
Dissecting Multiple Pathways in the Relaxation Dynamics of Helix <==> Coil Transitions with Optimum Dimensionality Reduction


Abstract
:
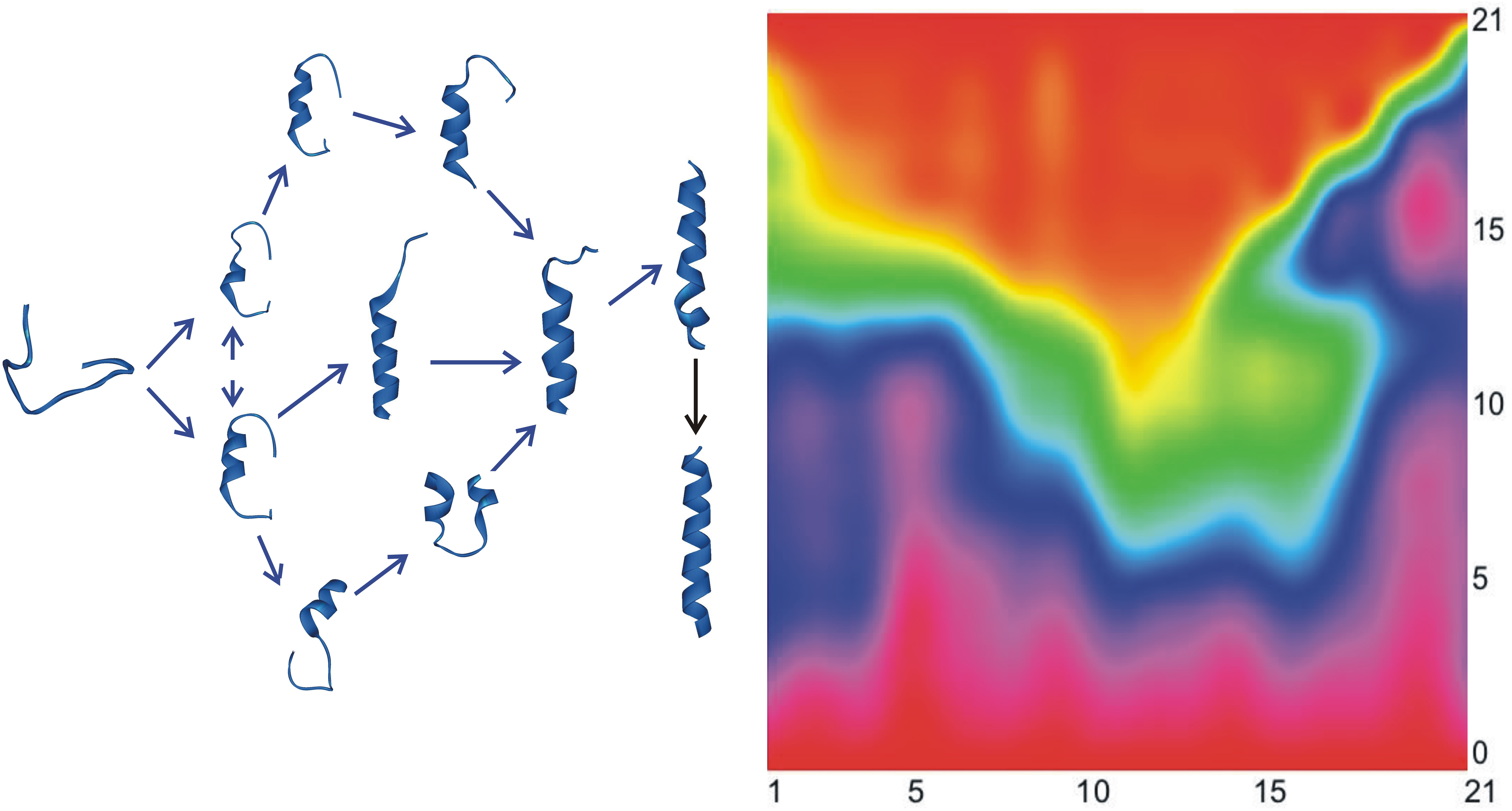{kind=link}
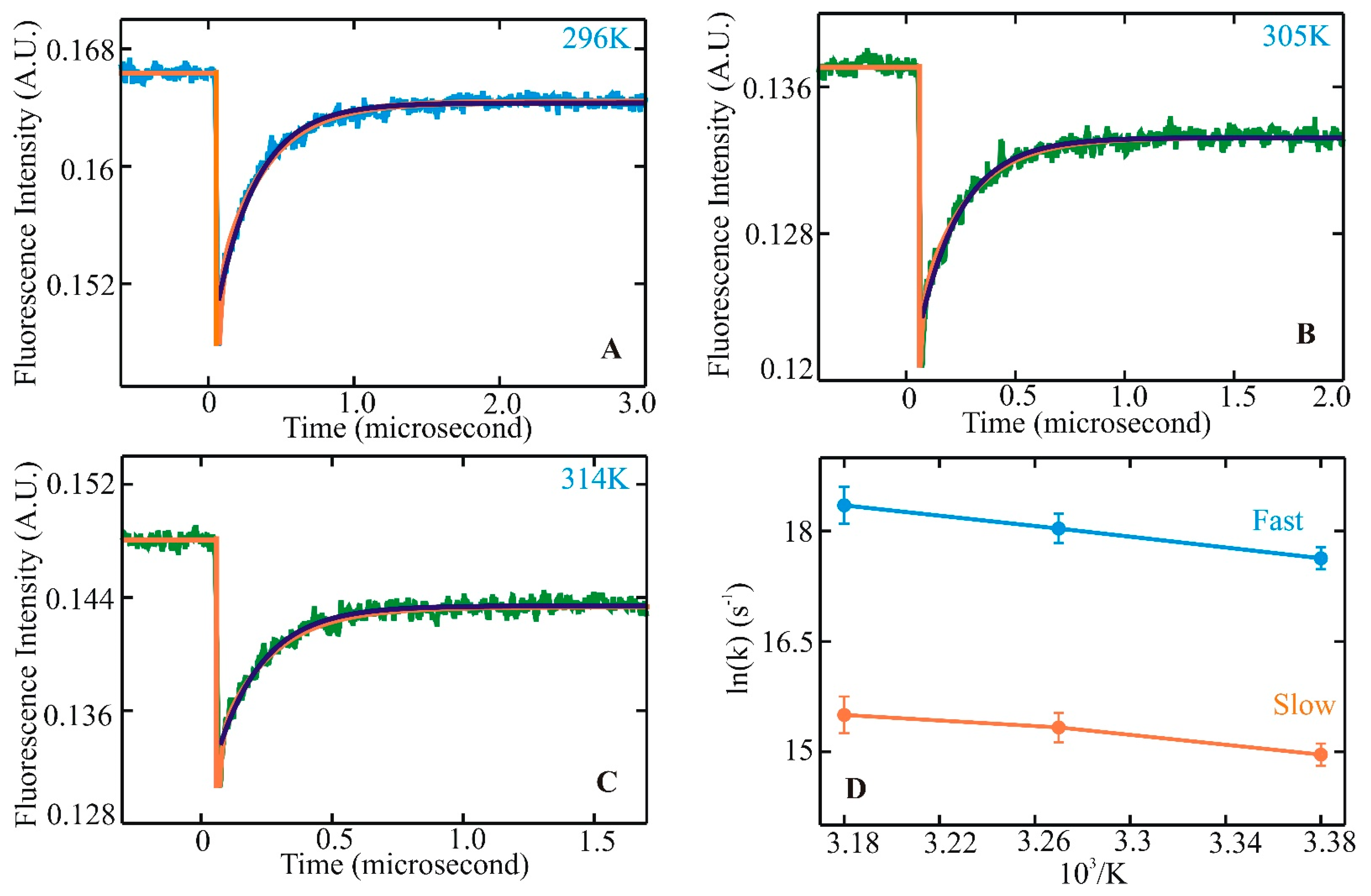{kind=link}
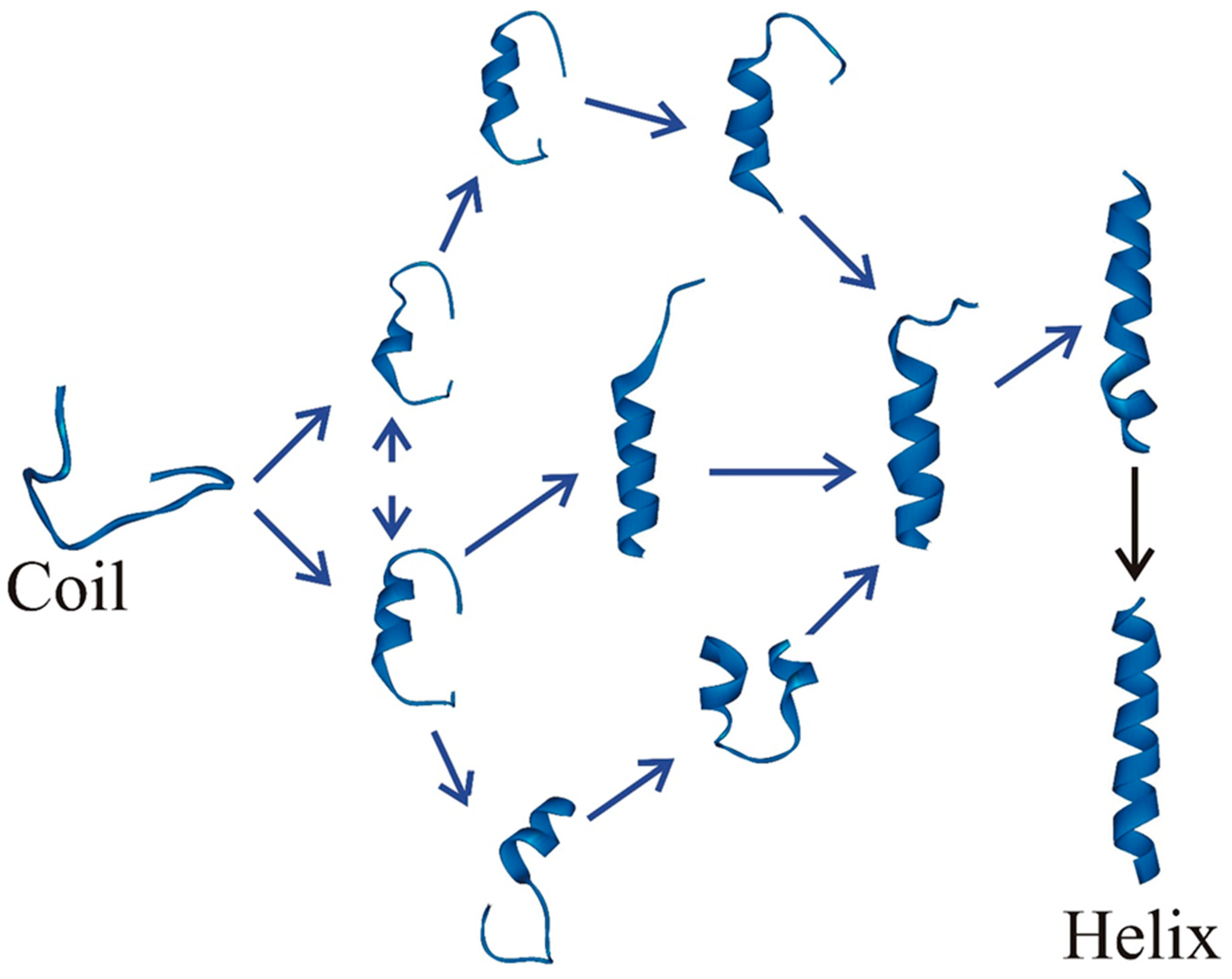{kind=link}
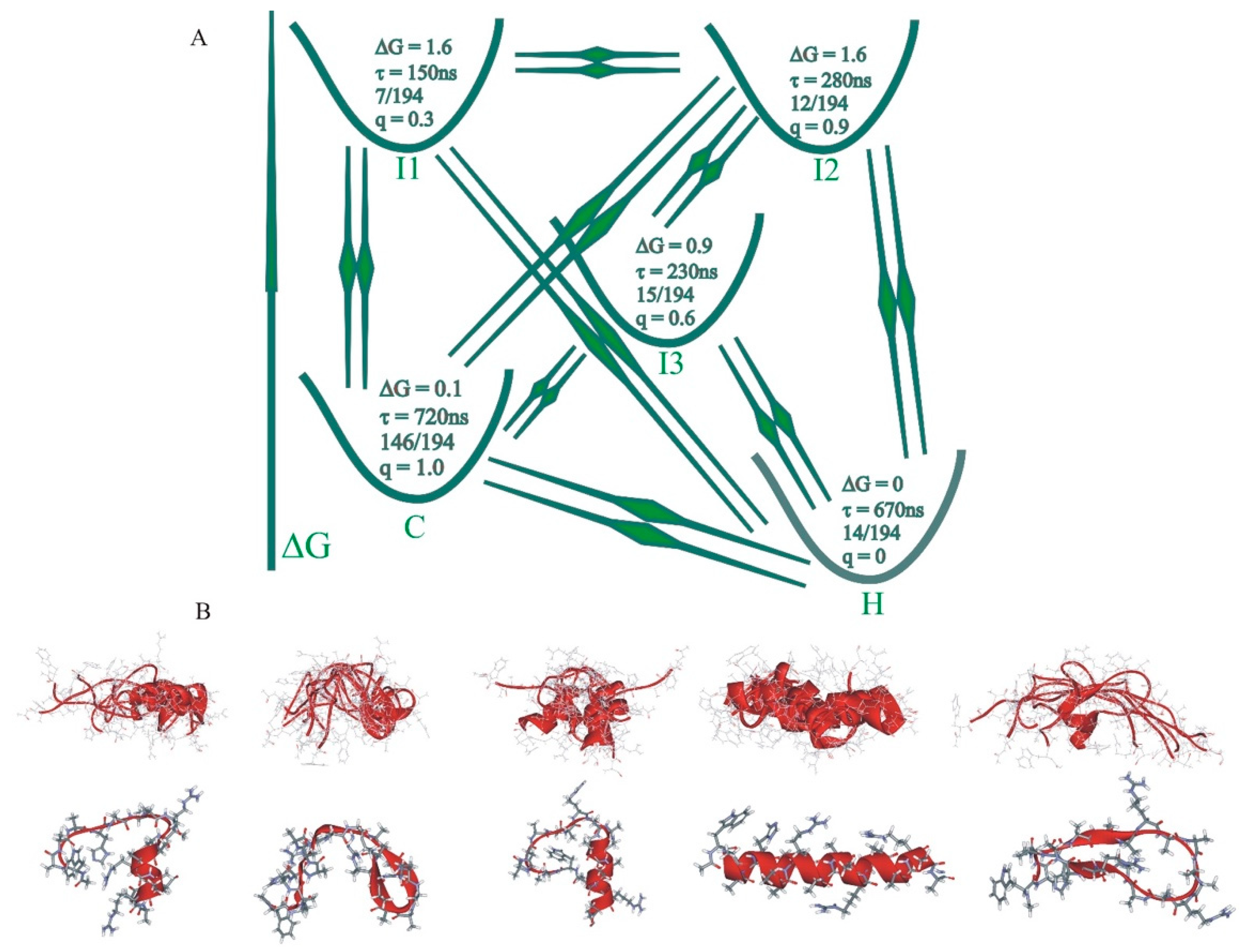{kind=link}
{kind=link}
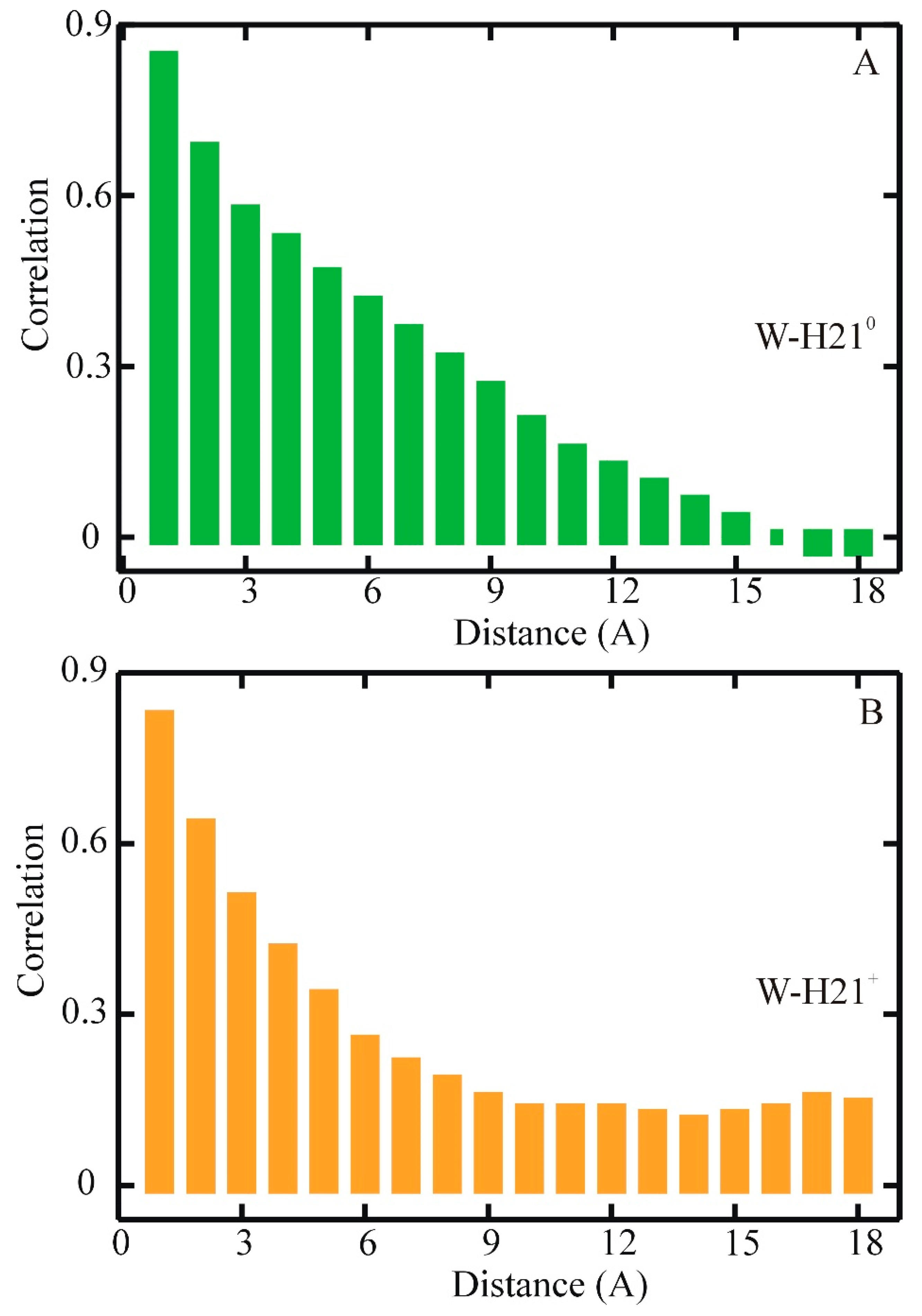{kind=link}
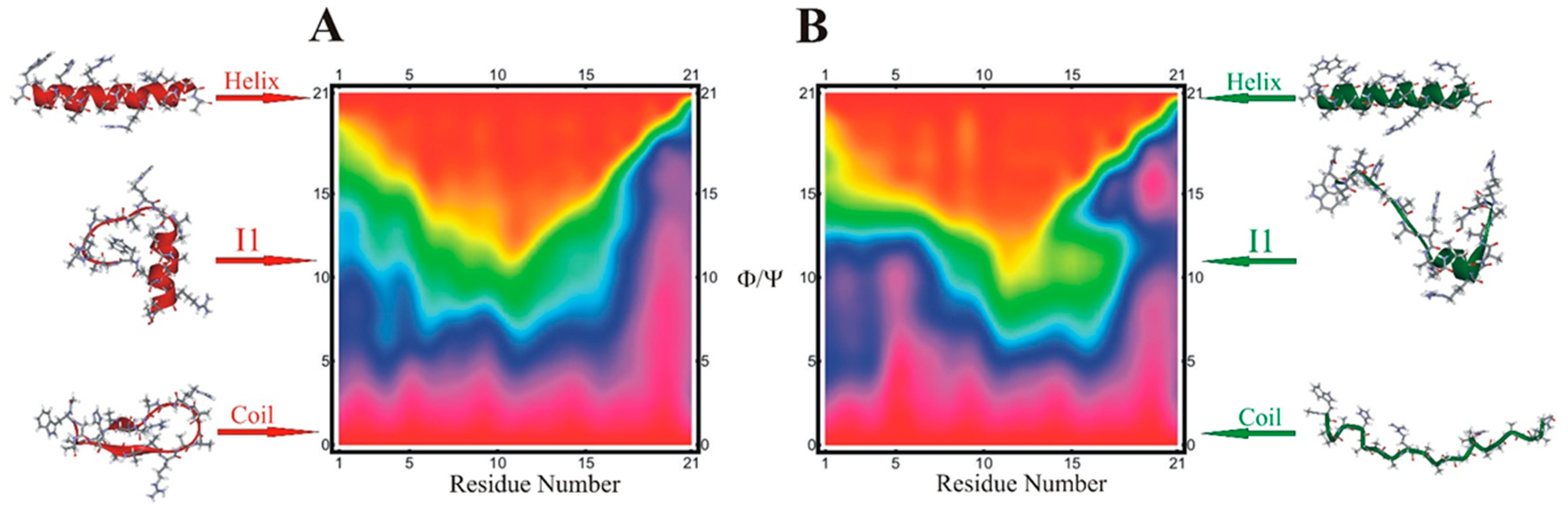{kind=link}
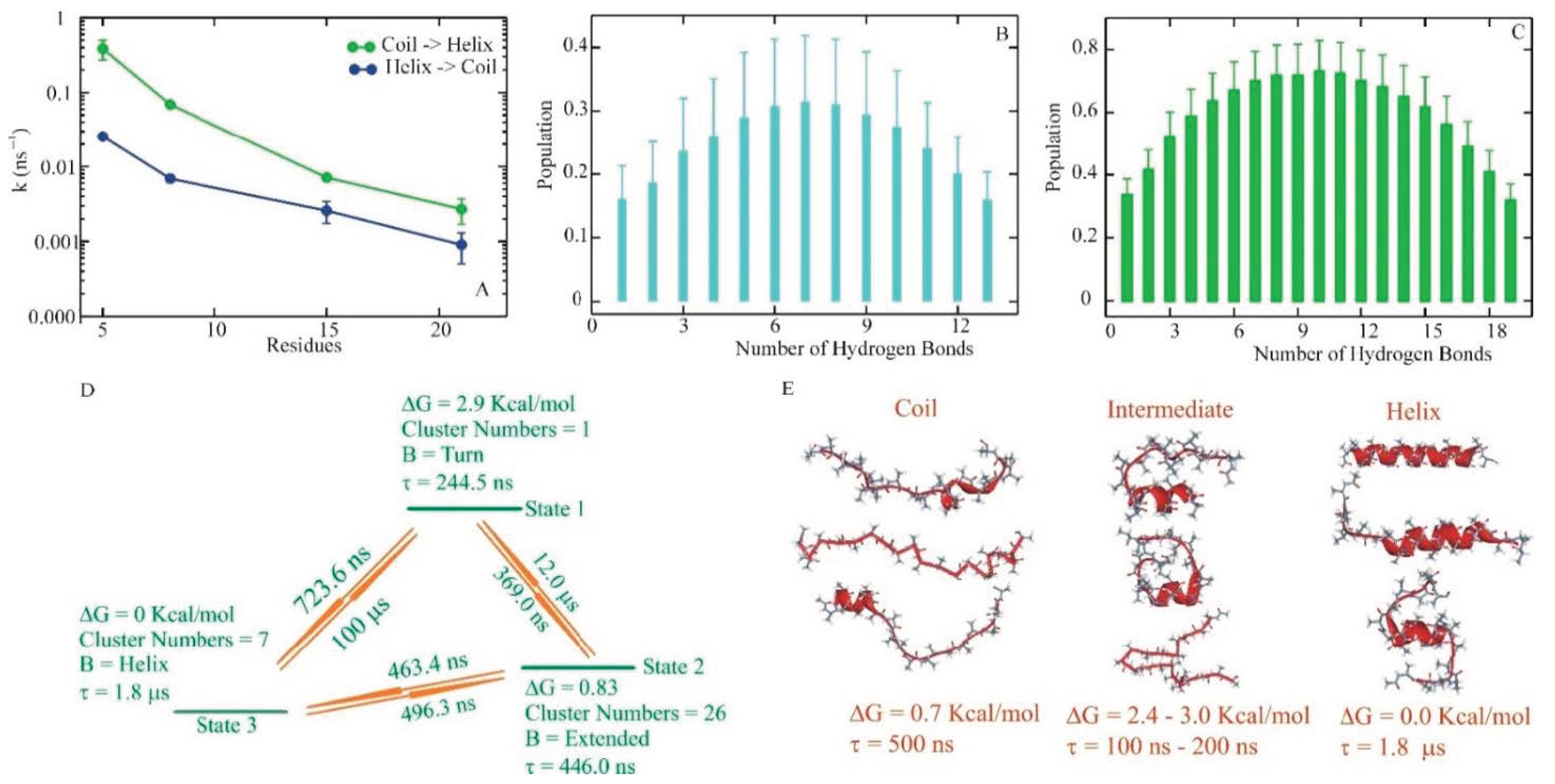{kind=link}
1. Introduction
2. Helix–Coil Kinetics
3. Multiple Pathways
4. Optimum Dimensionality Reduction
5. Summary
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bryngelson, J.D.; Wolynes, P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar] [CrossRef] [Green Version]
- Bryngelson, J.D.; Wolynes, P.G. Intermediates and barrier crossing in a random energy model (with applications to protein folding). J. Phys. Chem. 1989, 93, 6902–6915. [Google Scholar] [CrossRef]
- Alm, E.; Baker, D. Matching theory and experiment in protein folding. Curr. Opin. Struct. Biol. 1999, 9, 189–196. [Google Scholar] [CrossRef]
- Alm, E.; Baker, D. Prediction of protein-folding mechanisms from free-energy landscapes derived from native structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11305–11310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allew, R.M.; Sabelko, J.; Gruebele, M. Direct observation of fast protein folding: The initial collapse of apomyoglobin. Proc. Natl. Acad. Sci. USA 1996, 93, 5759–5764. [Google Scholar] [CrossRef] [Green Version]
- Ballew, R.M.; Sabelko, J.; Gruebele, M. Observation of distinct nanosecond and microsecond protein folding events. Nat. Struct. Biol. 1996, 3, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Bieri, O.; Wirz, J.; Hellrung, B.; Schutkowski, M.; Drewello, M.; Kiefhaber, T. The speed limit for protein folding measured by triplet-triplet energy transfer. Proc. Natl. Acad. Sci. USA 1999, 96, 9597–9601. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.J.; Rivas, G.; Serrano, L. A short linear peptide that folds into a β-hairpin in aqueous solution. Nat. Struct. Biol. 1994, 1, 584–590. [Google Scholar] [CrossRef]
- Brooks, C.L. Simulations of protein folding and unfolding. Curr. Opin. Struct. Biol. 1998, 8, 222–226. [Google Scholar] [CrossRef]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins: Struct. Funct. Bioinform. 1995, 21, 167–195. [Google Scholar] [CrossRef] [Green Version]
- Buckler, D.R.; Haas, E.; Scheraga, H.A. Analysis of the Structure of Ribonuclease A in Native and Partially Denatured States by Time-Resolved Nonradiative Dynamic Excitation Energy Transfer between Site-Specific Extrinsic Probes. Biochemistry 1995, 34, 15965–15978. [Google Scholar] [CrossRef] [PubMed]
- Burton, R.E.; Huang, G.S.; Daugherty, M.A.; Calderone, T.L.; Oas, T.G. The energy landscape of a fast-folding protein mapped by Ala-->Gly substitutions. Nat. Struct. Biol. 1997, 4, 305–310. [Google Scholar] [CrossRef]
- Burton, R.E.; Huang, G.S.; Daugherty, M.A.; Fullbright, P.W.; Oas, T.G. Microsecond protein folding through a compact transition state. J. Mol. Biol. 1996, 263, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Callender, R.H.; Dyer, R.; Gilmanshin, R.; Woodruff, W.H. FAST EVENTS IN PROTEIN FOLDING: The Time Evolution of Primary Processes. Annu. Rev. Phys. Chem. 1998, 49, 173–202. [Google Scholar] [CrossRef]
- Camacho, J.; Thirumalai, D. Theoretical predictions of folding pathways by using the proximity rule, with applications to bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA 1995, 92, 1277–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabartty, A.; Baldwin, R.L. Stability of α-helices. Adv. Prot. Chem. 1995, 46, 141–176. [Google Scholar]
- Chan, C.K.; Hofrichter, J.; Eaton, W.A.; Winkler, J.R.; Gray, H.B. Optical Triggers of Protein Folding. Science 1996, 274, 628–629. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-K.; Hu, Y.; Takahashi, S.; Rousseau, D.L.; Eaton, W.A.; Hofrichter, J. Submillisecond protein folding kinetics studied by ultrarapid mixing. Proc. Natl. Acad. Sci. USA 1997, 94, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.S.; Dill, K.A. Protein folding in the landscape perspective: Chevron plots and non-arrhenius kinetics. Proteins 1998, 30, 2–33. [Google Scholar] [CrossRef]
- Chen, E.; Wittung-Stafshede, P.; Kliger, D.S. Far-UV time-resolved circular dichroism detection of electron-transfer-triggered cytochrome c folding. J. Am. Chem. Soc. 1999, 121, 3811–3817. [Google Scholar] [CrossRef]
- Clarke, D.T.; Doig, A.J.; Stapley, B.J.; Jones, G.R. The α-helix folds on the millisecond time scale. Proc. Natl. Acad. Sci. USA 1999, 96, 7232–7237. [Google Scholar] [CrossRef] [Green Version]
- Daggett, V.; Levitt, M. Molecular-dynamics simulation of helix denaturation. J. Mol. Biol. 1992, 223, 1121–1138. [Google Scholar] [CrossRef]
- Daura, X.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol. 1998, 280, 925–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daura, X.; van Gunsteren, W.F.; Mark, A.E. Folding-unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins 1999, 34, 269–280. [Google Scholar] [CrossRef]
- Dill, K.A.; Shortle, D. Denatured states of proteins. Annu. Rev. Biochem. 1991, 60, 795–825. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Stigter, D. Modeling protein stability as heteropolymer collapse. Adv. Prot. Chem. 1995, 46, 59–104. [Google Scholar]
- Dinner, A.R.; Lazaridis, T.; Karplus, M. Understanding β-hairpin formation. Proc. Natl. Acad. Sci. USA 1999, 96, 9068–9073. [Google Scholar] [CrossRef] [Green Version]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Dobson, C.M.; Sali, A.; Karplus, M. Protein folding: A perspective from theory and experiment. Angew. Chem. Int. Edit. 1998, 37, 868–893. [Google Scholar] [CrossRef]
- Duan, Y.; Kollman, P.A. Pathways to a protein folding intermediate observed in 1-microsecond simulation in aqueous solution. Science 1998, 282, 740–744. [Google Scholar] [CrossRef] [Green Version]
- Dyer, R.B.; Gai, F.; Woodruff, W.H.; Gilmanshin, R.; Callender, R.H. Infrared studies of fast events in protein folding. Acc. Chem. Res. 1998, 31, 709–716. [Google Scholar] [CrossRef]
- Eaton, W.A. Commentary: Searching for “downhill scenarios” in protein folding. Proc. Natl. Acad. Sci. USA 1999, 96, 5897–5899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elove, G.A.; Bhuyan, A.K.; Roder, H. Kinetic mechanism of cytochrome c folding: Involvement of the heme and its ligands. Biochemistry 1994, 33, 6925–6935. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; Freeman: San Francisco, CA, USA, 1998. [Google Scholar]
- Fersht, A.R.; Matouschek, A.; Serrano, L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J. Mol. Biol. 1992, 224, 771–782. [Google Scholar] [CrossRef]
- Mayor, U.; Guydosh, N.R.; Johnson, C.M.; Grossmann, J.G.; Sato, S.; Jas, G.S.; Freund, S.M.V.; Alonso, D.O.V.; Daggett, V.; Fersht, A.R. The complete folding pathway of a protein from nanoseconds to microseconds. Nature 2003, 421, 863–867. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Itzhaki, L.S.; Otzen, D.E.; Fersht, A.R. The structure of the transition state for folding of chymotrypsin inhibitor-2 analyzed by protein engineering methods evidence for a nucleation condensation mechanism for protein folding. J. Mol. Biol. 1995, 254, 260–288. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.E. How do small singledomain proteins fold? Fold. Des. 1998, 3, R81–R91. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.E.; Fersht, A.R. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 1991, 30, 10428–10435. [Google Scholar] [CrossRef]
- Williams, S.; Causgrove, T.P.; Gilmanshin, R.; Fang, K.S.; Callender, R.H.; Woodruff, A.W.H.; Dyer, R. Fast Events in Protein Folding: Helix Melting and Formation in a Small Peptide. Biochemistry 1996, 35, 691–697. [Google Scholar] [CrossRef]
- Thompson, P.A.; Eaton, W.A.; Hofrichter, J. Laser temperature jump study of the helix <==> coil kinetics of an alanine peptide interpreted with a “kinetic zipper” model. Biochemistry 1997, 36, 9200–9210. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Munoz, V.; Jas, G.S.; Henry, E.R.; Eaton, W.A.; Hofrichter, J. The helix-coil kinetics of a heteropeptide. J. Phys. Chem. B 2000, 104, 378–389. [Google Scholar] [CrossRef]
- Mohammed, O.F.; Jas, G.S.; Lin, M.M.; Zewail, A.H. Primary Peptide Folding Dynamics Observed with Ultrafast Temperature Jump. Angew. Chem. Int. Edit. 2009, 48, 5628–5632. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.M.; Mohammed, O.F.; Jas, G.S.; Zewail, A.H. Speed limit of protein folding evidenced in secondary structure dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 16622–16627. [Google Scholar] [CrossRef] [Green Version]
- Jas, G.S.; Kuczera, K. Computer simulations of helix folding in homo- and heteropeptides. Mol. Simulat. 2012, 38, 682–694. [Google Scholar] [CrossRef]
- Kuczera, K.; Jas, G.S.; Elber, R. Kinetics of Helix Unfolding: Molecular Dynamics Simulations with Milestoning. J. Phys. Chem. A 2009, 113, 7461–7473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzer, S.M.; Elber, R.; Moon, T.J. Early Events in Helix Unfolding under External Forces: A Milestoning Analysis. J. Phys. Chem. B 2012, 116, 8662–8691. [Google Scholar] [CrossRef] [Green Version]
- Jas, G.S.; Middaugh, C.R.; Kuczera, K. Non-Exponential Kinetics and a Complete Folding Pathway of an α-Helical Heteropeptide: Direct Observation and Comprehensive Molecular Dynamics. J. Phys. Chem. B 2014, 118, 639–647. [Google Scholar] [CrossRef]
- Onuchic, J.; Luthey-Schulten, A.; Wolynes, P.G. Theory of protein folding: The energy landscape perspective. Annu. Rev.Phys. Chem. 1997, 48, 545–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onuchic, J.; Socci, N.D.; Luthey-Schulten, Z.; Wolynes, P.G. Protein folding funnels: The nature of the transition state ensemble. Fold. Des. 1996, 1, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Pande, V.S.; Grosberg, A.Y.; Tanaka, T.; Rokhsar, D.S. Pathways for protein folding: Is a new view needed? Curr. Opin. Struct. Biol. 1998, 8, 68–79. [Google Scholar] [CrossRef]
- Pande, V.S.; Rokhsar, D.S. Is the molten globule a third phase of proteins? Proc. Natl. Acad. Sci. USA 1998, 95, 1490–1494. [Google Scholar] [CrossRef] [Green Version]
- Pande, V.S.; Rokhsar, D.S. Molecular dynamics simulations of unfolding and refolding of a β-hairpin fragment of protein G. Proc. Natl. Acad. Sci. USA 1999, 96, 9062–9067. [Google Scholar] [CrossRef] [Green Version]
- Portman, J.J.; Takada, S.; Wolynes, P.G. Variational theory for site resolved protein folding free energy surfaces. Phys. Rev. Lett. 1998, 81, 5237–5240. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, V.; Thompson, P.; Hofrichter, J.; Eaton, W.A. Folding dynamics and mechanism of β-hairpin formation. Nature 1997, 390, 196–199. [Google Scholar] [CrossRef]
- Jas, G.S.; Eaton, W.A.; Hofrichter, J. Effect of viscosity on the kinetics of α-helix and β-hairpin formation. J. Phys. Chem. B 2001, 105, 261–272. [Google Scholar] [CrossRef]
- Hegefeld, W.A.; Chen, S.E.; DeLeon, K.Y.; Kuczera, K.; Jas, G.S. Helix Formation in a Pentapeptide Experiment and Force-field Dependent Dynamics. J. Phys. Chem. A 2010, 114, 12391–12402. [Google Scholar] [CrossRef]
- Doig, A.J. The alpha-helix as the simplest protein model: Helix-Coil Theory, Stability and Design. In Protein Folding, Misfolding and Aggregation: Classical Themes and Novel Approaches; Muñoz, V., Ed.; Royal Society of Chemistry: London, UK, 2008. [Google Scholar]
- Huo, S.; Straub, J.E. Direct Computation of Long Time Processes in Peptides and Proteins: Reaction Path Study of the Coil-to-Helix Transition in Polyalanine. Proteins 1999, 36, 249–261. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 1987. [Google Scholar]
- Allen, R.J.; Frenkel, D.; ten Wolde, P.R. Forward flux sampling-type schemes for simulating rare events: Efficiency analysis. J. Chem. Phys. 2006, 124, 194111. [Google Scholar] [CrossRef] [Green Version]
- Aristoff, D.; Bello-Rivas, J.M.; Elber, R. A mathematical framework for exact milestoning. Multiscale Model. Simul. 2016, 14, 301–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [Green Version]
- Bello-Rivas, J.M.; Elber, R. Exact milestoning. J. Chem. Phys. 2015, 142, 94102. [Google Scholar] [CrossRef] [Green Version]
- Bello-Rivas, J.M.; Elber, R. Simulations of thermodynamics and kinetics on rough energy landscapes with milestoning. J. Comput. Chem. 2016, 37, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Bolhuis, P.G.; Chandler, D.; Dellago, C.; Geissler, P.L. Transition path sampling: Throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002, 53, 291–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, G.R.; Pande, V.S. An Introduction to Markov State Models and Their Applications to Long Timescale Molecular Simulations; Springer: Berlin, Germany, 2014. [Google Scholar]
- Cardenas, A.E.; Elber, R. Computational study of peptide permeation through membrane: Searching for hidden slow variables. Mol. Phys. 2013, 111, 3565–3578. [Google Scholar] [CrossRef] [Green Version]
- Cardenas, A.E.; Jas, G.S.; DeLeon, K.Y.; Hegefeld, W.A.; Kuczera, K.; Elber, R. Unassisted transport of N-acetyl-L-tryptophanamide through membrane: Experiment and simulation of kinetics. J. Phys. Chem. B 2012, 116, 2739–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, A.E.; Elber, R. Modeling kinetics and equilibrium of membranes with fields: Milestoning analysis and implication to permeation. J. Chem. Phys. 2014, 141, 54101. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. A Computational Mi-croscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [Green Version]
- Bowman, G.R. An Overview and Practical Guide to Building Markov State Models. In An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation; Bowman, G.R., Pande, V.S., Noe, F., Eds.; Springer: Heidelberg, Germany, 2014; pp. 7–22. [Google Scholar]
- Kube, S.; Weber, M. A coarse graining method for the identification of transition rates between molecular conformations. J. Chem. Phys. 2007, 126, 24103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummer, G.; Szabo, A. Optimal Dimensionality Reduction of Multistate Kinetic and Markov-State Models. J. Phys. Chem. B 2015, 119, 9029–9037. [Google Scholar] [CrossRef] [Green Version]
- Jas, G.S.; Vallejo-Calzada, R.; Johnson, C.K.; Kuczera, K. Dynamic elements and kinetics: Most favorable conformations of peptides in solution with measurements and simulations. J. Chem. Phys. 2019, 151, 225102. [Google Scholar] [CrossRef] [PubMed]
- Jas, G.S.; Kuczera, K. Helix-Coil Transition Courses through Multiple Pathways and Intermediates: Fast Kinetic Measurements and Dimensionality Reduction. J. Phys. Chem. B 2018, 122, 10806–10816. [Google Scholar] [CrossRef] [PubMed]
- Senne, M.; Trendelkamp-Schroer, B.; Mey, A.S.J.S.; Schutte, C.; Noe, F. EMMA: A Software Package for Markov Model Building and Analysis. J. Chem. Theory Comput. 2012, 8, 2223–2238. [Google Scholar] [CrossRef]
- Trendelkamp-Schroer, B.; Wu, H.; Paul, F.; Noe, F. Estimation’ and Uncertainty of Reversible Markov Models. J. Chem. Phys. 2015, 143, 174101. [Google Scholar] [CrossRef] [PubMed]
- Jas, G.S.; Childs, E.W.; Kuczera, K. Kinetic pathway analysis of an α-helix in two protonation states: Direct observation and optimal dimensionality reduction. J. Chem. Phys. 2019, 150, 74902. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L.; MacKerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Krokhotin, A.; Liwo, A.; Maisuradze, G.; Niemi, J.; Scheraga, H.A. Kinks, loops and protein folding, with protein A as an example. J. Chem. Phys. 2014, 140, 25101. [Google Scholar] [CrossRef] [Green Version]
- Maisuradze, G.G.; Liwo, A.; Senet, P.; Scheraga, H.A. Local vs. global motions in protein folding. J. Chem. Theory Comput. 2013, 9, 2907–2921. [Google Scholar] [CrossRef] [Green Version]
- Kuczera, K.; Szoszkiewicz, R.; He, J.; Jas, G.S. Length Dependent Folding Kinetics of Alanine-Based Helical Peptides from Optimal Dimensionality Reduction. Life 2021, 11, 385. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Gasparotto, P. Using Dimensionality Reduction to Analyze Protein Trajectories. Front. Mol. Biosci. 2019, 6, 1–11. [Google Scholar] [CrossRef]
- de Souza, V.C.; Goliatt, L.; Capriles, P.V.Z. Insight About Nonlinear Dimensionality Reduction Methods Applied to Protein Molecular Dynamics. In Bioinformatics and Biomedical Engineering; Rojas, I., Venzuela, O., Rojas, F., Ortuno, F., Eds.; Springer: Heidelberg, Germany, 2019; pp. 219–230. [Google Scholar]
- Stamati, H.; Clementi, C.; Kavrak, L.E. Application of nonlinear dimensionality reduction to characterize the conformational landscape of small peptides. Proteins 2010, 78, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jas, G.S.; Hegefeld, W.A.; Majek, P.; Kuczera, K.; Elber, R. Experiments and Comprehensive Simulations of the Formation of a Helical Turn. J. Phys. Chem. B 2012, 116, 6598–6610. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jas, G.S.; Childs, E.W.; Middaugh, C.R.; Kuczera, K. Dissecting Multiple Pathways in the Relaxation Dynamics of Helix <==> Coil Transitions with Optimum Dimensionality Reduction. Biomolecules 2021, 11, 1351. https://doi.org/10.3390/biom11091351
Jas GS, Childs EW, Middaugh CR, Kuczera K. Dissecting Multiple Pathways in the Relaxation Dynamics of Helix <==> Coil Transitions with Optimum Dimensionality Reduction. Biomolecules. 2021; 11(9):1351. https://doi.org/10.3390/biom11091351
Chicago/Turabian StyleJas, Gouri S., Ed W. Childs, C. Russell Middaugh, and Krzysztof Kuczera. 2021. "Dissecting Multiple Pathways in the Relaxation Dynamics of Helix <==> Coil Transitions with Optimum Dimensionality Reduction" Biomolecules 11, no. 9: 1351. https://doi.org/10.3390/biom11091351