
(−)-Epigallocatechin-3-Gallate Diminishes Intra-and Extracellular Amyloid-Induced Cytotoxic Effects on Cholinergic-like Neurons from Familial Alzheimer’s Disease PSEN1 E280A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cholinergic-like Neuron (ChLN) Differentiation
2.2. Astrocyte-like Cells (ALCs) Differentiation
2.3. Assay Protocol
2.4. Immunofluorescence Analysis
2.5. Flow Cytometry Analysis
2.6. Western Blot Analysis
2.7. Evaluation of Intracellular Hydrogen Peroxide (H2O2) by Fluorescence Microscopy
2.8. Analysis of Mitochondrial Membrane Potential (ΔΨm) by Fluorescence Microscopy
2.9. Measurement of Extracellular (e)Aβ42 Peptide in Culture Medium
2.10. Intracellular Calcium Imaging
2.11. Measurement of Interleukin-6 (IL-6) in Culture Medium
2.12. Photomicrography and Image Analysis
2.13. Data Analysis
3. Results
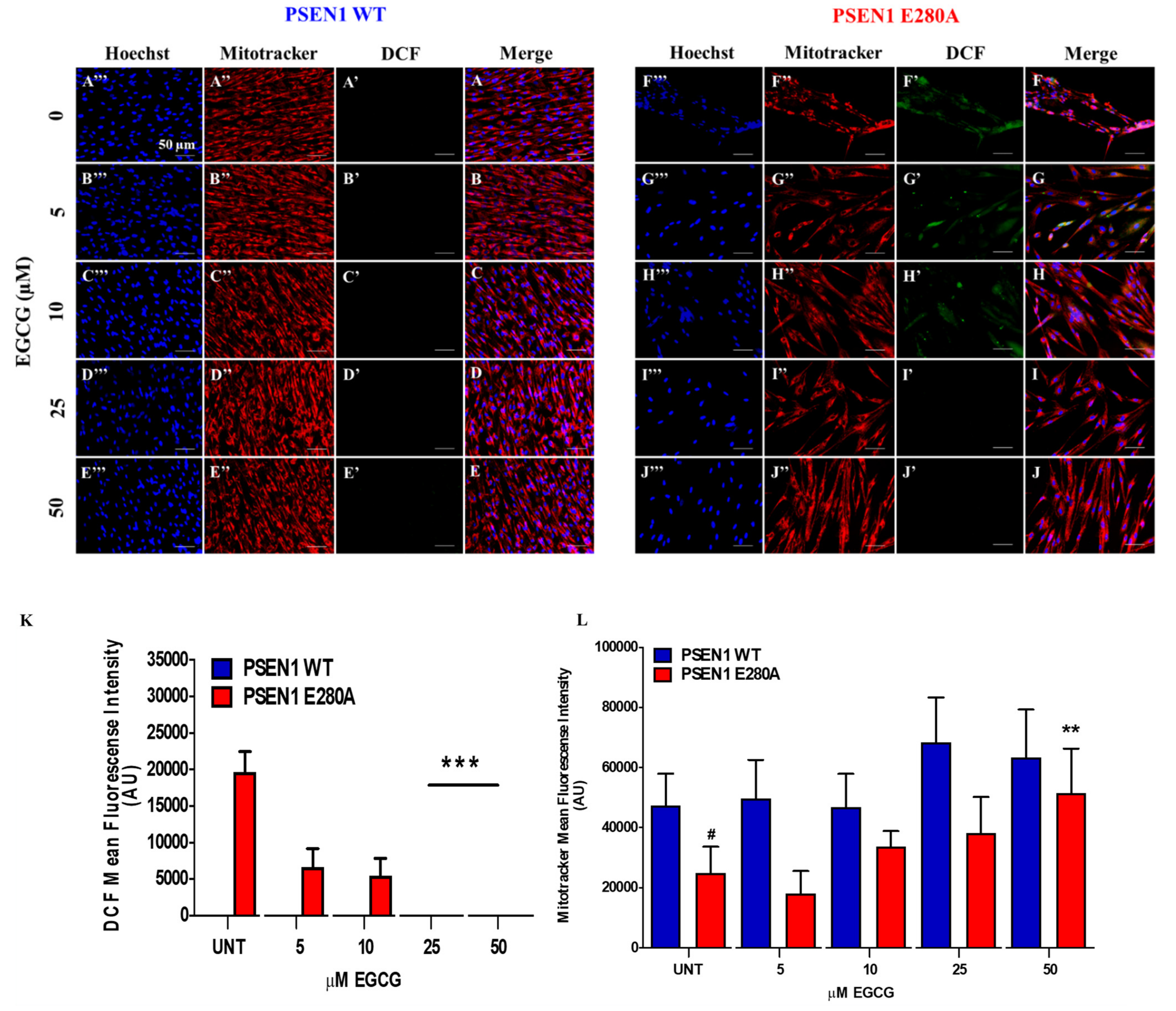
3.1. EGCG Scavenges Reactive Oxygen Species (ROS) and Reestablishes the Mitochondrial Membrane Potential (ΔΨm) in PSEN1 E280A ChLNs
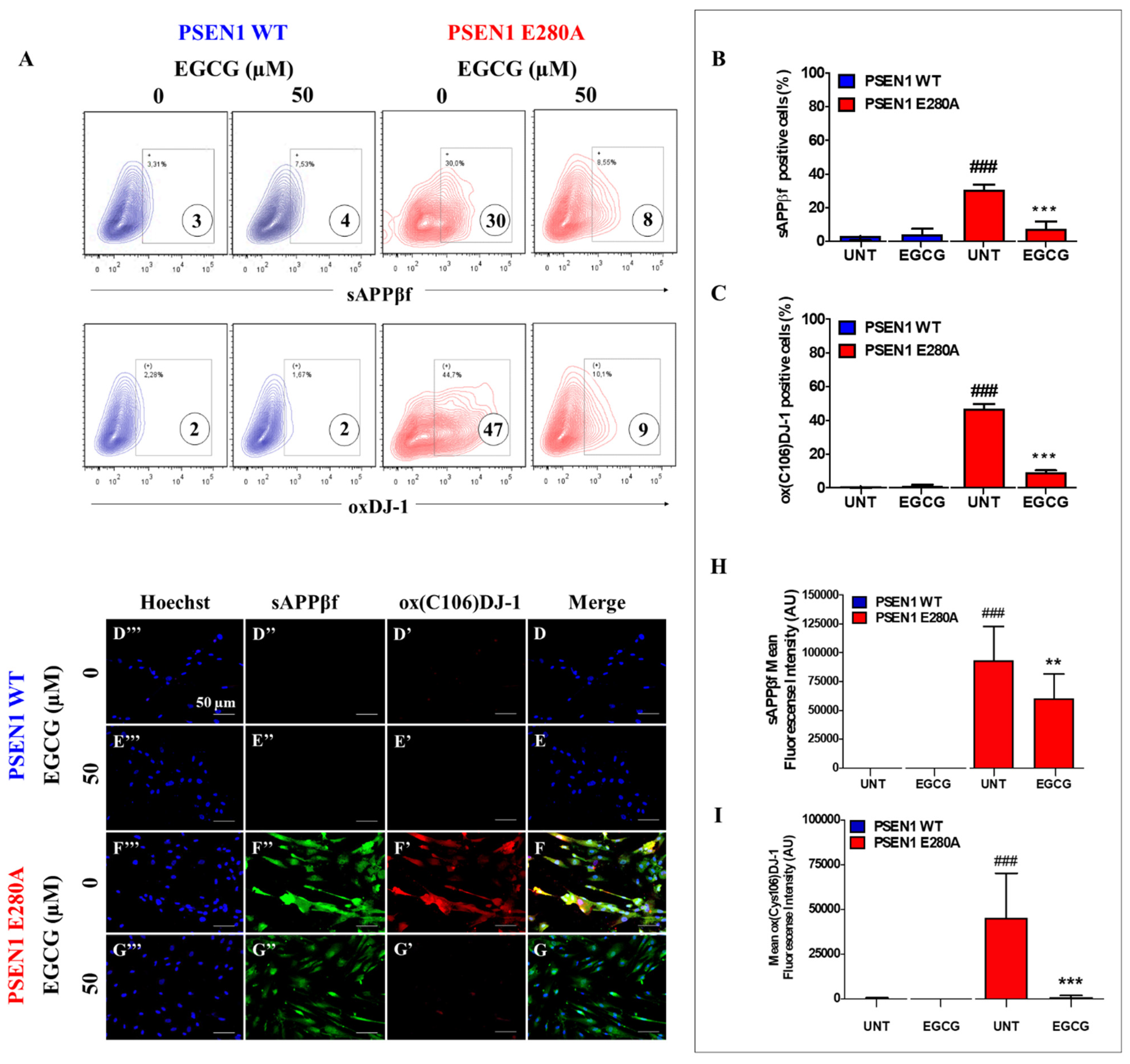
3.2. EGCG Partially Reduces Intracellular sAPPβf Aggregation but Completely Inhibits Oxidized DJ-1 in PSEN1 E280A ChLNs
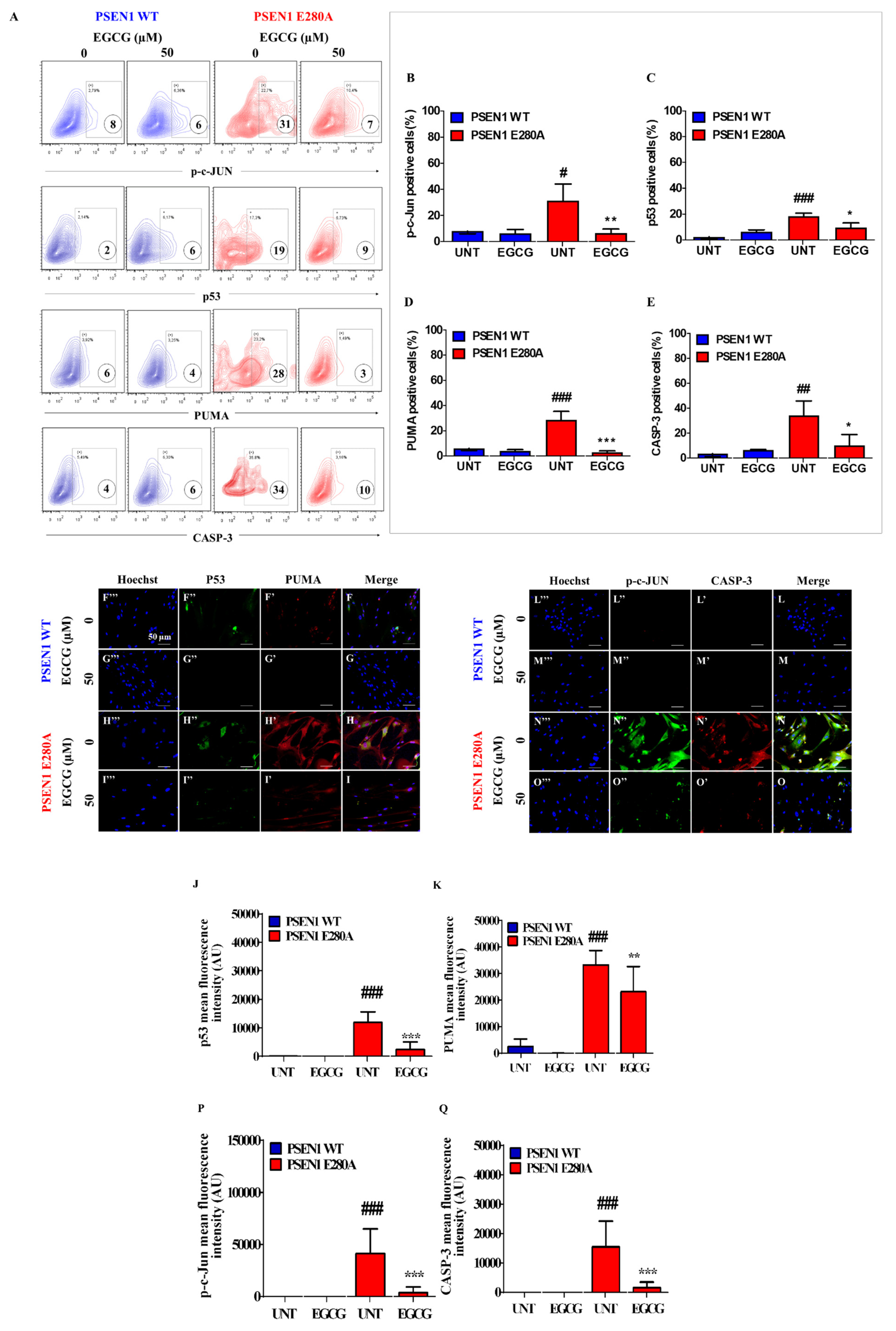
3.3. EGCG Blocks Apoptosis Signaling in PSEN1 E280A ChLNs
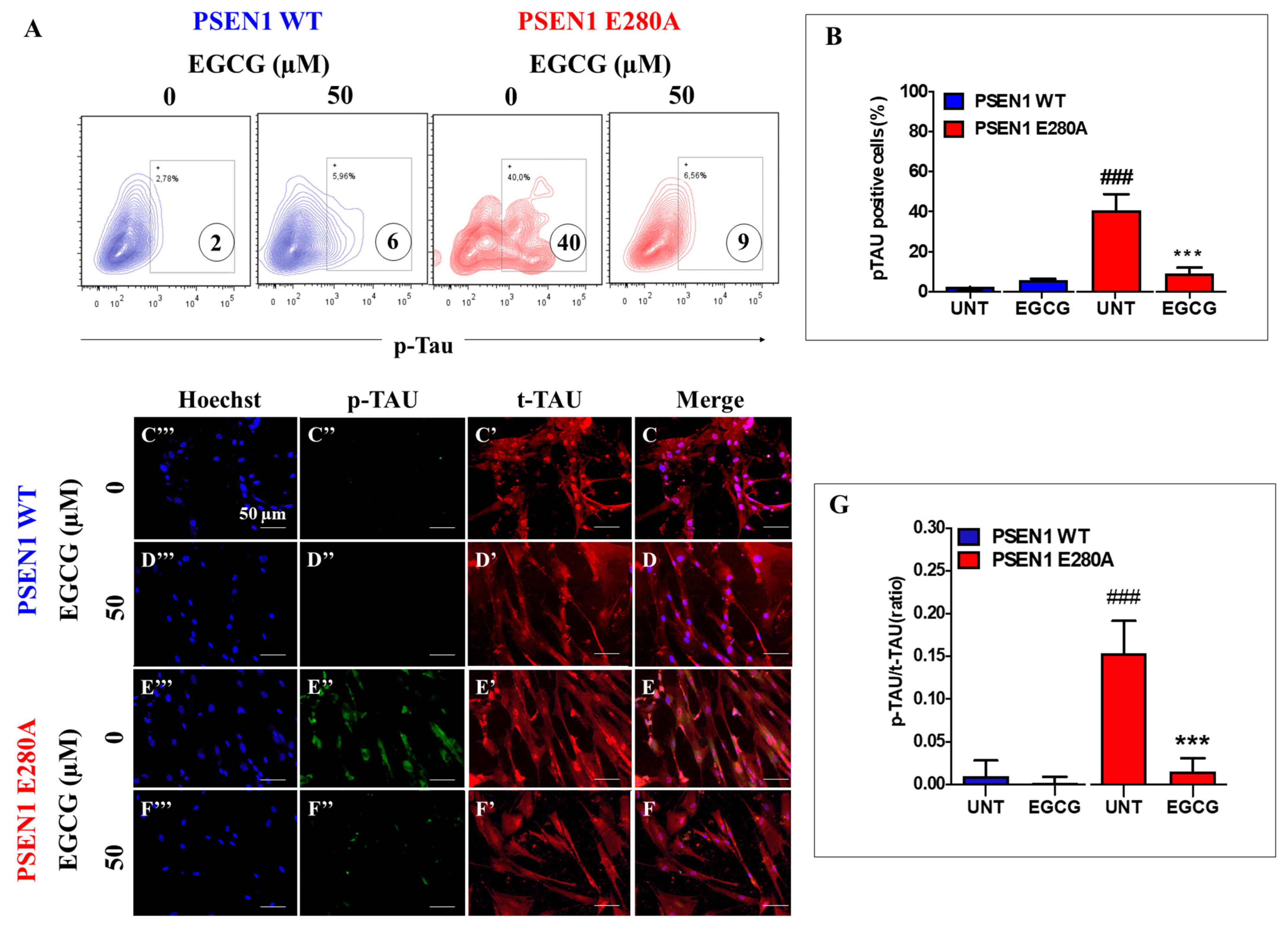
3.4. EGCG Inhibits TAU Phosphorylation in PSEN1 E280A ChLNs
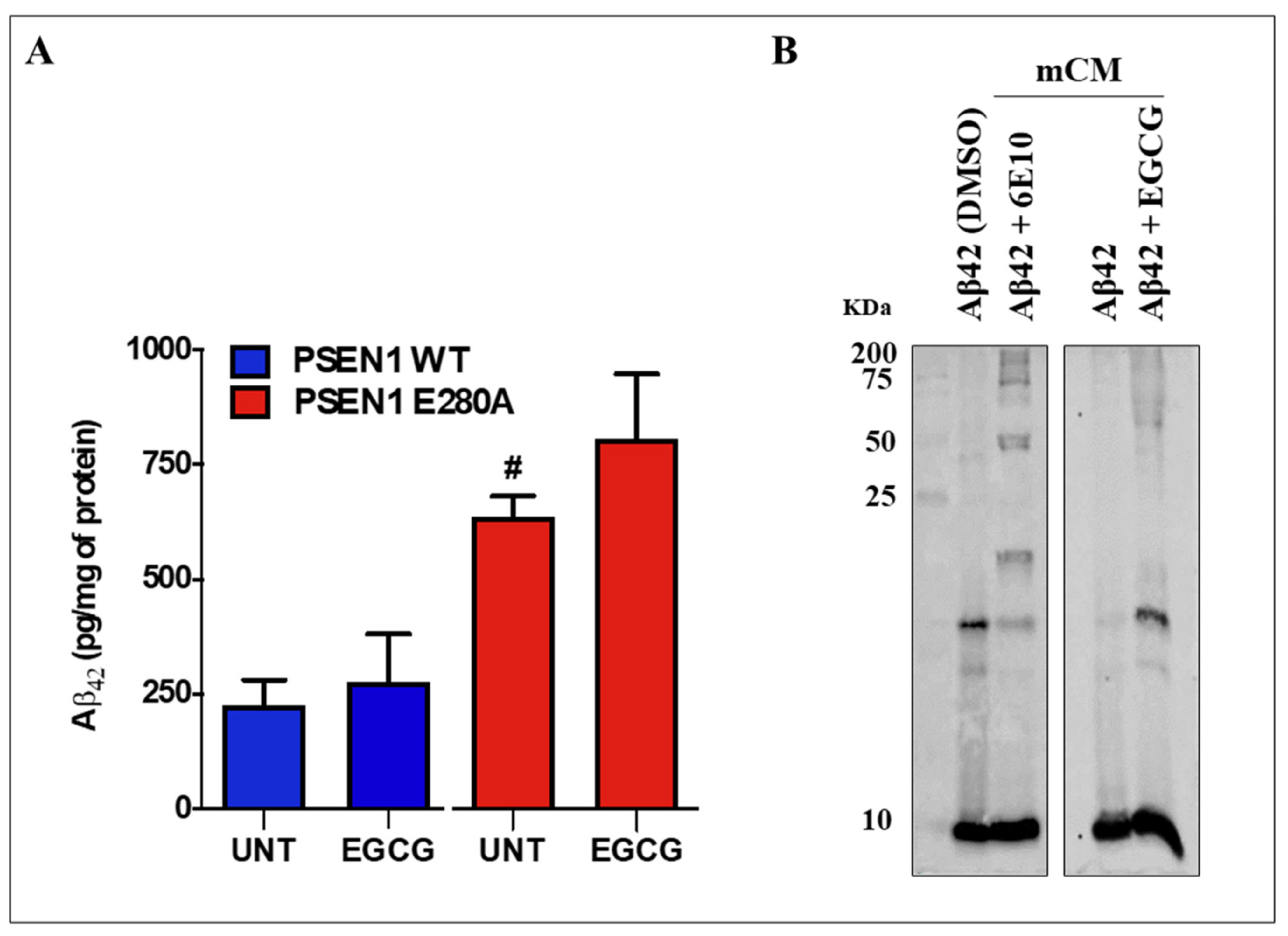
3.5. EGCG Does Not Reduce the Levels of Extracellular Aβ42 (eAβ42) Protein Fragment in PSEN 1 E280A ChLNs
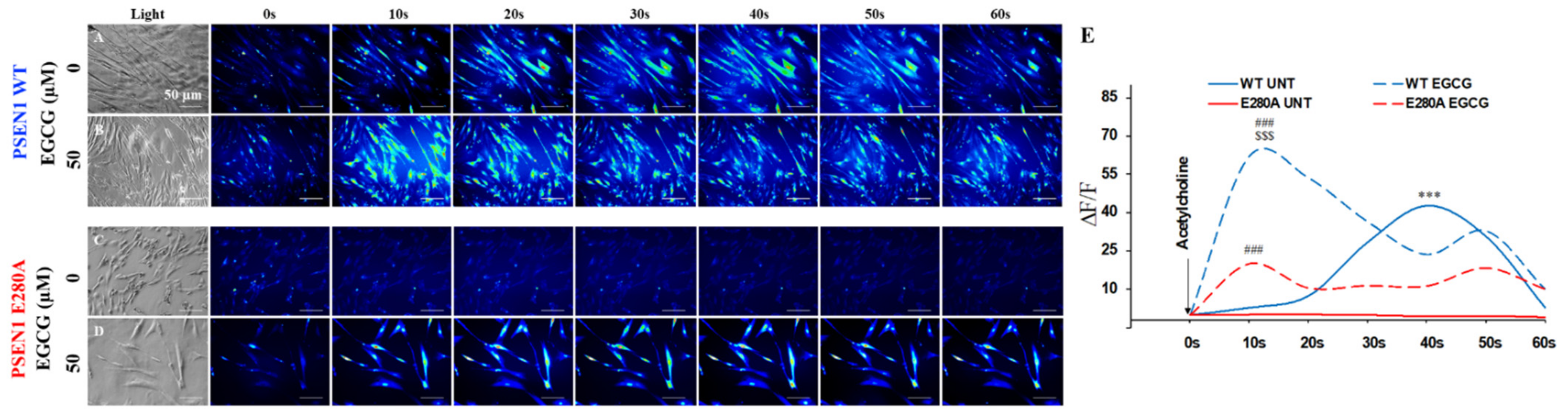
3.6. EGCG Recovers Ca2+ Dysregulation in PSEN1 E280A ChLNs
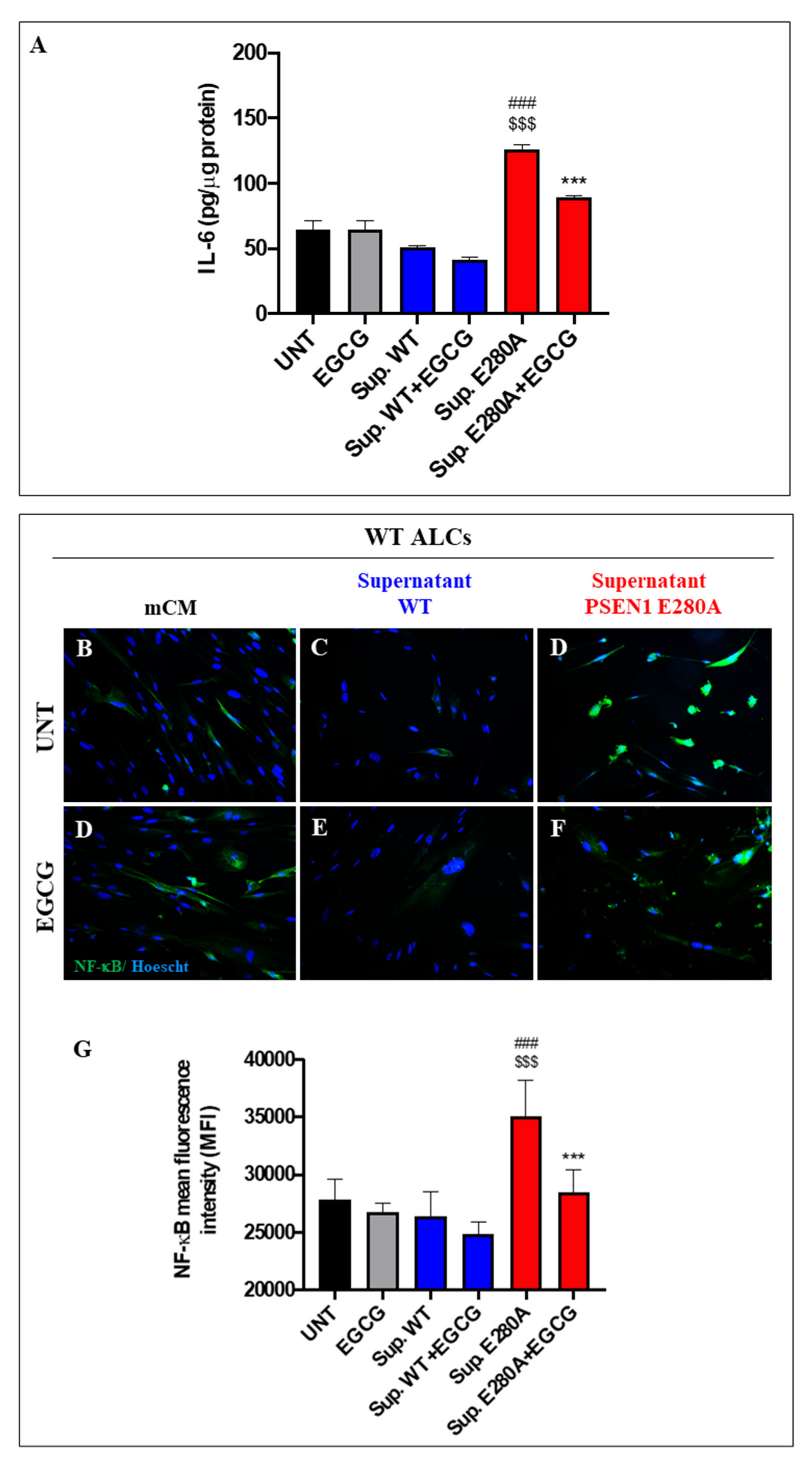
3.7. EGCG Dramatically Reduces Reactive Astrocyte-like Cells Derived from Menstrual Stromal Cells (MenSCs) Exposed to ChLNs’ Supernatant Culture Medium
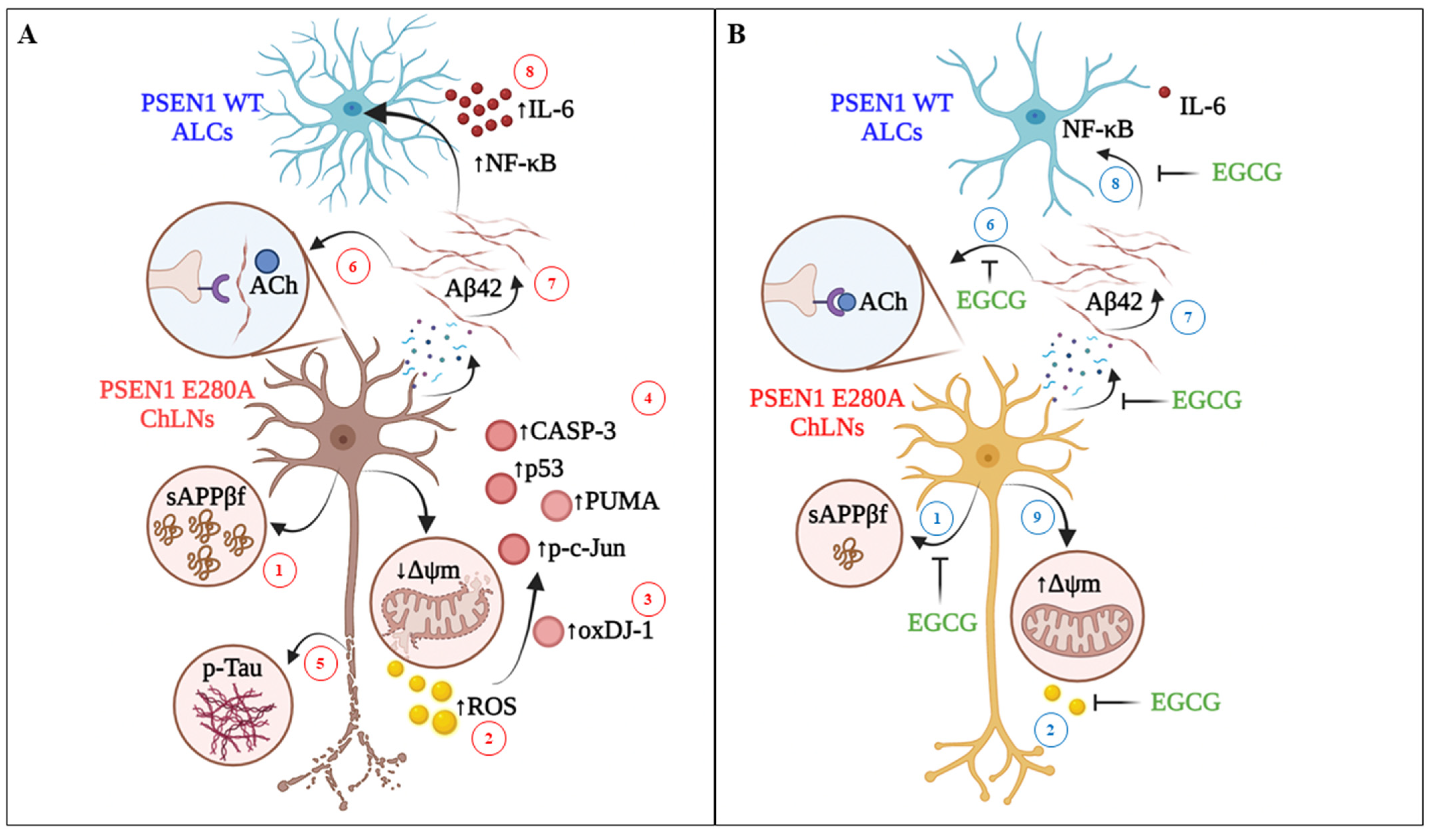
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Bekdash, R.A. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1273. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Tan, J.Z.A.; Gleeson, P.A. The role of membrane trafficking in the processing of amyloid precursor protein and production of amyloid peptides in Alzheimer’s disease. Biochim. Biophys. Acta Biomembr. 2019, 1861, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure and Function of the γ-Secretase Complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [Green Version]
- Lopera, F. Clinical Features of Early-Onset Alzheimer Disease in a Large Kindred With an E280A Presenilin-1 Mutation. JAMA 1997, 277, 793. [Google Scholar] [CrossRef]
- Lalli, M.A.; Cox, H.C.; Arcila, M.L.; Cadavid, L.; Moreno, S.; Garcia, G.; Madrigal, L.; Reiman, E.M.; Arcos-Burgos, M.; Bedoya, G.; et al. Origin of the PSEN1 E280A mutation causing early-onset Alzheimer’s disease. Alzheimers Dement. 2014, 10 (Suppl. S5), S277–S283. [Google Scholar] [CrossRef] [Green Version]
- Fuller, J.; Cronin-Golomb, A.; Gatchel, J.R.; Norton, D.J.; Guzmán-Vélez, E.; Jacobs, H.; Hanseeuw, B.; Pardilla-Delgado, E.; Artola, A.; Baena, A.; et al. Biological And Cognitive Markers Of Presenilin1 E280a Autosomal Dominant Alzheimer’s Disease: A Comprehensive Review Of The Colombian Kindred. J. Prev. Alzheimers Dis. 2019, 6, 112–120. [Google Scholar] [CrossRef]
- Tariot, P.N.; Lopera, F.; Langbaum, J.B.; Thomas, R.G.; Hendrix, S.; Schneider, L.S.; Rios-Romenets, S.; Giraldo, M.; Acosta, N.; Tobon, C.; et al. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. 2018, 4, 150–160. [Google Scholar]
- Rios-Romenets, S.; Lopera, F.; Sink, K.M.; Hu, N.; Lian, Q.; Guthrie, H.; Smith, J.; Cho, W.; Mackey, H.; Langbaum, J.B.; et al. Baseline demographic, clinical, and cognitive characteristics of the Alzheimer’s Prevention Initiative (API) Autosomal-Dominant Alzheimer’s Disease Colombia Trial. Alzheimers Dement. 2020, 16, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, Y.T.; Sperling, R.A.; Norton, D.J.; Baena, A.; Arboleda-Velasquez, J.F.; Cosio, D.; Schultz, A.; LaPoint, M.; Guzman-Velez, E.; Miller, J.B.; et al. Association Between Amyloid and Tau Accumulation in Young Adults With Autosomal Dominant Alzheimer Disease. JAMA Neurol. 2018, 75, 548–556. [Google Scholar] [CrossRef]
- Sanchez, J.S.; Hanseeuw, B.J.; Lopera, F.; Sperling, R.A.; Baena, A.; Bocanegra, Y.; Aguillon, D.; Guzmán-Vélez, E.; Pardilla-Delgado, E.; Ramirez-Gomez, L.; et al. Longitudinal amyloid and tau accumulation in autosomal dominant Alzheimer’s disease: Findings from the Colom-bia-Boston (COLBOS) biomarker study. Alzheimers Res. Ther. 2021, 13, 27. [Google Scholar] [CrossRef]
- Soto-Mercado, V.; Mendivil-Perez, M.; Velez-Pardo, C.; Lopera, F.; Jimenez-Del-Rio, M. Cholinergic-like neurons carrying PSEN1 E280A mutation from familial Alzheimer’s disease reveal in-traneuronal sAPPβ fragments accumulation, hyperphosphorylation of TAU, oxidative stress, apoptosis and Ca2+ dysregulation: Thera-peutic implications. PLoS ONE 2020, 15, e0221669. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline. Alzheimers Dement. 2020, 6, e12050. [Google Scholar]
- Soto-Mercado, V.; Mendivil-Perez, M.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Multi-Target Effects of the Cannabinoid CP55940 on Familial Alzheimer’s Disease PSEN1 E280A Choliner-gic-Like Neurons: Role of CB1 Receptor. J. Alzheimers Dis. 2021, 82, S359–S378. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef]
- Lakey-Beitia, J.; Burillo, A.M.; La Penna, G.; Hegde, M.L.; Rao, K. Polyphenols as Potential Metal Chelation Compounds Against Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 82, S335–S357. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Ahn, N.-H.; Choi, S.-B.; Kwon, Y.; Yang, S.-H. Natural Products Targeting Amyloid Beta in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2341. [Google Scholar] [CrossRef]
- Andrich, K.; Bieschke, J. The Effect of (−)-Epigallo-catechin-(3)-gallate on Amyloidogenic Proteins Suggests a Common Mechanism. Adv. Exp. Med. Biol. 2015, 863, 139–161. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.R.; Choi, H.J.; Kang, Y.G.; Kim, J.K.; Shin, J.-W. In vitro study on anti-inflammatory effects of epigallocatechin-3-gallate-loaded nano- and microscale particles. Int. J. Nanomed. 2017, 12, 7007–7013. [Google Scholar] [CrossRef] [Green Version]
- Pagano, K.; Tomaselli, S.; Molinari, H.; Ragona, L. Natural Compounds as Inhibitors of Aβ Peptide Aggregation: Chemical Requirements and Molecular Mechanisms. Front. Neurosci. 2020, 14, 619667. [Google Scholar] [CrossRef]
- Fernandes, L.; Cardim-Pires, T.R.; Foguel, D.; Palhano, F.L. Green Tea Polyphenol Epigallocatechin-Gallate in Amyloid Aggregation and Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 718188. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhang, N.; Guan, P.; Chen, P.; Ding, S.; Hou, T.; Hu, X.; Wang, J.; Wang, C. Drug-based magnetic imprinted nanoparticles: Enhanced lysozyme amyloid fibrils cleansing and anti-amyloid fibrils toxicity. Int. J. Biol. Macromol. 2020, 153, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Bansal, Y.; Kumar, R.; Bansal, G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorganic Med. Chem. 2020, 28, 115327. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Per-spective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintero-Espinosa, D.; Soto-Mercado, V.; Quintero-Quinchia, C.; Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Latent Tri-lineage Potential of Human Menstrual Blood–Derived Mesenchymal Stromal Cells Revealed by Specific In Vitro Culture Conditions. Mol. Neurobiol. 2021, 58, 5194–5209. [Google Scholar] [CrossRef]
- Carrero, I.; Gonzalo, M.R.; Martin, B.; Sanz-Anquela, J.M.; Arevalo-Serrano, J.; Gonzalo-Ruiz, A. Oligomers of β-amyloid protein (Aβ1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1β, tumour necrosis factor-α, and a nuclear factor κ-B mechanism in the rat brain. Exp. Neurol. 2012, 236, 215–227. [Google Scholar] [CrossRef]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Direct transdifferentiation of human Wharton’s jelly mesenchymal stromal cells into cholinergic-like neurons. J. Neurosci. Methods 2018, 312, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Armijo, E.; Gonzalez, C.; Shahnawaz, M.; Flores, A.; Davis, B.; Soto, C. Increased susceptibility to Abeta toxicity in neuronal cultures derived from familial Alzheimer’s disease (PSEN1-A246E) induced pluripotent stem cells. Neurosci. Lett. 2017, 639, 74–81. [Google Scholar] [CrossRef]
- Pap, P.; Kőszeghy, A.; Szűcs, G.; Rusznák, Z. Cytoplasmic Ca2+ concentration changes evoked by cholinergic stimulation in primary astrocyte cultures prepared from the rat cochlear nucleus. Hear. Res. 2009, 255, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi-Tonosaki, M.; Obata, M.; Haruki, A.; Himi, T.; Kosaka, J. Acetylcholine induces Ca2+ signaling in chicken retinal pigmented epithelial cells during dedifferentiation. Am. J. Physiol. Cell Physiol. 2009, 296, C1195–C1206. [Google Scholar] [CrossRef] [Green Version]
- Lazic, S.E.; Clarke-Williams, C.J.; Munafo, M.R. What exactly is ‘N’ in cell culture and animal experiments? PLoS Biol. 2018, 16, e2005282. [Google Scholar] [CrossRef] [Green Version]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the Molecular Mechanism(s) by Which EGCG Treatment Remodels Mature Amyloid Fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, L.; Messias, B.; Pereira-Neves, A.; Azevedo, E.P.; Araújo, J.; Foguel, D.; Palhano, F.L. Green Tea Polyphenol Microparticles Based on the Oxidative Coupling of EGCG Inhibit Amyloid Aggrega-tion/Cytotoxicity and Serve as a Platform for Drug Delivery. ACS Biomater. Sci. Eng. 2020, 6, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, S.K.; Chidambaram, H.; Boral, D.; Gorantla, N.V.; Balmik, A.A.; Dangi, A.; Ramasamy, S.; Marelli, U.K.; Chinnathambi, S. EGCG impedes human Tau aggregation and interacts with Tau. Sci. Rep. 2020, 10, 12579. [Google Scholar] [CrossRef]
- Andrich, K.; Hegenbart, U.; Kimmich, C.; Kedia, N.; Bergen, H.R.; Schönland, S.; Wanker, E.; Bieschke, J. Aggregation of Full-length Immunoglobulin Light Chains from Systemic Light Chain Amyloidosis (AL) Patients Is Remodeled by Epigallocatechin-3-gallate. J. Biol. Chem. 2017, 292, 2328–2344. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, J.; Derreumaux, P.; Mu, Y. Molecular mechanism of the inhibition of EGCG on the Alzheimer Aβ(1-42) dimer. J. Phys. Chem. B 2013, 117, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Truong, D.T.; Tam, N.M.; Nguyen, M.T. EGCG inhibits the oligomerization of amyloid beta (16-22) hexamer: Theoretical studies. J. Mol. Graph. Model. 2017, 76, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Stockmann, J.; Beyer, L.; Rudack, T.; Nabers, A.; Gumbart, J.C.; Gerwert, K.; Batista, V.S. The Effect of (−)-Epigallocatechin-3-Gallate on the Amyloid-β Secondary Structure. Biophys. J. 2020, 119, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Epigallocatechin-3-gallate and curcumin suppress amyloid beta-induced beta-site APP cleaving enzyme-1 upregu-lation. Neuroreport 2008, 19, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Guéroux, M.; Fleau, C.; Slozeck, M.; Laguerre, M.; Pianet, I. Epigallocatechin 3-Gallate as an Inhibitor of Tau Phosphorylation and Aggregation: A Molecular and Structural Insight. J. Prev. Alzheimers Dis. 2017, 4, 218–225. [Google Scholar]
- He, J.; Xu, L.; Yang, L.; Wang, X. Epigallocatechin Gallate Is the Most Effective Catechin against Antioxidant Stress via Hydrogen Peroxide and Radical Scavenging Activity. Med. Sci. Monit. 2018, 24, 8198–8206. [Google Scholar] [CrossRef]
- Guo, Q.; Zhao, B.; Shen, S.; Hou, J.; Hu, J.; Xin, W. ESR study on the structure–antioxidant activity relationship of tea catechins and their epimers. Biochim. Biophys. Acta (BBA) Gen. Subj. 1999, 1427, 13–23. [Google Scholar] [CrossRef]
- Valcic, S.; Muders, A.; Jacobsen, N.E.; Liebler, D.C.; Timmermann, B.N. Antioxidant chemistry of green tea catechins. Identification of products of the reaction of (−)-epigallocatechin gallate with peroxyl radicals. Chem. Res. Toxicol. 1999, 12, 382–386. [Google Scholar] [CrossRef]
- Pal, S.; Dey, S.K.; Saha, C. Inhibition of Catalase by Tea Catechins in Free and Cellular State: A Biophysical Approach. PLoS ONE 2014, 9, e102460. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, M.R.; Nabavi, S.F.; Daglia, M.; Rastrelli, L.; Nabavi, S.M. Epigallocatechin gallate and mitochondria-A story of life and death. Pharmacol. Res. 2016, 104, 70–85. [Google Scholar] [CrossRef]
- Yao, K.; Ye, P.; Zhang, L.; Tan, J.; Tang, X.; Zhang, Y. Epigallocatechin gallate protects against oxidative stress-induced mitochondria-dependent apoptosis in human lens epi-thelial cells. Mol. Vis. 2008, 14, 217–223. [Google Scholar] [PubMed]
- Srividhya, R.; Kalaiselvi, P. Neuroprotective potential of epigallo catechin-3-gallate in PC-12 cells. Neurochem. Res. 2012, 38, 486–493. [Google Scholar] [CrossRef]
- Koh, S.-H.; Kim, S.H.; Kwon, H.; Park, Y.; Kim, K.S.; Song, C.W.; Kim, J.; Kim, M.-H.; Yu, H.-J.; Henkel, J.S.; et al. Epigallocatechin gallate protects nerve growth factor differentiated PC12 cells from oxidative-radical-stress-induced apoptosis through its effect on phosphoinositide 3-kinase/Akt and glycogen synthase kinase-3. Mol. Brain Res. 2003, 118, 72–81. [Google Scholar] [CrossRef]
- Jimenez-Del-Rio, M.; Velez-Pardo, C. Alzheimer’s Disease, Drosophila melanogaster and Polyphenols. Adv. Exp. Med. Biol. 2015, 863, 21–53. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ju, Y.; Asahi, T.; Sawamura, N. Arctic mutant Abeta40 aggregates on alpha7 nicotinic acetylcholine receptors and inhibits their functions. J. Neurochem. 2014, 131, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhu, Q.; Chen, J.Y.; OuYang, D.; Lu, J.H. The pharmacological activity of epigallocatechin-3-gallate (EGCG) on Alzheimer’s disease animal model: A systematic review. Phytomedicine 2020, 79, 153316. [Google Scholar] [CrossRef]
- Hu, J.; Webster, D.; Cao, J.; Shao, A. The safety of green tea and green tea extract consumption in adults—Results of a systematic review. Regul. Toxicol. Pharmacol. 2018, 95, 412–433. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Bimonte, S.; Muzio, M.R.; Schiavone, V.; Cuomo, A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agents Cancer 2017, 12, 36. [Google Scholar] [CrossRef]
- Hoyos-Ceballos, G.P.; Sánchez-Giraldo, V.; Mendivil-Perez, M.; Jimenez-Del-Rio, M.; Sierra-Garcia, L.; Vélez-Pardo, C.; López-Osorio, B.L. Design of epigallocatechin gallate loaded PLGA/PF127 nanoparticles and their effect upon an oxidative stress model. J. Drug Deliv. Sci. Technol. 2018, 48, 152–160. [Google Scholar] [CrossRef]
- Sánchez-Giraldo, V.; Monsalve, Y.; Palacio, J.; Mendivil-Perez, M.; Sierra, L.; Velez-Pardo, C.; López, B.L.; Jiménez-Del-Rio, M. Role of a novel (−)-epigallocatechin-3-gallate delivery system on the prevention against oxidative stress damage in vitro and in vivo model of Parkinson’s disease. J. Drug Deliv. Sci. Technol. 2020, 55, 101466. [Google Scholar] [CrossRef]
- Ding, S.; Khan, A.I.; Cai, X.; Song, Y.; Lyu, Z.; Du, D.; Dutta, P.; Lin, Y. Overcoming blood–brain barrier transport: Advances in nanoparticle-based drug delivery strategies. Mater. Today 2020, 37, 112–125. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto-Mercado, V.; Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. (−)-Epigallocatechin-3-Gallate Diminishes Intra-and Extracellular Amyloid-Induced Cytotoxic Effects on Cholinergic-like Neurons from Familial Alzheimer’s Disease PSEN1 E280A. Biomolecules 2021, 11, 1845. https://doi.org/10.3390/biom11121845
Soto-Mercado V, Mendivil-Perez M, Velez-Pardo C, Jimenez-Del-Rio M. (−)-Epigallocatechin-3-Gallate Diminishes Intra-and Extracellular Amyloid-Induced Cytotoxic Effects on Cholinergic-like Neurons from Familial Alzheimer’s Disease PSEN1 E280A. Biomolecules. 2021; 11(12):1845. https://doi.org/10.3390/biom11121845
Chicago/Turabian StyleSoto-Mercado, Viviana, Miguel Mendivil-Perez, Carlos Velez-Pardo, and Marlene Jimenez-Del-Rio. 2021. "(−)-Epigallocatechin-3-Gallate Diminishes Intra-and Extracellular Amyloid-Induced Cytotoxic Effects on Cholinergic-like Neurons from Familial Alzheimer’s Disease PSEN1 E280A" Biomolecules 11, no. 12: 1845. https://doi.org/10.3390/biom11121845