
Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration

 , , and
, , and
Abstract
:1. Introduction
2. Mitostasis in Neurons
2.1. Mitochondrial Trafficking
2.2. Mitochondrial Anchoring
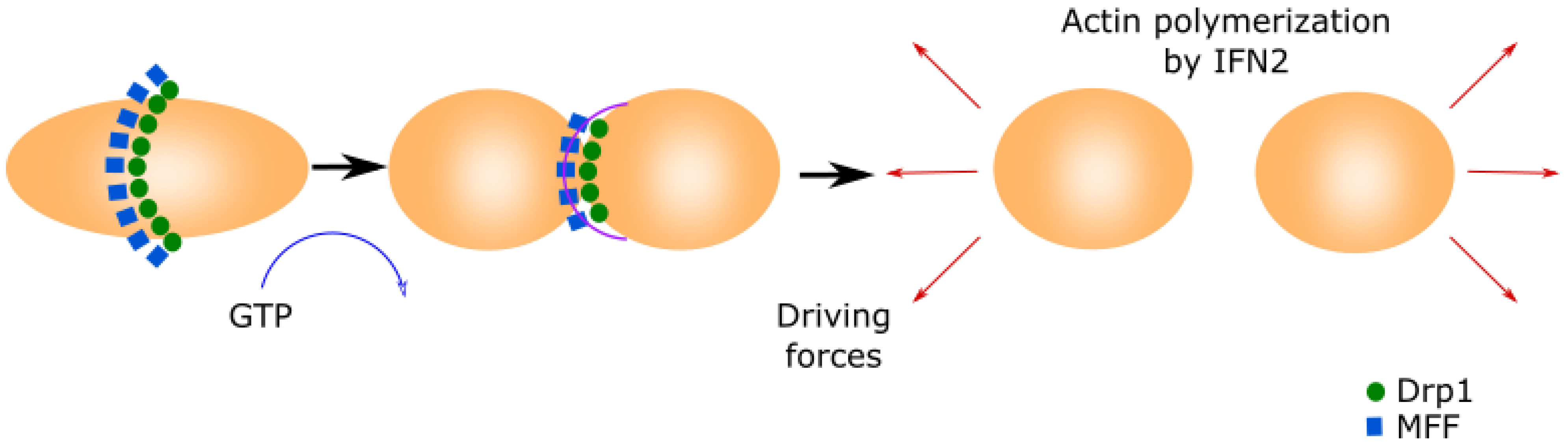
2.3. Mitochondrial Fission
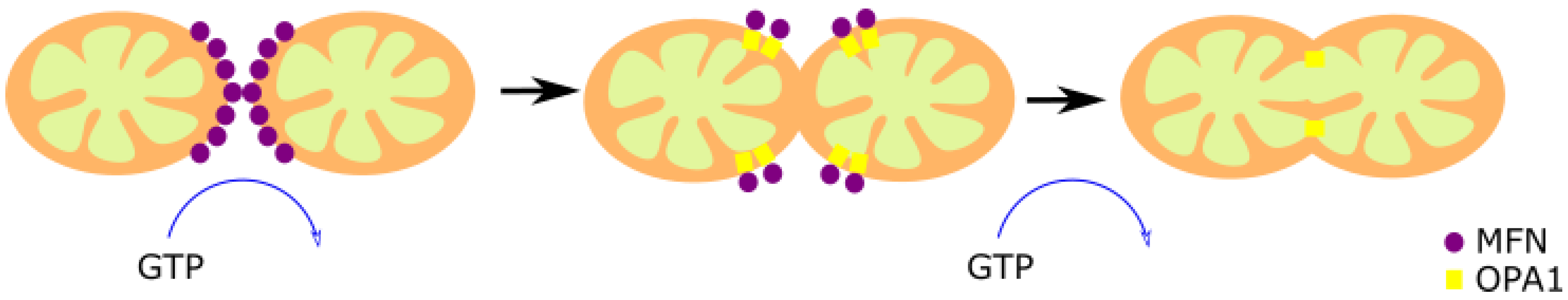
2.4. Mitochondrial Fusion
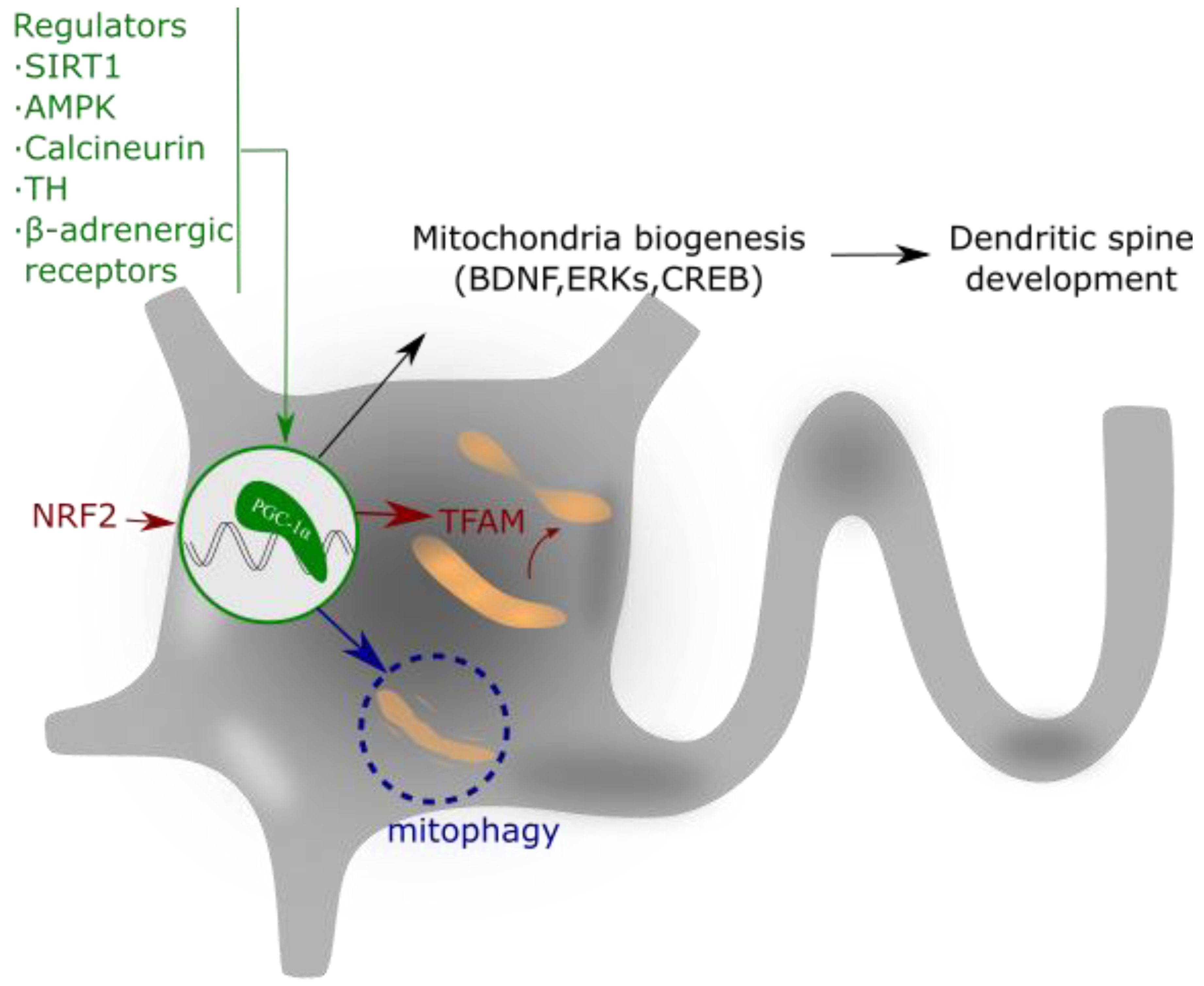
2.5. The regulator of Mitochondrial Biogenesis PGC-1α
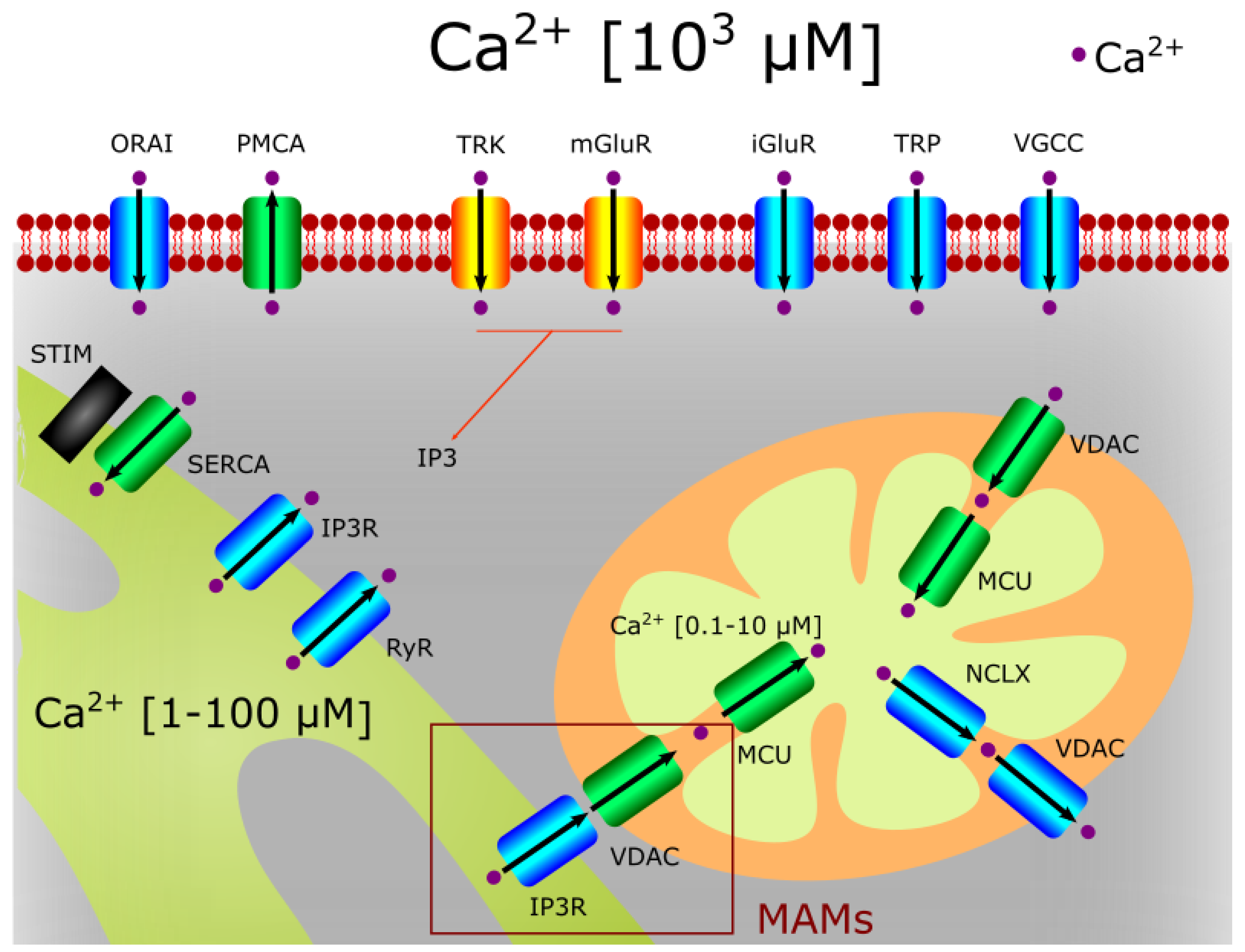
3. The Role of Mitochondria in Calcium Homeostasis
3.1. Calcium Entrance to Mitochondria
3.2. Calcium Functions in Mitochondria
3.3. The Role of the MAMs
3.4. The Role of Calcium in mPTP Opening
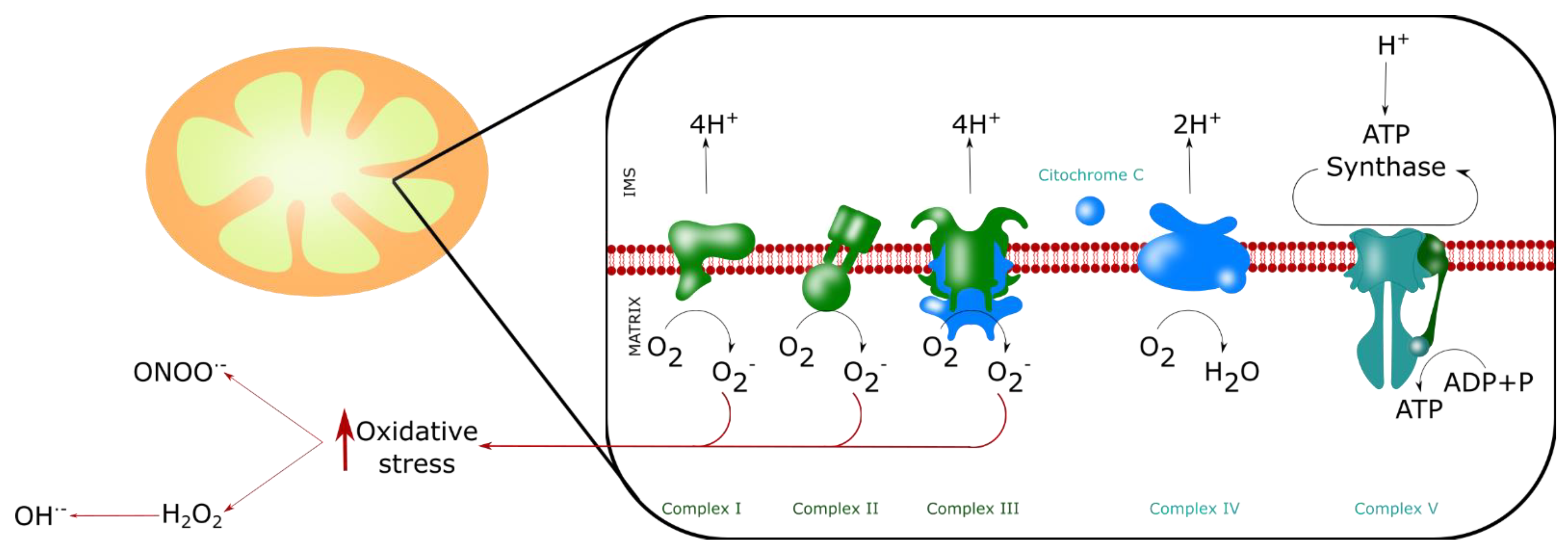
4. ROS Production by Mitochondria
5. Nitric Oxide and Mitochondria
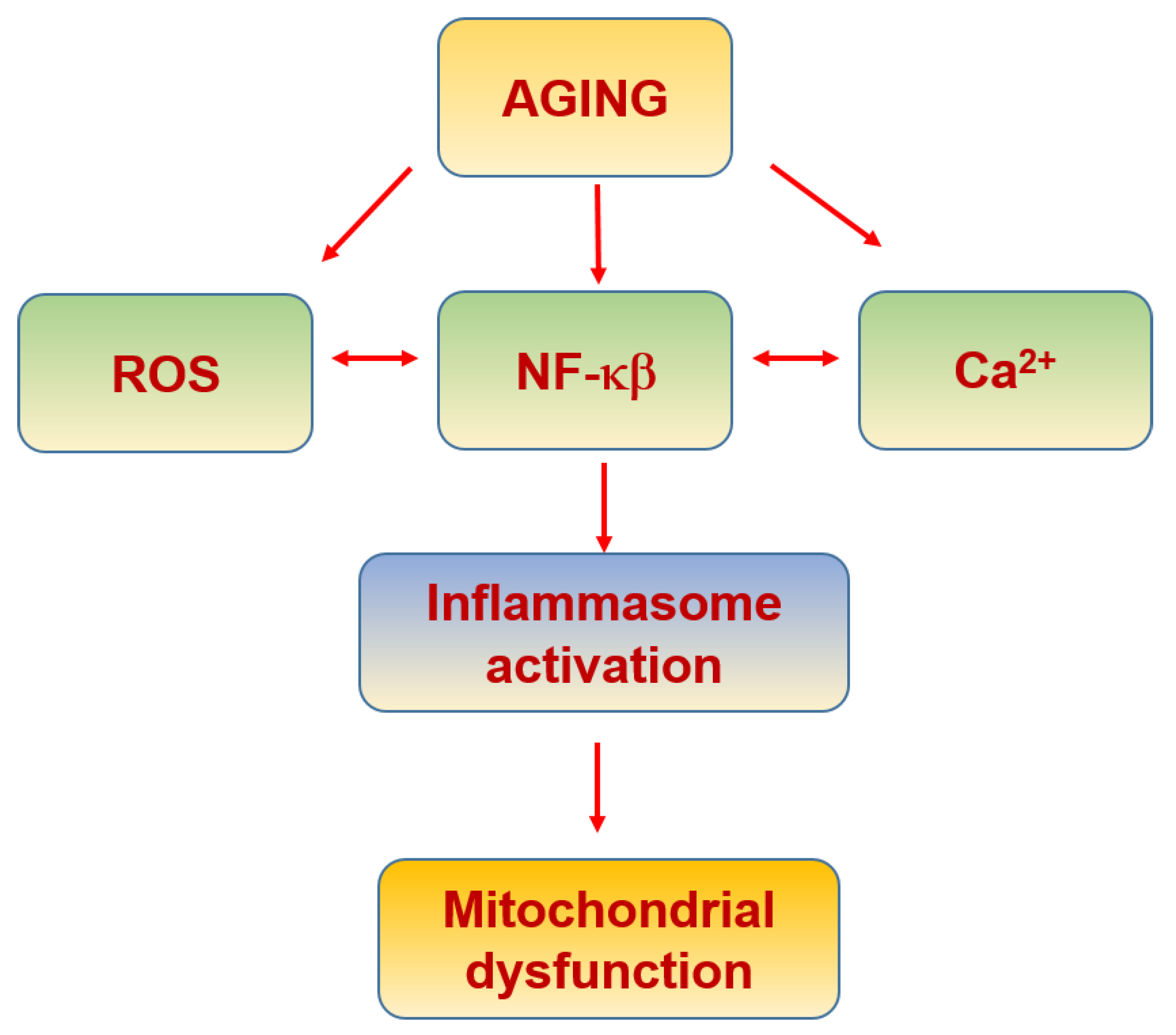
6. Mitochondria in Aging and Neurodegeneration
6.1. Physiological Aging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Function | Ref. |
|---|---|---|
| DNA polymerase γ (Polγ) | Mitochondrial DNA polymerase | [189] |
| Hsp70/Hsp90 molecular chaperones | Protection and interaction with protein import receptor Tom70 | [190] |
| Lon Protease | Involved in drug-induced mitochondrial dysfunction and endoplasmic reticulum stress | [191] |
| Transcription Factor ATF5 | Mitochondrial unfolded protein response (mtUPR) | [192] |
| Estrogen receptor | Involved in mtUPR | [193] |
| α-ketoglutarate dehydrogenase, and pyruvate dehydrogenase | Mitochondrial respiration | [194] |
| DRP1 | Replication machinery | [195] |
6.2. Inflammaging
| Molecule | Function | Ref. |
|---|---|---|
| Cyclic GMP-AMP (cGAS) and the cyclic GMP-AMP receptor stimulator of interferon genes (STING), (cGAS-STING) | Inflammatory response | [212] |
| Mitochondria-associated viral sensor (MAVS) | Mitochondrial adaptor | [213] |
| Retinoic acid-inducible gene I (RIG-I) | RNA helicase | [214] |
| NOD-like receptor family member X1 (NLRX1) | Regulators of MAVS function | [215] |
| TNF-receptor-associated factor (TRAF) | Type I IFN production | [216] |
| Succinate dehydrogenase | Inflammatory response | [217] |
| mtDNA | Dysfunctionby oxidation | [218] |
| Cardiolipin | Nlrp3 activation | [219] |
| N-formyl peptides | Proinflammatory signalling | [220] |
| TFAM transcription factor | Inflammatory response | [221] |
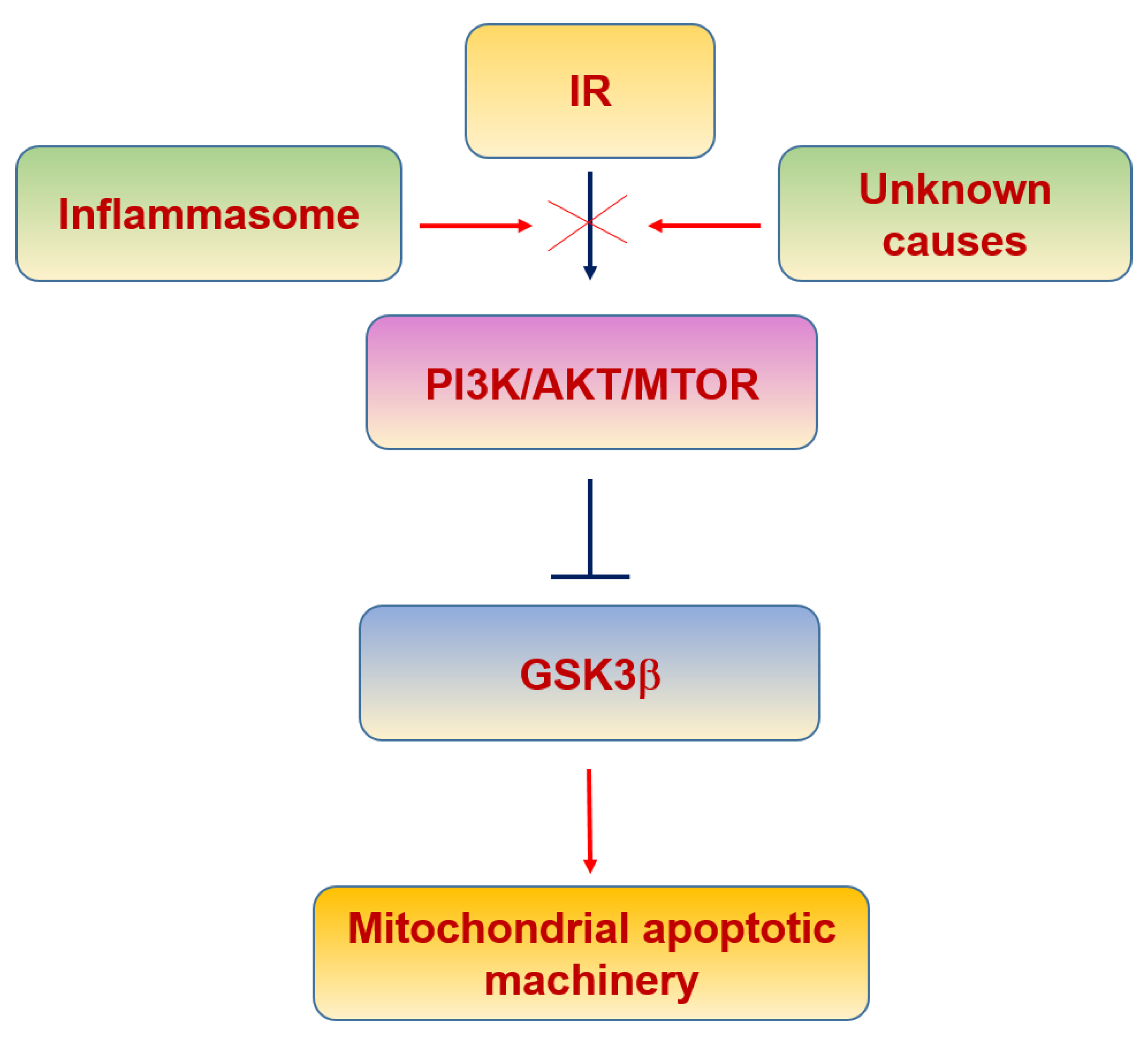
6.3. Insulin Resistance
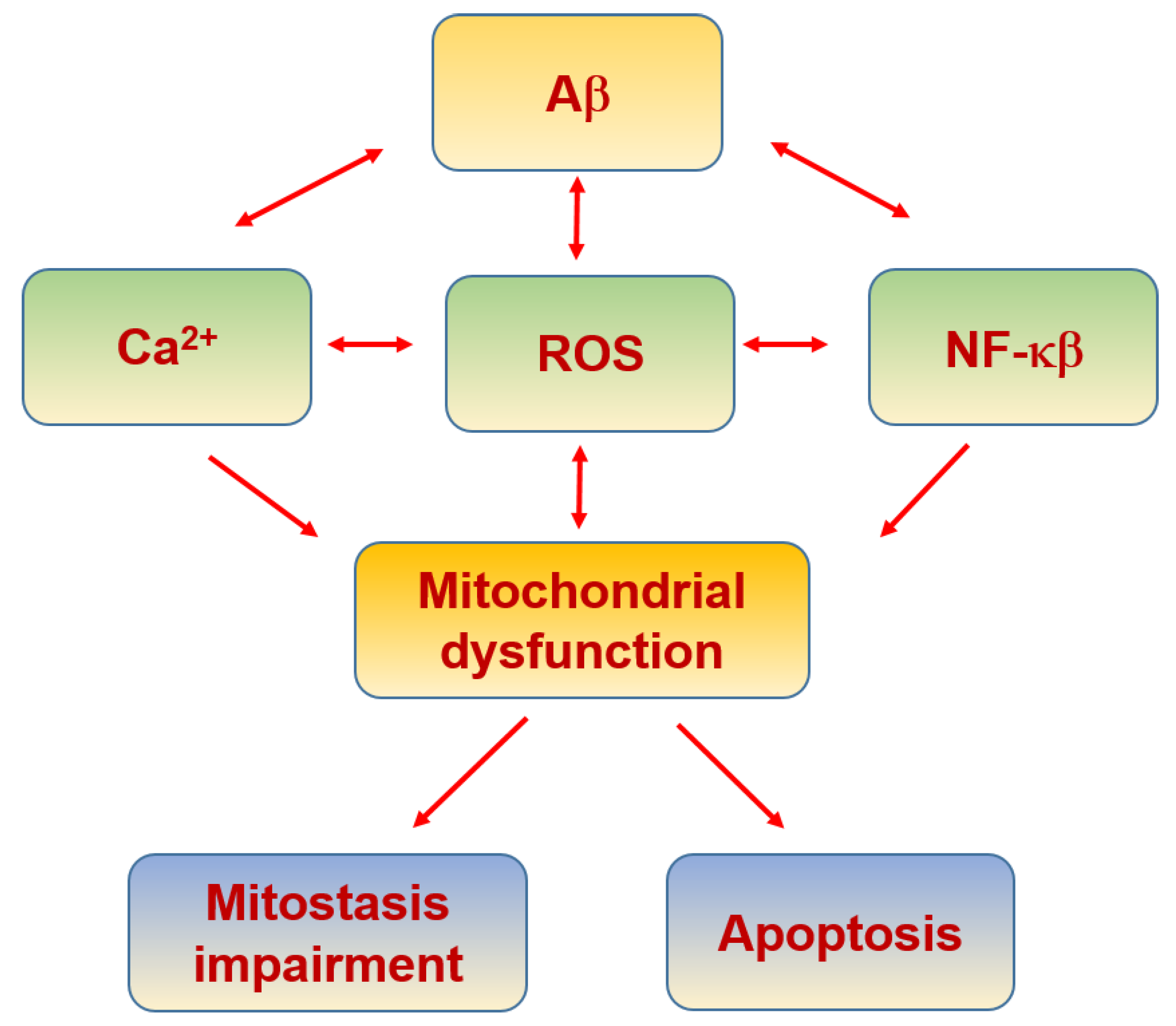
6.4. Alzheimer’s Disease
| Molecule | Function | Ref. |
|---|---|---|
| Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) | MRC | [297] |
| ERK | Oxidative stress-mediated ERK signal | [298] |
| Amyloid beta (Aβ) binding alcohol dehydrogenase (ABAD) | Promotes Aβ-mediated mitochondrial a dysfunction | [299] |
| mtNADH dehydrogenase 1 (mt-Nd1) | MRC | [300] |
| OPA1, Mfn1, Mfn2, DRP1 and Fis1 | Mitochondrial fusion | [269] |
| MT-ND2 and MT-ND5 | MRC | [301] |
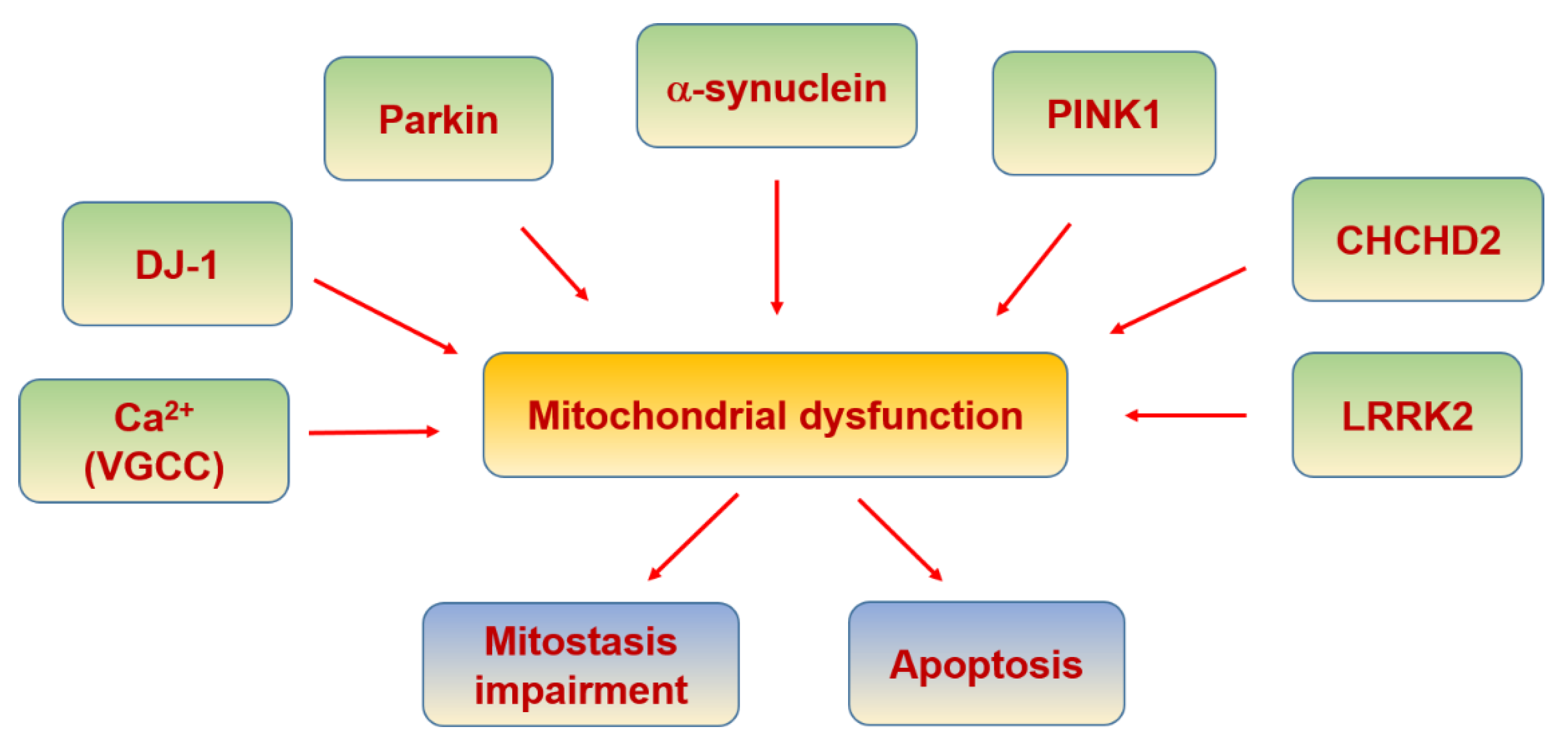
6.5. Parkinson’s Disease
| Molecule | Function | Ref. |
|---|---|---|
| MITOL/MARCH5 | Recruitment to dysfunctional mitochondria | [319] |
| p97 (AAA + ATPase) | Degradation of ubiquitinated Mfn1 | [320] |
| Tim23 complex | Translocation of the positively charged MTS across the IMM | [321] |
| HTRA2/Omi protease | Regulated by PINK 1 | [322] |
| TRAP1 | Regulated by PINK 1 in IMS | [323] |
| complex I subunit NDUFA10 | Regulated by PINK 1 in IMM | [324] |
| PINK1/Parkin | Mutated PINK1 activates Parkin causing mitophagy | [325] |
| PGC-1α | Blocked by mutated Parkin | [308] |
| L- and N-type VGCC | Increase of cytosolic calcium | [310] |
| α-synuclein | MAMs disruption and complex I from MRC inhibition when it is mutated | [312,314] |
| DJ-1 | Oxidative stress sensor and neuroprotective | [326] |
| LRRK2 | Its mutation reduces mitophagy through its disrupted interaction with Miro and a blockade of PINK1/Parkin-dependent mitophagy. | [317,327] |
| CHCHD2 | Regulation of complex IV activity | [318] |
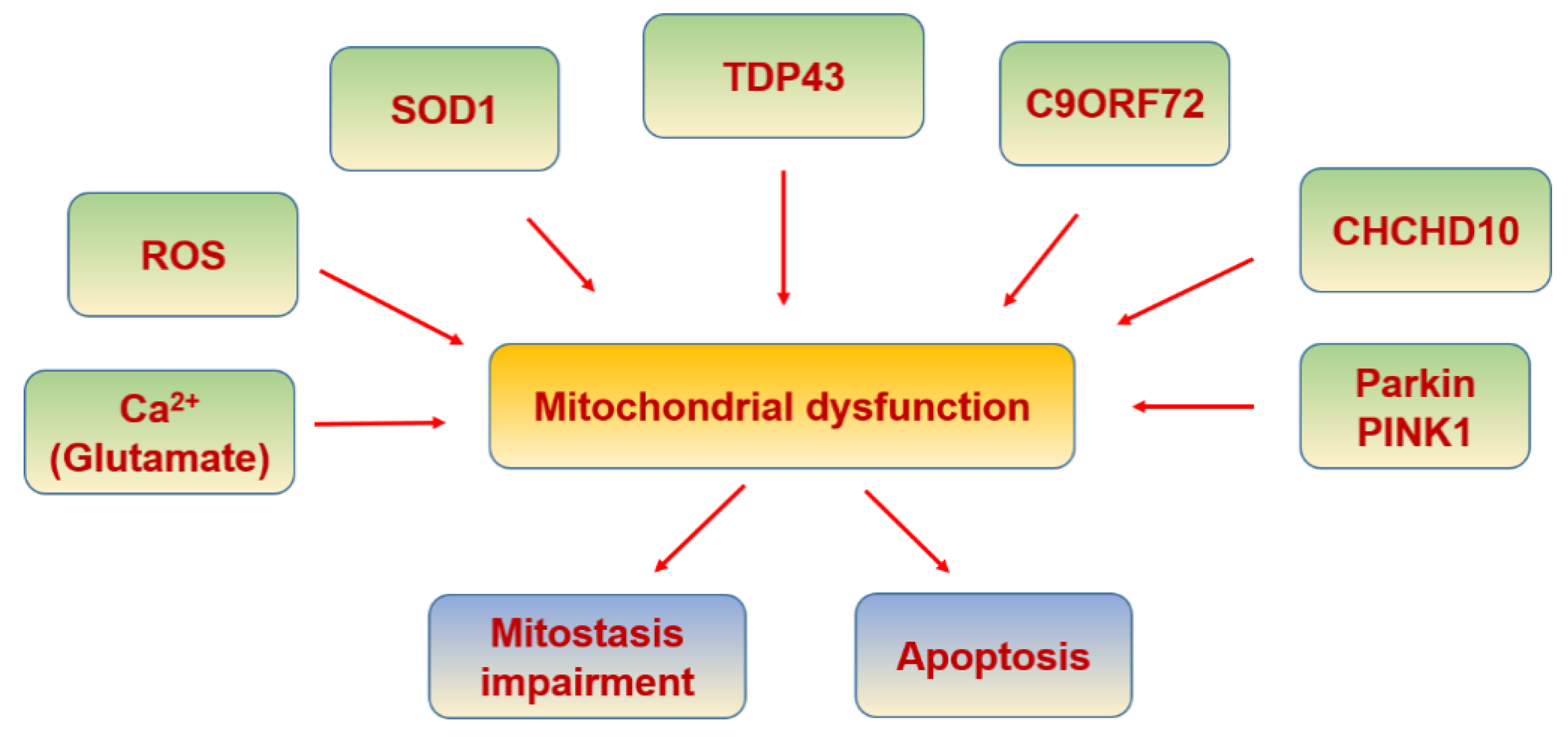
6.6. Amyotrophic Lateral Sclerosis
| Molecule | Function | Ref. |
|---|---|---|
| SOD-1 | Its aggregation impairs proper mitophagy by optineurin sequestration, and axonal transport of mitochondria | [345] |
| TDP-43 | Involved in interaction with mitochondrial proteins | [349] |
| TIM22 | TDP-43 gains access to the mitochondrial matrix via the mitochondrial import inner membrane | [350] |
| CHCHD10 | Involved in mitochondrial defects | [337] |
| Dbr1 | suppresses TDP-43 toxicity | [351] |
| Ataxin 2 | Increased risk for ALS | [352] |
| GSK-3β | disrupt the VAPB PTPIP51 interaction and ER–mitochondria associations | [353] |
| Alsin | Increased susceptibility to oxidative stress due to a defect in Rab5 relocation to mitochondria | [354] |
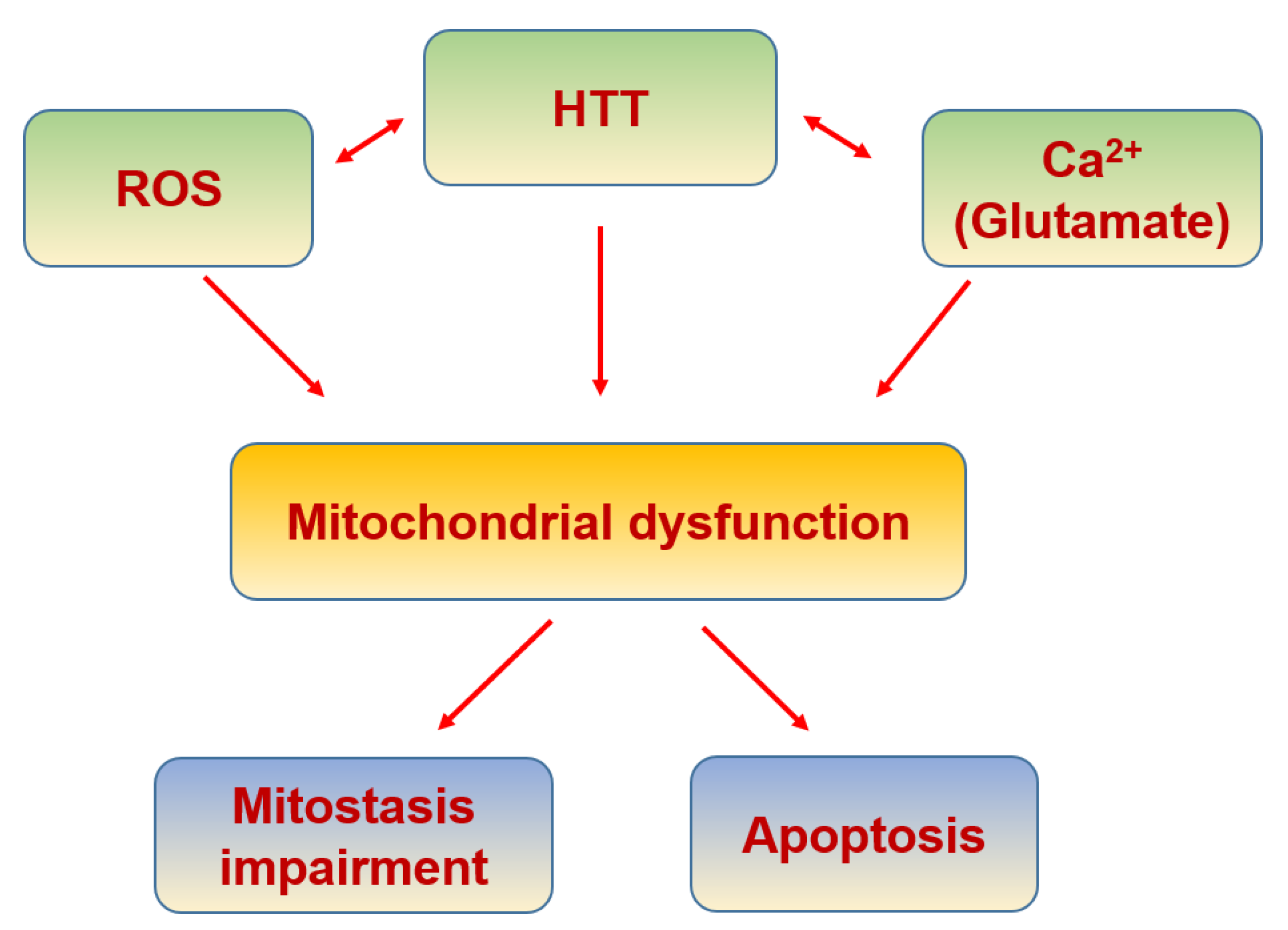
6.7. Huntington’s Disease
| Molecule | Function | Ref. |
|---|---|---|
| Huntingtin | Its aggregation increases mitochondrial calcium concentration and ROS production | [356] |
| Ubiquitin-like protein 1 (NUB1) | Suppressor of mutant Huntington toxicity via enhanced protein clearance | [367] |
| Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (BNip3) | Evidence of an abnormal activation in cells expressing mutant Huntingtin. | [368] |
| p53 | Regulates the levels of huntingtin gene expression | [369] |
| BDNF | Enhancing BDNF vesicular transport along microtubules | [370] |
| Alpha-tubulin deacetylase (HDAC) | Pharmacological inhibition of the HDAC6 deacetylase activity increased acetylated alpha-tubulin levels, and induced mitochondrial motility and fusion in striatal neurons | [371] |
| PGC1a | Transcriptional repression of PGC-1alpha by mutant huntingtin | [372] |
| Drp1 | Its interaction with mutant HTT impairs mitochondrial biogenesis, causing defective mitochondria axonal transport and synaptic degeneration | [366] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| AMPA | α-amino-3 hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| AMPK | adenosine monophosphate-activated protein kinase |
| ANT | adenine nucleotide translocase |
| APAF-1 | apoptosis protease-activating factor-1 |
| ASC | adaptor protein |
| ATP | adenosine triphosphate |
| Aβ | amyloid beta-peptide |
| BAP31 | B-cell receptor-associated protein 31 |
| BDNF | brain-derived neurotrophic factor |
| BER | Base excision repair |
| β/cAMP | β-adrenergic signalling |
| Ca2+ | Calcium |
| CAMKII | Ca2+/calmodulin-dependent protein kinase |
| CAT | Catalase |
| CDKs | cyclin-dependent kinases |
| CICR | Ca2+-induced Ca2+ release |
| CHCHD2 | coiled-coil-helix-coiled-coil-helix domain containing 2 |
| CHCHD10 | coiled-coil-helix-coiled-coil-helix domain-containing 10 |
| CREB | cAMP response element-binding protein |
| Cybrid | cytoplasmatic hybrids |
| CypD | cyclophilin D |
| CytC | cytochrome C |
| Drp1 | dynamin-related protein 1 |
| EMRE | essential MCU regulator |
| eNOS | endothelial NOS |
| ER | endoplasmic reticulum |
| ERKs | extracellular signal-regulated kinases |
| Fis1 | mitochondrial fission 1 |
| FUS | fused in sarcoma |
| FZ | frizzled receptor |
| GABA | γ-aminobutyric acid |
| GPx | glutathione peroxidase |
| GSH | Glutathione |
| Grp75 | mitochondrial chaperone glucose-regulated protein 75 |
| GSK3β | glycogen synthase kinase-3β |
| H2O2 | hydrogen peroxide |
| HD | Huntington’s disease |
| HKII | hexokinase II |
| HTT | Huntingtin |
| IKK | IkB kinase |
| IL-18 | interleukin 18 |
| IL-1β | interleukin 1β |
| IL-6 | interleukin 6 |
| IMM | inner mitochondrial membrane |
| INF2 | inverted formin 2 |
| IP3R | inositol triphosphate receptor |
| IR | insulin receptor |
| IRS | insulin receptor substrate |
| JNK | c-Jun N-terminal kinase |
| LRP5/6 | low-density lipoprotein receptor-related protein 5/6 |
| LRRK | Leucine Rich Repeat Kinase 2 |
| LTP | long-term potentiation |
| MAM | mitochondria associated membranes |
| MAPK | mitogen activated protein kinases |
| MCI | mild cognitive impairment |
| MCU | mitochondrial Ca2+ uniporter |
| MCUb | Mitochondrial Calcium Uniporter Dominant Negative Subunit Beta |
| MCUR1 | MCU regulator 1 |
| MFF | mitochondrial fission factor |
| MFN | Mitofusin |
| MFN1 | mitofusin protein 1 |
| MFN2 | mitofusin protein 2 |
| MICU1 | mitochondrial calcium uptake 1 |
| MICU2 | mitochondrial calcium uptake 2 |
| MiD49 | mitochondrial dynamic protein 49 |
| MiD50 | mitochondrial dynamic protein 50 |
| Miro | Mitochondrial Rho GTPase |
| MNRR1 | Mitochondria Nuclear Retrograde Regulator 1 |
| mPTP | mitochondrial permeability transition pore |
| MPTP | 1-methyl4-phenyl-1,2,3,4-tetrahydropyridine |
| MRC | mitochondrial respiratory chain |
| mtDNA | mitochondrial DNA |
| mtNOS | mitochondrial NOS |
| mTOR | mechanistic target of rapamycin kinase |
| Myo19 | myosin19 |
| NAD+/NADH | nicotinamide adenine dinucleotide |
| NCLX | Li+-permeable Na+/Ca2+ exchanger |
| NFAT | nuclear factor of activated T-cells |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | Nod-like receptor 3 |
| NMDAR | N-methyl-D-aspartate receptor |
| nNOS | neuronal NOS |
| NO | Nitric oxide |
| NOS | constitutive NO-synthase |
| NRF1 | nuclear respiratory factor 1 |
| NRF2 | nuclear respiratory factor 2 |
| O2·− | superoxide anion |
| OH.− | hydroxyl radical |
| OMM | outer mitochondrial membrane |
| ONOO− | Peroxynitrite |
| OPA1 | protein optic atrophy 1 |
| p38α | p38 mitogen-activated protein kinase alpha |
| PACS-2 | phosphofurin acidic cluster sorting protein 2 |
| PD | Parkinson’s disease |
| PGC-1α | peroxisome proliferator-activated receptor-γ (PPARγ) coactivator-1α |
| PGC1β | peroxisome proliferator-activated receptor gamma coactivator 1-beta |
| PI3K | phospho-inositol-3 kinase |
| PINK1 | PTEN-induced kinase 1 |
| PIP | phosphatidylinositol phosphate |
| PKC | protein kinase C |
| PRC | PGC-related coactivator |
| PS2 | presenilin 2 |
| pS302 | phosphorylation of insulin receptor at serine-302 |
| pS307 | phosphorylation of insulin receptor at serine-307 |
| PTPIP51 | tyrosine phosphatase-interacting protein-51 |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| SERCA | sarco/endoplasmic reticulum Ca2+-ATPase |
| SIRT | silent mating type information regulation 2 homolog |
| SOC | store-operated Ca2+ channel |
| SOCE | store operated Ca2+ entry |
| SOCS | suppressor cytokine signalling |
| SOD | antioxidant enzymes superoxide dismutase |
| SOD1 | Cu/Zn superoxide dismutase 1 |
| SLC25A23 | solute carrier 25A23 |
| STIM | stromal interaction molecule |
| T2D | type 2 diabetes |
| SNPC | substantia nigra pars compacta |
| SNPH | Syntaphilin |
| TCA | tricarboxylic acid cycle |
| TARDBP/TDP-43 | transactive response DNA binding protein 43 |
| TFAM | mitochondrial transcription factor A |
| TFB1M | cofactor mitochondrial transcription factor B |
| TH | thyroid hormone |
| TNF-α | tumor necrosis factor α |
| TOMM 40 | Translocase of the OMM 40 |
| TRAK | trafficking kinesin protein |
| TRP | transient receptor potential |
| VAPB | vesicle-associated membrane protein-B |
| VDAC | voltage dependent anion channel 1 |
| VGCC | voltage-gated Ca2+ channels |
| WISP3/CCN6 | Wnt-induced signaling protein 3 |
| Wnt | Wnt signaling pathway |
| ΔΨm | mitochondrial membrane potential. |
References
- Erbslöh, F.; Bernsmeier, A.; Hillesheim, H.R. Der Glucoseverbrauch des Gehirns und seine Abhängigkeit von der Leber. Arch. für Psychiatr. und Nervenkrankheiten Ver. mit Zeitschrift für die Gesamte Neurol. und Psychiatr. 1958, 196, 611–626. [Google Scholar] [CrossRef]
- Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999, 24, 321–329. [Google Scholar] [CrossRef]
- Lin, M.Y.; Sheng, Z.H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Misgeld, T.; Schwarz, T.L. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and calcium in Alzheimer’s disease: From cell signaling to neuronal cell death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Kruman, I.I.; Mattson, M.P. Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J. Neurochem. 1999, 72, 529–540. [Google Scholar] [CrossRef]
- Braschi, E.; McBride, H.M. Mitochondria and the culture of the Borg. BioEssays 2010, 32, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, E.; Tricarico, R.; Savage, M.; Golemis, E.A.; Hall, M.J. Disease-associated genetic variation in human mitochondrial protein import. Am. J. Hum. Genet. 2019, 104, 784–801. [Google Scholar] [CrossRef] [Green Version]
- Connerth, M.; Tatsuta, T.; Haag, M.; Klecker, T.; Westermann, B.; Langer, T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science (80-. ). 2012, 338, 815–818. [Google Scholar] [CrossRef]
- Vicario-Orri, E.; Opazo, C.M.; Muñoz, F.J. The pathophysiology of axonal transport in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 43, 1097–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase dMiro is required for axonal transport of mitochondria to drosophila synapses. Neuron 2005, 47, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Brickley, K.; Stephenson, F.A. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J. Biol. Chem. 2011, 286, 18079–18092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowers, R.S.; Megeath, L.J.; Górska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron 2002, 36, 1063–1077. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M.; Sato-Yoshitake, R.; Okada, Y.; Noda, Y.; Takemura, R.; Yamazaki, H.; Hirokawa, N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 1994, 79, 1209–1220. [Google Scholar] [CrossRef]
- Fransson, Å.; Ruusala, A.; Aspenström, P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 2006, 344, 500–510. [Google Scholar] [CrossRef]
- Van Spronsen, M.; Mikhaylova, M.; Lipka, J.; Schlager, M.A.; van den Heuvel, D.J.; Kuijpers, M.; Wulf, P.S.; Keijzer, N.; Demmers, J.; Kapitein, L.C.; et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 2013, 77, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [Green Version]
- Kapitein, L.C.; Schlager, M.A.; Kuijpers, M.; Wulf, P.S.; van Spronsen, M.; MacKintosh, F.C.; Hoogenraad, C.C. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 2010, 20, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintero, O.A.; DiVito, M.M.; Adikes, R.C.; Kortan, M.B.; Case, L.B.; Lier, A.J.; Panaretos, N.S.; Slater, S.Q.; Rengarajan, M.; Feliu, M.; et al. Human Myo19 is a novel myosin that associates with mitochondria. Curr. Biol. 2009, 19, 2008–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Doménech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Morris, R.L.; Hollenbeck, P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993, 104, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Kraft, L.M.; Lackner, L.L. Mitochondrial anchors: Positioning mitochondria and more. Biochem. Biophys. Res. Commun. 2018, 500, 2–8. [Google Scholar] [CrossRef]
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.C.; Zhang, C.L.; Babujee, L.; Vevea, J.D.; August, B.K.; Sheng, Z.H.; Chapman, E.R.; Gomez, T.M.; Chiu, S.Y. Inappropriate intrusion of an axonal mitochondrial anchor into dendrites causes neurodegeneration. Cell Rep. 2019, 29, 685–696.e5. [Google Scholar] [CrossRef] [Green Version]
- Fukumitsu, K.; Hatsukano, T.; Yoshimura, A.; Heuser, J.; Fujishima, K.; Kengaku, M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol. Cell. Neurosci. 2016, 71, 56–65. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; Van Der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; Van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Hales, K.G.; Fuller, M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 1997, 90, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Zorzano, A.; Pich, S. What is the biological significance of the two mitofusin proteins present in the outer mitochondrial membrane of mammalian cells? IUBMB Life 2006, 58, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Cosson, P.; Marchetti, A.; Ravazzola, M.; Orci, L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: An ultrastructural study. PLoS One 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef] [Green Version]
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtdna stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, V.; Shadel, G.S. Human mitochondrial transcription factor B1 interacts with the C-terminal activation region of h-mtTFA and stimulates transcription independently of Its RNA methyltransferase activity. Mol. Cell. Biol. 2003, 23, 5816–5824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Zheng, L.; Liu, W.; Wang, X.; Wang, Z.; Wang, Z.; French, A.J.; Kang, D.; Chen, L.; Thibodeau, S.N.; et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011, 71, 2978–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell 2007, 18, 3225–3236. [Google Scholar] [CrossRef]
- Schilling, J.; Kelly, D.P. The PGC-1 cascade as a therapeutic target for heart failure. J. Mol. Cell. Cardiol. 2011, 51, 578–583. [Google Scholar] [CrossRef] [Green Version]
- McClure, T.D.; Young, M.E.; Taegtmeyer, H.; Ning, X.-H.; Buroker, N.E.; López-Guisa, J.; Portman, M.A. Thyroid hormone interacts with PPARα and PGC-1 during mitochondrial maturation in sheep heart. Am. J. Physiol. Circ. Physiol. 2005, 289, H2258–H2264. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Gurd, B.J.; Yoshida, Y.; McFarlan, J.T.; Holloway, G.P.; Moyes, C.D.; Heigenhauser, G.J.F.; Spriet, L.; Bonen, A. Nuclear SIRT1 activity, but not protein content, regulates mitochondrial biogenesis in rat and human skeletal muscle. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 2011, 301. [Google Scholar] [CrossRef]
- Schaeffer, P.J.; Wende, A.R.; Magee, C.J.; Neilson, J.R.; Leone, T.C.; Chen, F.; Kelly, D.P. Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J. Biol. Chem. 2004, 279, 39593–39603. [Google Scholar] [CrossRef] [Green Version]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M.; Wang, S.C.; Shirai, M.; Scaglia, F.; Xie, M.; Sakai, S.; Tanaka, T.; Kulkarni, P.A.; Barger, P.M.; Youker, K.A.; et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004, 23, 3559–3569. [Google Scholar] [CrossRef] [Green Version]
- Brandt, N.; Nielsen, L.; Thiellesen Buch, B.; Gudiksen, A.; Ringholm, S.; Hellsten, Y.; Bangsbo, J.; Pilegaard, H. Impact of β-adrenergic signaling in PGC-1α-mediated adaptations in mouse skeletal muscle. Am. J. Physiol. Metab. 2018, 314, E1–E20. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Fan, M.; Rhee, J.; St-Pierre, J.; Handschin, C.; Puigserver, P.; Lin, J.; Jäeger, S.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1α: Modulation by p38 MAPK. Genes Dev. 2004, 18, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta - Biomembr. 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Gincel, D.; Silberberg, S.D.; Shoshan-Barmatz, V. Modulation of the voltage-dependent anion channel (VDAC) by glutamate. J. Bioenerg. Biomembr. 2000, 32, 571–583. [Google Scholar] [CrossRef]
- Rostovtseva, T.; Colombini, M. VDAC channels mediate and gate the flow of ATP: Implications for the regulation of mitochondrial function. Biophys. J. 1997, 72, 1954–1962. [Google Scholar] [CrossRef] [Green Version]
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta - Biomembr. 2007, 1768, 2510–2515. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Malliankaraman, K.; Guo, S.; Rajan, S.; Elrod, J.W.; et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol. Biol. Cell 2014, 25, 936–947. [Google Scholar] [CrossRef]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (80-. ). 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.W.; Wu, Y.; Williams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 2016, 5. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for mcu-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamer, K.J.; Mootha, V.K. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 2014, 15, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca2+ binding by MICU 1– MICU 2 serves as an on–off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 serves as a molecular gatekeeper to prevent In vivo mitochondrial calcium overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J.K. MICU2 restricts spatial crosstalk between InsP3R and MCU channels by regulating threshold and gain of MICU1-mediated inhibition and activation of MCU. Cell Rep. 2017, 21, 3141–3154. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; De la Fuente, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; VecellioReane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; DeStefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Gilabert, J.; Parekh, A. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J. 2000, 19, 6401–6407. [Google Scholar] [CrossRef]
- Pacher, P.; Thomas, A.P.; Hajnóczky, G. Ca2+ marks: Miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 2380–2385. [Google Scholar] [CrossRef] [Green Version]
- Nita, I.I.; Hershfinkel, M.; Fishman, D.; Ozeri, E.; Rutter, G.A.; Sensi, S.L.; Khananshvili, D.; Lewis, E.C.; Sekler, I. The mitochondrial Na +/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Nita, I.I.; Caspi, Y.; Gudes, S.; Fishman, D.; Lev, S.; Hersfinkel, M.; Sekler, I.; Binshtok, A.M. Privileged crosstalk between TRPV1 channels and mitochondrial calcium shuttling machinery controls nociception. Biochim. Biophys. Acta - Mol. Cell Res. 2016, 1863, 2868–2880. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- Arnold, S.; Kadenbach, B. Cell respiration is controlled by ATP, an allosteric inhibitor of cytochrome-C oxidase. Eur. J. Biochem. 1997, 249, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Scaduto, R.C. Influence of calcium on NADH and succinate oxidation by rat heart submitochondrial particles. Arch. Biochem. Biophys. 1995, 316, 815–820. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. Intracellular calcium ions and intramitochondrial Ca2+ in the regulation of energy metabolism in mammalian tissues. Proc. Nutr. Soc. 1990, 49, 57–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Territo, P.R.; Mootha, V.K.; French, S.A.; Balaban, R.S. Ca2+ activation of heart mitochondrial oxidative phosphorylation: Role of the F0/F1-ATPase. Am. J. Physiol. Cell Physiol. 2000, 278. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Takacs, K.; Kövér, K.; Adam-Vizi, V. Stimulation of H2O2 generation by calcium in brain mitochondria respiring on α-glycerophosphate. J. Neurosci. Res. 2007, 85, 3471–3479. [Google Scholar] [CrossRef] [PubMed]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Dev. 2016, 143, 1981–1992. [Google Scholar] [CrossRef] [Green Version]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Berman, S.B.; Hastings, T.G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: Implications for Parkinson’s disease. J. Neurochem. 1999, 73, 1127–1137. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; A Sosunov, A.; M McKhann, G.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M. The mitochondrial permeabitity transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Achleitner, G.; Gaigg, B.; Krasser, A.; Kainersdorfer, E.; Kohlwein, S.D.; Perktold, A.; Zellnig, G.; Daum, G. Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur. J. Biochem. 1999, 264, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Eisner, V.; Csordás, G.; Hajnóczky, G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle-pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 2013, 126, 2965–2978. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Witherspoon, M.; Sandu, D.; Lu, C.; Wang, K.; Edwards, R.; Yeung, A.; Gelincik, O.; Manfredi, G.; Gross, S.; Kopelovich, L.; et al. ETHE1 overexpression promotes SIRT1 and PGC1α mediated aerobic glycolysis, oxidative phosphorylation, mitochondrial biogenesis and colorectal cancer. Oncotarget 2019, 10, 4004–4017. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [Green Version]
- De vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Fujimoto, M. Detergent-resistant microdomains determine the localization of σ-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 2010, 77, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arasaki, K.; Shimizu, H.; Mogari, H.; Nishida, N.; Hirota, N.; Furuno, A.; Kudo, Y.; Baba, M.; Baba, N.; Cheng, J.; et al. A Role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev. Cell 2015, 32, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Qian, T.; Herman, B.; Lemasters, J.J. The mitochondrial permeability transition mediates both necrotic and apoptotic death of hepatocytes exposed to Br-A23187. Toxicol. Appl. Pharmacol. 1999, 154, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerella, C.; Diederich, M.; Ghibelli, L. The dual role of calcium as messenger and stressor in cell damage, death, and survival. Int. J. Cell Biol. 2010, 2010, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Guix, F.X.; Clarimón, J.; Miscione, G.P.; Boada, M.; et al. Methylglyoxal produced by amyloid-β peptide-induced nitrotyrosination of triosephosphate isomerase triggers neuronal death in alzheimer’s disease. J. Alzheimer’s Dis. 2014, 41, 273–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Bosch-Morató, M.; Valls-Comamala, V.; Muñoz, F.J. Methylglyoxal reduces mitochondrial potential and activates Bax and caspase-3 in neurons: Implications for Alzheimer’s disease. Neurosci. Lett. 2014, 580, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henshall, D.C.; Bonislawski, D.P.; Skradski, S.L.; Araki, T.; Lan, J.Q.; Schindler, C.K.; Meller, R.; Simon, R.P. Formation of the Apaf-1/cytochrome c complex precedes activation of caspase-9 during seizure-induced neuronal death. Cell Death Differ. 2001, 8, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; de Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J. Mitochondrial dynamics in mitochondrial diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 186–197. [Google Scholar] [CrossRef]
- Keep, M.; Elmér, E.; Fong, K.S.K.; Csiszar, K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001, 894, 327–331. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Ultrastructural change of synapses of Betz cells in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 268, 29–32. [Google Scholar] [CrossRef]
- Thomas, B.; Banerjee, R.; Starkova, N.N.; Zhang, S.F.; Calingasan, N.Y.; Yang, L.; Wille, E.; Lorenzo, B.J.; Ho, D.J.; Beal, M.F.; et al. Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson’s disease. Antioxidants Redox Signal. 2012, 16, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, R.A.; Johnson, G.V.W. Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res. Bull. 2009, 80, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, Y.S.; Johnson, G.V.W.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef]
- Miranda, S.; Opazo, C.; Larrondo, L.F.; Muñoz, F.J.; Ruiz, F.; Leighton, F.; Inestrosa, N.C. The role of oxidative stress in the toxicity induced by amyloid β-peptide in Alzheimer’s disease. Prog. Neurobiol. 2000, 62, 633–648. [Google Scholar] [CrossRef]
- Szalai, G.; Krishnamurthy, R.; Hajnóczky, G. Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef] [PubMed]
- Sharov, V.S.; Dremina, E.S.; Galeva, N.A.; Williams, T.D.; Schöneich, C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: Selective protein oxidation during biological aging. Biochem. J. 2006, 394, 605–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Zhang, X.; Xu, J.; Jian, C.; Huang, Z.; Ye, T.; Hu, K.; Zheng, M.; Gao, F.; Wang, X.; et al. Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. J. Biol. Chem. 2013, 288, 4602–4612. [Google Scholar] [CrossRef] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/antioxidant imbalance in Alzheimer’s disease: Therapeutic and diagnostic prospects. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Yegla, B.; Foster, T.C. Redox signaling in neurotransmission and cognition during aging. Antioxidants Redox Signal. 2018, 28, 1724–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Potus, F.; Wu, D.; Dasgupta, A.; Chen, K.-H.; Mewburn, J.; Lima, P.; Archer, S.L. Increased Drp1-mediated mitochondrial fission promotes proliferation and collagen production by right ventricular fibroblasts in experimental pulmonary arterial hypertension. Front. Physiol. 2018, 9, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jendrach, M.; Mai, S.; Pohl, S.; Vöth, M.; Bereiter-Hahn, J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 2008, 8, 293–304. [Google Scholar] [CrossRef]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Muñoz, F.J. The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta - Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef]
- Brodsky, S.V.; Gao, S.; Li, H.; Goligorsky, M.S. Hyperglycemic switch from mitochondrial nitric oxide to superoxide production in endothelial cells. Am. J. Physiol. - Hear. Circ. Physiol. 2002, 283. [Google Scholar] [CrossRef] [Green Version]
- Dzurik, R.; Krivosikova, Z.; Stefikova, K.; Spustova, V. Mitochondria and mitochondrial nitric oxide synthase alterations participate in energetical dysbalance, aging and age-related diseases—PubMed. Bratisl. Lek. Listy 2006, 107, 405–411. [Google Scholar]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Valdez, L.B.; Zaobornyj, T.; Boveris, A. Oxygen dependence of mitochondrial nitric oxide synthase activity. Biochem. Biophys. Res. Commun. 2003, 305, 771–775. [Google Scholar] [CrossRef]
- Sarti, P.; Giuffré, A.; Forte, E.; Mastronicola, D.; Barone, M.C.; Brunori, M. Nitric oxide and cytochrome C oxidase: Mechanisms of inhibition and NO degradation. Biochem. Biophys. Res. Commun. 2000, 274, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Padilla Salinas, E.; Darley-Usmar, K.; Eiserich, J.P.; Freeman, B.A.; Darley-Usmar, V.M.; Anderson, P.G. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J. Biol. Chem. 2000, 275, 20474–20479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ILL-Raga, G.; Palomer, E.; Ramos-Fernández, E.; Guix, F.X.; Bosch-Morató, M.; Guivernau, B.; Tajes, M.; Valls-Comamala, V.; Jiménez-Conde, J.; Ois, A.; et al. Fibrinogen nitrotyrosination after ischemic stroke impairs thrombolysis and promotes neuronal death. Biochim. Biophys. Acta - Mol. Basis Dis. 2015, 1852, 421–428. [Google Scholar] [CrossRef]
- Tajes, M.; Ill-Raga, G.; Palomer, E.; Ramos-Fernández, E.; Guix, F.X.; Bosch-Morató, M.; Guivernau, B.; Jiménez-Conde, J.; Ois, A.; Pérez-Asensio, F.; et al. Nitro-oxidative stress after neuronal ischemia induces protein nitrotyrosination and cell death. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, F.J.; Opazo, C.; Gil-Gómez, G.; Tapia, G.; Fernández, V.; Valverde, M.A.; Inestrosa, N.C. Vitamin E but not 17β-estradiol protects against vascular toxicity induced by β-amyloid wild type and the dutch amyloid variant. J. Neurosci. 2002, 22, 3081–3089. [Google Scholar] [CrossRef]
- Coma, M.; Guix, F.X.; Uribesalgo, I.; Espuña, G.; Solé, M.; Andreu, D.; Muñoz, F.J.; Muñoz, F.J. Lack of oestrogen protection in amyloid-mediated endothelial damage due to protein nitrotyrosination. Brain 2005, 128, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; Meechan, D.W.; Karpinski, B.A.; Paronett, E.M.; Bryan, C.A.; Rutz, H.L.; Radin, E.A.; Lubin, N.; Bonner, E.R.; Popratiloff, A.; et al. Mitochondrial dysfunction leads to cortical under-connectivity and cognitive impairment. Neuron 2019, 102, 1127–1142.e3. [Google Scholar] [CrossRef]
- Netto, M.B.; de Oliveira Junior, A.N.; Goldim, M.; Mathias, K.; Fileti, M.E.; da Rosa, N.; Laurentino, A.O.; de Farias, B.X.; Costa, A.B.; Rezin, G.T.; et al. Oxidative stress and mitochondrial dysfunction contributes to postoperative cognitive dysfunction in elderly rats. Brain. Behav. Immun. 2018, 73, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Cabeza, R. Hemispheric asymmetry reduction in older adults: The HAROLD model. Psychol. Aging 2002, 17, 85–100. [Google Scholar] [CrossRef]
- Park, D.C.; Reuter-Lorenz, P. The adaptive brain: Aging and neurocognitive scaffolding. Annu. Rev. Psychol. 2009, 60, 173–196. [Google Scholar] [CrossRef] [Green Version]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Andrews-Hanna, J.R.; Snyder, A.Z.; Vincent, J.L.; Lustig, C.; Head, D.; Raichle, M.E.E.; Buckner, R.L. Disruption of large-scale brain systems in advanced aging. Neuron 2007, 56, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyango, I.G.; Lu, J.; Rodova, M.; Lezi, E.; Crafter, A.B.; Swerdlow, R.H. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Ojaimi, J.; Masters, C.L.; Opeskin, K.; McKelvie, P.; Byrne, E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 1999, 111, 39–47. [Google Scholar] [CrossRef]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial dysfunction in rat brain with aging. Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Kaur Sandhu, S.; Kaur, G. Mitochondrial electron transport chain complexes in aging rat brain and lymphocytes. Biogerontology 2003, 4, 19–29. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Maldonado, E.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Schampel, A.; Kuerten, S. Danger: High voltage—the role of Voltage-Gated Calcium Channels in central nervous system pathology. Cells 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaguni, L. DNA polymerase gamma, the mitochondrial replicase. Annu. Rev. Biochem. 2004, 73, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, P.; Hutter-Paier, B.; Havas, D.; Windisch, M.; Winblad, B. Development of GMP-1 a molecular chaperone network modulator protecting mitochondrial function and its assessment in fly and mice models of Alzheimer’s disease. J. Cell. Mol. Med. 2018, 22, 3464–3474. [Google Scholar] [CrossRef] [PubMed]
- Polo, M.; Alegre, F.; Moragrega, A.; Gibellini, L.; Marti-Rodrigo, A.; Blas-Garcia, A.; Esplugues, J.; Apostolova, N. Lon protease: A novel mitochondrial matrix protein in the interconnection between drug-induced mitochondrial dysfunction and endoplasmic reticulum stress. Br. J. Pharmacol. 2017, 174, 4409–4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorese, C.; Schulz, A.; Lin, Y.; Rosin, N.; Pellegrino, M.; Haynes, C. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.A.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 2007, 21, 3244–3257. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, Y.; Yan, K.; Chen, L.; Chen, X.R.; Li, P.; Chen, F.F.; Jiang, X.D. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflamm. 2013, 10, 871. [Google Scholar] [CrossRef] [Green Version]
- Tilstra, J.S.; Clauson, C.L.; Niedernhofer, L.J.; Robbins, P.D. NF-κB in aging and disease. Aging Dis. 2011, 2(6), 449–465. [Google Scholar]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Swacha, P.; Gekara, N.O.; Erttmann, S.F. Biochemical and microscopic analysis of inflammasome complex formation. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 625, pp. 287–298. ISBN 9780128183595. [Google Scholar]
- Yuk, J.M.; Silwal, P.; Jo, E.K. Inflammasome and mitophagy connection in health and disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yin, N.; Liu, W.; Cui, X.; Chen, S.; Wang, E. Curcumin ameliorates diabetic nephropathy by suppressing NLRP3 inflammasome signaling. Biomed Res. Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta - Bioenerg. 2014, 1837, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Schrader, M.; Costello, J.L.; Godinho, L.F.; Azadi, A.S.; Islinger, M. Proliferation and fission of peroxisomes - An update. Biochim. Biophys. Acta - Mol. Cell Res. 2016, 1863, 971–983. [Google Scholar] [CrossRef]
- De Mario, A.; Quintana-Cabrera, R.; Martinvalet, D.; Giacomello, M. (Neuro)degenerated mitochondria-ER contacts. Biochem. Biophys. Res. Commun. 2017, 483, 1096–1109. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Denis Alexander, H.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Chung, K.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e5. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Sun, L.; Ea, C.; Chen, Z. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Moore, C.; Bergstralh, D.; Duncan, J.; Lei, Y.; Morrison, T.; Zimmermann, A.; Accavitti-Loper, M.; Madden, V.; Sun, L.; Ye, Z.; et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008, 451, 573–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oganesyan, G.; Saha, S.; Guo, B.; He, J.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Kelly, B.; Logan, A.; Costa, A.; Varma, M.; Bryant, C.; Tourlomousis, P.; Däbritz, J.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.; Hajizadeh, S.; Holme, E.; Jonsson, I.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Iyer, S.; He, Q.; Janczy, J.; Elliott, E.; Zhong, Z.; Olivier, A.; Sadler, J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Wenceslau, C.; McCarthy, C.; Goulopoulou, S.; Szasz, T.; NeSmith, E.; Webb, R. Mitochondrial-derived N-formyl peptides: Novel links between trauma, vascular collapse and sepsis. Med. Hypotheses 2013, 81, 532–535. [Google Scholar] [CrossRef]
- Julian, M.; Shao, G.; Vangundy, Z.; Papenfuss, T.; Crouser, E. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFα release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M.J. Concentrations of insulin and of insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J. Clin. Invest. 1979, 64, 636–642. [Google Scholar] [CrossRef]
- Park, C.R.; Seeley, R.J.; Craft, S.; Woods, S.C. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol. Behav. 2000, 68, 509–514. [Google Scholar] [CrossRef]
- Van Der Heide, L.P.; Kamal, A.; Artola, A.; Gispen, W.H.; Ramakers, G.M.J. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-D-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J. Neurochem. 2005, 94, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.R.; Ko, H.W.; Jou, I.; Noh, J.S.; Gwag, B.J. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J. Neurobiol. 1999, 39, 536–546. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Bruce, C.R.; Beale, S.M.; Hoehn, K.L.; So, T.; Rolph, M.S.; Cooney, G.J. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: Evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 2007, 56, 2085–2092. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; De La Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; De La Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease - Is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.M.; Strittmatter, S.M. Binding sites for amyloid-β oligomers and synaptic toxicity. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Wardelmann, K.; Blümel, S.; Rath, M.; Alfine, E.; Chudoba, C.; Schell, M.; Cai, W.; Hauffe, R.; Warnke, K.; Flore, T.; et al. Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 2019, 21, 68–81. [Google Scholar] [CrossRef]
- Okouchi, M.; Okayama, N.; Steven Alexander, J.; Yee Aw, T. NRF2-dependent glutamate-L-cysteine ligase catalytic subunit expression mediates insulin protection against hyperglycemia-induced brain endothelial cell apoptosis. Curr. Neurovasc. Res. 2006, 3, 249–261. [Google Scholar] [CrossRef]
- Melser, S.; Lavie, J.; Bénard, G. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2812–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-α-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Pagel-Langenickel, I.; Bao, J.; Joseph, J.J.; Schwartz, D.R.; Mantell, B.S.; Xu, X.; Raghavachari, N.; Sack, M.N. PGC-1α integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J. Biol. Chem. 2008, 283, 22464–22472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Wong, R.; Rajapakse, N.; Murphy, E.; Steenbergen, C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 2008, 103, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The key roles of GSK-3β in regulating mitochondrial activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Kuno, A.; Ishikawa, S.; Miki, T.; Kouzu, H.; Yano, T.; Murase, H.; Tobisawa, T.; Ogasawara, M.; Horio, Y.; et al. Translocation of glycogen synthase kinase-3β (GSK-3β), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent anion channel 2 (VDAC2). J. Biol. Chem. 2014, 289, 29285–29296. [Google Scholar] [CrossRef] [Green Version]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3β mediates convergence of protection signalling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 2004, 113, 1535–1549. [Google Scholar] [CrossRef]
- Nishihara, M.; Miura, T.; Miki, T.; Tanno, M.; Yano, T.; Naitoh, K.; Ohori, K.; Hotta, H.; Terashima, Y.; Shimamoto, K. Modulation of the mitochondrial permeability transition pore complex in GSK-3β-mediated myocardial protection. J. Mol. Cell. Cardiol. 2007, 43, 564–570. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, X.; Zuberi, A.; Hwang, D.; Quon, M.J.; Lefevre, M.; Ye, J. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol. Endocrinol. 2004, 18, 2024–2034. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, D.; Gao, Y.; Yang, R.; Guan, T.; Hong, J.S.; Gao, H.M. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J. Neuroinflammation 2019, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronn, S.G.; Billestrup, N.; Mandrup-Poulsen, T. Diabetes and Suppressors of Cytokine Signaling Proteins. Diabetes 2007, 56, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.; He, J.; Menshikova, E.; Ritov, V. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Cummins, T.; Holden, C.; Sansbury, B.; Gibb, A.; Shah, J.; Zafar, N.; Tang, Y.; Hellmann, J.; Rai, S.; Spite, M.; et al. Metabolic remodeling of white adipose tissue in obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 307. [Google Scholar] [CrossRef]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Mattingly, K.; Ivanova, M.; Riggs, K.; Wickramasinghe, N.; Barch, M.; Klinge, C. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Guo, S.; Copps, K.; Dong, X.; Kollipara, R.; Rodgers, J.; Depinho, R.; Puigserver, P.; White, M. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor-gamma on brain and peripheral inflammation. Cell. Mol. Neurobiol. 2018, 38, 121. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picón-Pagès, P.; Bonet, J.; García-García, J.; Garcia-Buendia, J.; Gutierrez, D.; Valle, J.; Gómez-Casuso, C.E.S.; Sidelkivska, V.; Alvarez, A.; Perálvarez-Marín, A.; et al. Human albumin impairs amyloid β-peptide fibrillation through its C-terminus: From docking modeling to protection against neurotoxicity in Alzheimer’s disease. Comput. Struct. Biotechnol. J. 2019, 17, 963–971. [Google Scholar] [CrossRef]
- Valls-Comamala, V.; Guivernau, B.; Bonet, J.; Puig, M.; Perálvarez-Marín, A.; Palomer, E.; Fernàndez-Busquets, X.; Altafaj, X.; Tajes, M.; Puig-Pijoan, A.; et al. The antigen-binding fragment of human gamma immunoglobulin prevents amyloid β-peptide folding into β-sheet to form oligomers. Oncotarget 2017, 8, 41154–41165. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Gutiérrez, D.A.; Barranco-Almohalla, A.; Crepin, G.; Tajes, M.; Ill-Raga, G.; Guix, F.X.; Menéndez, S.; Arumí-Uría, M.; Vicente, R.; et al. Amyloid beta-peptide increases BACE1 translation through the phosphorylation of the eukaryotic initiation factor-2 α. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Guix, F.X.; Wahle, T.; Vennekens, K.; Snellinx, A.; Chávez-Gutiérrez, L.; Ill-Raga, G.; Ramos-Fernandez, E.; Guardia-Laguarta, C.; Lleó, A.; Arimon, M.; et al. Modification of γ-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer’s disease. EMBO Mol. Med. 2012, 4, 660–673. [Google Scholar] [CrossRef]
- Ill-Raga, G.; Tajes, M.; Busquets-García, A.; Ramos-Fernández, E.; Vargas, L.M.; Bosch-Morató, M.; Guivernau, B.; Valls-Comamala, V.; Eraso-Pichot, A.; Guix, F.X.; et al. Physiological control of nitric oxide in neuronal BACE1 translation by heme-regulated eIF2α kinase HRI induces synaptogenesis. Antioxidants Redox Signal. 2015, 22, 1295–1307. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Fernández, E.; Tajes, M.; Palomer, E.; Ill-Raga, G.; Bosch-Morató, M.; Guivernau, B.; Román-Dégano, I.; Eraso-Pichot, A.; Alcolea, D.; Fortea, J.; et al. Posttranslational nitro-glycative modifications of albumin in alzheimer’s disease: Implications in cytotoxicity and amyloid-β peptide aggregation. J. Alzheimer’s Dis. 2014, 40, 643–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajes, M.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Palomer, E.; Guix, F.X.; Muñoz, F.J. The pathophysiology of triose phosphate isomerase dysfunction in alzheimer’s disease. Histol. Histopathol. 2013, 28, 43–51. [Google Scholar] [PubMed]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Fiala, J.C.; Feinberg, M.; Peters, A.; Barbas, H. Mitochondrial degeneration in dystrophic neurites of senile plaques may lead to extracellular deposition of fine filaments. Brain Struct. Funct. 2007, 212, 195–207. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. NeuroMolecular Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; Lezi, E.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.D.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. On the role of Mitofusin 2 in endoplasmic reticulum-mitochondria tethering. Proc. Natl. Acad. Sci. USA 2017, 114, E2266–E2267. [Google Scholar] [CrossRef] [Green Version]
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER–mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 2016, 20, 1686–1695. [Google Scholar] [CrossRef]
- Wang, R.; Li, J.J.; Diao, S.; Kwak, Y.D.; Liu, L.; Zhi, L.; Büeler, H.; Bhat, N.R.; Williams, R.W.; Park, E.A.; et al. Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 2013, 17, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, P.; Tang, Z.; Yin, J.; Maalouf, M.; Beach, T.G.; Reiman, E.M.; Shi, J. Pituitary adenylate cyclase-activating polypeptide protects against β-amyloid toxicity. Neurobiol. Aging 2014, 35, 2064–2071. [Google Scholar] [CrossRef]
- Khanna, A.; Acharjee, P.; Acharjee, A.; Trigun, S.K. Mitochondrial SIRT3 and neurodegenerative brain disorders. J. Chem. Neuroanat. 2019, 95, 43–53. [Google Scholar] [CrossRef]
- Silva-Alvarez, C.; Arrázola, M.S.; Godoy, J.A.; Ordenes, D.; Inestrosa, N.C. Canonical Wnt signaling protects hippocampal neurons from Aβ oligomers: Role of non-canonical Wnt-5a/Ca2+ in mitochondrial dynamics. Front. Cell. Neurosci. 2013, 7, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrázola, M.S.; Silva-Alvarez, C.; Inestrosa, N.C. How the Wnt signaling pathway protects from neurodegeneration: The mitochondrial scenario. Front. Cell. Neurosci. 2015, 9, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennica, D.; Swanson, T.A.; Welsh, J.W.; Roy, M.A.; Lawrence, D.A.; Lee, J.; Brush, J.; Taneyhill, L.A.; Deuel, B.; Lew, M.; et al. WISP genes are members of the connective tissue growth factor family that are up-regulated in Wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc. Natl. Acad. Sci. USA 1998, 95, 14717–14722. [Google Scholar] [CrossRef] [Green Version]
- Patra, M.; Mahata, S.K.; Padhan, D.K.; Sen, M. CCN6 regulates mitochondrial function. J. Cell Sci. 2016, 129, 2841–2851. [Google Scholar] [CrossRef] [Green Version]
- Arrázola, M.S.; Ramos-Fernández, E.; Cisternas, P.; Ordenes, D.; Inestrosa, N.C. Wnt signaling prevents the aβ oligomer-induced mitochondrial permeability transition pore opening preserving mitochondrial structure in hippocampal neurons. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Gavello, D.; Calorio, C.; Franchino, C.; Cesano, F.; Carabelli, V.; Carbone, E.; Marcantoni, A. Early alterations of hippocampal neuronal firing induced by Abeta42. Cereb. Cortex 2018, 28, 433–446. [Google Scholar] [CrossRef]
- Guivernau, B.; Bonet, J.; Valls-Comamala, V.; Bosch-Morató, M.; Godoy, J.A.; Inestrosa, N.C.; Perálvarez-Marín, A.; Fernández-Busquets, X.; Andreu, D.; Oliva, B.; et al. Amyloid-β peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J. Neurosci. 2016, 36, 11693–11703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oulès, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of alzheimer disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: Physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch-Morató, M.; Iriondo, C.; Guivernau, B.; Valls-Comamala, V.; Vidal, N.; Olivé, M.; Querfurth, H.; Muñoz, F.J. Increased amyloid β-peptide uptake in skeletal muscle is induced by hyposialylation and may account for apoptosis in GNE myopathy. Oncotarget 2016, 7, 13354–13371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Moscardo, F.; Setó-Salvia, N.; Pera, M.; Bosch-Morató, M.; Plata, C.; Belbin, O.; Gené, G.; Dols-Icardo, O.; Ingelsson, M.; Helisalmi, S.; et al. Rare Variants in Calcium Homeostasis Modulator 1 (CALHM1) Found in Early Onset Alzheimer’s Disease Patients Alter Calcium Homeostasis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Supnet, C.; Bezprozvanny, I. Presenilins function in ER calcium leak and Alzheimer’s disease pathogenesis. Cell Calcium 2011, 50, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Ill-Raga, G.; Ramos-Fernández, E.; Guix, F.X.; Tajes, M.; Bosch-Morató, M.; Palomer, E.; Godoy, J.; Belmar, S.; Cerpa, W.; Simpkins, J.W.; et al. Amyloid-β peptide fibrils induce nitro-oxidative stress in neuronal cells. J. Alzheimer’s Dis. 2010, 22, 641–652. [Google Scholar] [CrossRef] [Green Version]
- Guix, F.X.; Ill-Raga, G.; Bravo, R.; Nakaya, T.; De Fabritiis, G.; Coma, M.; Miscione, G.P.; Vill-Freixa, J.; Suzuki, T.; Fernandez-Busquets, X.; et al. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain 2009, 132, 1335–1345. [Google Scholar] [CrossRef] [Green Version]
- Coma, M.; Guix, F.X.; Ill-Raga, G.; Uribesalgo, I.; Alameda, F.; Valverde, M.A.; Muñoz, F.J. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol. Aging 2008, 29, 969–980. [Google Scholar] [CrossRef]
- Kang, D.; Hamasaki, N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: Overview of its multiple roles. Ann. N. Y. Acad. Sci. 2005, 1042, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Schurman, S.H.; Harboe, C.; De Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwinska, M.W.; Violante, I.R.; Wise, R.J.S.; Leech, R.; Devlin, J.T.; Geranmayeh, F.; Hampshire, A. Stimulating multiple-demand cortex enhances vocabulary learning. J. Neurosci. 2017, 37, 7606–7618. [Google Scholar] [CrossRef] [PubMed]
- Lillenes, M.S.; Rabano, A.; Støen, M.; Riaz, T.; Misaghian, D.; Møllersen, L.; Esbensen, Y.; Günther, C.C.; Selnes, P.; Stenset, V.T.V.; et al. Altered DNA base excision repair profile in brain tissue and blood in Alzheimer’s disease. Mol. Brain 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.; Murri, L.; Rapoport, S.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Gan, X.; Huang, S.; Wu, L.; Wang, Y.; Hu, G.; Li, G.; Zhang, H.; Yu, H.; Swerdlow, R.; Chen, J.; et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta 2014, 1842, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Valaasani, K.; Sun, Q.; Hu, G.; Li, J.; Du, F.; Guo, Y.; Carlson, E.; Gan, X.; Yan, S. Identification of human ABAD inhibitors for rescuing Aβ-mediated mitochondrial dysfunction. Curr. Alzheimer Res. 2014, 11, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Cieślik, M.; Czapski, G.; Wójtowicz, S.; Wieczorek, I.; Wencel, P.; Strosznajder, R.; Jaber, V.; Lukiw, W.; Strosznajder, J. Alterations of Transcription of Genes Coding Anti-oxidative and Mitochondria-Related Proteins in Amyloid β Toxicity: Relevance to Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1374–1388. [Google Scholar] [CrossRef] [Green Version]
- Dzitoyeva, S.; Chen, H.; Manev, H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging 2012, 33, 2881–2891. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langston, J.; Ballard, P.; Tetrud, J.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akundi, R.S.; Zhi, L.; Sullivan, P.G.; Büeler, H. Shared and cell type-specific mitochondrial defects and metabolic adaptations in primary cells from PINK1-deficient mice. Neurodegener. Dis. 2013, 12, 136–149. [Google Scholar] [CrossRef]
- Büeler, H. Mitochondrial dynamics, cell death and the pathogenesis of Parkinson’s disease. Apoptosis 2010, 15, 1336–1353. [Google Scholar] [CrossRef] [PubMed]
- Ciron, C.; Zheng, L.; Bobela, W.; Knott, G.W.; Leone, T.C.; Kelly, D.P.; Schneider, B.L. PGC-1α activity in nigral dopamine neurons determines vulnerability to α-synuclein. Acta Neuropathol. Commun. 2015, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.K.; Cookson, M.R. Mitochondrial quality control and dynamics in Parkinson’s disease. Antioxidants Redox Signal. 2012, 16, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1α dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta - Mol. Basis Dis. 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, M. The changing landscape of Voltage-Gated Calcium Channels in neurovascular disorders and in neurodegenerative diseases. Curr. Neuropharmacol. 2013, 11, 276–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Miró-Casas, E.; Abellán, A.; Soler-Soler, J. Mitochondrial Ca2+ uptake during simulated ischemia does not affect permeability transition pore opening upon simulated reperfusion. Cardiovasc. Res. 2006, 71, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Thoudam, T.; Jeon, J.-H.; Ha, C.-M.; Lee, I.-K. Role of mitochondria-associated endoplasmic reticulum membrane in inflammation-mediated metabolic diseases. Mediators Inflamm. 2016, 2016. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; da Cruz, A.B.; Burbulla, L.F.; St. Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in Miro degradation and mitophagy Is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Hüttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Koyano, F.; Yamano, K.; Kosako, H.; Tanaka, K.; Matsuda, N. Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J. Biol. Chem. 2019, 294, 10300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovich, E.; Kerem, A.; Fröhlich, K.; Diamant, N.; Bar-Nun, S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 2002, 22, 626–634. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Youle, R. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16. [Google Scholar] [CrossRef] [Green Version]
- Plun-Favreau, H.; Klupsch, K.; Moisoi, N.; Gandhi, S.; Kjaer, S.; Frith, D.; Harvey, K.; Deas, E.; Harvey, R.J.; McDonald, N.; et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 2007 911 2007, 9, 1243–1252. [Google Scholar] [CrossRef]
- Pridgeon, J.; Olzmann, J.; Chin, L.; Li, L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007, 5, 1494–1503. [Google Scholar] [CrossRef]
- Morais, V.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgacheva, L.; Berezhnov, A.; Fedotova, E.; Zinchenko, V.; Abramov, A. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonello, F.; Hassoun, S.; Mouton-Liger, F.; Shin, Y.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.C.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; MacKenzie, I.R.A. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; de Munain, A.L. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 2019, 100. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.J.; Bredvik, K.; Burstein, S.R.; Davis, C.; Meadows, S.M.; Dash, J.; Case, L.; Milner, T.A.; Kawamata, H.; Zuberi, A.; et al. ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 2019, 138. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Woo, J.A.A.; Bukhari, M.Z.; LePochat, P.; Chacko, A.; Selenica, M.L.B.; Yan, Y.; Kotsiviras, P.; Buosi, S.C.; Zhao, X.; et al. CHCHD10-regulated OPA1-mitofilin complex mediates TDP-43-induced mitochondrial phenotypes associated with frontotemporal dementia. FASEB J. 2020, 34, 8493–8509. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Semmler, S.; Broom, H.R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N.R.; Vande Velde, C. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 2008, 17, 3303–3317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crociara, P.; Chieppa, M.N.; Vallino Costassa, E.; Berrone, E.; Gallo, M.; Lo Faro, M.; Pintore, M.D.; Iulini, B.; D’Angelo, A.; Perona, G.; et al. Motor neuron degeneration, severe myopathy and TDP-43 increase in a transgenic pig model of SOD1-linked familiar ALS. Neurobiol. Dis. 2019, 124, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired mitophagy plays a role in denervation of neuromuscular junctions in ALS mice. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Tak, Y.J.; Park, J.H.; Rhim, H.; Kang, S. ALS-related mutant SOD1 aggregates interfere with mitophagy by sequestering the autophagy receptor optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta - Mol. Basis Dis. 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.; Fujioka, H.; Liang, J.; Jiang, S.; X, M.; Jiang, Z.; da Rocha, E.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Armakola, M.; Higgins, M.; Figley, M.; Barmada, S.; Scarborough, E.; Diaz, Z.; Fang, X.; Shorter, J.; Krogan, N.; Finkbeiner, S.; et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat. Genet. 2012, 44, 1302–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nat. 2010 4667310 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.; Lau, D.; Gray, E.; Sancho, R.; Vizcay-Barrena, G.; De Vos, K.; Shaw, C.; et al. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Hsu, F.; Spannl, S.; Ferguson, C.; Hyman, A.; Parton, R.; Zerial, M. Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. Elife 2018, 7. [Google Scholar] [CrossRef]
- Strong, T.V.; Tagle, D.A.; Valdes, J.M.; Elmer, L.W.; Boehm, K.; Swaroop, M.; Kaatz, K.W.; Collins, F.S.; Albin, R.L. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet. 1993, 5, 259–265. [Google Scholar] [CrossRef]
- Chen, N.; Luo, T.; Wellington, C.; Metzler, M.; McCutcheon, K.; Hayden, M.R.; Raymond, L.A. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J. Neurochem. 1999, 72, 1890–1898. [Google Scholar] [CrossRef]
- Ramos-Fernández, E.; Tajes, M.; ILL-Raga, G.; Vargas, L.; Busquets-García, A.; Bosch-Morató, M.; Guivernau, B.; Valls-Comamala, V.; Gomis, M.; Grau, C.; et al. Glutamatergic stimulation induces GluN2B translation by the nitric oxide-Heme-Regulated eIF2alpha kinase in cortical neurons. Oncotarget 2016, 7, 58876–58892. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, F.J.; Godoy, J.A.; Cerpa, W.; Poblete, I.M.; Huidobro-Toro, J.P.; Inestrosa, N.C. Wnt-5a increases NO and modulates NMDA receptor in rat hippocampal neurons. Biochem. Biophys. Res. Commun. 2014, 444, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Ivanova, E.; Lee, J.M.; Choo, Y.S.; Fossale, E.; Anderson, M.A.; Gusella, J.F.; Laramie, J.M.; Myers, R.H.; Lesort, M.; et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005, 14, 2871–2880. [Google Scholar] [CrossRef]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.A.; Leavitt, B.R.; Lepiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Hands, S.; Sajjad, M.U.; Newton, M.J.; Wyttenbach, A. In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J. Biol. Chem. 2011, 286, 44512–44520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firdaus, W.J.J.; Wyttenbach, A.; Giuliano, P.; Kretz-Remy, C.; Currie, R.W.; Arrigo, A.P. Huntingtin inclusion bodies are iron-dependent centers of oxidative events. FEBS J. 2006, 273, 5428–5441. [Google Scholar] [CrossRef]
- Ayala-Peña, S. Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic. Biol. Med. 2013, 62, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s disease and mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Gemba, H.; Kyuhou, S.; Matsuzaki, R.; Amino, Y. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s Disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef]
- Lu, B.; Al-Ramahi, I.; Valencia, A.; Wang, Q.; Berenshteyn, F.; Yang, H.; Gallego-Flores, T.; Ichcho, S.; Lacoste, A.; Hild, M.; et al. Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat. Neurosci. 2013, 16, 562–570. [Google Scholar] [CrossRef]
- Sassone, J.; Colciago, C.; Marchi, P.; Ascardi, C.; Alberti, L.; Pardo, A.D.; Zippel, R.; Sipione, S.; Silani, V.; Ciammola, A. Mutant Huntingtin induces activation of the Bcl-2/adenovirus E1B 19-kDa interacting protein (BNip3). Cell Death Dis. 2010, 1, e7. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Jin, S.; Zupnick, A.; Hoh, J.; de Stanchina, E.; Lowe, S.; Prives, C.; Levine, A. p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene 2006, 25, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.; Charrin, B.; Borrell-Pagès, M.; Dompierre, J.; Rangone, H.; Cordelières, F.; De Mey, J.; MacDonald, M.; Lessmann, V.; Lessmann, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Guedes-Dias, P.; de Proença, J.; Soares, T.; Leitão-Rocha, A.; Pinho, B.; Duchen, M.; Oliveira, J. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim. Biophys. Acta 2015, 1852, 2484–2493. [Google Scholar] [CrossRef]
- Jesse, S.; Bayer, H.; Alupei, M.C.; Zügel, M.; Mulaw, M.; Tuorto, F.; Malmsheimer, S.; Singh, K.; Steinacker, J.; Schumann, U.; et al. Ribosomal transcription is regulated by PGC-1alpha and disturbed in Huntington’s disease. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]











| Molecule | Function | Ref. |
|---|---|---|
| NADH:O2 oxidoreductase | Oxidoreductase activity | [246] |
| NADH dehydrogenase NDUFB8, succinate dehydrogenase SDHB and COX4I1 | Encoding a complex I accessory subunit to reducemitochondrial activity | [247] |
| Nitric oxide (NO) | Mitochondrial biogenesis at the transcriptional level through activation of cGMP and PGC1α | [161] |
| Hydrogen sulfide (H2S) | Preserves mitochondrial function via increased AKT phosphorylation, increased nuclear localization of NRF1/2, and increased mitochondrial biogenesis | [248] |
| Estrogen 17beta-estradiol | Transcription of nuclear respiratory factor-1 and increases mitocondrial biogenesis. | [249] |
| FOXO 1 | Mitochondrial function | [250] |
| PPARγ | Glucose metabolism and regulation of inflammatory response | [251] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration. Biomolecules 2021, 11, 1012. https://doi.org/10.3390/biom11071012
Godoy JA, Rios JA, Picón-Pagès P, Herrera-Fernández V, Swaby B, Crepin G, Vicente R, Fernández-Fernández JM, Muñoz FJ. Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration. Biomolecules. 2021; 11(7):1012. https://doi.org/10.3390/biom11071012
Chicago/Turabian StyleGodoy, Juan A., Juvenal A. Rios, Pol Picón-Pagès, Víctor Herrera-Fernández, Bronte Swaby, Giulia Crepin, Rubén Vicente, Jose M. Fernández-Fernández, and Francisco J. Muñoz. 2021. "Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration" Biomolecules 11, no. 7: 1012. https://doi.org/10.3390/biom11071012