated Diabetes Mellitus. Gastroenterology 2008, 134, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Sah, R.P.; Nagpal, S.J.S.; Mukhopadhyay, D.; Chari, S.T. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G. Diabetes and Pancreatic Cancer—A Dangerous Liaison Relying on Carbonyl Stress. Cancers 2021, 13, 313. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bian, X.; Wei, S.; He, M.; Yang, Y. The relationship between pancreatic cancer and type 2 diabetes: Cause and consequence. Cancer Manag. Res. 2019, 11, 8257–8268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chari, S.T.; Andersen, D.K. Metabolic Surveillance for Those at High Risk for Developing Pancreatic Cancer. Gastroenterology 2021, 161, 1379–1380. [Google Scholar] [CrossRef]
- Salvatore, T.; Marfella, R.; Rizzo, M.R.; Sasso, F.C. Pancreatic cancer and diabetes: A two-way relationship in the perspective of diabetologist. Int. J. Surg. 2015, 21, S72–S77. [Google Scholar] [CrossRef]
- Bonelli, L.; Aste, H.; Bovo, P.; Cavallini, G.; Felder, M.; Gusmaroli, R. Exocrine Pancreatic Cancer, Cigarette Smoking, and Diabetes Mellitus: A Case-Control Study in Northern Italy. Pancreas 2003, 27, 143–149. [Google Scholar] [CrossRef]
- Khan, S.; Safarudin, R.F.; Kupec, J.T. Validation of the ENDPAC model: Identifying new-onset diabetics at risk of pancreatic cancer. Pancreatology 2021, 21, 550–555. [Google Scholar] [CrossRef]
- Sharma, A.; Kandlakunta, H.; Nagpal, S.J.S.; Feng, Z.; Hoos, W.; Petersen, G.M.; Chari, S.T. Model to Determine Risk of Pancreatic Cancer in Patients With New-Onset Diabetes. Gastroenterology 2018, 155, 730–739.e3. [Google Scholar] [CrossRef]
- Chen, W.; Butler, R.K.; Lustigova, E.; Chari, S.T.; Wu, B.U. Validation of the Enriching New-Onset Diabetes for Pancreatic Cancer Model in a Diverse and Integrated Healthcare Setting. Dig. Dis. Sci. 2021, 66, 78–87. [Google Scholar] [CrossRef]
- Mueller, A.M.; Meier, C.R.; Jick, S.S.; Schneider, C. Characterization of the deterioration of diabetes control in patients with a subsequent diagnosis of pancreatic cancer: A descriptive study. Pancreatology 2022, 22, 387–395. [Google Scholar] [CrossRef]
- Lee, S.; Hwang, H.K.; Kang, C.M.; Lee, W.J. Adverse Oncologic Impact of New-Onset Diabetes Mellitus on Recurrence in Resected Pancreatic Ductal Adenocarcinoma: A Comparison With Long-standing and Non–Diabetes Mellitus Patients. Pancreas 2018, 47, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Kartsonaki, C.; Guo, Y.; Bragg, F.; Yang, L.; Bian, Z. Diabetes, plasma glucose and incidence of pancreatic cancer: A prospective study of 0.5 million C hinese adults and a meta-analysis of 22 cohort studies. Int. J. Cancer 2017, 140, 1781–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, S.J.S.; Kandlakunta, H.; Her, T.; Sharma, A.; Sannapaneni, S.; Smyrk, T.C.; Velamala, P.; Garg, S.K.; Rakshit, K.; Majumder, S.; et al. Pancreatic ductal adenocarcinoma is associated with a unique endocrinopathy distinct from type 2 diabetes mellitus. Pancreatology 2020, 20, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Smyrk, T.C.; Levy, M.J.; Topazian, M.A.; Chari, S.T. Fasting Blood Glucose Levels Provide Estimate of Duration and Progression of Pancreatic Cancer Before Diagnosis. Gastroenterology 2018, 155, 490–500.e2. [Google Scholar] [CrossRef] [PubMed]
- Morales-Oyarvide, V.; Mino-Kenudson, M.; Ferrone, C.R.; Sahani, D.V.; Pergolini, I.; Negreros-Osuna, A.A.; Warshaw, A.L.; Lillemoe, K.D.; Castillo, C.F.-D. Diabetes mellitus in intraductal papillary mucinous neoplasm of the pancreas is associated with high-grade dysplasia and invasive carcinoma. Pancreatology 2017, 17, 920–926. [Google Scholar] [CrossRef]
- Pergolini, I.; Jäger, C.; Safak, O.; Göß, R.; Novotny, A.; Ceyhan, G.O.; Friess, H.; Demir, I.E. Diabetes and Weight Loss Are Associated With Malignancies in Patients With Intraductal Papillary Mucinous Neoplasms. Clin. Gastroenterol. Hepatol. 2021, 19, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Pergolini, I.; Schorn, S.; Jäger, C.; Göß, R.; Novotny, A.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Diabetes mellitus in intraductal papillary mucinous neoplasms: A systematic review and meta-analysis. Surgery 2021, 169, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular Mechanism of Pancreatic Stellate Cells Activation in Chronic Pancreatitis and Pancreatic Cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Kim, J.; Park, H.; Lee, E.; Yoon, K. Pancreatic stellate cells in the islets as a novel target to preserve the pancreatic β-cell mass and function. J. Diabetes Investig. 2020, 11, 268–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korc, M. Pathogenesis of Pancreatic Cancer-Related Diabetes Mellitus: Quo Vadis? Pancreas 2019, 48, 594–597. [Google Scholar] [CrossRef]
- Nunemaker, C.S. Considerations for Defining Cytokine Dose, Duration, and Milieu That Are Appropriate for Modeling Chronic Low-Grade Inflammation in Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ni, Q.; Sun, J.; Xu, M.; Xie, J.; Zhang, J.; Fang, Y.; Ning, G.; Wang, Q. Paraneoplastic β Cell Dedifferentiation in Nondiabetic Patients with Pancreatic Cancer. J. Clin. Endocrinol. Metab. 2020, 105, e1489–e1503. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhou, Y.; Li, Q.; Zhou, Q.; Tan, L.; Song, Y.; Zhao, X.; Yu, M.; Zheng, S.; Ye, H.; et al. Analysis of global gene expression profiles suggests a role of acute inflammation in type 3C diabetes mellitus caused by pancreatic ductal adenocarcinoma. Diabetologia 2015, 58, 835–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javeed, N.; Sagar, G.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Bhattacharya, S. Pancreatic Cancer–Derived Exosomes Cause Paraneoplastic β-cell Dysfunction. Clin Cancer Res. 2015, 21, 1722–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, G.; Ramachandran, V.; Javeed, N.; Arumugam, T.; Dutta, S.; Klee, G.G.; Klee, E.W.; Smyrk, T.C.; Bamlet, W.; Han, J.J.; et al. Adrenomedullin is Up-regulated in Patients With Pancreatic Cancer and Causes Insulin Resistance in β Cells and Mice. Gastroenterology 2012, 143, 1510–1517.e1. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Knezetic, J.A.; Strömmer, L.; Permert, J.; Larsson, J.; Adrian, T.E. The Intracellular Mechanism of Insulin Resistance in Pancreatic Cancer Patients 1. J. Clin. Endocrinol. Metab. 2000, 85, 1232–1238. [Google Scholar] [CrossRef]
- Hirano, K.; Isogawa, A.; Tada, M.; Isayama, H.; Takahara, N.; Miyabayashi, K.; Mizuno, S.; Mohri, D.; Kawakubo, K.; Sasaki, T.; et al. Long-Term Prognosis of Autoimmune Pancreatitis in Terms of Glucose Tolerance. Pancreas 2012, 41, 691–695. [Google Scholar] [CrossRef]
- Ito, T.; Nakamura, T.; Fujimori, N.; Niina, Y.; Igarashi, H.; Oono, T. Characteristics of pancreatic diabetes in patients with autoimmune pancreatitis: Pancreatic diabetes in AIP. J. Dig. Dis. 2011, 12, 210–216. [Google Scholar] [CrossRef]
- Ito, T.; Kawabe, K.; Arita, Y.; Hisano, T.; Igarashi, H.; Funakoshi, A.; Sumii, T.; Yamanaka, T.; Takayanagi, R. Evaluation of Pancreatic Endocrine and Exocrine Function in Patients With Autoimmune Pancreatitis. Pancreas 2007, 34, 254–259. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cell Type | Hormone Released | Action of the Hormone | Effect of Hormone Loss |
|---|---|---|---|
| α-cell | glucagon |
|
|
| β-cell | insulin |
|
|
| δ-cell | somatostatin |
|
|
| PP-cell | pancreatic polypeptide |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciochina, M.; Balaban, D.V.; Manucu, G.; Jinga, M.; Gheorghe, C. The Impact of Pancreatic Exocrine Diseases on the β-Cell and Glucose Metabolism—A Review with Currently Available Evidence. Biomolecules 2022, 12, 618. https://doi.org/10.3390/biom12050618
Ciochina M, Balaban DV, Manucu G, Jinga M, Gheorghe C. The Impact of Pancreatic Exocrine Diseases on the β-Cell and Glucose Metabolism—A Review with Currently Available Evidence. Biomolecules. 2022; 12(5):618. https://doi.org/10.3390/biom12050618
Chicago/Turabian StyleCiochina, Marina, Daniel Vasile Balaban, George Manucu, Mariana Jinga, and Cristian Gheorghe. 2022. "The Impact of Pancreatic Exocrine Diseases on the β-Cell and Glucose Metabolism—A Review with Currently Available Evidence" Biomolecules 12, no. 5: 618. https://doi.org/10.3390/biom12050618