
Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis
by
 , and
, and
Yifan Tai
1,†, 
Emma L. Woods
2,3,†,
Jordanna Dally
2,3,
Deling Kong
1,
Robert Steadman
2,
Ryan Moseley
3,* and
Adam C. Midgley
1,* 1
Key Laboratory of Bioactive Materials for the Ministry of Education, College of Life Sciences, Nankai University, Tianjin 300071, China
2
Welsh Kidney Research Unit, Division of Infection and Immunity, Cardiff Institute of Tissue Engineering and Repair (CITER), School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK
3
Regenerative Biology Group, Oral and Biomedical Sciences, Cardiff Institute of Tissue Engineering and Repair (CITER), School of Dentistry, Cardiff University, Heath Park, Cardiff CF14 4XY, UK
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomolecules 2021, 11(8), 1095; https://doi.org/10.3390/biom11081095
Submission received: 3 July 2021
/
Revised: 16 July 2021
/
Accepted: 20 July 2021
/
Published: 23 July 2021
(This article belongs to the Special Issue Cellular and Molecular Mechanisms of Wound Healing)
Abstract
:Myofibroblasts are contractile, α-smooth muscle actin-positive cells with multiple roles in pathophysiological processes. Myofibroblasts mediate wound contractions, but their persistent presence in tissues is central to driving fibrosis, making them attractive cell targets for the development of therapeutic treatments. However, due to shared cellular markers with several other phenotypes, the specific targeting of myofibroblasts has long presented a scientific and clinical challenge. In recent years, myofibroblasts have drawn much attention among scientific research communities from multiple disciplines and specialisations. As further research uncovers the characterisations of myofibroblast formation, function, and regulation, the realisation of novel interventional routes for myofibroblasts within pathologies has emerged. The research community is approaching the means to finally target these cells, to prevent fibrosis, accelerate scarless wound healing, and attenuate associated disease-processes in clinical settings. This comprehensive review article describes the myofibroblast cell phenotype, their origins, and their diverse physiological and pathological functionality. Special attention has been given to mechanisms and molecular pathways governing myofibroblast differentiation, and updates in molecular interventions.
1. Myofibroblasts
In 1971, Gabbiani et al. identified large fibroblast-like cells within granulation tissue that had 40–80 A° diameter filamentous fibres traversing their entire cytoplasm. Since similar features are typical of smooth-muscle cells, the term ‘myofibroblast’ was coined (essentially, muscle-fibroblast intermediate cells) and it was proposed that these cells were implicated in wound contraction [1,2]. Further characterisation identified that the filamentous fibres were actin-based, with incorporated myosin and α-smooth muscle actin (α-SMA) proteins. Therefore, the myofibroblast’s ability to exert contractile force and to contract the wound edge was explained [3,4,5]. Myofibroblasts are morphologically enlarged and irregular (star or web-shaped) fusiform cells with well-developed cell–matrix focal interactions and intracellular gap junctions [6,7]. The incorporation of α-SMA into actin stress fibres grants the myofibroblast contractile power, approximately 2-fold that of the force of fibroblasts, when cultured on substrates with high elastomer stiffness [8,9,10]. Increased production of extracellular matrix (ECM) components: type I and type III fibrillar collagens, hyaluronan (HA), fibronectin (FN), and extra domain A fibronectin (EDA–FN) distinguish the hallmarks of myofibroblasts [11]. This elevated ECM content is not always causally linked, driving the myofibroblast differentiation process. Rather, there is a prominent interplay between ECM composition/arrangement and myofibroblast formation/function [12,13]. Following the delineation of the roles of myofibroblasts in wound contracture, they were quickly established as key drivers of progressive organ fibrosis [14], and have since been implicated in tumour development and metastasis [15,16]. More recently, studies describing an array of functions and regulatory factors exhibited by myofibroblasts have suggested roles beyond wound contraction and scar formation, including macrophage-like phagocytosis [17], immunomodulation [18,19], and autophagy [20]. In this review, we focus on the best described roles of myofibroblasts during wound healing and fibrosis. In addition, we summarise recent advancements towards targeting these cells with therapeutic molecular interventions.
2. Myofibroblast Origins
Multiple cell types are suggested to give rise to myofibroblasts, seemingly dependent on the tissue type. The heterogenous origins of myofibroblasts implies that these cells can form inside almost every tissue within the human body. In addition to resident fibroblasts and pericytes, their cellular origins include circulating bone-marrow-derived fibrocytes, tissue-derived mesenchymal stem cells, local epithelial and endothelial cells, de-differentiated smooth muscle cells, other hepatic stellate cells, mesangial cells, Schwann cells, and even monocytes and macrophages [21,22,23,24].
Differentiation from resident fibroblasts, present within most organs and connective tissues, is the best described process of myofibroblast development. Resting (inactivated) fibroblasts produce ECM and matrix proteases required for homeostatic turnover [25]. Upon activation, fibroblasts become highly migratory, proliferative, and increase the production of ECM, enzymes, and cytokines [26,27]; this transitional, activated state is termed the proto-myofibroblast. Despite limited information available in the literature regarding this phenotype, key features of the activated fibroblast, or proto-myofibroblast, have been described [28]. The rearrangement of the actin cytoskeleton from largely membrane-associated monomeric G-actin to polymerised cytoplasmic F-actin stress filaments, which traverse the length of the widened cell, is a hallmark feature [10]. These stress fibres allow junction formation with ECM components and other cells via integrin-containing complexes at the cell membrane and cadherin-type adherens junctions, respectively [7,29]. The absence of α-SMA within these stress fibres allows for proto-myofibroblast distinction from myofibroblasts [28]. Maturation into myofibroblasts can be determined by the neo-expression of α-SMA-positive stress fibres [30].
3. Myofibroblasts in Skin Fibrosis
The fibrotic process is characterised by chronic inflammation; altered epithelial–mesenchymal interactions; fibroblast proliferation, and fibroblast–myofibroblast differentiation. The latter feature (differentiation of fibroblasts into myofibroblasts) is central to the dysregulated and excessive production of collagen-rich ECM, otherwise called scar tissue. Myofibroblasts are thought to be terminally differentiated cells that typically undergo apoptosis [31,32,33,34] after wound contraction, as they are rarely found in non-pathological situations. Upon tissue trauma, myofibroblasts contribute to excessive ECM production for rapid, albeit dysfunctional, tissue repair. The tissue defect is repaired, but at the cost of function, as the organised spatial arrangement of specialised cells and ECM constituents is replaced by disorganised, abundant fibrous ECM. In this regard, the myofibroblast could be considered the primordial emergency repair cell. The aberrant persistence and chronic activation of myofibroblasts can lead to the development of pathological healing called fibrosis, which afflicts most tissues within the body. Progressive fibrosis leading to organ failure is considered the end-stage pathology of multiple diseases affecting the major organs, including but not limited to myocardial fibrosis [35], pulmonary fibrosis [36], liver cirrhosis [37], and chronic kidney disease [38]. Skin fibrosis (cicatrix) is an umbrella term for a large, heterogeneous spectrum of pathological conditions that affect the skin. Examples of skin conditions in which myofibroblast activity is central include hypertrophic and keloid scars, scleroderma, Dupuytren’s contracture, eosinophilic fasciitis, and chronic graft-versus-host disease [39,40,41,42,43,44]. The shared features of the abnormal and excessive accumulation of ECM constituents, particularly collagens, HA, and FN, in these diseases are governed by the myofibroblast phenotype. Infiltration of immune cells into fibrotic tissue also plays a key role in amplifying the fibrotic response, by secreting several cytokines and chemokines responsible for fibroblast–myofibroblast differentiation, the stimulation of ECM deposition, and the further recruitment of immune cells [45].
4. Mechanisms of Myofibroblast Formation
At least three local events are needed to generate mature α-SMA-positive, fully differentiated myofibroblasts: (i) biologically active TGF-β1; (ii) extracellular stress, arising from the mechanical properties of the ECM (particularly collagen) and EDA–FN/integrin interactions; and (iii) the precursory production of phenotypic modulators (EDA–FN, HA) following activation (Figure 1). Increasing evidence strongly supports the role of inflammatory cell interactions in promoting myofibroblast development. Regardless of the origin, the resultant myofibroblast cells share the same properties and signalling cascade events that led to their formation.
4.1. The Canonical Pathway: TGF-β1/Smad
In classical cutaneous wound healing, tissue injury leads to TGF-β1 release from keratinocytes, macrophages and degranulating platelets [46]. In turn, TGF-β1 binds to and induces TGF-β receptor (TGFβR) I and II association on the fibroblast cell membrane, enabling TGFβRII phosphorylation of the TGFβRI kinase domain. In-turn, TGFβRI phosphorylates Smad2 and Smad3, which subsequently oligomerize with Smad4, forming trimeric protein complexes. These complexes are translocated to the nucleus, where they act as the transcription or co-transcription factors in the induction or repression of gene expression [47] (Figure 2A). Ultimately, TGF-β1/Smad pathway activation in fibroblasts leads to their differentiation into myofibroblasts [48]. TGF-β1 can also induce Smad-independent and co-receptor signalling pathways, such as mitogen-activated protein kinase (MAPK), p42/p44 extracellular signal regulated kinase (ERK1/2), Rho/Rho-associated protein kinase (ROCK), phosphatidylinositol-3-kinase (PI3K)/AKT, protein phosphatase 2A (PP2A), p38/c-Jun N-terminal kinase (JNK), protein kinase C (PKC), and tumour necrosis factor receptor associated factor (TRAF)-4/6 [49,50,51,52,53]. The tightly controlled TGF-β1/Smad-driven signalling events have been extensively researched within the context of myofibroblast differentiation and fibrosis, as highlighted in an eloquent review by Frangogiannis [54].
4.2. The Non-Canonical Pathway: HA/CD44/EGFR
A principal mediator of a non-canonical differentiation pathway in human myofibroblasts is HA, a glycosaminoglycan (GAG) formed of repeating d-glucuronic acid and N-acetyl-glucosamine disaccharide units. HA is synthesised by three HA-synthase (HAS) isoenzymes (HAS1, HAS2, HAS3) [55], and is metabolised by hyaluronidase (HYAL) enzymes (HYAL1, HYAL2, HYAL4) [56]. HA accumulation occurs simultaneously with upregulated HYAL1 and HYAL2 expression, but both these HA degrading enzymes have diminished expression in myofibroblasts. A subsequent decrease in HA degradation may also favour the accumulation of HA in fibrotic disease [57]. HAS2-synthesised HA is widely considered to be an important mediator of fibroblast–myofibroblast differentiation and EMT processes [58,59,60,61,62]. HAS2-synthesised linear HA forms a thickened pericellular coat that surrounds the activated fibroblast. This crosslinked and thickened HA pericellular coat, or glycocalyx, has been shown to be an essential structural element for human fibroblast–myofibroblast differentiation and phenotypic maintenance [59,63]. The HA pericellular coat remains thin in fibroblast phenotypes resistant to TGF-β1-driven differentiation, such as oral mucosal fibroblasts [64] and aged/senescent fibroblasts [58]. Interestingly, an enriched pericellular HA microenvironment is synthesised by foetal fibroblasts during foetal wound healing.
The HA pericellular coat is anchored to the fibroblast cell surface by the CD44 receptor [65,66], and crosslinked by hyaladherins [59]. Association with CD44 and other hyaladherins orchestrates the HA pericellular coat and results in the activation of downstream extracellular–intracellular signal cascades, shown to be prominent in disease pathogenesis including fibrosis, cancers, and inflammatory diseases [67,68]. CD44 exists as multiple alternatively spliced variants and all CD44 variants (CD44v), as well as the standard form (CD44s), possess HA binding motifs within their extracellular head domain. CD44v have variability within alternatively spliced exons of the extracellular stem region [69,70]. Studies have shown that silencing CD44 using small interfering RNA (siRNA) prevented the downstream intracellular signalling required for human fibroblast–myofibroblast differentiation and inhibited HA pericellular coat formation [71,72]. These studies concluded that HA/CD44 association was essential for human myofibroblast differentiation, with recent research showing that CD44s expressed on the surface of human fibroblasts was the dominant player in regulating HA coat-mediated myofibroblast differentiation [73]. HA-crosslinking hyaladherins include inter-α-inhibitor (IαI) heavy chains (HC), which are localised by association with the tumour necrosis factor-stimulated gene (TSG)-6 [74,75]. The increased presence of HA-associated HC and TSG-6 complexes have been associated with the pathology of multiple inflammatory diseases including arthritis, asthma, and some cancers [76]. Martin et al. identified that IαI-HC5 was covalently bound to chondroitin sulphate on bikunin and was essential for the formation of HA pericellular coats in myofibroblasts. Increased expression of TSG-6 following TGF-β1 activation of fibroblasts facilitated the transfer of HC5 to HA by TSG-6 catalysis, in a metal ion-dependent manner [77].
HA binding to CD44 plays a pivotal role in the intracellular signalling required for human fibroblast–myofibroblast differentiation [58,63,72]. CD44 was shown to be present throughout the membranes of fibroblasts but clustered into localised populations within myofibroblast membranes. These distinct areas were identified to be cholesterol-rich lipid rafts, and ‘locked’ clusters of CD44 within lipid rafts were shown to be dependent on HA binding and crosslinking into pericellular coats [72]. HA coat removal released CD44 from lipid rafts and re-enabled their movement through the membrane, but also inhibited downstream signalling cascades [63]. Further investigation revealed that CD44 was co-localised with epidermal growth factor (EGFR) within membrane lipid raft domains, which resulted in EGFR activation, and the phosphorylation of downstream effectors and transcription factors, ERK1/2 and calcium calmodulin kinase (CaMK)-II (CaMKII). The chemical inhibition of ERK1/2 or CaMKII prevented myofibroblast differentiation, and only when TGF-βRII and CD44/EGFR signalling were active could myofibroblast differentiation occur [72]. Both ERK1/2 and CaMKII were phosphorylated in a biphasic manner, with early and late-phase activation. Early activation of ERK1/2 (<5 min) was thought to be associated with eventual fibroblast–myofibroblast differentiation, whereas later activation (>30 min) was suggested to play a role mediating cellular proliferation responses [72] (Figure 2B). However, signalling functions appear to be cell-dependent as early, but not late, ERK1/2 signalling peaks were observed in TGF-β1-stimulated non-scarring oral mucosal fibroblasts; these cells exhibited an anti-proliferative response in a HA/CD44-independent manner [65].
4.3. The Wnt/β-Catenin Pathway
The Wnt signalling pathway is well described in its vital roles during embryogenesis and determination of cell fate, differentiation, proliferation, and apoptosis. We refer interested readers to a thorough review article on Wnt signalling functions [78]. Carefully orchestrated Wnt signalling has been found to be essential for tissue homeostasis, whilst the dysregulation of the Wnt pathway can result in pathogenesis [79,80]. Wnt signals simultaneously through the co-association of Frazzled receptors and low-density lipid protein co-receptors (LRP). The fate of β-catenin, an important regulator of Wnt signalling, is regulated by Dickkopf-related protein-1 (DKK1). DKK1 inhibits Wnt signalling by activating downstream signalling complexes, and results in β-catenin degradation. When DKK1 is not present, Wnt signalling leads to β-catenin translocation to the nucleus, its association with co-transcription factors, and the subsequent activation of Wnt-associated pro-fibrotic genes [78,79,81] (Figure 2C). The activation of Wnt signalling was implicated in the fibrogenesis of multiple organs [82]. Wnt-1 and Wnt-10b were noted to be overexpressed, whereas DKK1 was decreased, which led to the increased nuclear accumulation of β-catenin observed in human tissue samples from systemic scleroderma (SSc), idiopathic pulmonary fibrosis (IPF), and liver cirrhosis [82]. Wnt signalling stimulated by TGF-β1-induced inhibition of DKK1 was shown to induce myofibroblast differentiation, up-regulate the release of ECM components (notably collagens) and induce fibrosis [82,83,84]. In addition, crosstalk between the TGF-β/Smad pathway and the Wnt/β-catenin pathway is suggested to be a prominent mechanism in the development of hypertrophic and keloid scars [85].
4.4. Mechanotransduction
Mechanotransduction is the ability of stress force to convert extracellular to intracellular signalling. Fibroblasts can perceive external forces (mechanoperception) through their fibronexus structures in vivo or mature focal adhesion (FA) structures in vitro [86,87]. ECM rigidity determines the size of the cell’s FAs, or ‘anchors’, which in turn limit the level of tension generated within intracellular stress fibres. Only when substrate stiffness permits the formation of mature FAs (8–30 μm), and the generation of approximately four-fold greater stress compared with usual FAs (2–6 μm), does α-SMA become incorporated into pre-existing cytoplasmic β-actin stress fibres [88]. Thus, the myofibroblast phenotype is mechanosensitive. The transition of fibroblasts to the proto-myofibroblast state was suggested to be related to increased microenvironment stiffness [28,88]. TGF-β1 is locked within the ECM by latency-associated peptide (LAP) and latent TGF-β1-binding protein (LTBP), and is released by proteolysis or integrin-dependent mechanotransduction [89]; resulting in a feedback loop of pro-fibrotic fibroblast activity and increased ECM stiffness [29,90,91,92]. In a recent study, the balance of ECM composition, elasticity, and TGF-β1 signalling was shown to govern fibroblast phenotypic heterogeneity and give rise to distinct, but overlapping, fibroblast subsets [93]. Indeed, Kollmannsberger and colleagues showed that within 3D microtissues grown in vitro, fibroblasts transitioned to proliferative myofibroblasts at the growth front, where a high degree of tensile force and stretched FN fibres were present. As the tissue matrix matured into collagen-rich ECM with low FN fibre tension, more fibroblasts were present, suggesting that the myofibroblast phenotype stabilised by tensile forces could revert to a fibroblast phenotype by low tensile force [94]. These studies suggest that substrate stiffness is a key determinant of fibroblast and myofibroblast plasticity, and that reducing excessive mechanotransduction may be an option in controlling myofibroblast presence.
Myofibroblasts generate force through stress fibre contraction, and this is transmitted to the ECM via FAs containing transmembrane receptors, typically integrins [87,95], as summarised in Figure 3. Integrins mediate cell–cell, and especially, cell–ECM interactions, and are prominently involved in the initiation, maintenance, and resolution of fibrosis. EDA–FN is synthesised by fibroblasts and various other cell types [96,97]. The EDA–FN splice variant can only be detected during tissue repair [98,99], fibrosis [100], tumour development [101,102,103], and transiently during embryogenesis [104]. The accumulation of EDA–FN has been observed in multiple fibrotic disorders, including lung, liver, and skin [100,105,106,107]. EDA–FN promotes myofibroblast differentiation by orchestrating LAP/LTBP release and activation of TGF-β1 [108], increasing matrix stress–strain tension [109,110], and by activating mechanotransducer-integrin signalling via FAK activation [111,112,113,114]. Research by Shinde et al. [115] and Kohan et al. [112] demonstrated that fibroblast-expressed integrins, α4β1 (VLA-4) and α4β7 (LPAM-1), mediate different roles in myofibroblast formation, respectively. Integrin α4β1/EDA–FN promoted ECM synthesis and stiffening [115], whereas integrin α4β7/EDA–FN induced myofibroblast stress fibre formation and contractility [112]. The combined exclusivity of EDA–FN expression and the prominent roles it plays in driving the myofibroblast phenotype have highlighted the protein as an attractive target for anti-fibrotic interventional therapies.
Discoidin domain receptor (DDR)1 is a mediator of stromal–epithelial interactions, and belongs to the family of collagen receptors [116] overexpressed in pro-fibrotic keloid fibroblasts [117]. DDR1 mediates collagen contraction and stiffness, thereby indirectly mediating pro-fibrogenic responses that are largely independent from collagen binding to integrins [118]. Additionally, the spatial and structural properties of the local 3D collagen microarchitecture can distinctly affect fibroblast and myofibroblast activity. Collagen fibril alignment and diameter were shown to affect fibroblast contractility and migration via alteration in integrin clustering and the stability of adhesion sites [119]. Seo and colleagues suggested that collagen fibre thickness held more pertinence over dictating myofibroblast differentiation independently from collagen quantity [120]. Furthermore, collagen-rich ECM stiffening can also be potentiated by the myofibroblast and inflammatory cell production of collagen crosslinking enzymes, such as lysyl oxidases [11,90]. CD147, also known as extracellular matrix metalloproteinase inducer (EMMPRIN), is a well-established matrix metalloproteinase (MMP)-inducer that is rapidly becoming understood to mediate multiple cellular responses. The linked association of CD147 with integrin α6β1 was observed during increased metastasis in human hepatoma cells [121], suggesting a role for CD147 in modulating integrin α6β1 associations, with matricellular protein CCN1, and mechano-sensitivity to senescence or apoptosis [122]. Interestingly, the nullification of the extracellular domain of CD147 reversed associations with integrin β subunits and FA sites, suggesting that the extracellular domain of CD147 binds to integrins to regulate the mechanical tension of the ECM [121,123]. The roles of these mechanotransducer proteins in stress relay, which is involved in myofibroblast differentiation, will become clearer with continued investigation.
Increasing evidence suggests that myofibroblasts have the capacity for classical CaMK-myosin light chain kinase (MLCK)-dependent smooth muscle cell contraction mechanisms [124] and contractile activation by the RhoA/ROCK/myosin light chain phosphatase (MLCP) pathway [28,125,126], highlighting a key difference between the smooth muscle cell and myofibroblast contractile mechanisms. It was recently discovered that HYAL2, previously thought to only possess HA catalytic activity, was shown to have non-enzymatic functions [69,127,128]. Following TGF-β1 activation, enhanced HAS2 expression by fibroblasts leads to increased HA accumulation within the ECM observed in 3D matrices in vitro [129] and in fibrosis progression in vivo [130]. However, HYAL2 was shown to re-localize to the cytoplasm and align along the actin cytoskeleton in myofibroblasts [128], which may contribute to increasing extracellular HA accumulation. The cytoskeleton-aligned HYAL2 was associated with α-SMA, RhoA, and MLCK. The silencing of HYAL2 did not prevent but only delayed myofibroblast differentiation. The presence of HYAL2 was shown to accelerate RhoA/MLCK phosphorylation and cellular contractility, suggesting a role in the mechanotransduction and the orchestration of key cytoskeletal and FA-related proteins [128]. These early findings suggest that there may be complex orchestration roles that HYAL2 plays in cytoskeletal reassembly.
Other mediators of mechanotransduction and promising targets in mediating fibroblast–myofibroblast differentiation include myocardin-related transcription factor (MRTF) [131,132], Yes-associated protein (YAP) [133,134,135,136,137], cadherins [138], and Notch [139]. In the case of these multifunctional proteins, further exploratory research will help establish the feasibility of targeting their activity to interfere with myofibroblast differentiation or function.
5. Interventional Strategies to Target Myofibroblasts for Scarless Skin Healing
There are currently no clinically approved anti-scarring therapies specifically designed to limit or prevent dermal fibrosis. Several wound management strategies completed phase II trials, but did not advance to phase III trials in recent years. The clinical studies database, https://clinicaltrials.gov (‘Fibrosis, Skin’ searched 25 June 2021), showed 10 registered clinical trials that have either completed phase III or are currently in progress (Table 1). The proposed therapies include a range of treatment modes, including antibody and drug therapies, scar resection or reduction, and tissue-engineering strategies.
Rituximab is a mouse/human chimeric antibody that binds CD20 on the surface of B-lymphocytes and initiates cell apoptosis, complement activation, and cell-mediated cytotoxicity. B-cell depletion by rituximab was shown to reduce fibrosis serum markers and myofibroblast activation in IgG4-related inflammatory diseases [140]. The results from the phase II/III trial are yet to be released; however, rituximab was shown to reduce B-cell presence in autoimmune scleroderma skin tissues, but had no discernible benefits to skin scar tissue histology after 6 months of treatment [141], which suggests that rituximab may be more effective as a pre-emptive measure. The anti-fibrotic effect of kynurenic acid was also proposed to occur via the induction of apoptosis in leukocytes and a reduction in inflammation, but with the additional regulation of the fibroblast production of MMPs and collagens [142]. Pirfenidone has received extensive attention for its efficacious action in the treatment of IPF [143,144]. Recently, Chen et al. used in situ self-assembling HA lattice nanostructure spray-on dressings loaded with pirfenidone to treat deep partial-thickness burn injuries in mouse models. The addition of pirfenidone to the wound dressing reduced collagen accumulation and myofibroblast presence within wound sites [145]. Multiple ongoing clinical trials focus on the management of established scars, with the aim to reduce scar appearance or to induce physiological healing after scar resection. The combinatory treatment of radiation-induced fibrosis with pentoxifylline (a hemorheological agent) and vitamin E was suggested to exhibit synergistic improvement in scar appearance and ECM remodelling, through actions of increased tissue perfusion and free radical scavenging [146]. Carbon dioxide (CO2) ablative fractional laser therapy has emerged as a promising low-invasiveness therapy for the induction of collagen remodelling, resulting in scar revision and reduced hypertrophic scar appearance after multiple sessions [147]. Integra is a biosynthetic dermal substitute wound dressing widely used in acute wound management; the ongoing clinical trial aims to perform scar resection and uses Integra to encourage attenuated scar healing, in a similar manner to previously shown outcomes of Integra-treated post-burn scar resections [148]. Autologous skin cells were harvested at the point of graft surgery, using ReCell technology [149]. The cells were applied to chronic wounds prior to the grafting of autologous donor skin, which resulted in remarkable healing results [150]. The ongoing clinical trial will attempt to recapitulate this improved healing response following scar resection in patients with hypertrophic scars.
There are numerous records of clinical trials aimed at one or more of the multifaceted aspects of skin fibrosis. However, progression to phase III trials appears to be rarely achieved, perhaps owing to the limited experimental evidence of myofibroblast-specific targets. Below, we highlight progresses in the experimental and pre-clinical evaluation of candidate molecular-based therapies that target key myofibroblast functions or activities, to prevent skin fibrosis.
5.1. Experimental and Pre-Clinical Interventions for Myofibroblast Differentiation
Popularised pre-clinical strategies for preventing myofibroblast formation focus on inhibition of the canonical TGF-β1/Smad signalling pathway using cytokines, antibodies, and regulators of gene expression. Demonstrating the clinical efficacy of anti-TGF-β1 therapies has been hindered by the cytokine’s pleiotropism and ubiquitous presence, as well as the heterogenous processes of healing and fibrosis. Consequently, therapeutic targeting of TGF-β and its specific isoforms has proved challenging. Available clinical data are limited to early phase trials and studies with conflicting outcomes [54]. Recombinant human TGF-β3 (Avotermin, Juvista) was developed by Renovo and hailed as a potential novel inhibitor of dermal scar formation, based on its elevated presence and roles in non-scarring tissues compared to TGF-β1, such as early gestational foetal skin and the oral mucosa [151,152,153,154]. Phase I/II clinical trials concluded that the intradermal application of Juvista yielded both short- and long-term improvements in scar appearance, compared to placebo and standard wound care management [155,156]. Unfortunately, Juvista did not meet the necessary primary or secondary endpoints required by phase III trials, failing to demonstrate reduced scarring after excisional surgery. Fresolimumab is a neutralising antibody that targets all TGF-β isoforms; the effects of Fresolimumab on cutaneous SSc revealed reduced expression of fibrotic biomarkers and diminished myofibroblasts [157]. This suggested that a shotgun approach to TGF-β inhibition had anti-fibrotic effects, whilst neutralising TGF-β1 alone in SSc failed to demonstrate clinical efficacy [158]. An apparent trend has emerged, wherein anti-scarring interventions that target an individual aspect or factor of fibrotic conditions generally fail to realise clinical benefit. Moreover, the efficacious index of growth factors is limited by their low stability, short half-life in vivo, adverse effects arising from elevated local and/or systemic concentrations, and their non-specific nature (multiple protein-binding partners) [159].
MicroRNAs (miRNA) are short non-coding RNAs (18–25 nucleotides) that bind the 3′ untranslated region of mRNA, thereby inhibiting mRNA translation or promoting mRNA degradation. Multiple studies have shown the regulatory roles of miRNA in pathological wound healing and skin fibrosis. Notable miRNA targets upregulated in skin fibrosis include miRNA (miR)-21 [160] and miR-130b [161], whereas miR-29 [162], miR-129-5p [163], and miR-7 [164,165] were shown to be negative regulators of myofibroblast activity and fibrosis. The miRNAs, miR-21 and miR-17-5p, have been shown to be directly implicated in TGF-β1/Smad pathway promotion. Smad2 activity was potentiated by these miRNAs through their inhibition of Smad7 [166,167]. Thus, inhibiting the activity of these miRNAs has arisen as a potential avenue in gene therapies to mitigate TGF-β1/Smad pathway activity and myofibroblast differentiation. We refer readers to an elegantly written review of miRNAs in various fibrotic diseases for a more in-depth discussion of the miRNAs involved in modulating TGF-β1 signalling [168].
Bone morphogenetic protein-7 (BMP7) exerted potent anti-fibrotic actions in vitro [169], and in pre-clinical fibrosis models [170,171,172,173]. BMP7 activation of Smad1/5/8 results in the competitive binding of Smad4, thereby inhibiting the TGF-β1/Smad2/3 pathway’s transcriptional activities [173]. Early reports indicated that BMP7 failed to prevent bleomycin-induced skin fibrosis [174]. However, a later study showed that BMP7 treatment at the point of thermal injury prevented hypertrophic scar formation [173]. Thus, BMP7 therapies require further optimization and testing to determine their effectiveness in the prevention of dermal fibrosis. Interferon (IFN)-γ induced the expression of Smad7, which antagonised interactions between Smad2/3 and TGFβRI/II [175]. The effects of IFN-γ on fibroblasts in vitro are conflicting; exogenous IFN-γ was suggested to abrogate TGF-β1-induced proliferation, migration, and differentiation to myofibroblasts [176]; but T-cell secretion of IFN-γ, found to be prominent in SSc, aggravated fibrosis by promoting fibroblast proliferation and collagen synthesis [177]. Early pre-clinical and small clinical studies suggested that IFN-γ was effective in the attenuation of lung, renal, and liver fibrosis [178,179,180], and oral submucosal fibrosis [178,179,180,181]. There is a current lack of studies describing definitive IFN-γ mechanisms involved in the regulation of myofibroblasts. Thus, whether IFN-γ delivery would be effective in attenuating dermal fibrosis remains unclear.
Alternative approaches include molecular inhibitors of non-canonical and mechanotransduction pathways to regulate ECM composition and disrupt the myofibroblast phenotype. Hepatocyte growth factor (HGF) was first implicated in liver regeneration [182], but has since been shown to prevent fibrosis initiation and progression in animal models [183,184]. HGF treatment was shown to attenuate collagen production by fibroblasts in multiple tissues [185,186,187]. Elevated HGF expression in fibrosis [188,189,190,191], but also in differentiation-resistant fibroblasts [192,193], suggests a duality of HGF actions which may be a consequence of truncated isoforms possessing a variable number of kringle domains (HGF/NK1-4) with differential signalling activities. Indeed, human oral mucosal fibroblast resistance to TGF-β1-induced differentiation was dependent on heightened HGF and HGF/NK1 expression, whereas HGF/NK2 was preferentially expressed by dermal fibroblasts [194]. Recently, HGF/NK1 gene therapy was shown to exert potent anti-fibrotic effects through attenuated collagen types I, III, and IV deposition in mouse models of renal fibrosis [171]. In addition, the HGF inhibition of collagen synthesis and promotion of MMP production has shown promise for therapeutic applications in reducing the pro-fibrotic activity of keloid fibroblasts in vitro and in a keloid heterograft mouse model [195,196,197]. Small molecule inhibitors of DDR1 have shown promise in reducing collagen types I and IV deposition in bleomycin-induced renal and lung fibrosis [198,199,200]. Ongoing research into DDR1-specific inhibitors have yielded promising results in models of renal fibrosis [199].
HA bioactivity is dependent on its molecular weight, enzymatic synthesis, and endogenous versus exogenous application [55,201,202]. The disruption or prevention of HA pericellular coat synthesis using hyaluronidase enzymes [203], HAS inhibitors [204,205] or exogenous HA oligosaccharides [59] results in the failure of TGF-β1-stimulated myofibroblast phenotypic acquisition. The inhibition of HAS2 activity [206] or global HAS synthesis of HA [204,205] have demonstrated preventative effects in various models of fibrosis. However, given the ubiquitous role of HAS-synthesised HA in tissues, and the lack of specific HAS isoenzyme inhibitors, it is unclear at present whether the inhibition of HA synthesis would be beneficial in a clinical setting. An additional action of BMP7 involved the induction of the nuclear translocation of HYAL2 and the subsequent splicing of CD44 mRNA, which resulted in the upregulated expression of the variant isoform, CD44v7/8. Fibroblasts with upregulated expression of CD44v7/8 exhibited ‘HA-phage’ activity, wherein the HA pericellular coat was rapidly internalised and broken down, resulting in the destabilization of the myofibroblast and phenotypic reversion to fibroblasts [69,169]. Whether CD44v7/8-dependent actions convey anti-fibrotic effects in dermal fibrosis is currently unknown.
The most promising integrin targets that have demonstrated roles in the pathogenesis of fibrosis are α4-containing integrins (α4β1/7) [112,115] and αv-containing integrins (αvβ1/3/5/6) [207,208,209]. Mechanotransduction by integrin αvβ6 promoted traction from proliferating liver cholangiocytes to FN/LAP, which subsequently released active TGF-β1 and initiated the differentiation of surrounding hepatic stellate cells to myofibroblasts [207,210]. A small peptidomimetic, EMD527040, mimics the RGD-binding sites of αvβ6 and αvβ1; orally administered EMD527040 attenuated biliary and non-biliary fibrogenesis [207]. The expression of integrin αvβ6 was elevated in keratinocytes during wound healing and fibrosis [211], but the detailed mechanistic roles of integrin αvβ6 and epithelial–mesenchymal crosstalk during dermal fibrosis have yet to be elucidated. Research has identified that integrin interactions with the EDGIHEL motif of EDA–FN was causative of downstream profibrotic responses [109,113,115]. The IST-9 [109,110], F8 [212,213], and vaccine-generated [214] antibodies that target EDA–FN or integrin α4 have demonstrated prevention of fibroblast–myofibroblast differentiation [109,110]. Therapeutic applications of antibodies can be limited by the cost and complexity of production, off-target immune activation or unspecific protein masking of critical protein–protein interactions. To address these issues, Zhang et al. developed a small blocking polypeptide to bind and block EDA–FN interactions with the α4β1 binding cleft, with high specificity [114]. The polypeptide, AF38Pep, was designed to mimic the integrin α4β1 receptor site for the EDGIHEL motif of EDA–FN and was shown to interfere with TGF-β1-stimulated fibroblast–myofibroblast formation by the specific blockade of integrin α4β1 signalling, the inhibition of FAK activation, and the prevention of profibrotic gene transcription [114]. The aforementioned first generation of small blocking polypeptides have revealed the renewed promise of the specific interruption of integrin-mediated mechanotransduction and myofibroblast formation. The implementation of integrin receptor peptidomimetics for the prevention of skin fibrosis will become more apparent in the coming years when the in vitro research progresses into pre-clinical models.
Another potential peptide therapy is the N-terminal amino acid sequence of α-SMA, Ac-EEED, which is important for the tropomyosin-1.6/7-stabilized incorporation of α-SMA into cytoplasmic stress fibres [215]. Interestingly, the cytoplasmic delivery of the Ac-EEED peptide resulted in the loss of α-SMA from β-cytoplasmic actin stress fibres and inhibited G-actin polymerization into F-actin [216,217], thereby reducing myofibroblast contraction in wound healing [218]. Ac-EEED peptide therapy has yet to be evaluated in skin fibrosis models, which may be related to the current lack of available dermal myofibroblast-specific targeting moieties that operate by endosomal uptake and the cytoplasmic release of payloads.
5.2. Immunomodulating Biomolecules for Fibrosis Attenuation
Immunoregulatory interventions aim to control the inflammatory phase in postnatal skin healing to attenuate scar formation [219,220]. Studies in transgenic mice provided early indicators that the absence of neutrophils and macrophages led to scar-free healing [221]. Targeted repression of the gap junction and inflammatory mediator protein, connexin-43, supported these findings [220,222]. Additionally, connexin-43 was shown to mediate cardiac fibroblast–myofibroblast differentiation [223] and promote aberrant cardiomyocyte–myofibroblast functional coupling [224]. Certain interleukin (IL) cytokines are implicated in the activation of inflammatory cascades. IL-8 production is a chemoattractant to neutrophils, whereas IL-6 secretion by fibroblasts activates macrophages and monocyte chemotaxis. Both IL-6 and IL-8 exhibit the rapid induction of expression following tissue injury, resulting in the recruitment of circulating inflammatory cells. The expression levels of IL-6 and IL-8 are elevated and maintained for longer in adult skin, compared to scarless scar-free foetal repair. The inhibition of phosphodiesterase 4 (PDE4) reduced scar formation in skin fibrosis models by interfering with the release of IL-6 from M2 macrophages [225]. Thus, IL-6 and IL-8 are considered pro-fibrotic mediators, whereas IL-10 antagonised their activity [219,226]. IL-10 gene therapy resulted in reduced inflammation and the promotion of scarless healing in mouse wound healing studies [227]. The exogenous addition and macrophage paracrine production of IL-10 were shown to induce myofibroblast reversal to fibroblastic phenotypes in vitro [228,229]. More recently, IL-10 induced myofibroblast–fibroblast dedifferentiation was shown to alter dynamic interactions with the surrounding fibrillar matrix with a demonstratable loss of contractility in IL-10 treated myofibroblasts [230]. Research into inflammation-induced fibrosis has revealed additional potential candidate ILs that may be targetable in skin fibrosis, including IL-11 [35], IL-16 [231], and IL-33 [232]. Keratinocytes have suggested roles in the regulation of the myofibroblast phenotype and profibrotic ECM during wound healing [233,234,235]. More recently, the keratinocyte secretion of IL-1α was demonstrated to restrain the myofibroblast phenotype, dependent on fibroblast integrin α4β1 expression and Cox-2/Nrf2 signalling [236,237].
The importance and therapeutic potential of macrophages in the wound healing process has been highlighted in recent years [6,238,239]. Crosstalk between myofibroblasts and macrophages during skin repair was reported [240]. Growth factors secreted by CD301b+ M2-type macrophages were shown to selectively stimulate the proliferation of adipocyte precursor (AP)-derived myofibroblasts only. In aged mice wounds and experimentally induced mouse skin fibrosis, AP-derived myofibroblasts and CD301b+ macrophages were reduced, and a CD29+ myofibroblast pool was increased. In keloids, CD301b+ macrophages and AP-derived myofibroblasts were also increased [241]. In fibrotic lung tissues, cadherin-11 mediated the adhesion between macrophages and myofibroblasts, promoting pro-fibrotic myofibroblast activity via the paracrine release of TGF-β1 by the macrophages [242]. Experimental research has suggested roles of mast cells in scar formation [243]; reduced numbers of activated mast cells were reported to improve healing and minimize scarring [244,245,246], whereas mast cell hyperplasia was causally linked to myofibroblast hyperplasia [247,248,249]. Increased neuropeptide activity and the presence of substance-P (SP) were found in hypertrophic scar samples [250,251]. Mast cells were previously suggested to be a major source of neuropeptide SP-stimulated inflammation and increased myofibroblast activity [252]. These studies showed that myofibroblast activity could be mediated through regulated leukocyte–myofibroblast interactions and suggest that targeting certain leukocyte subpopulations may serve as anti-fibrotic strategies.
Targeting inflammatory meditators has shown promise in alleviating the magnitude of fibrosis in various animal models. Follistatin-like-1 (FSTL1) was found to be elevated in serum from patients with silicosis and in mouse lung fibrosis models. FSTL1-induced IL-1β production by macrophages and positively regulated TGF-β1 signalling in fibroblasts. The inhibition of FSTL1 expression or activity protected against lung injury and fibrosis [253,254]. Recently, FSTL1 neutralizing antibodies were shown to exhibit potent anti-inflammatory actions, attenuate bleomycin-induced IPF and dermal fibrosis in vivo, and downregulate TGF-β1-driven fibrosis in human skin ex vivo [255].
The transmembrane serine protease and collagenase, fibroblast activation protein (FAP) is prominently expressed by activated fibroblasts and myofibroblasts during tissue remodelling and fibrosis [256]. FAP-cleaved collagen binds to the scavenger receptor (SR)-A, recruiting SR-A+ macrophages to sites of collagen turnover [257]. The liver expression of FAP in cirrhosis was shown to correlate with the severity of fibrosis but was not exclusively expressed by α-SMA+ myofibroblasts, suggesting that FAP marks a differentially activated fibroblast state [258]. Lines of research have started to establish the mechanistic actions of FAP in fibroblast heterogeneity and governance over the pro-fibrotic ECM [93]. Treatment with anti-FAP antibody reduced collagen type I production by fibro-stenotic intestinal myofibroblasts [259]. The FAP and dipeptidyl peptidase IV inhibitor, talabostat mesylate (PT100), was used to treat bleomycin-induced IPF murine models. Treatment with PT100 showed anti-fibro-proliferative activity but increased macrophage activation, with no effect on collagen expression [260]. Therefore, the present scope for specific FAP inhibition in dermal fibrosis models remains unclear, until more mechanistic information is reported.
5.3. Targeted Myofibroblast Apoptosis
In physiological wound healing, myofibroblasts disappeared following wound closure and resolution [122,261], predominantly by apoptosis [33]. Despite the elevated production of reactive oxygen species (ROS) by myofibroblasts during fibrosis, the persistence of TGF-β1 expression, ECM deposition, and accumulative stress-induced FAK promotes pro-survival and anti-apoptotic myofibroblast phenotypes [31,262]. The susceptibility of myofibroblasts to nitric oxide (NO)-induced apoptosis has been reported in vitro [263]. Therefore, a combination of reduced profibrotic growth factor expression, increased ECM turnover, and increased NO generation may set the stage for triggering myofibroblast apoptosis during the resolution of tissue repair and remodelling [264,265].
A single chain antibody (C1-3) specifically targets synaptophysin+ liver myofibroblasts without co-localising with liver monocytes or macrophages [266], thus demonstrating that the identification of unique markers of myofibroblasts in fibrosis could serve as targeting devices. The researchers showed that C1-3-gliotoxin conjugates induced non-parenchymal cell apoptosis and depleted liver myofibroblasts without affecting monocytes or macrophages, resulting in the reduced severity of fibrosis [266]. The anticancer drug, Elesclomol, was found to selectively induce apoptosis in activated fibroblasts and myofibroblasts isolated from scar tissue samples. Elesclomol upregulated intracellular levels of ROS, caspase-3, and cytochrome-c proteins, resulting in reduced myofibroblast numbers and a lower scar elevation in vivo [267].
The mechanical tension-stimulated myofibroblast differentiation increased mitochondrial priming and death signalling proteins, such as the pro-apoptotic BH3-only protein BIM [268,269]. The anti-apoptotic protein BCL-XL sequesters BIM and ensures myofibroblast survival. Lagares et al. showed that myofibroblasts were susceptible to apoptosis induced by the BCL-2 inhibitor and the BH3 mimetic drug, ABT-263 (Navitoclax), which inhibited BCL-XL and allowed BIM to activate myofibroblast apoptosis in mouse models of scleroderma dermal fibrosis [32]. These results were recapitulated in rabbit ear hypertrophic scar models, wherein ABT-263 improved scar appearance and collagen arrangement [40]. Future studies into the physiological triggers for time-appropriate myofibroblast apoptosis could potentially lead to the identification of novel treatments with improved therapeutic indexes for scarless wound healing.
5.4. Antioxidant Therapeutics
Another proposed regulator of normal and pathological scarring in numerous tissues is oxidative stress, referring to the overproduction of ROS via such mechanisms as NADPH oxidases (NOXs) and the mitochondria, at the expense of cellular and tissue antioxidant defences [270,271,272,273,274]. The induction of the myofibroblast phenotype is accompanied by depleted cellular antioxidants, leading to ROS generation and the implication of ROS in multiple signalling pathways associated with myofibroblast differentiation. Thus, an emergent area of research is focusing on evaluating the efficacious index of antioxidants against fibrosis and in restoring cellular redox balance. The liposomal delivery of copper/zinc (Cu/Zn) superoxide dismutase (SOD)-attenuated TGF-β1, α-SMA and collagen type I expression in dermal fibroblasts, although myofibroblast apoptosis remained unaffected [275]. Similarly, SOD1-containing fusion proteins alleviated oxidative stress in cardiac myofibroblasts via the reduced expression of TGF-β1, α-SMA, and collagen types I and III—whilst restoring MMP-1 and attenuating MMP inhibitor (TIMP-1) secretion [276]. Small molecule inhibitors of NOXs, such as GKT136901 and GKT137831, also exhibited therapeutic potential by reducing murine liver fibrosis [277,278]. In addition to the progress made with such promising findings, the development of nanoparticles with inherent antioxidant activities, such as cerium oxide, fullerene, and mesoporous silica, have also been explored as potential therapeutic options for fibrosis, which may also be combined with payloads of pharmaceuticals, genes, or proteins [279].
Natural compounds have a long history of use in wound healing, and the cellular mechanisms of action are beginning to be delineated as active components are extracted and assessed [280,281,282]. The exploitation of the aromatic nature and antioxidant capabilities of various naturally sourced polyphenolic compounds and their extracts have been demonstrated in numerous in vitro and in vivo systems, with desirable biocompatible and anti-fibrotic effects in models of pulmonary [283,284,285]; renal [286,287]; myocardial [288,289]; and skin fibrosis [290,291]. The clinical potential of natural compound-based therapies is often restricted by the lack of clarity in their multifaceted bioactivities. Hence, delineating cellular responses to the more specific and potent actions of natural compound extracts has become a popularised concept towards their clinical translation.
6. Future Perspectives for the Discovery of Novel Therapeutics
The complex multistage process of wound healing is vulnerable to dysregulation by a plethora of factors that can initiate and maintain myofibroblast differentiation. Failure of timely wound resolution and myofibroblast apoptosis can inevitably result in the excessive and disorganised deposition of the collagen-rich ECM that is characteristic of fibrosis. The growing knowledge base surrounding myofibroblast cell formation, function, and profibrotic regulators of differentiation has revealed promising candidate therapies that target the myofibroblast phenotype and functions at various stages of wound healing, with the aim to achieve attenuated scar formation. In the wake of unsatisfactory pre-clinical and clinical trial results from the inhibition of TGF-β1, researchers have sought alternative routes to preventing myofibroblast differentiation. Fibroblasts resistant to TGF-β1-stimulated myofibroblast differentiation, such as embryonic fibroblasts, oral mucosal fibroblasts, and aged/senescent fibroblasts, which continue to be extensively studied to uncover important targets, including cytokines, ECM components, receptors, and intracellular signalling cascades involved in resistance to myofibroblast phenotypic acquisition. Fundamental research into molecular-based interventions have given rise to an abundance of candidate treatment modalities, but a key limitation remains: namely a deficit of cell-specific targeted therapies due to the current lack of identified markers unique to myofibroblasts, perhaps as a result of their heterogenous origins. Biomaterials and drug carrier technologies may offer circumvention by facilitating the localised release of bioactive molecules within the microenvironment and the vicinity of myofibroblasts and are likely to take precedence in the sophisticated and controlled spatiotemporal delivery of therapeutic agents to wound sites. The progress and increased accessibility of single cell analytics and multi-omics may help to further identify subsets of dermal fibroblasts (e.g., fibroblasts from papillary or reticular dermis) or myofibroblasts that have more prominent roles in driving fibrosis, and this may facilitate the discovery of uniquely expressed cell surface receptors, proteins, or other targetable moieties. An alternative strategy only briefly touched upon here includes the regulation of cell–cell dynamics. Research into direct heterogenous cell binding, cell paracrine activity, and influence on surrounding cells may provide further clues towards myofibroblast regulation (e.g., leukocyte, stem cell, keratinocyte/epithelial cell, or endothelial cell regulation of fibroblasts and vice versa). Regardless of the afflicted tissue, the underlying cellular aetiology of fibrosis is largely similar. This undoubtedly implies that the progress of research in organ fibrosis will also provide applicable therapies for scarless wound healing. Here, we provided a comprehensive overview of promising candidate molecular-based therapies, which have been directly inspired by the discovery of key regulatory mechanisms implicated in myofibroblast formation and function. Certainly, continued research into myofibroblast biology will usher in a new era of novel therapeutics that will, in-turn, contribute to the knowledge pool. This review highlights the current literature and understanding of cellular mechanisms and interventions of myofibroblasts in the context of reduced scar skin repair, and our hope is that it serves as a reference guide for ongoing and planned research.
Author Contributions
Conceptualization, A.C.M., R.M., and R.S.; writing—original draft preparation, Y.T., E.L.W., J.D., and A.C.M.; editing, D.K., R.S., R.M., and A.C.M. All authors have read and agreed to the published version of the manuscript.
Funding
The authors thank the following funding bodies for supporting the writing of this review; National Natural Science Foundation of China (NSFC) (82050410449; 81921004; 8130060); QBiotics Group (Australia); and The Dunhill Medical Trust (UK, RPGF2006\248).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gabbiani, G.; Ryan, G.B.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Majno, G.; Gabbiani, G.; Hirschel, B.J.; Ryan, G.B.; Statkov, P.R. Contraction of granulation tissue in vitro: Similarity to smooth muscle. Science 1971, 173, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Skalli, O.; Ropraz, P.; Trzeciak, A.; Benzonana, G.; Gillessen, D.; Gabbiani, G. A monoclonal antibody against alpha-smooth muscle actin: A new probe for smooth muscle differentiation. J. Cell Biol. 1986, 103, 2787–2796. [Google Scholar] [CrossRef]
- Darby, I.; Skalli, O.; Gabbiani, G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab. Investig. 1990, 63, 21–29. [Google Scholar] [PubMed]
- Serini, G.; Gabbiani, G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999, 250, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Pittet, P.; Smith-Clerc, J.; Chaponnier, C.; Meister, J.J. Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol. Biol. Cell 2004, 15, 4310–4320. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, L.K.; Fray, T.R.; Molloy, J.E.; Adams, J.J.; Armitage, M.P.; Sparrow, J.C. Contractility of single human dermal myofibroblasts and fibroblasts. Cell Motil. Cytoskelet. 2002, 52, 82–90. [Google Scholar] [CrossRef]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [Green Version]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Lampi, M.C.; Reinhart-King, C.A. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Sapudom, J.; Rubner, S.; Martin, S.; Thoenes, S.; Anderegg, U.; Pompe, T. The interplay of fibronectin functionalization and TGF-beta1 presence on fibroblast proliferation, differentiation and migration in 3D matrices. Biomater. Sci. 2015, 3, 1291–1301. [Google Scholar] [CrossRef] [Green Version]
- Gabbiani, G. The myofibroblast: A key cell for wound healing and fibrocontractive diseases. Prog. Clin. Biol. Res. 1981, 54, 183–194. [Google Scholar]
- Lieubeau, B.; Garrigue, L.; Barbieux, I.; Meflah, K.; Gregoire, M. The role of transforming growth factor beta 1 in the fibroblastic reaction associated with rat colorectal tumor development. Cancer Res. 1994, 54, 6526–6532. [Google Scholar]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Watari, K.; Tajima, M.; Nakaya, T.; Matsuda, S.; Ohara, H.; Nishihara, H.; Yamaguchi, H.; Hashimoto, A.; Nishida, M.; et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Investig. 2017, 127, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Gargus, M.; Niu, C.; Vallone, J.G.; Binkley, J.; Rubin, D.C.; Shaker, A. Human esophageal myofibroblasts secrete proinflammatory cytokines in response to acid and Toll-like receptor 4 ligands. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G904–G923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, M.R.; Giembycz, M.A.; McKay, D.M. Bidirectional crosstalk via IL-6, PGE2 and PGD2 between murine myofibroblasts and alternatively activated macrophages enhances anti-inflammatory phenotype in both cells. Br. J. Pharmacol. 2016, 173, 899–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, M.; Dieude, M.; Yang, B.; Hamelin, K.; Underwood, K.; Hebert, M.J. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy 2014, 10, 2193–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vierhout, M.; Ayoub, A.; Naiel, S.; Yazdanshenas, P.; Revill, S.D.; Reihani, A.; Dvorkin-Gheva, A.; Shi, W.; Ask, K. Monocyte and macrophage derived myofibroblasts: Is it fate? A review of the current evidence. Wound Repair Regen. 2021. [Google Scholar] [CrossRef]
- Schuster, R.; Rockel, J.S.; Kapoor, M.; Hinz, B. The inflammatory speech of fibroblasts. Immunol. Rev. 2021. [Google Scholar] [CrossRef]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Laurent, G.J.; Chambers, R.C.; Hill, M.R.; McAnulty, R.J. Regulation of matrix turnover: Fibroblasts, forces, factors and fibrosis. Biochem. Soc. Trans. 2007, 35, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.J.; Rognoni, E. Dissecting Fibroblast Heterogeneity in Health and Fibrotic Disease. Curr. Rheumatol. Rep. 2020, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Martin, P. Wound repair: A showcase for cell plasticity and migration. Curr. Opin. Cell Biol. 2016, 42, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Duscher, D.; Maan, Z.N.; Wong, V.W.; Rennert, R.C.; Januszyk, M.; Rodrigues, M.; Hu, M.; Whitmore, A.J.; Whittam, A.J.; Longaker, M.T.; et al. Mechanotransduction and fibrosis. J. Biomech. 2014, 47, 1997–2005. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B. Masters and servants of the force: The role of matrix adhesions in myofibroblast force perception and transmission. Eur. J. Cell Biol. 2006, 85, 175–181. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- Lagares, D.; Santos, A.; Grasberger, P.E.; Liu, F.; Probst, C.K.; Rahimi, R.A.; Sakai, N.; Kuehl, T.; Ryan, J.; Bhola, P.; et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Desmouliere, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar] [PubMed]
- Grinnell, F.; Zhu, M.; Carlson, M.A.; Abrams, J.M. Release of mechanical tension triggers apoptosis of human fibroblasts in a model of regressing granulation tissue. Exp. Cell Res. 1999, 248, 608–619. [Google Scholar] [CrossRef] [Green Version]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, S.U.; Li, Y.H.; Roy, J.; Zhang, F.; Puchulu-Campanella, E.; Lindeman, S.D.; Srinivasarao, M.; Tsoyi, K.; Liang, X.; Ayaub, E.A.; et al. Targeted inhibition of PI3 kinase/mTOR specifically in fibrotic lung fibroblasts suppresses pulmonary fibrosis in experimental models. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, X.; Zhang, R.; Cao, T.; Cai, S.Y.; Boyer, J.L.; Zhang, X.; Li, D.; Huang, Y. A Positive Feedback Loop of TET3 and TGF-beta1 Promotes Liver Fibrosis. Cell Rep. 2020, 30, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Patón, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Nangole, F.W.; Agak, G.W. Keloid pathophysiology: Fibroblast or inflammatory disorders? JPRAS Open 2019, 22, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xiao, Y.; Zhong, C.; Shu, F.; Xiao, S.; Zheng, Y.; Xia, Z. ABT-263 Reduces Hypertrophic Scars by Targeting Apoptosis of Myofibroblasts. Front. Pharmacol. 2020, 11, 615505. [Google Scholar] [CrossRef] [PubMed]
- Canady, J.; Karrer, S.; Fleck, M.; Bosserhoff, A.K. Fibrosing connective tissue disorders of the skin: Molecular similarities and distinctions. J. Dermatol. Sci. 2013, 70, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Oh, Y.; Park, Y.J.; Park, O.; Yang, H.; Slania, S.; Hummers, L.K.; Shah, A.A.; An, H.T.; Jang, J.; et al. Targeting of dermal myofibroblasts through death receptor 5 arrests fibrosis in mouse models of scleroderma. Nat. Commun. 2019, 10, 1128. [Google Scholar] [CrossRef]
- Musumeci, M.; Vadala, G.; Russo, F.; Pelacchi, F.; Lanotte, A.; Denaro, V. Dupuytren’s disease therapy: Targeting the vicious cycle of myofibroblasts? Expert Opin. Ther. Targets 2015, 19, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, E.; Spiropoulos, A.; Theodosaki, M.; Stefanaki, K.; Petrakou, E.; Graphakos, S. Myofibroblasts generated in culture from sclerotic skin lesions of a patient with extensive chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation are of recipient origin. Stem Cells Dev. 2010, 19, 1285–1287. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.A.; Tian, Y.A.C.; Steadman, R.; Phillips, A.O. TGF-β1-mediated fibroblast–myofibroblast terminal differentiation—the role of smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal transduction by the TGF-beta superfamily. Science 2002, 296, 1646–1647. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, A.; Samso, P.; Fontova, P.; Simon-Molas, H.; Manzano, A.; Castano, E.; Rosa, J.L.; Martinez-Outshoorn, U.; Ventura, F.; Navarro-Sabate, A.; et al. TGF-beta1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017, 284, 3437–3454. [Google Scholar] [CrossRef] [Green Version]
- Samuel, G.H.; Bujor, A.M.; Nakerakanti, S.S.; Hant, F.N.; Trojanowska, M. Autocrine transforming growth factor beta signaling regulates extracellular signal-regulated kinase 1/2 phosphorylation via modulation of protein phosphatase 2A expression in scleroderma fibroblasts. Fibrogenesis Tissue Repair 2010, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Vander Ark, A.; Cao, J.; Li, X. TGF-beta receptors: In and beyond TGF-beta signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three Isoforms of Mammalian Hyaluronan Synthases Have Distinct Enzymatic Properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, P.M.; Rochester, C.L.; Freundlich, B.; Elias, J.A. Cytokine regulation of human lung fibroblast hyaluronan (hyaluronic acid) production. Evidence for cytokine-regulated hyaluronan (hyaluronic acid) degradation and human lung fibroblast-derived hyaluronidase. J. Clin. Investig. 1992, 90, 1492–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.H.; Thomas, G.J.; Williams, J.D.; Steadman, R. Myofibroblastic differentiation leads to hyaluronan accumulation through reduced hyaluronan turnover. J. Biol. Chem. 2004, 279, 41453–41460. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.M.; Meran, S.; Thomas, D.; Stephens, P.; Bowen, T.; Steadman, R.; Phillips, A. Age-related changes in pericellular hyaluronan organization leads to impaired dermal fibroblast to myofibroblast differentiation. Am. J. Pathol. 2009, 175, 1915–1928. [Google Scholar] [CrossRef] [Green Version]
- Webber, J.; Jenkins, R.H.; Meran, S.; Phillips, A.; Steadman, R. Modulation of TGF beta 1-Dependent Myofibroblast Differentiation by Hyaluronan. Am. J. Pathol. 2009, 175, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Bommaya, G.; Meran, S.; Krupa, A.; Phillips, A.O.; Steadman, R. Tumour necrosis factor-stimulated gene (TSG)-6 controls epithelial–mesenchymal transition of proximal tubular epithelial cells. Int. J. Biochem. Cell Biol. 2011, 43, 1739–1746. [Google Scholar] [CrossRef]
- Porsch, H.; Bernert, B.; Mehić, M.; Theocharis, A.D.; Heldin, C.H.; Heldin, P. Efficient TGFβ-induced epithelial–mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 2013, 32, 4355–4365. [Google Scholar] [CrossRef]
- Preca, B.-T.; Bajdak, K.; Mock, K.; Lehmann, W.; Sundararajan, V.; Bronsert, P.; Matzge-Ogi, A.; Orian-Rousseau, V.; Brabletz, S.; Brabletz, T.; et al. A novel ZEB1/HAS2 positive feedback loop promotes EMT in breast cancer. Oncotarget 2017, 8, 11530–11543. [Google Scholar] [CrossRef] [Green Version]
- Webber, J.; Meran, S.; Steadman, R.; Phillips, A. Hyaluronan Orchestrates Transforming Growth Factor-β1-dependent Maintenance of Myofibroblast Phenotype*. J. Biol. Chem. 2009, 284, 9083–9092. [Google Scholar] [CrossRef] [Green Version]
- Meran, S.; Thomas, D.W.; Stephens, P.; Enoch, S.; Martin, J.; Steadman, R.; Phillips, A.O. Hyaluronan Facilitates Transforming Growth Factor-β1-mediated Fibroblast Proliferation*. J. Biol. Chem. 2008, 283, 6530–6545. [Google Scholar] [CrossRef] [Green Version]
- Meran, S.; Thomas, D.; Stephens, P.; Martin, J.; Bowen, T.; Phillips, A.; Steadman, R. Involvement of hyaluronan in regulation of fibroblast phenotype. J. Biol. Chem. 2007, 282, 25687–25697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meran, S.; Martin, J.; Luo, D.D.; Steadman, R.; Phillips, A. Interleukin-1β Induces Hyaluronan and CD44-Dependent Cell Protrusions That Facilitate Fibroblast-Monocyte Binding. Am. J. Pathol. 2013, 182, 2223–2240. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Arif, A.A.; Lee-Sayer, S.S.M.; Dong, Y. Hyaluronan and Its Interactions with Immune Cells in the Healthy and Inflamed Lung. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley, A.C.; Oltean, S.; Hascall, V.; Woods, E.L.; Steadman, R.; Phillips, A.O.; Meran, S. Nuclear hyaluronidase 2 drives alternative splicing of CD44 pre-mRNA to determine profibrotic or antifibrotic cell phenotype. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Meran, S.; Luo, D.D.; Simpson, R.; Martin, J.; Wells, A.; Steadman, R.; Phillips, A.O. Hyaluronan facilitates transforming growth factor-beta1-dependent proliferation via CD44 and epidermal growth factor receptor interaction. J. Biol. Chem. 2011, 286, 17618–17630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley, A.C.; Rogers, M.; Hallett, M.B.; Clayton, A.; Bowen, T.; Phillips, A.O.; Steadman, R. Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, E.L.; Grigorieva, I.V.; Midgley, A.C.; Brown, C.V.M.; Lu, Y.; Phillips, A.O.; Bowen, T.; Meran, S.; Steadman, R. CD147 mediates the CD44s-dependent differentiation of myofibroblasts driven by transforming growth factor-β1. J. Biol. Chem. 2021, in press. [Google Scholar]
- Selbi, W.; de la Motte, C.; Hascall, V.C.; Day, A.J.; Bowen, T.; Phillips, A.O. Characterization of hyaluronan cable structure and function in renal proximal tubular epithelial cells. Kidney Int. 2006, 70, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, L.; Kimata, K. Structure and function of inter-alpha-trypsin inhibitor heavy chains. Connect Tissue Res. 2008, 49, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Melrose, J.; Day, A.J.; Whitelock, J.M. The Inter-α-Trypsin Inhibitor Family: Versatile Molecules in Biology and Pathology. J. Histochem. Cytochem. 2020, 68, 907–927. [Google Scholar] [CrossRef]
- Martin, J.; Midgley, A.; Meran, S.; Woods, E.; Bowen, T.; Phillips, A.O.; Steadman, R. Tumor Necrosis Factor-stimulated Gene 6 (TSG-6)-mediated Interactions with the Inter-α-inhibitor Heavy Chain 5 Facilitate Tumor Growth Factor β1 (TGFβ1)-dependent Fibroblast to Myofibroblast Differentiation. J. Biol. Chem. 2016, 291, 13789–13801. [Google Scholar] [CrossRef] [Green Version]
- Tepekoy, F.; Akkoyunlu, G.; Demir, R. The role of Wnt signaling members in the uterus and embryo during pre-implantation and implantation. J. Assist. Reprod. Genet. 2015, 32, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Folke, J.; Pakkenberg, B.; Brudek, T. Impaired Wnt Signaling in the Prefrontal Cortex of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 873–891. [Google Scholar] [CrossRef]
- Wanitsuwan, W.; Kanngurn, S.; Boonpipattanapong, T.; Sangthong, R.; Sangkhathat, S. Overall expression of beta-catenin outperforms its nuclear accumulation in predicting outcomes of colorectal cancers. World J. Gastroenterol. 2008, 14, 6052–6059. [Google Scholar] [CrossRef]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [Green Version]
- Hamburg-Shields, E.; DiNuoscio, G.J.; Mullin, N.K.; Lafyatis, R.; Atit, R.P. Sustained beta-catenin activity in dermal fibroblasts promotes fibrosis by up-regulating expression of extracellular matrix protein-coding genes. J. Pathol. 2015, 235, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Tong, B.-D.; Wu, Y.-J.; Xiong, W. MicroRNA-29 mediates TGFβ1-induced extracellular matrix synthesis by targeting wnt/β-catenin pathway in human orbital fibroblasts. Int. J. Clin. Exp. Pathol. 2014, 7, 7571–7577. [Google Scholar] [PubMed]
- Sun, Q.; Guo, S.; Wang, C.C.; Sun, X.; Wang, D.; Xu, N.; Jin, S.F.; Li, K.Z. Cross-talk between TGF-beta/Smad pathway and Wnt/beta-catenin pathway in pathological scar formation. Int. J. Clin. Exp. Pathol. 2015, 8, 7631–7639. [Google Scholar] [PubMed]
- Singer, I.I.; Kawka, D.W.; Kazazis, D.M.; Clark, R.A. In vivo co-distribution of fibronectin and actin fibers in granulation tissue: Immunofluorescence and electron microscope studies of the fibronexus at the myofibroblast surface. J. Cell Biol. 1984, 98, 2091–2106. [Google Scholar] [CrossRef] [Green Version]
- Dugina, V.; Fontao, L.; Chaponnier, C.; Vasiliev, J.; Gabbiani, G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J. Cell Sci. 2001, 114, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Goffin, J.M.; Pittet, P.; Csucs, G.; Lussi, J.W.; Meister, J.J.; Hinz, B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J. Cell Biol. 2006, 172, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Water, L.; Varney, S.; Tomasek, J.J. Mechanoregulation of the Myofibroblast in Wound Contraction, Scarring, and Fibrosis: Opportunities for New Therapeutic Intervention. Adv. Wound Care 2013, 2, 122–141. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.R.; Shimbori, C.; Bellaye, P.S.; Inman, M.; Obex, S.; Fatima, S.; Jenkins, G.; Gauldie, J.; Ask, K.; Kolb, M. Stretch-induced Activation of Transforming Growth Factor-beta1 in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 84–96. [Google Scholar] [CrossRef]
- Klingberg, F.; Chow, M.L.; Koehler, A.; Boo, S.; Buscemi, L.; Quinn, T.M.; Costell, M.; Alman, B.A.; Genot, E.; Hinz, B. Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J. Cell Biol. 2014, 207, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Pure, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Kollmannsberger, P.; Bidan, C.M.; Dunlop, J.W.C.; Fratzl, P.; Vogel, V. Tensile forces drive a reversible fibroblast-to-myofibroblast transition during tissue growth in engineered clefts. Sci. Adv. 2018, 4, eaao4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochaton-Piallat, M.L.; Gabbiani, G.; Hinz, B. The myofibroblast in wound healing and fibrosis: Answered and unanswered questions. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Schwarzbauer, J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Sens, C.; Huck, K.; Pettera, S.; Uebel, S.; Wabnitz, G.; Moser, M.; Nakchbandi, I.A. Fibronectins containing extradomain A or B enhance osteoblast differentiation via distinct integrins. J. Biol. Chem. 2017, 292, 7745–7760. [Google Scholar] [CrossRef] [Green Version]
- Jarnagin, W.R.; Rockey, D.C.; Koteliansky, V.E.; Wang, S.S.; Bissell, D.M. Expression of variant fibronectins in wound healing: Cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J. Cell Biol. 1994, 127, 2037–2048. [Google Scholar] [CrossRef]
- Muro, A.F.; Chauhan, A.K.; Gajovic, S.; Iaconcig, A.; Porro, F.; Stanta, G.; Baralle, F.E. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J. Cell Biol. 2003, 162, 149–160. [Google Scholar] [CrossRef]
- Deng, N.; Sanchez, C.G.; Lasky, J.A.; Zhu, D. Detecting splicing variants in idiopathic pulmonary fibrosis from non-differentially expressed genes. PLoS ONE 2013, 8, e68352. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Xie, G.; Ou, J.; Wei, X.; Pan, F.; Liang, H. The extra domain A of fibronectin increases VEGF-C expression in colorectal carcinoma involving the PI3K/AKT signaling pathway. PLoS ONE 2012, 7, e35378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.; Deng, J.; Wei, X.; Xie, G.; Zhou, R.; Yu, L.; Liang, H. Fibronectin extra domain A (EDA) sustains CD133(+)/CD44(+) subpopulation of colorectal cancer cells. Stem Cell Res. 2013, 11, 820–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossnagl, S.; Altrock, E.; Sens, C.; Kraft, S.; Rau, K.; Milsom, M.D.; Giese, T.; Samstag, Y.; Nakchbandi, I.A. EDA-Fibronectin Originating from Osteoblasts Inhibits the Immune Response against Cancer. PLoS Biol. 2016, 14, e1002562. [Google Scholar] [CrossRef] [PubMed]
- Ffrench-Constant, C.; Van de Water, L.; Dvorak, H.F.; Hynes, R.O. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J. Cell Biol. 1989, 109, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.; Shen, X.D.; Gao, F.; Busuttil, R.W.; Coito, A.J. Fibronectin-alpha4beta1 integrin interactions regulate metalloproteinase-9 expression in steatotic liver ischemia and reperfusion injury. Am. J. Pathol. 2007, 170, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelsh, R.M.; McKeown-Longo, P.J.; Clark, R.A.F. EDA Fibronectin in Keloids Create a Vicious Cycle of Fibrotic Tumor Formation. J. Investig. Derm. 2015, 135, 1714–1718. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Tamaki, Z.; Wang, W.; Hinchcliff, M.; Hoover, P.; Getsios, S.; White, E.S.; Varga, J. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci. Transl. Med. 2014, 6, 232ra250. [Google Scholar] [CrossRef] [Green Version]
- Klingberg, F.; Chau, G.; Walraven, M.; Boo, S.; Koehler, A.; Chow, M.L.; Olsen, A.L.; Im, M.; Lodyga, M.; Wells, R.G.; et al. The fibronectin ED-A domain enhances recruitment of latent TGF-beta-binding protein-1 to the fibroblast matrix. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Serini, G.; Bochaton-Piallat, M.L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Mastrangelo, D.; Iselin, C.E.; Chaponnier, C.; Gabbiani, G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 2001, 159, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.F.; Gotwals, P.J.; Koteliansky, V.E.; Sheppard, D.; Van De Water, L. The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J. Biol. Chem. 2002, 277, 14467–14474. [Google Scholar] [CrossRef] [Green Version]
- Kohan, M.; Muro, A.F.; White, E.S.; Berkman, N. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. 2010, 24, 4503–4512. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Bystroff, C.; Wang, C.; Vogelezang, M.G.; Vincent, P.A.; Hynes, R.O.; Van De Water, L. Identification of the peptide sequences within the EIIIA (EDA) segment of fibronectin that mediate integrin alpha9beta1-dependent cellular activities. J. Biol. Chem. 2008, 283, 2858–2870. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yan, H.; Tai, Y.; Xue, Y.; Wei, Y.; Wang, K.; Zhao, Q.; Wang, S.; Kong, D.; Midgley, A.C. Design and Evaluation of a Polypeptide that Mimics the Integrin Binding Site for EDA Fibronectin to Block Profibrotic Cell Activity. Int. J. Mol. Sci. 2021, 22, 1575. [Google Scholar] [CrossRef]
- Shinde, A.V.; Kelsh, R.; Peters, J.H.; Sekiguchi, K.; Van De Water, L.; McKeown-Longo, P.J. The alpha4beta1 integrin and the EDA domain of fibronectin regulate a profibrotic phenotype in dermal fibroblasts. Matrix Biol. 2015, 41, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Vogel, W.F.; Aszodi, A.; Alves, F.; Pawson, T. Discoidin domain receptor 1 tyrosine kinase has an essential role in mammary gland development. Mol. Cell Biol. 2001, 21, 2906–2917. [Google Scholar] [CrossRef] [Green Version]
- Cario, M. DDR1 and DDR2 in skin. Cell Adh. Migr. 2018, 12, 386–393. [Google Scholar] [CrossRef]
- Coelho, N.M.; Wang, A.; McCulloch, C.A. Discoidin domain receptor 1 interactions with myosin motors contribute to collagen remodeling and tissue fibrosis. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118510. [Google Scholar] [CrossRef]
- Doyle, A.D.; Carvajal, N.; Jin, A.; Matsumoto, K.; Yamada, K.M. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun. 2015, 6, 8720. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.R.; Chen, X.; Ling, L.; Song, Y.H.; Shimpi, A.A.; Choi, S.; Gonzalez, J.; Sapudom, J.; Wang, K.; Andresen Eguiluz, R.C.; et al. Collagen microarchitecture mechanically controls myofibroblast differentiation. Proc. Natl. Acad. Sci. USA 2020, 117, 11387–11398. [Google Scholar] [CrossRef]
- Dai, J.-Y.; Dou, K.-F.; Wang, C.-H.; Zhao, P.; Lau, W.B.; Tao, L.; Wu, Y.-M.; Tang, J.; Jiang, J.-L.; Chen, Z.-N. The interaction of HAb18G/CD147 with integrin α6β1 and its implications for the invasion potential of human hepatoma cells. BMC Cancer 2009, 9, 337. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.C.; Petley-Ragan, L.M.; Das, M.; Auld, V.J. Basigin Associates with Integrin in Order to Regulate Perineurial Glia and Drosophila Nervous System Morphology. J. Neurosci. 2020, 40, 3360. [Google Scholar] [CrossRef] [PubMed]
- Arnoldi, R.; Chaponnier, C.; Gabbiani, G.; Hinz, B. Chapter 88—Heterogeneity of Smooth Muscle. In Muscle; Hill, J.A., Olson, E.N., Eds.; Academic Press: Cambridge, MA, USA, 2012; pp. 1183–1195. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Vaughan, M.B.; Kropp, B.P.; Gabbiani, G.; Martin, M.D.; Haaksma, C.J.; Hinz, B. Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair Regen. 2006, 14, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; DiCesare, L.; Tan, I.; Leung, T.; SundarRaj, N. Rho-mediated assembly of stress fibers is differentially regulated in corneal fibroblasts and myofibroblasts. Exp. Cell Res. 2004, 298, 574–583. [Google Scholar] [CrossRef]
- Duterme, C.; Mertens-Strijthagen, J.; Tammi, M.; Flamion, B. Two novel functions of hyaluronidase-2 (Hyal2) are formation of the glycocalyx and control of CD44-ERM interactions. J. Biol. Chem. 2009, 284, 33495–33508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley, A.C.; Woods, E.L.; Jenkins, R.H.; Brown, C.; Khalid, U.; Chavez, R.; Hascall, V.; Steadman, R.; Phillips, A.O.; Meran, S. Hyaluronidase-2 Regulates RhoA Signaling, Myofibroblast Contractility, and Other Key Profibrotic Myofibroblast Functions. Am. J. Pathol. 2020, 190, 1236–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapudom, J.; Muller, C.D.; Nguyen, K.T.; Martin, S.; Anderegg, U.; Pompe, T. Matrix Remodeling and Hyaluronan Production by Myofibroblasts and Cancer-Associated Fibroblasts in 3D Collagen Matrices. Gels 2020, 6, 33. [Google Scholar] [CrossRef]
- Albeiroti, S.; Soroosh, A.; de la Motte, C.A. Hyaluronan’s Role in Fibrosis: A Pathogenic Factor or a Passive Player? BioMed Res. Int. 2015, 2015, 790203. [Google Scholar] [CrossRef] [Green Version]
- Haak, A.J.; Tsou, P.S.; Amin, M.A.; Ruth, J.H.; Campbell, P.; Fox, D.A.; Khanna, D.; Larsen, S.D.; Neubig, R.R. Targeting the myofibroblast genetic switch: Inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J. Pharmacol. Exp. 2014, 349, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Rodansky, E.S.; Haak, A.J.; Larsen, S.D.; Neubig, R.R.; Higgins, P.D. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis. 2014, 20, 154–165. [Google Scholar] [CrossRef]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Ooki, T.; Murata-Kamiya, N.; Takahashi-Kanemitsu, A.; Wu, W.; Hatakeyama, M. High-Molecular-Weight Hyaluronan Is a Hippo Pathway Ligand Directing Cell Density-Dependent Growth Inhibition via PAR1b. Dev. Cell 2019, 49, 590–604. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; de Rond, S.; Werker, P.M.; Boo, S.; Hinz, B.; van Beuge, M.M.; Bank, R.A. YAP1 Is a Driver of Myofibroblast Differentiation in Normal and Diseased Fibroblasts. Am. J. Pathol. 2015, 185, 3326–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Xia, H.; Xu, H.; Ma, J.; Zhou, S.; Hou, W.; Tang, Q.; Gong, Q.; Nie, Y.; Bi, F. Standard CD44 modulates YAP1 through a positive feedback loop in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 103, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Black, M.; Milewski, D.; Le, T.; Ren, X.; Xu, Y.; Kalinichenko, V.V.; Kalin, T.V. FOXF1 Inhibits Pulmonary Fibrosis by Preventing CDH2-CDH11 Cadherin Switch in Myofibroblasts. Cell Rep. 2018, 23, 442–458. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, A.G.; El Hachem, M.; Zambruno, G.; Nystrom, A.; Candi, E.; Castiglia, D. Notch-ing up knowledge on molecular mechanisms of skin fibrosis: Focus on the multifaceted Notch signalling pathway. J. Biomed. Sci. 2021, 28, 36. [Google Scholar] [CrossRef]
- Della-Torre, E.; Feeney, E.; Deshpande, V.; Mattoo, H.; Mahajan, V.; Kulikova, M.; Wallace, Z.S.; Carruthers, M.; Chung, R.T.; Pillai, S.; et al. B-cell depletion attenuates serological biomarkers of fibrosis and myofibroblast activation in IgG4-related disease. Ann. Rheum. Dis. 2015, 74, 2236–2243. [Google Scholar] [CrossRef] [Green Version]
- Lafyatis, R.; Kissin, E.; York, M.; Farina, G.; Viger, K.; Fritzler, M.J.; Merkel, P.A.; Simms, R.W. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2009, 60, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Dolivo, D.M.; Larson, S.A.; Dominko, T. Tryptophan metabolites kynurenine and serotonin regulate fibroblast activation and fibrosis. Cell. Mol. Life Sci. 2018, 75, 3663–3681. [Google Scholar] [CrossRef]
- Fang, C.; Huang, H.; Guo, J.; Ferianc, M.; Xu, Z. Real-world experiences: Efficacy and tolerability of pirfenidone in clinical practice. PLoS ONE 2020, 15, e0228390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Zhao, Y.; Li, Z.; Kang, J. Real-life experiences in a single center: Efficacy of pirfenidone in idiopathic pulmonary fibrosis and fibrotic idiopathic non-specific interstitial pneumonia patients. Adv. Respir. Dis. 2020, 14. [Google Scholar] [CrossRef]
- Chen, J.; Wang, H.; Mei, L.; Wang, B.; Huang, Y.; Quan, G.; Lu, C.; Peng, T.; Pan, X.; Wu, C. A pirfenidone loaded spray dressing based on lyotropic liquid crystals for deep partial thickness burn treatment: Healing promotion and scar prophylaxis. J. Mater. Chem. B 2020, 8, 2573–2588. [Google Scholar] [CrossRef]
- Kaidar-Person, O.; Marks, L.B.; Jones, E.L. Pentoxifylline and vitamin E for treatment or prevention of radiation-induced fibrosis in patients with breast cancer. Breast J. 2018, 24, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Nguyen, H.V.; Mannschreck, D.; Redett, R.J.; Puttgen, K.B.; Stewart, F.D. Fractional CO2 Laser Treatment Outcomes for Pediatric Hypertrophic Burn Scars. J. Burn Care Res. 2019, 40, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, D.; Schiestl, C.; Meuli, M. Integra Artificial Skin for burn scar revision in adolescents and children. Burns 2010, 36, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Wood, F.M.; Giles, N.; Stevenson, A.; Rea, S.; Fear, M. Characterisation of the cell suspension harvested from the dermal epidermal junction using a ReCell(R) kit. Burns 2012, 38, 44–51. [Google Scholar] [CrossRef]
- Hu, Z.C.; Chen, D.; Guo, D.; Liang, Y.Y.; Zhang, J.; Zhu, J.Y.; Tang, B. Randomized clinical trial of autologous skin cell suspension combined with skin grafting for chronic wounds. Br. J. Surg. 2015, 102, e117–e123. [Google Scholar] [CrossRef]
- Shah, M.; Foreman, D.M.; Ferguson, M.W. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995, 108, 985–1002. [Google Scholar] [CrossRef]
- Occleston, N.L.; O’Kane, S.; Laverty, H.G.; Cooper, M.; Fairlamb, D.; Mason, T.; Bush, J.A.; Ferguson, M.W. Discovery and development of avotermin (recombinant human transforming growth factor beta 3): A new class of prophylactic therapeutic for the improvement of scarring. Wound Repair Regen. 2011, 19 (Suppl. 1), s38–s48. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.W.; O’Kane, S. Scar-free healing: From embryonic mechanisms to adult therapeutic intervention. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Eslami, A.; Gallant-Behm, C.L.; Hart, D.A.; Wiebe, C.; Honardoust, D.; Gardner, H.; Hakkinen, L.; Larjava, H.S. Expression of integrin alphavbeta6 and TGF-beta in scarless vs. scar-forming wound healing. J. Histochem. Cytochem. 2009, 57, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Durani, P.; Occleston, N.; O’Kane, S.; Ferguson, M.W. Avotermin: A novel antiscarring agent. Int. J. Low Extrem. Wounds 2008, 7, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.W.; Duncan, J.; Bond, J.; Bush, J.; Durani, P.; So, K.; Taylor, L.; Chantrey, J.; Mason, T.; James, G.; et al. Prophylactic administration of avotermin for improvement of skin scarring: Three double-blind, placebo-controlled, phase I/II studies. Lancet 2009, 373, 1264–1274. [Google Scholar] [CrossRef]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef]
- Denton, C.P.; Merkel, P.A.; Furst, D.E.; Khanna, D.; Emery, P.; Hsu, V.M.; Silliman, N.; Streisand, J.; Powell, J.; Akesson, A.; et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007, 56, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hwang, S.R.; Yoon, I.S. Advanced Growth Factor Delivery Systems in Wound Management and Skin Regeneration. Molecules 2017, 22, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, J.; Lei, Y.; Lyu, L.; Zuo, R.; Chen, T. MicroRNA-21 in Skin Fibrosis: Potential for Diagnosis and Treatment. Mol. Diagn. 2017, 21, 633–642. [Google Scholar] [CrossRef]
- Luo, H.; Zhu, H.; Zhou, B.; Xiao, X.; Zuo, X. MicroRNA-130b regulates scleroderma fibrosis by targeting peroxisome proliferator-activated receptor gamma. Mod. Rheumatol. 2015, 25, 595–602. [Google Scholar] [CrossRef]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Jinnin, M.; Yamane, K.; Honda, N.; Kajihara, I.; Makino, T.; Masuguchi, S.; Fukushima, S.; Okamoto, Y.; Hasegawa, M.; et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J. Immunol. 2012, 188, 3573–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley, A.C.; Bowen, T.; Phillips, A.O.; Steadman, R. MicroRNA-7 inhibition rescues age-associated loss of epidermal growth factor receptor and hyaluronan-dependent differentiation in fibroblasts. Aging Cell 2014, 13, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley, A.C.; Morris, G.; Phillips, A.O.; Steadman, R. 17beta-estradiol ameliorates age-associated loss of fibroblast function by attenuating IFN-gamma/STAT1-dependent miR-7 upregulation. Aging Cell 2016, 15, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell Physiol. Biochem. 2017, 42, 2207–2219. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Guo, Y.; Chen, B.; Dong, P.; Zheng, J. MicroRNA-17-5p activates hepatic stellate cells through targeting of Smad7. Lab. Investig. 2015, 95, 781–789. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, S. MicroRNAs in fibrosis: Opportunities and challenges. Arthritis Res. 2016, 18, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley, A.C.; Duggal, L.; Jenkins, R.; Hascall, V.; Steadman, R.; Phillips, A.O.; Meran, S. Hyaluronan regulates bone morphogenetic protein-7-dependent prevention and reversal of myofibroblast phenotype. J. Biol. Chem. 2015, 290, 11218–11234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, D.F.; Ewart, L.M.; Masterson, E.; Tennant, S.; Grebnev, G.; Prunotto, M.; Pomposiello, S.; Conde-Knape, K.; Martin, F.M.; Godson, C. BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- Midgley, A.C.; Wei, Y.; Zhu, D.; Gao, F.; Yan, H.; Khalique, A.; Luo, W.; Jiang, H.; Liu, X.; Guo, J.; et al. Multifunctional Natural Polymer Nanoparticles as Antifibrotic Gene Carriers for CKD Therapy. J. Am. Soc. Nephrol. 2020, 31, 2292–2311. [Google Scholar] [CrossRef]
- Tsujimura, T.; Idei, M.; Yoshikawa, M.; Takase, O.; Hishikawa, K. Roles and regulation of bone morphogenetic protein-7 in kidney development and diseases. World J. Stem Cells 2016, 8, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Lin, Q.; Shao, Y.; Rong, L.; Zhang, D. BMP7 suppresses excessive scar formation by activating the BMP7/Smad1/5/8 signaling pathway. Mol. Med. Rep. 2017, 16, 1957–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, L.A.; Hackett, T.L.; Warner, S.M.; Shaheen, F.; Argentieri, R.L.; Dudas, P.; Farrell, F.X.; Knight, D.A. BMP-7 does not protect against bleomycin-induced lung or skin fibrosis. PLoS ONE 2008, 3, e4039. [Google Scholar] [CrossRef]
- Ulloa, L.; Doody, J.; Massague, J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 1999, 397, 710–713. [Google Scholar] [CrossRef]
- Vu, T.N.; Chen, X.; Foda, H.D.; Smaldone, G.C.; Hasaneen, N.A. Interferon-gamma enhances the antifibrotic effects of pirfenidone by attenuating IPF lung fibroblast activation and differentiation. Respir. Res. 2019, 20, 206. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Li, A.; Tan, H.; Zhou, Y. IFN-gamma(+) IL-17(+) Th17 cells regulate fibrosis through secreting IL-21 in systemic scleroderma. J. Cell Mol. Med. 2020, 24, 13600–13608. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, S.D.; Thomas, G.L.; Gabbiani, G.; El Nahas, A.M. Interferon-gamma inhibits experimental renal fibrosis. Kidney Int. 1999, 56, 2116–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, H.L.; Feng, D.C.; Radaeva, S.; Kong, X.N.; Wang, L.; Liu, Y.; Li, Q.; Shen, H.; Gao, Y.P.; Mullenbach, R.; et al. IFN-gamma inhibits liver progenitor cell proliferation in HBV-infected patients and in 3,5-diethoxycarbonyl-1,4-dihydrocollidine diet-fed mice. J. Hepatol. 2013, 59, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Fusiak, T.; Smaldone, G.C.; Condos, R. Pulmonary Fibrosis Treated with Inhaled Interferon-gamma (IFN-gamma). J. Aerosol Med. Pulm. Drug Deliv. 2015, 28, 406–410. [Google Scholar] [CrossRef]
- Haque, M.F.; Meghji, S.; Nazir, R.; Harris, M. Interferon gamma (IFN-gamma) may reverse oral submucous fibrosis. J. Oral Pathol. Med. 2001, 30, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar] [CrossRef]
- Liu, Y. Hepatocyte growth factor in kidney fibrosis: Therapeutic potential and mechanisms of action. Am. J. Physiol. Ren. Physiol. 2004, 287, F7–F16. [Google Scholar] [CrossRef] [Green Version]
- Okayama, K.; Azuma, J.; Dosaka, N.; Iekushi, K.; Sanada, F.; Kusunoki, H.; Iwabayashi, M.; Rakugi, H.; Taniyama, Y.; Morishita, R. Hepatocyte growth factor reduces cardiac fibrosis by inhibiting endothelial-mesenchymal transition. Hypertension 2012, 59, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Mou, S.; Wang, Q.; Shi, B.; Gu, L.; Ni, Z. Hepatocyte growth factor suppresses transforming growth factor-beta-1 and type III collagen in human primary renal fibroblasts. Kaohsiung J. Med. Sci. 2009, 25, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Wang, Z.; Jiang, D.; Qu, L.; Guo, J.; Li, Z. HGF inhibits TGF-beta1-induced myofibroblast differentiation and ECM deposition via MMP-2 in Achilles tendon in rat. Eur. J. Appl. Physiol. 2011, 111, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Jiang, Z.; Han, F.; Zhang, Y.; Li, Z. HGF suppresses the production of collagen type III and alpha-SMA induced by TGF-beta1 in healing fibroblasts. Eur. J. Appl. Physiol. 2008, 103, 489–493. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Nguyen, Q.D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Atta, H.; El-Rehany, M.; Hammam, O.; Abdel-Ghany, H.; Ramzy, M.; Roderfeld, M.; Roeb, E.; Al-Hendy, A.; Raheim, S.A.; Allam, H.; et al. Mutant MMP-9 and HGF gene transfer enhance resolution of CCl4-induced liver fibrosis in rats: Role of ASH1 and EZH2 methyltransferases repression. PLoS ONE 2014, 9, e112384. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Sheng, G.G.; Mifflin, R.C.; Powell, D.W.; Sheng, H. Roles of myofibroblasts in prostaglandin E2-stimulated intestinal epithelial proliferation and angiogenesis. Cancer Res. 2006, 66, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Kajihara, I.; Jinnin, M.; Makino, T.; Masuguchi, S.; Sakai, K.; Fukushima, S.; Maruo, K.; Inoue, Y.; Ihn, H. Overexpression of hepatocyte growth factor receptor in scleroderma dermal fibroblasts is caused by autocrine transforming growth factor beta signaling. Biosci. Trends 2012, 6, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Qin, Z.; Yang, Y.; Fisher, G.J.; Quan, T. Age-related elevation of HGF is driven by the reduction of fibroblast size in a YAP/TAZ/CCN2 axis-dependent manner. J. Dermatol. Sci. 2021, 102, 36–46. [Google Scholar] [CrossRef]
- Qin, Z.; Worthen, C.A.; Quan, T. Cell-size-dependent upregulation of HGF expression in dermal fibroblasts: Impact on human skin connective tissue aging. J. Dermatol. Sci. 2017, 88, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dally, J.; Khan, J.S.; Voisey, A.; Charalambous, C.; John, H.L.; Woods, E.L.; Steadman, R.; Moseley, R.; Midgley, A.C. Hepatocyte Growth Factor Mediates Enhanced Wound Healing Responses and Resistance to Transforming Growth Factor-beta(1)-Driven Myofibroblast Differentiation in Oral Mucosal Fibroblasts. Int. J. Mol. Sci. 2017, 18, 1843. [Google Scholar] [CrossRef]
- Lee, W.J.; Park, S.E.; Rah, D.K. Effects of hepatocyte growth factor on collagen synthesis and matrix metalloproteinase production in keloids. J. Korean Med. Sci. 2011, 26, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eto, H.; Suga, H.; Aoi, N.; Kato, H.; Doi, K.; Kuno, S.; Tabata, Y.; Yoshimura, K. Therapeutic potential of fibroblast growth factor-2 for hypertrophic scars: Upregulation of MMP-1 and HGF expression. Lab. Investig. 2012, 92, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, Y.R.; Ahn, H.M.; Choi, I.K.; Yun, C.O.; Rah, D.K.; Lew, D.H.; Lee, W.J. Hepatocyte growth factor-expressing adenovirus upregulates matrix metalloproteinase-1 expression in keloid fibroblasts. Int. J. Dermatol. 2016, 55, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Guerrot, D.; Kerroch, M.; Placier, S.; Vandermeersch, S.; Trivin, C.; Mael-Ainin, M.; Chatziantoniou, C.; Dussaule, J.C. Discoidin domain receptor 1 is a major mediator of inflammation and fibrosis in obstructive nephropathy. Am. J. Pathol. 2011, 179, 83–91. [Google Scholar] [CrossRef]
- Moll, S.; Desmouliere, A.; Moeller, M.J.; Pache, J.C.; Badi, L.; Arcadu, F.; Richter, H.; Satz, A.; Uhles, S.; Cavalli, A.; et al. DDR1 role in fibrosis and its pharmacological targeting. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118474. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Bartual, S.G.; Luo, J.; Xu, T.; Du, W.; Xun, Q.; Tu, Z.; Brekken, R.A.; Ren, X.; et al. Tetrahydroisoquinoline-7-carboxamide Derivatives as New Selective Discoidin Domain Receptor 1 (DDR1) Inhibitors. ACS Med. Chem. Lett. 2017, 8, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, J.M.; Trempus, C.S.; Garantziotis, S. Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int. J. Cell Biol. 2015, 2015, 563818. [Google Scholar] [CrossRef] [Green Version]
- Qin, K.; Wang, F.; Simpson, R.M.L.; Zheng, X.; Wang, H.; Hu, Y.; Gao, Z.; Xu, Q.; Zhao, Q. Hyaluronan promotes the regeneration of vascular smooth muscle with potent contractile function in rapidly biodegradable vascular grafts. Biomaterials 2020, 257, 120226. [Google Scholar] [CrossRef]
- Skurikhin, E.G.; Pershina, O.V.; Reztsova, A.M.; Ermakova, N.N.; Khmelevskaya, E.S.; Krupin, V.A.; Stepanova, I.E.; Artamonov, A.V.; Bekarev, A.A.; Madonov, P.G.; et al. Modulation of bleomycin-induced lung fibrosis by pegylated hyaluronidase and dopamine receptor antagonist in mice. PLoS ONE 2015, 10, e0125065. [Google Scholar] [CrossRef] [Green Version]
- Andreichenko, I.N.; Tsitrina, A.A.; Fokin, A.V.; Gabdulkhakova, A.I.; Maltsev, D.I.; Perelman, G.S.; Bulgakova, E.V.; Kulikov, A.M.; Mikaelyan, A.S.; Kotelevtsev, Y.V. 4-methylumbelliferone Prevents Liver Fibrosis by Affecting Hyaluronan Deposition, FSTL1 Expression and Cell Localization. Int. J. Mol. Sci. 2019, 20, 6301. [Google Scholar] [CrossRef] [Green Version]
- Collum, S.D.; Chen, N.Y.; Hernandez, A.M.; Hanmandlu, A.; Sweeney, H.; Mertens, T.C.J.; Weng, T.; Luo, F.; Molina, J.G.; Davies, J.; et al. Inhibition of hyaluronan synthesis attenuates pulmonary hypertension associated with lung fibrosis. Br. J. Pharmacol. 2017, 174, 3284–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liang, J.; Yang, T.; Monterrosa Mena, J.; Huan, C.; Xie, T.; Kurkciyan, A.; Liu, N.; Jiang, D.; Noble, P.W. Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol. 2016, 55, 35–48. [Google Scholar] [CrossRef]
- Patsenker, E.; Popov, Y.; Stickel, F.; Jonczyk, A.; Goodman, S.L.; Schuppan, D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology 2008, 135, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Bagnato, G.L.; Irrera, N.; Pizzino, G.; Santoro, D.; Roberts, W.N.; Bagnato, G.; Pallio, G.; Vaccaro, M.; Squadrito, F.; Saitta, A.; et al. Dual alphavbeta3 and alphavbeta5 blockade attenuates fibrotic and vascular alterations in a murine model of systemic sclerosis. Clin. Sci. 2018, 132, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivisto, L.; Bi, J.; Hakkinen, L.; Larjava, H. Integrin alphavbeta6: Structure, function and role in health and disease. Int. J. Biochem. Cell Biol. 2018, 99, 186–196. [Google Scholar] [CrossRef]
- Villa, A.; Trachsel, E.; Kaspar, M.; Schliemann, C.; Sommavilla, R.; Rybak, J.N.; Rosli, C.; Borsi, L.; Neri, D. A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int. J. Cancer 2008, 122, 2405–2413. [Google Scholar] [CrossRef]
- Ziffels, B.; Grotsch, A.; Al-Bayati, L.; Neri, D. Targeted delivery of calreticulin to ED-A fibronectin leads to tumor-growth retardation. J. Biotechnol. 2019, 290, 53–58. [Google Scholar] [CrossRef]
- Femel, J.; Huijbers, E.J.; Saupe, F.; Cedervall, J.; Zhang, L.; Roswall, P.; Larsson, E.; Olofsson, H.; Pietras, K.; Dimberg, A.; et al. Therapeutic vaccination against fibronectin ED-A attenuates progression of metastatic breast cancer. Oncotarget 2014, 5, 12418–12427. [Google Scholar] [CrossRef] [Green Version]
- Prunotto, M.; Bruschi, M.; Gunning, P.; Gabbiani, G.; Weibel, F.; Ghiggeri, G.M.; Petretto, A.; Scaloni, A.; Bonello, T.; Schevzov, G.; et al. Stable incorporation of alpha-smooth muscle actin into stress fibers is dependent on specific tropomyosin isoforms. Cytoskeleton 2015, 72, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Caballero, S.; Yang, R.; Grant, M.B.; Chaqour, B. Selective blockade of cytoskeletal actin remodeling reduces experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Hinz, B.; Dugina, V.; Gabbiani, G.; Chaponnier, C. The N-terminal Ac-EEED sequence plays a role in alpha-smooth-muscle actin incorporation into stress fibers. J. Cell Sci. 2005, 118, 1395–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Gabbiani, G.; Chaponnier, C. The NH2-terminal peptide of alpha-smooth muscle actin inhibits force generation by the myofibroblast in vitro and in vivo. J. Cell Biol. 2002, 157, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Liechty, K.W.; Crombleholme, T.M.; Cass, D.L.; Martin, B.; Adzick, N.S. Diminished interleukin-8 (IL-8) production in the fetal wound healing response. J. Surg. Res. 1998, 77, 80–84. [Google Scholar] [CrossRef]
- Gawronska-Kozak, B.; Bogacki, M.; Rim, J.S.; Monroe, W.T.; Manuel, J.A. Scarless skin repair in immunodeficient mice. Wound Repair Regen. 2006, 14, 265–276. [Google Scholar] [CrossRef]
- Redd, M.J.; Cooper, L.; Wood, W.; Stramer, B.; Martin, P. Wound healing and inflammation: Embryos reveal the way to perfect repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.Y.; Martin, P.; Green, C.R.; Becker, D.L. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 2003, 13, 1697–1703. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Chen, Y.; Lu, L.; Liu, Y.; Wang, Y.; Fan, J.; Yin, Y. Angiotensin II upregulates fibroblast-myofibroblast transition through Cx43-dependent CaMKII and TGF-beta1 signaling in neonatal rat cardiac fibroblasts. Acta Biochim. Biophys. Sin. 2018, 50, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Schultz, F.; Swiatlowska, P.; Alvarez-Laviada, A.; Sanchez-Alonso, J.L.; Song, Q.; de Vries, A.A.F.; Pijnappels, D.A.; Ongstad, E.; Braga, V.M.M.; Entcheva, E.; et al. Cardiomyocyte-myofibroblast contact dynamism is modulated by connexin-43. FASEB J. 2019, 33, 10453–10468. [Google Scholar] [CrossRef]
- Maier, C.; Ramming, A.; Bergmann, C.; Weinkam, R.; Kittan, N.; Schett, G.; Distler, J.H.W.; Beyer, C. Inhibition of phosphodiesterase 4 (PDE4) reduces dermal fibrosis by interfering with the release of interleukin-6 from M2 macrophages. Ann. Rheum. Dis. 2017, 76, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Liechty, K.W.; Kim, H.B.; Adzick, N.S.; Crombleholme, T.M. Fetal wound repair results in scar formation in interleukin-10-deficient mice in a syngeneic murine model of scarless fetal wound repair. J. Pediatr. Surg. 2000, 35, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Kozin, E.D.; Keswani, S.G.; Vaikunth, S.S.; Katz, A.B.; Zoltick, P.W.; Favata, M.; Radu, A.P.; Soslowsky, L.J.; Herlyn, M.; et al. Permissive environment in postnatal wounds induced by adenoviral-mediated overexpression of the anti-inflammatory cytokine interleukin-10 prevents scar formation. Wound Repair Regen. 2008, 16, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Sapudom, J.; Wu, X.; Chkolnikov, M.; Ansorge, M.; Anderegg, U.; Pompe, T. Fibroblast fate regulation by time dependent TGF-beta1 and IL-10 stimulation in biomimetic 3D matrices. Biomater. Sci. 2017, 5, 1858–1867. [Google Scholar] [CrossRef]
- Ullm, F.; Riedl, P.; Machado de Amorim, A.; Patzschke, A.; Weiss, R.; Hauschildt, S.; Franke, K.; Anderegg, U.; Pompe, T. 3D Scaffold-Based Macrophage Fibroblast Coculture Model Reveals IL-10 Dependence of Wound Resolution Phase. Adv. Biosyst. 2020, 4, e1900220. [Google Scholar] [CrossRef] [PubMed]
- Riedl, P.; Pompe, T. Functional label-free assessment of fibroblast differentiation in 3D collagen-I-matrices using particle image velocimetry. Biomater. Sci. 2021. [Google Scholar] [CrossRef]
- Tamaki, S.; Mano, T.; Sakata, Y.; Ohtani, T.; Takeda, Y.; Kamimura, D.; Omori, Y.; Tsukamoto, Y.; Ikeya, Y.; Kawai, M.; et al. Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLoS ONE 2013, 8, e68893. [Google Scholar] [CrossRef] [Green Version]
- Kotsiou, O.S.; Gourgoulianis, K.I.; Zarogiannis, S.G. IL-33/ST2 Axis in Organ Fibrosis. Front. Immunol. 2018, 9, 2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, B.; Brembilla, N.C.; Chizzolini, C. Interplay between Keratinocytes and Fibroblasts: A Systematic Review Providing a New Angle for Understanding Skin Fibrotic Disorders. Front. Immunol. 2020, 11, 648. [Google Scholar] [CrossRef] [PubMed]
- Koskela, A.; Engstrom, K.; Hakelius, M.; Nowinski, D.; Ivarsson, M. Regulation of fibroblast gene expression by keratinocytes in organotypic skin culture provides possible mechanisms for the antifibrotic effect of reepithelialization. Wound Repair Regen. 2010, 18, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Ghahary, A.; Ghaffari, A. Role of keratinocyte-fibroblast cross-talk in development of hypertrophic scar. Wound Repair Regen 2007, 15 (Suppl. 1), S46–S53. [Google Scholar] [CrossRef]
- Zheng, R.; Longmate, W.M.; DeFreest, L.; Varney, S.; Wu, L.; DiPersio, C.M.; Van De Water, L. Keratinocyte Integrin alpha3beta1 Promotes Secretion of IL-1alpha to Effect Paracrine Regulation of Fibroblast Gene Expression and Differentiation. J. Investig. Dermatol. 2019, 139, 2029–2038. [Google Scholar] [CrossRef]
- Zheng, R.; Varney, S.D.; Wu, L.; DiPersio, C.M.; Van De Water, L. Integrin alpha4beta1 is required for IL-1alpha- and Nrf2-dependent, Cox-2 induction in fibroblasts, supporting a mechanism that suppresses alpha-SMA expression. Wound Repair Regen. 2021, 29, 597–601. [Google Scholar] [CrossRef]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; Lopez-Giraldez, F.; Dash, B.C.; Munoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Wang, X.; Zhu, M.; Qu, M.; Bogari, M.; Lin, L.; Mar Aung, Z.; Chen, W.; Chen, X.; Chai, G.; et al. Expansion of CD26 positive fibroblast population promotes keloid progression. Exp. Cell Res. 2017, 356, 104–113. [Google Scholar] [CrossRef]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-beta. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Khomtchouk, K.; Santa Maria, P.L. A Review of the Contribution of Mast Cells in Wound Healing: Involved Molecular and Cellular Mechanisms. Clin. Rev. Allergy Immunol. 2020, 58, 298–312. [Google Scholar] [CrossRef]
- Wulff, B.C.; Parent, A.E.; Meleski, M.A.; DiPietro, L.A.; Schrementi, M.E.; Wilgus, T.A. Mast cells contribute to scar formation during fetal wound healing. J. Investig. Dermatol. 2012, 132, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Glim, J.E.; Beelen, R.H.; Niessen, F.B.; Everts, V.; Ulrich, M.M. The number of immune cells is lower in healthy oral mucosa compared to skin and does not increase after scarring. Arch. Oral Biol. 2015, 60, 272–281. [Google Scholar] [CrossRef]
- Tellechea, A.; Bai, S.; Dangwal, S.; Theocharidis, G.; Nagai, M.; Koerner, S.; Cheong, J.E.; Bhasin, S.; Shih, T.Y.; Zheng, Y.; et al. Topical Application of a Mast Cell Stabilizer Improves Impaired Diabetic Wound Healing. J. Investig. Dermatol. 2020, 140, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Ozbilgin, M.K.; Inan, S. The roles of transforming growth factor type beta3 (TGF-beta3) and mast cells in the pathogenesis of scleroderma. Clin. Rheumatol. 2003, 22, 189–195. [Google Scholar] [CrossRef]
- Irani, A.M.; Gruber, B.L.; Kaufman, L.D.; Kahaleh, M.B.; Schwartz, L.B. Mast cell changes in scleroderma. Presence of MCT cells in the skin and evidence of mast cell activation. Arthritis Rheum. 1992, 35, 933–939. [Google Scholar] [CrossRef]
- Monument, M.J.; Hart, D.A.; Salo, P.T.; Befus, A.D.; Hildebrand, K.A. Neuroinflammatory Mechanisms of Connective Tissue Fibrosis: Targeting Neurogenic and Mast Cell Contributions. Adv. Wound Care 2015, 4, 137–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, I.S.; Choi, Y.H.; Jang, Y.C.; Lee, Y.K. Immunohistochemical analysis of neuropeptides (protein gene product 9.5, substance P and calcitonin gene-related peptide) in hypertrophic burn scar with pain and itching. Burns 2014, 40, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Lebonvallet, N.; Laverdet, B.; Misery, L.; Desmouliere, A.; Girard, D. New insights into the roles of myofibroblasts and innervation during skin healing and innovative therapies to improve scar innervation. Exp. Dermatol. 2018, 27, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, K.A.; Zhang, M.; Befus, A.D.; Salo, P.T.; Hart, D.A. A myofibroblast-mast cell-neuropeptide axis of fibrosis in post-traumatic joint contractures: An in vitro analysis of mechanistic components. J. Orthop. Res. 2014, 32, 1290–1296. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, S.; Li, X.; Jiang, F.; Ye, Q.; Ning, W. Follistatin like-1 aggravates silica-induced mouse lung injury. Sci. Rep. 2017, 7, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Fang, Y.; Zhang, S.; Li, L.; Wang, L.; Zhang, A.; Yuan, Z.; Wang, P.; Zhou, H.; Cui, W.; et al. Haplodeletion of Follistatin-Like 1 Attenuates Radiation-Induced Pulmonary Fibrosis in Mice. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, Y.; Jiang, D.; Dong, Y.; Liu, Y.; Zhang, S.; Guo, J.; Qi, C.; Zhao, C.; Jiang, F.; et al. Targeting FSTL1 for Multiple Fibrotic and Systemic Autoimmune Diseases. Mol. Ther. 2021, 29, 347–364. [Google Scholar] [CrossRef]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020, 39, 783–803. [Google Scholar] [CrossRef]
- Mazur, A.; Holthoff, E.; Vadali, S.; Kelly, T.; Post, S.R. Cleavage of Type I Collagen by Fibroblast Activation Protein-alpha Enhances Class A Scavenger Receptor Mediated Macrophage Adhesion. PLoS ONE 2016, 11, e0150287. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.T.; McCaughan, G.W.; Abbott, C.A.; Park, J.E.; Cunningham, A.M.; Muller, E.; Rettig, W.J.; Gorrell, M.D. Fibroblast activation protein: A cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology 1999, 29, 1768–1778. [Google Scholar] [CrossRef]
- Truffi, M.; Sorrentino, L.; Monieri, M.; Fociani, P.; Mazzucchelli, S.; Bonzini, M.; Zerbi, P.; Sampietro, G.M.; Di Sabatino, A.; Corsi, F. Inhibition of Fibroblast Activation Protein Restores a Balanced Extracellular Matrix and Reduces Fibrosis in Crohn’s Disease Strictures Ex Vivo. Inflamm. Bowel Dis. 2018, 24, 332–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egger, C.; Cannet, C.; Gerard, C.; Suply, T.; Ksiazek, I.; Jarman, E.; Beckmann, N. Effects of the fibroblast activation protein inhibitor, PT100, in a murine model of pulmonary fibrosis. Eur. J. Pharmacol. 2017, 809, 64–72. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- Varney, S.D.; Betts, C.B.; Zheng, R.; Wu, L.; Hinz, B.; Zhou, J.; Van De Water, L. Hic-5 is required for myofibroblast differentiation by regulating mechanically dependent MRTF-A nuclear accumulation. J. Cell Sci. 2016, 129, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Bae, H.; Kim, T.; Lim, I. Effects of nitric oxide on apoptosis and voltage-gated calcium channels in human cardiac myofibroblasts. Clin. Exp. Pharmacol. Physiol. 2020, 47, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Forte, A.; Della Corte, A.; De Feo, M.; Cerasuolo, F.; Cipollaro, M. Role of myofibroblasts in vascular remodelling: Focus on restenosis and aneurysm. Cardiovasc. Res. 2010, 88, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Midgley, A.C.; Wei, Y.; Li, Z.; Kong, D.; Zhao, Q. Nitric-Oxide-Releasing Biomaterial Regulation of the Stem Cell Microenvironment in Regenerative Medicine. Adv. Mater. 2020, 32, e1805818. [Google Scholar] [CrossRef] [PubMed]
- Douglass, A.; Wallace, K.; Parr, R.; Park, J.; Durward, E.; Broadbent, I.; Barelle, C.; Porter, A.J.; Wright, M.C. Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury. J. Hepatol. 2008, 49, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wu, J.J.; Sun, Z.L.; Liu, S.Y.; Zou, M.L.; Yuan, Z.D.; Yu, S.; Lv, G.Z.; Yuan, F.L. Targeted apoptosis of myofibroblasts by elesclomol inhibits hypertrophic scar formation. EBioMedicine 2020, 54, 102715. [Google Scholar] [CrossRef]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Huang, X.; Hecker, L.; Kurundkar, D.; Kurundkar, A.; Liu, H.; Jin, T.H.; Desai, L.; Bernard, K.; Thannickal, V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Investig. 2013, 123, 1096–1108. [Google Scholar] [CrossRef]
- Shroff, A.; Mamalis, A.; Jagdeo, J. Oxidative Stress and Skin Fibrosis. Curr. Pathobiol. Rep. 2014, 2, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Siani, A.; Tirelli, N. Myofibroblast differentiation: Main features, biomedical relevance, and the role of reactive oxygen species. Antioxid. Redox. Signal. 2014, 21, 768–785. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, F.J.; Chandel, N.S.; Jain, M.; Budinger, G.R.S. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl. Res. 2017, 190, 61–68. [Google Scholar] [CrossRef]
- Su, H.; Wan, C.; Song, A.; Qiu, Y.; Xiong, W.; Zhang, C. Oxidative Stress and Renal Fibrosis: Mechanisms and Therapies. Adv. Exp. Med. Biol. 2019, 1165, 585–604. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef] [PubMed]
- Vozenin-Brotons, M.C.; Sivan, V.; Gault, N.; Renard, C.; Geffrotin, C.; Delanian, S.; Lefaix, J.L.; Martin, M. Antifibrotic action of Cu/Zn SOD is mediated by TGF-beta1 repression and phenotypic reversion of myofibroblasts. Free Radic. Biol. Med. 2001, 30, 30–42. [Google Scholar] [CrossRef]
- Tan, L.G.; Xiao, J.H.; Yu, D.L.; Zhang, L.; Zheng, F.; Guo, L.Y.; Yang, J.Y.; Tang, J.M.; Chen, S.Y.; Wang, J.N. PEP-1-SOD1 fusion proteins block cardiac myofibroblast activation and angiotensin II-induced collagen production. BMC Cardiovasc. Disord. 2015, 15, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef] [PubMed]
- Nishio, T.; Hu, R.; Koyama, Y.; Liang, S.; Rosenthal, S.B.; Yamamoto, G.; Karin, D.; Baglieri, J.; Ma, H.Y.; Xu, J.; et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J. Hepatol. 2019, 71, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef]
- Celiksoy, V.; Moses, R.L.; Sloan, A.J.; Moseley, R.; Heard, C.M. Evaluation of the In Vitro Oral Wound Healing Effects of Pomegranate (Punica granatum) Rind Extract and Punicalagin, in Combination with Zn (II). Biomolecules 2020, 10, 1234. [Google Scholar] [CrossRef]
- Moses, R.L.; Dally, J.; Lundy, F.T.; Langat, M.; Kiapranis, R.; Tsolaki, A.G.; Moseley, R.; Prescott, T.A.K. Lepiniopsis ternatensis sap stimulates fibroblast proliferation and down regulates macrophage TNF-alpha secretion. Fitoterapia 2020, 141, 104478. [Google Scholar] [CrossRef]
- Moses, R.L.; Fang, R.; Dally, J.; Briggs, M.; Lundy, F.T.; Kiapranis, R.; Moseley, R.; Prescott, T.A.K. Evaluation of Cypholophus macrocephalus sap as a treatment for infected cutaneous ulcers in Papua New Guinea. Fitoterapia 2020, 143, 104554. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xu, H.; Zhang, L.; Zhang, C.; Yang, L.; Ma, E.; Liu, L.; Li, Y. Salvianolic acid B inhibits myofibroblast transdifferentiation in experimental pulmonary fibrosis via the up-regulation of Nrf2. Biochem. Biophys. Res. Commun. 2018, 495, 325–331. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, H.; Yu, X.; Zhu, J. Fucoidan attenuates hyperoxia-induced lung injury in newborn rats by mediating lung fibroblasts differentiate into myofibroblasts. Ann. Transl. Med. 2020, 8, 1501. [Google Scholar] [CrossRef]
- Tavares, L.A.; Rezende, A.A.; Santos, J.L.; Estevam, C.S.; Silva, A.M.O.; Schneider, J.K.; Cunha, J.L.S.; Droppa-Almeida, D.; Correia-Neto, I.J.; Cardoso, J.C.; et al. Cymbopogon winterianus Essential Oil Attenuates Bleomycin-Induced Pulmonary Fibrosis in a Murine Model. Pharmaceutics 2021, 13, 679. [Google Scholar] [CrossRef]
- Wang, W.; Ma, B.L.; Xu, C.G.; Zhou, X.J. Dihydroquercetin protects against renal fibrosis by activating the Nrf2 pathway. Phytomedicine 2020, 69, 153185. [Google Scholar] [CrossRef] [PubMed]
- Veeren, B.; Bringart, M.; Turpin, C.; Rondeau, P.; Planesse, C.; Ait-Arsa, I.; Gimie, F.; Marodon, C.; Meilhac, O.; Gonthier, M.P.; et al. Caffeic Acid, One of the Major Phenolic Acids of the Medicinal Plant Antirhea borbonica, Reduces Renal Tubulointerstitial Fibrosis. Biomedicines 2021, 9, 358. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, G.N.; Xie, J.; Li, R.; Chen, Q.H.; Chen, J.Z.; Wei, Z.H.; Kang, L.N.; Xu, B. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-beta/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc. Disord. 2016, 16, 5. [Google Scholar] [CrossRef] [Green Version]
- Lyu, L.; Chen, J.; Wang, W.; Yan, T.; Lin, J.; Gao, H.; Li, H.; Lv, R.; Xu, F.; Fang, L.; et al. Scoparone alleviates Ang II-induced pathological myocardial hypertrophy in mice by inhibiting oxidative stress. J. Cell Mol. Med. 2021, 25, 3136–3148. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Motegi, S.I.; Fujiwara, C.; Yamazaki, S.; Inoue, Y.; Uchiyama, A.; Akai, R.; Iwawaki, T.; Ishikawa, O. Inhibitory effect of kaempferol on skin fibrosis in systemic sclerosis by the suppression of oxidative stress. J. Dermatol. Sci. 2019, 96, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Deng, Y.; Li, W.; Lu, Y. Arctigenin suppresses fibroblast activity and extracellular matrix deposition in hypertrophic scarring by reducing inflammation and oxidative stress. Mol. Med. Rep. 2020, 22, 4783–4791. [Google Scholar] [CrossRef]
Figure 1.
Fibroblast–myofibroblast differentiation. The process of fibroblast–myofibroblast differentiation requires: (i) the activation of the TGF-β1/Smad signalling pathway; (ii) cell–ECM mechanotransduction signalling; and (iii) the synthesis of modulators that promote and maintain the myofibroblast phenotype, such as EDA–FN and HAS2-synthesised HA. The intermediate stage is known as the proto-myofibroblast, and can be distinguished by increased proliferation, migration, and the rearrangement of cortical, membrane-associated actin into cytoplasmic filamentous actin stress fibres, which form focal adhesion sites at membrane–ECM junctions. The mature myofibroblast exhibits thick and crosslinked HA pericellular coats, and contains α-SMA+ cytoplasmic actin stress fibres that form mature focal adhesion plaques at membrane–ECM junctions and grant increased contractile strength.
Figure 1.
Fibroblast–myofibroblast differentiation. The process of fibroblast–myofibroblast differentiation requires: (i) the activation of the TGF-β1/Smad signalling pathway; (ii) cell–ECM mechanotransduction signalling; and (iii) the synthesis of modulators that promote and maintain the myofibroblast phenotype, such as EDA–FN and HAS2-synthesised HA. The intermediate stage is known as the proto-myofibroblast, and can be distinguished by increased proliferation, migration, and the rearrangement of cortical, membrane-associated actin into cytoplasmic filamentous actin stress fibres, which form focal adhesion sites at membrane–ECM junctions. The mature myofibroblast exhibits thick and crosslinked HA pericellular coats, and contains α-SMA+ cytoplasmic actin stress fibres that form mature focal adhesion plaques at membrane–ECM junctions and grant increased contractile strength.

Figure 2.
Canonical and non-canonical pathways implicated in fibroblast–myofibroblast differentiation. Three of the best described pathways involved in myofibroblast phenotypic acquisition are the TGF-β1/Smad; HA/CD44/EGFR; and Wnt/β-catenin pathways: (A) TGF-β1 is released from the LAP complex and binds to TGFβRII dimers, enabling association with and the activation of TGFβRI. Smad2 and Smad3 are subsequently phosphorylated and co-associate with Smad4 to initiate the transcription of pro-fibrotic genes; (B) TGF-β1 induced the upregulation of HAS2 synthesised linear HA and hyaladherin crosslinkers, such as TSG-6 and HC5, resulting in HA interactions with CD44s and the thickening of a crosslinked HA pericellular coat. HA interactions with CD44s induce clustering of CD44s within lipid rafts, wherein the co-receptor EGFR is activated and initiates biphasic signalling via ERK1/2 and CaMKII. Early signalling was suggested to promote differentiation-associated gene expression, whereas late signalling was suggested to promote proliferation-associated gene expression; and (C) the Wnt ligand binding to the transmembrane Frazzled receptor (FZD), in the absence of DKK1, which induces localisation with and the activation of LRP5 and LRP6. The Wnt/FZD/LRP signalling drives the dishevelled-mediated attenuation of the β-catenin inhibitory complex (CK-1/Axin/APC/GSK-3), facilitating the accumulation of β-catenin and its translocation to the nucleus, wherein it associates with co-transcription factors, TLF and LEF, to induce pro-fibrotic gene expression.
Figure 2.
Canonical and non-canonical pathways implicated in fibroblast–myofibroblast differentiation. Three of the best described pathways involved in myofibroblast phenotypic acquisition are the TGF-β1/Smad; HA/CD44/EGFR; and Wnt/β-catenin pathways: (A) TGF-β1 is released from the LAP complex and binds to TGFβRII dimers, enabling association with and the activation of TGFβRI. Smad2 and Smad3 are subsequently phosphorylated and co-associate with Smad4 to initiate the transcription of pro-fibrotic genes; (B) TGF-β1 induced the upregulation of HAS2 synthesised linear HA and hyaladherin crosslinkers, such as TSG-6 and HC5, resulting in HA interactions with CD44s and the thickening of a crosslinked HA pericellular coat. HA interactions with CD44s induce clustering of CD44s within lipid rafts, wherein the co-receptor EGFR is activated and initiates biphasic signalling via ERK1/2 and CaMKII. Early signalling was suggested to promote differentiation-associated gene expression, whereas late signalling was suggested to promote proliferation-associated gene expression; and (C) the Wnt ligand binding to the transmembrane Frazzled receptor (FZD), in the absence of DKK1, which induces localisation with and the activation of LRP5 and LRP6. The Wnt/FZD/LRP signalling drives the dishevelled-mediated attenuation of the β-catenin inhibitory complex (CK-1/Axin/APC/GSK-3), facilitating the accumulation of β-catenin and its translocation to the nucleus, wherein it associates with co-transcription factors, TLF and LEF, to induce pro-fibrotic gene expression.

Figure 3.
Mechanisms of mechanotransduction implicated in myofibroblast differentiation and function. TGF-β1 stimulates an increased expression of EDA–FN. The EDA segment contains the EDGIHEL motif, which binds to the integrin α4β1/α4β7 receptor cleft, initiating signalling through RhoA/MLCK and FAK. RhoA signalling results in myosin light chain (MLC) promotion of myosin (MYH)/actin assembly and stress fibre polymerisation, which is augmented by HYAL2 that relocates from the membrane to the cytoplasm. RGD motif containing ligands, such as FN and collagens, bind integrins and facilitate the formation of focal adhesion sites, increasing contractile force and α-SMA incorporation into actin stress fibres. In addition, the increased matrix stress generated by EDA–FN and increased MMP activation from CD147 recruitment exacerbates active TGF-β1 release from the extracellular LAP complex.
Figure 3.
Mechanisms of mechanotransduction implicated in myofibroblast differentiation and function. TGF-β1 stimulates an increased expression of EDA–FN. The EDA segment contains the EDGIHEL motif, which binds to the integrin α4β1/α4β7 receptor cleft, initiating signalling through RhoA/MLCK and FAK. RhoA signalling results in myosin light chain (MLC) promotion of myosin (MYH)/actin assembly and stress fibre polymerisation, which is augmented by HYAL2 that relocates from the membrane to the cytoplasm. RGD motif containing ligands, such as FN and collagens, bind integrins and facilitate the formation of focal adhesion sites, increasing contractile force and α-SMA incorporation into actin stress fibres. In addition, the increased matrix stress generated by EDA–FN and increased MMP activation from CD147 recruitment exacerbates active TGF-β1 release from the extracellular LAP complex.


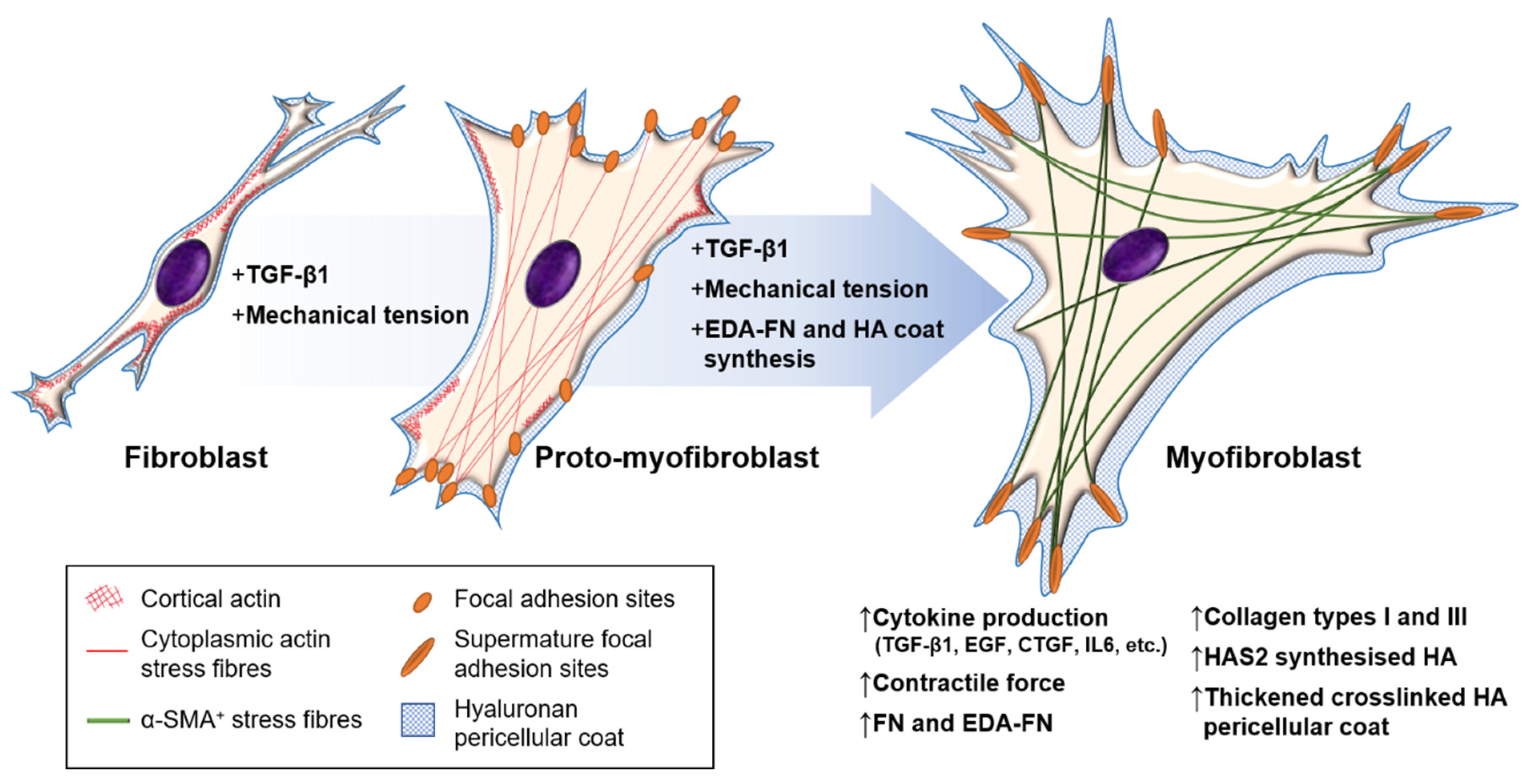{kind=link}
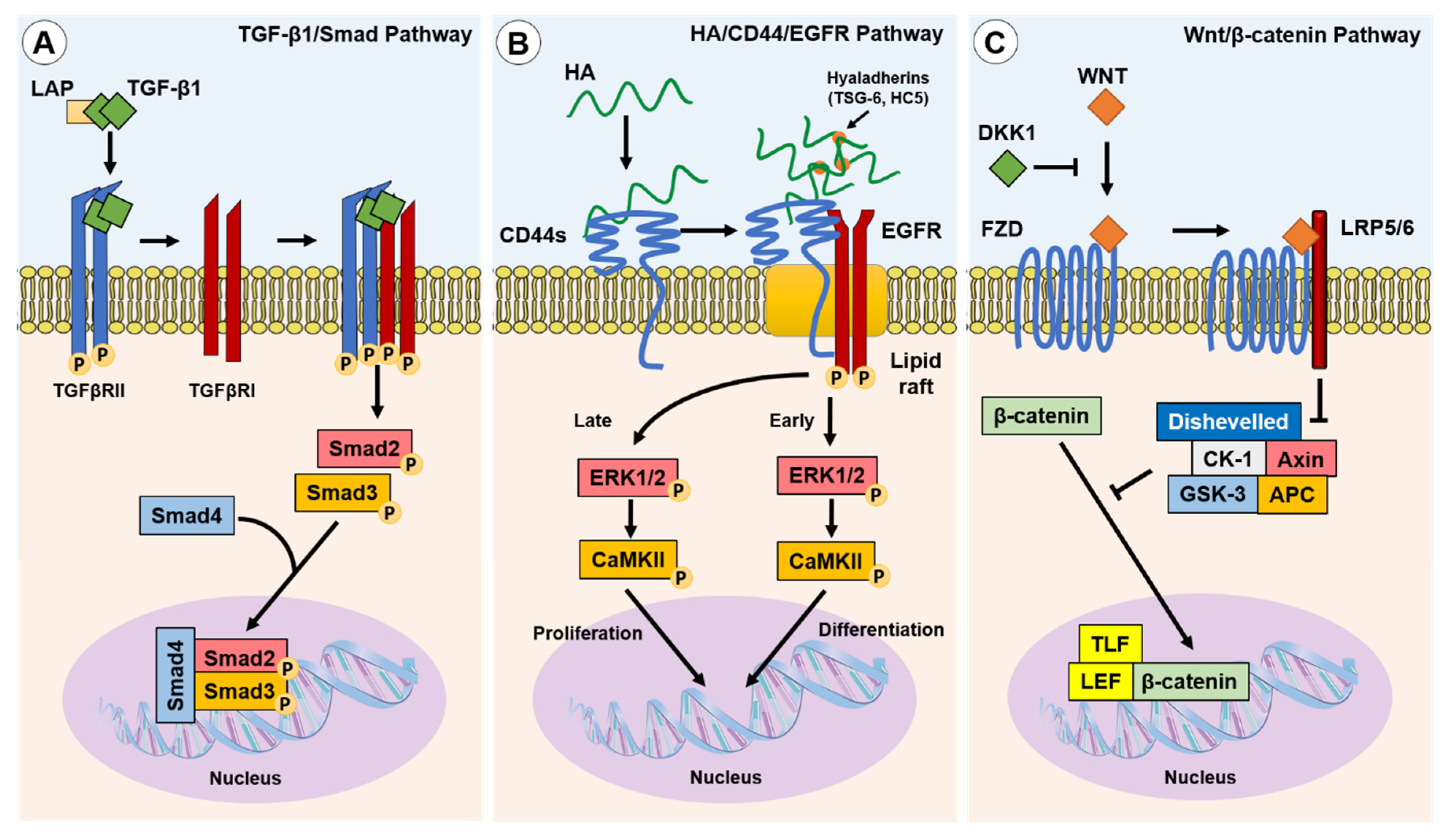{kind=link}
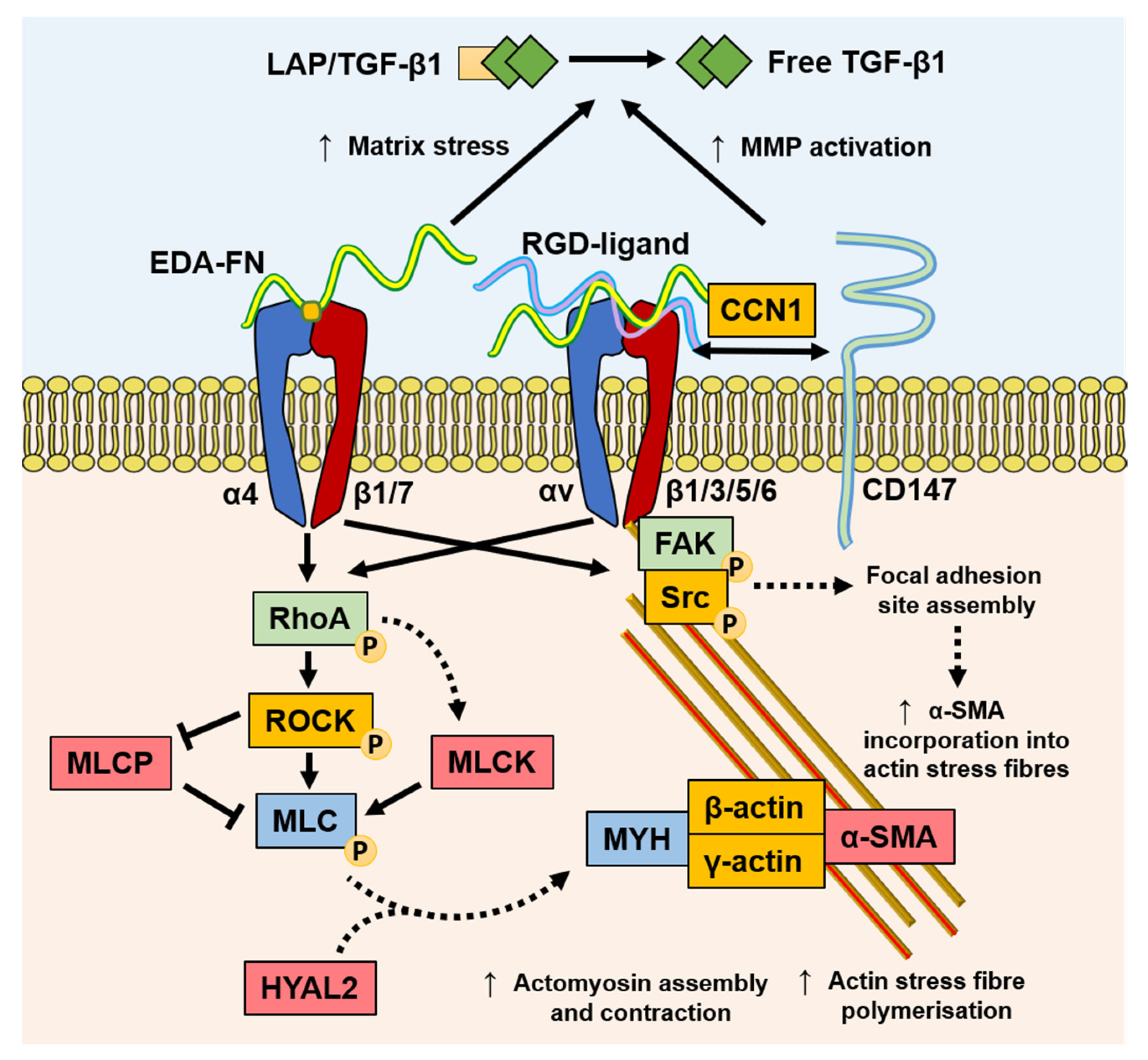{kind=link}
Table 1.
Recent clinical trials for attenuated scar skin repair (completed phase III or ongoing).
| Treatment (Mechanism) | Type of Skin Fibrosis | Phase Status | ClinicalTrials.gov ID |
|---|---|---|---|
| Rituximab/CD20 mAb (immunomodulation) | Scleroderma, SSc | Phase II/III (2019) | NCT04274257 |
| FS2/kynurenic acid (immunomodulation) | Scar, hypertrophic scar, graft scar, keloid | Phase II (ongoing) | NCT04186273 |
| Pirfenidone (unknown mechanism) | Scar | Phase II/III (ongoing) | NCT03068234 |
| Pentoxifylline, vitamin E (ECM remodelling) | Postoperative radiation-induced fibrosis | Phase III (ongoing) | NCT02898376 |
| Fractional CO2 laser (ECM remodelling) | Scar, burns, graft scars | N/A (ongoing) | NCT04456127 |
| Integra skin substitute (scar resection and healing) | Scar | N/A (ongoing) | NCT04420442 |
| Autologous cell dermal scaffolds (scar resection and healing) | Scar, hypertrophic scar | N/A (ongoing) | NCT04389164 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules 2021, 11, 1095. https://doi.org/10.3390/biom11081095
AMA Style
Tai Y, Woods EL, Dally J, Kong D, Steadman R, Moseley R, Midgley AC. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules. 2021; 11(8):1095. https://doi.org/10.3390/biom11081095
Chicago/Turabian StyleTai, Yifan, Emma L. Woods, Jordanna Dally, Deling Kong, Robert Steadman, Ryan Moseley, and Adam C. Midgley. 2021. "Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis" Biomolecules 11, no. 8: 1095. https://doi.org/10.3390/biom11081095
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.