
Harnessing the Physiological Functions of Cellular Prion Protein in the Kidneys: Applications for Treating Renal Diseases

Abstract
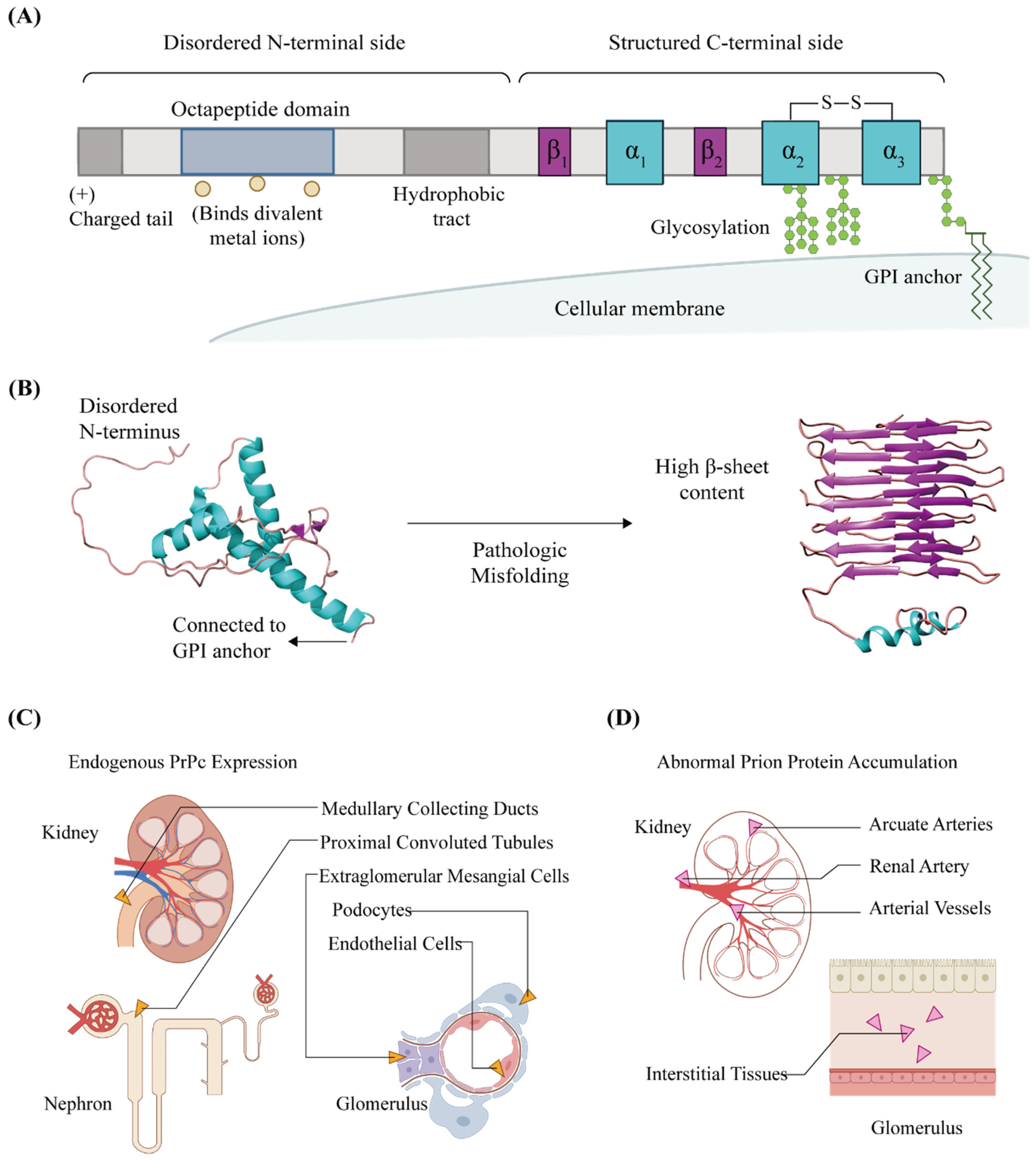
:1. General Characteristics of Cellular Prion Proteins in Kidneys
2. Physiological Functions of PrPC in the Kidneys
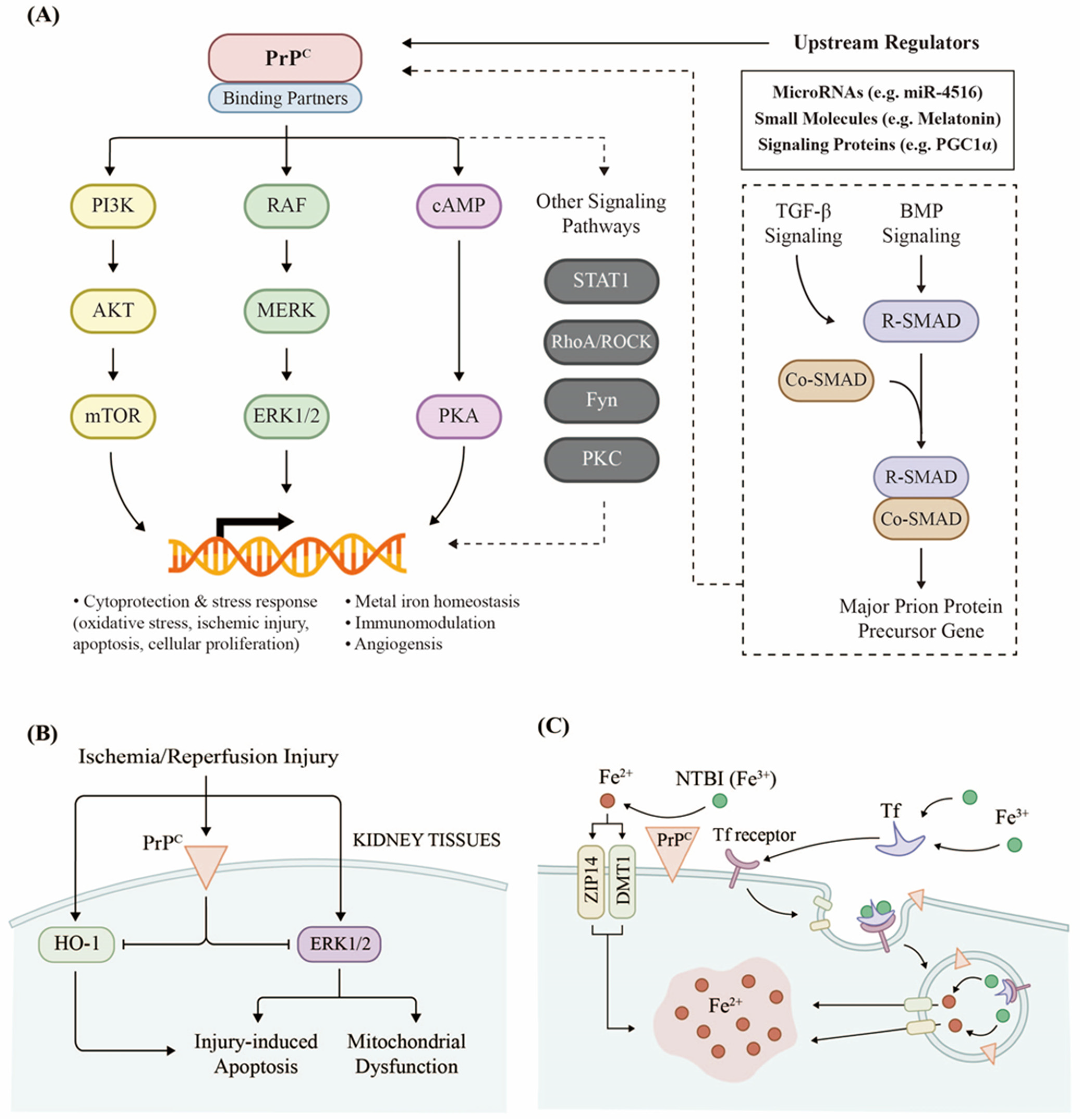
2.1. PrPC Regulates Renal Cellular Signaling
2.2. PrPC Responds to Kidney Injury
2.3. PrPC Promotes Iron Uptake in the Kidneys
3. PrPC and Kidney Disease
3.1. PrPC and AKI/CKD
3.2. PrPC and Renal Cancer
3.3. PrPC and Renal Fibrosis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Cell Biology of Prion Protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 57–82. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.N.; Giachin, G.; Wohlkonig, A.; Soror, S.H.; Pardon, E.; Legname, G.; Steyaert, J. Probing the N-terminal beta-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J. Am. Chem. Soc. 2014, 136, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 2012, 1, 15–31. [Google Scholar] [PubMed]
- Christen, B.; Damberger, F.F.; Perez, D.R.; Hornemann, S.; Wuthrich, K. Structural plasticity of the cellular prion protein and implications in health and disease. Proc. Natl. Acad. Sci. USA 2013, 110, 8549–8554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanusso, G.; Liu, D.; Ferrari, S.; Hegyi, I.; Yin, X.; Aguzzi, A.; Hornemann, S.; Liemann, S.; Glockshuber, R.; Manson, J.C.; et al. Prion protein expression in different species: Analysis with a panel of new mAbs. Proc. Natl. Acad. Sci. USA 1998, 95, 8812–8816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef]
- Spagnolli, G.; Rigoli, M.; Orioli, S.; Sevillano, A.M.; Faccioli, P.; Wille, H.; Biasini, E.; Requena, J.R. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019, 15, e1007864. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.J.; Millhauser, G.L. PrP overdrive: Does inhibition of alpha-cleavage contribute to PrP(C) toxicity and prion disease? Prion 2014, 8, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Robinson, S.M.; Diaz-Espinoza, R.; Urayama, A.; Soto, C. Transport of prion protein across the blood-brain barrier. Exp. Neurol 2009, 218, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, G.; Alejo Blanco, A.R.; Baybutt, H.; Horvat, S.; Manson, J.; Clinton, M. Embryonic activation and developmental expression of the murine prion protein gene. Gene Expr. 2003, 11, 1–12. [Google Scholar] [CrossRef]
- Tichopad, A.; Pfaffl, M.W.; Didier, A. Tissue-specific expression pattern of bovine prion gene: Quantification using real-time RT-PCR. Mol. Cell. Probes 2003, 17, 5–10. [Google Scholar] [CrossRef]
- Moudjou, M.; Frobert, Y.; Grassi, J.; La Bonnardiere, C. Cellular prion protein status in sheep: Tissue-specific biochemical signatures. J. Gen. Virol 2001, 82, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Amselgruber, W.M.; Steffl, M.; Didier, A.; Martlbauer, E.; Pfaff, E.; Buttner, M. Prion protein expression in bovine podocytes and extraglomerular mesangial cells. Cell Tissue Res. 2006, 324, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Peralta, O.A.; Eyestone, W.H. Quantitative and qualitative analysis of cellular prion protein (PrP(C)) expression in bovine somatic tissues. Prion 2009, 3, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Williams, W.M.; Stadtman, E.R.; Moskovitz, J. Ageing and exposure to oxidative stress in vivo differentially affect cellular levels of PrP in mouse cerebral microvessels and brain parenchyma. Neuropathol. Appl. Neurobiol. 2004, 30, 161–168. [Google Scholar] [CrossRef]
- Gasperini, L.; Legname, G. Prion protein and aging. Front. Cell Dev. Biol. 2014, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.A.; Lele, P.; Snider, W.D. Localization of the mRNA for a chicken prion protein by in situ hybridization. Proc. Natl. Acad. Sci. USA 1993, 90, 4309–4313. [Google Scholar] [CrossRef] [Green Version]
- Thumdee, P.; Ponsuksili, S.; Murani, E.; Nganvongpanit, K.; Gehrig, B.; Tesfaye, D.; Gilles, M.; Hoelker, M.; Jennen, D.; Griese, J.; et al. Expression of the prion protein gene (PRNP) and cellular prion protein (PrPc) in cattle and sheep fetuses and maternal tissues during pregnancy. Gene Expr. 2007, 13, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Parchi, P.; Castellani, R.; Cortelli, P.; Montagna, P.; Chen, S.G.; Petersen, R.B.; Manetto, V.; Vnencak-Jones, C.L.; McLean, M.J.; Sheller, J.R.; et al. Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann. Neurol 1995, 38, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10. [Google Scholar] [CrossRef]
- Notari, S.; Qing, L.; Pocchiari, M.; Dagdanova, A.; Hatcher, K.; Dogterom, A.; Groisman, J.F.; Lumholtz, I.B.; Puopolo, M.; Lasmezas, C.; et al. Assessing prion infectivity of human urine in sporadic Creutzfeldt-Jakob disease. Emerg. Infect. Dis. 2012, 18, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Brandel, J.P.; Knight, R. Variant Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Duque Velasquez, C.; Johnson, C.; Herbst, A.; Bolea, R.; Badiola, J.J.; Aiken, J.; McKenzie, D. Prion protein polymorphisms associated with reduced CWD susceptibility limit peripheral PrP(CWD) deposition in orally infected white-tailed deer. BMC Vet. Res. 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligios, C.; Cancedda, G.M.; Margalith, I.; Santucciu, C.; Madau, L.; Maestrale, C.; Basagni, M.; Saba, M.; Heikenwalder, M. Intraepithelial and interstitial deposition of pathological prion protein in kidneys of scrapie-affected sheep. PLoS ONE 2007, 2, e859. [Google Scholar] [CrossRef] [PubMed]
- Reichl, H.; Balen, A.; Jansen, C.A. Prion transmission in blood and urine: What are the implications for recombinant and urinary-derived gonadotrophins? Hum. Reprod 2002, 17, 2501–2508. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, J. Transmission of Creutzfeldt-Jakob disease from human blood and urine into mice. Lancet 1985, 2, 1074. [Google Scholar] [CrossRef]
- Gonzalez-Romero, D.; Barria, M.A.; Leon, P.; Morales, R.; Soto, C. Detection of infectious prions in urine. FEBS Lett. 2008, 582, 3161–3166. [Google Scholar] [CrossRef] [Green Version]
- Erana, H.; Charco, J.M.; Gonzalez-Miranda, E.; Garcia-Martinez, S.; Lopez-Moreno, R.; Perez-Castro, M.A.; Diaz-Dominguez, C.M.; Garcia-Salvador, A.; Castilla, J. Detection of Pathognomonic Biomarker PrP(Sc) and the Contribution of Cell Free-Amplification Techniques to the Diagnosis of Prion Diseases. Biomolecules 2020, 10, 469. [Google Scholar] [CrossRef] [Green Version]
- Takatsuki, H.; Fuse, T.; Nakagaki, T.; Mori, T.; Mihara, B.; Takao, M.; Iwasaki, Y.; Yoshida, M.; Murayama, S.; Atarashi, R.; et al. Prion-Seeding Activity Is widely Distributed in Tissues of Sporadic Creutzfeldt-Jakob Disease Patients. EBioMedicine 2016, 12, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Luk, C.; Jones, S.; Thomas, C.; Fox, N.C.; Mok, T.H.; Mead, S.; Collinge, J.; Jackson, G.S. Diagnosing Sporadic Creutzfeldt-Jakob Disease by the Detection of Abnormal Prion Protein in Patient Urine. JAMA Neurol. 2016, 73, 1454–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, M.; Mallucci, G.R. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology 2014, 76 Pt A, 169–174. [Google Scholar] [CrossRef]
- Go, G.; Lee, S.H. The Cellular Prion Protein: A Promising Therapeutic Target for Cancer. Int. J. Mol. Sci. 2020, 21, 9208. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Tumpach, C.; Drew, S.C.; Collins, S.J. The Prion Protein N1 and N2 Cleavage Fragments Bind to Phosphatidylserine and Phosphatidic Acid; Relevance to Stress-Protection Responses. PLoS ONE 2015, 10, e0134680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yoon, Y.M.; Han, Y.S.; Jung, S.K.; Lee, S.H. Melatonin protects mesenchymal stem cells from autophagy-mediated death under ischaemic ER-stress conditions by increasing prion protein expression. Cell Prolif. 2019, 52, e12545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachidi, W.; Vilette, D.; Guiraud, P.; Arlotto, M.; Riondel, J.; Laude, H.; Lehmann, S.; Favier, A. Expression of prion protein increases cellular copper binding and antioxidant enzyme activities but not copper delivery. J. Biol. Chem. 2003, 278, 9064–9072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti, F.; Carpi, A.; Menabo, R.; Giorgio, M.; Schulz, R.; Valen, G.; Baysa, A.; Massimino, M.L.; Sorgato, M.C.; Bertoli, A.; et al. The cellular prion protein counteracts cardiac oxidative stress. Cardiovasc. Res. 2014, 104, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, N.T.; Routledge, M.N.; Wild, C.P.; Hooper, N.M. Cellular prion protein protects against reactive-oxygen-species-induced DNA damage. Free Radic Biol. Med. 2007, 43, 959–967. [Google Scholar] [CrossRef]
- Miranda, A.; Pericuesta, E.; Ramirez, M.A.; Gutierrez-Adan, A. Prion protein expression regulates embryonic stem cell pluripotency and differentiation. PLoS ONE 2011, 6, e18422. [Google Scholar] [CrossRef]
- Terra-Granado, E.; Berbert, L.R.; de Meis, J.; Nomizo, R.; Martins, V.R.; Savino, W.; Silva-Barbosa, S.D. Is there a role for cellular prion protein in intrathymic T cell differentiation and migration? Neuroimmunomodulation 2007, 14, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Martins, V.R.; Jay, D.G.; Brentani, R.R. Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett. 2000, 482, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Fleisch, V.C.; Leighton, P.L.; Wang, H.; Pillay, L.M.; Ritzel, R.G.; Bhinder, G.; Roy, B.; Tierney, K.B.; Ali, D.W.; Waskiewicz, A.J.; et al. Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol. Dis. 2013, 55, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Ratte, S.; Vreugdenhil, M.; Boult, J.K.; Patel, A.; Asante, E.A.; Collinge, J.; Jefferys, J.G. Threshold for epileptiform activity is elevated in prion knockout mice. Neuroscience 2011, 179, 56–61. [Google Scholar] [CrossRef]
- Robinson, S.W.; Nugent, M.L.; Dinsdale, D.; Steinert, J.R. Prion protein facilitates synaptic vesicle release by enhancing release probability. Hum. Mol. Genet. 2014, 23, 4581–4596. [Google Scholar] [CrossRef] [Green Version]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.A.; Weis, J.; et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, 310–318. [Google Scholar] [CrossRef]
- Scalabrino, G.; Veber, D.; Tredici, G. Relationships between cobalamin, epidermal growth factor, and normal prions in the myelin maintenance of central nervous system. Int. J. Biochem. Cell Biol. 2014, 55, 232–241. [Google Scholar] [CrossRef]
- Radovanovic, I.; Braun, N.; Giger, O.T.; Mertz, K.; Miele, G.; Prinz, M.; Navarro, B.; Aguzzi, A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J. Neurosci. 2005, 25, 4879–4888. [Google Scholar] [CrossRef] [Green Version]
- Cagampang, F.R.; Whatley, S.A.; Mitchell, A.L.; Powell, J.F.; Campbell, I.C.; Coen, C.W. Circadian regulation of prion protein messenger RNA in the rat forebrain: A widespread and synchronous rhythm. Neuroscience 1999, 91, 1201–1204. [Google Scholar] [CrossRef]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rulicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef]
- Singh, A.; Mohan, M.L.; Isaac, A.O.; Luo, X.; Petrak, J.; Vyoral, D.; Singh, N. Prion protein modulates cellular iron uptake: A novel function with implications for prion disease pathogenesis. PLoS ONE 2009, 4, e4468. [Google Scholar] [CrossRef]
- Singh, A.; Kong, Q.; Luo, X.; Petersen, R.B.; Meyerson, H.; Singh, N. Prion protein (PrP) knock-out mice show altered iron metabolism: A functional role for PrP in iron uptake and transport. PLoS ONE 2009, 4, e6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, N.T.; Hooper, N.M. The prion protein and neuronal zinc homeostasis. Trends Biochem. Sci. 2003, 28, 406–410. [Google Scholar] [CrossRef]
- Isaacs, J.D.; Jackson, G.S.; Altmann, D.M. The role of the cellular prion protein in the immune system. Clin. Exp. Immunol. 2006, 146, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.H.; Whitehouse, I.J.; Hooper, N.M. Regulation of amyloid-beta production by the prion protein. Prion 2012, 6, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef] [Green Version]
- Didonna, A. Prion protein and its role in signal transduction. Cell Mol. Biol. Lett. 2013, 18, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; van der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Investig. 2014, 124, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, C.; Takeuchi, A.M.; Nishimura, T.; Haraguchi, K.; Kubosaki, A.; Matsumoto, Y.; Saeki, K.; Matsumoto, Y.; Yokoyama, T.; Itohara, S.; et al. Prions prevent neuronal cell-line death. Nature 1999, 400, 225–226. [Google Scholar] [CrossRef]
- Diarra-Mehrpour, M.; Arrabal, S.; Jalil, A.; Pinson, X.; Gaudin, C.; Pietu, G.; Pitaval, A.; Ripoche, H.; Eloit, M.; Dormont, D.; et al. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004, 64, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Bounhar, Y.; Zhang, Y.; Goodyer, C.G.; LeBlanc, A. Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 2001, 276, 39145–39149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.H.; Lee, H.G.; Choi, J.K.; Kim, J.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Brain Res. Mol. Brain Res. 2004, 124, 40–50. [Google Scholar] [CrossRef]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Shyu, W.C.; Lin, S.Z.; Chiang, M.F.; Ding, D.C.; Li, K.W.; Chen, S.F.; Yang, H.I.; Li, H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 2005, 25, 8967–8977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Bahr, M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006, 37, 1296–1300. [Google Scholar] [CrossRef] [Green Version]
- Haldar, S.; Tripathi, A.; Qian, J.; Beserra, A.; Suda, S.; McElwee, M.; Turner, J.; Hopfer, U.; Singh, N. Prion protein promotes kidney iron uptake via its ferrireductase activity. J. Biol. Chem. 2015, 290, 5512–5522. [Google Scholar] [CrossRef] [Green Version]
- Bignon, Y.; Poindessous, V.; Lazareth, H.; Passet, B.; Vilotte, J.L.; Djouadi, F.; Mouillet-Richard, S.; Pallet, N. The cellular prion protein is a stress protein secreted by renal tubular cells and a urinary marker of kidney injury. Cell Death Dis. 2020, 11, 243. [Google Scholar] [CrossRef]
- Yoon, Y.M.; Lee, J.H.; Song, K.H.; Noh, H.; Lee, S.H. Melatonin-stimulated exosomes enhance the regenerative potential of chronic kidney disease-derived mesenchymal stem/stromal cells via cellular prion proteins. J. Pineal. Res. 2020, 68, e12632. [Google Scholar] [CrossRef]
- Kobayashi, N.; Gao, S.Y.; Chen, J.; Saito, K.; Miyawaki, K.; Li, C.Y.; Pan, L.; Saito, S.; Terashita, T.; Matsuda, S. Process formation of the renal glomerular podocyte: Is there common molecular machinery for processes of podocytes and neurons? Anat. Sci. Int. 2004, 79, 1–10. [Google Scholar] [CrossRef]
- Rastaldi, M.P.; Armelloni, S.; Berra, S.; Calvaresi, N.; Corbelli, A.; Giardino, L.A.; Li, M.; Wang, G.Q.; Fornasieri, A.; Villa, A.; et al. Glomerular podocytes contain neuron-like functional synaptic vesicles. FASEB J. 2006, 20, 976–978. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Hu, R.; Sun, J.; Mao, X.; Zhao, Z.; Chen, Q.; Zhang, Z. The expression and significance of neuronal iconic proteins in podocytes. PLoS ONE 2014, 9, e93999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, F.A.; Lopes, M.H.; Hajj, G.N.; Machado, C.F.; Pinto Arantes, C.; Magalhaes, A.C.; Vieira Mde, P.; Americo, T.A.; Massensini, A.R.; Priola, S.A.; et al. Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 2008, 28, 6691–6702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Machado, C.F.; Roffe, M.; Hajj, G.N.; Lee, K.S.; Magalhaes, A.C.; Caetano, F.A.; Mancini, G.L.; et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011, 25, 265–279. [Google Scholar] [CrossRef]
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H.A. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef] [Green Version]
- Roffe, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Kuffer, A.; Lakkaraju, A.K.; Mogha, A.; Petersen, S.C.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef]
- Krebs, B.; Wiebelitz, A.; Balitzki-Korte, B.; Vassallo, N.; Paluch, S.; Mitteregger, G.; Onodera, T.; Kretzschmar, H.A.; Herms, J. Cellular prion protein modulates the intracellular calcium response to hydrogen peroxide. J. Neurochem. 2007, 100, 358–367. [Google Scholar] [CrossRef]
- Chen, R.J.; Chang, W.W.; Lin, Y.C.; Cheng, P.L.; Chen, Y.R. Alzheimer’s amyloid-beta oligomers rescue cellular prion protein induced tau reduction via the Fyn pathway. ACS Chem. Neurosci. 2013, 4, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Loubet, D.; Dakowski, C.; Pietri, M.; Pradines, E.; Bernard, S.; Callebert, J.; Ardila-Osorio, H.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O.; et al. Neuritogenesis: The prion protein controls beta1 integrin signaling activity. FASEB J. 2012, 26, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Pietri, M.; Pradines, E.; Loubet, D.; Launay, J.M.; Kellermann, O.; Mouillet-Richard, S. Understanding the neurospecificity of Prion protein signaling. Front. Biosci. (Landmark Ed.) 2011, 16, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 2008, 86, 1659–1669. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Cui, H.; Zhang, C.; Zhu, C.; Li, L. Apelin-13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci. Lett 2014, 568, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Zhou, Z.; Jackson, W.S.; Zhu, C.; Auluck, P.; Moskowitz, M.A.; Chesselet, M.F.; Lindquist, S. Context dependent neuroprotective properties of prion protein (PrP). Prion 2009, 3, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Cowden, D.; Zhang, F.; Yuan, J.; Siedlak, S.; Abouelsaad, M.; Zeng, L.; Zhou, X.; O’Toole, J.; Das, A.S.; et al. Prion Protein Protects against Renal Ischemia/Reperfusion Injury. PLoS ONE 2015, 10, e0136923. [Google Scholar] [CrossRef]
- Spudich, A.; Frigg, R.; Kilic, E.; Kilic, U.; Oesch, B.; Raeber, A.; Bassetti, C.L.; Hermann, D.M. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 2005, 20, 442–449. [Google Scholar] [CrossRef]
- Weise, J.; Doeppner, T.R.; Muller, T.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Witte, O.W.; Bahr, M. Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor. Neurol. Neurosci. 2008, 26, 57–64. [Google Scholar]
- Palmer, B.F. The renal tubule in the progression of chronic renal failure. J. Investig. Med. 1997, 45, 346–361. [Google Scholar]
- Thurman, J.M. Triggers of inflammation after renal ischemia/reperfusion. Clin. Immunol. 2007, 123, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Dudley, A.T.; Lyons, K.M.; Robertson, E.J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995, 9, 2795–2807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; Maresh, J.G.; Harris, S.E.; Hernandez, J.D.; Arar, M.; Olson, M.S.; Abboud, H.E. Expression of bone morphogenetic protein-7 mRNA in normal and ischemic adult rat kidney. Am. J. Physiol. 1999, 276, F382–F389. [Google Scholar] [CrossRef] [PubMed]
- Bosukonda, D.; Shih, M.S.; Sampath, K.T.; Vukicevic, S. Characterization of receptors for osteogenic protein-1/bone morphogenetic protein-7 (OP-1/BMP-7) in rat kidneys. Kidney Int. 2000, 58, 1902–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almanzar, M.M.; Frazier, K.S.; Dube, P.H.; Piqueras, A.I.; Jones, W.K.; Charette, M.F.; Paredes, A.L. Osteogenic protein-1 mRNA expression is selectively modulated after acute ischemic renal injury. J. Am. Soc. Nephrol. 1998, 9, 1456–1463. [Google Scholar] [CrossRef]
- Vukicevic, S.; Basic, V.; Rogic, D.; Basic, N.; Shih, M.S.; Shepard, A.; Jin, D.; Dattatreyamurty, B.; Jones, W.; Dorai, H.; et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J. Clin. Investig. 1998, 102, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Hruska, K.A.; Guo, G.; Wozniak, M.; Martin, D.; Miller, S.; Liapis, H.; Loveday, K.; Klahr, S.; Sampath, T.K.; Morrissey, J. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am. J. Physiol. Renal Physiol. 2000, 279, F130–F143. [Google Scholar] [CrossRef]
- Gould, S.E.; Day, M.; Jones, S.S.; Dorai, H. BMP-7 regulates chemokine, cytokine, and hemodynamic gene expression in proximal tubule cells. Kidney Int. 2002, 61, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.M.; Lee, J.H.; Yun, C.W.; Lee, S.H. Pioglitazone Improves the Function of Human Mesenchymal Stem Cells in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2019, 20, 2314. [Google Scholar] [CrossRef] [Green Version]
- Pease, D.; Scheckel, C.; Schaper, E.; Eckhardt, V.; Emmenegger, M.; Xenarios, I.; Aguzzi, A. Genome-wide identification of microRNAs regulating the human prion protein. Brain Pathol. 2019, 29, 232–244. [Google Scholar] [CrossRef] [Green Version]
- Neal, C.S.; Michael, M.Z.; Pimlott, L.K.; Yong, T.Y.; Li, J.Y.; Gleadle, J.M. Circulating microRNA expression is reduced in chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 3794–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnicki, M.; Perco, P.; D’haene, B.; Leierer, J.; Heinzel, A.; Muhlberger, I.; Schweibert, N.; Sunzenauer, J.; Regele, H.; Kronbichler, A.; et al. Renal microRNA- and RNA-profiles in progressive chronic kidney disease. Eur. J. Clin. Investig. 2016, 46, 213–226. [Google Scholar] [CrossRef]
- Assmann, T.S.; Recamonde-Mendoza, M.; de Souza, B.M.; Bauer, A.C.; Crispim, D. MicroRNAs and diabetic kidney disease: Systematic review and bioinformatic analysis. Mol. Cell. Endocrinol. 2018, 477, 90–102. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Shu, S.; Cai, J.; Tang, C.; Dong, Z. Non-coding RNAs in kidney injury and repair. Am. J. Physiol. Cell Physiol. 2019, 317, C177–C188. [Google Scholar] [CrossRef]
- Mitsios, N.; Saka, M.; Krupinski, J.; Pennucci, R.; Sanfeliu, C.; Miguel Turu, M.; Gaffney, J.; Kumar, P.; Kumar, S.; Sullivan, M.; et al. Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J. Neurosci. Res. 2007, 85, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Senator, A.; Rachidi, W.; Lehmann, S.; Favier, A.; Benboubetra, M. Prion protein protects against DNA damage induced by paraquat in cultured cells. Free Radic Biol. Med. 2004, 37, 1224–1230. [Google Scholar] [CrossRef]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.P.; Lee, W.K.; Haley, M.; Poulsen, S.B.; Thevenod, F.; Fenton, R.A. Proximal tubule transferrin uptake is modulated by cellular iron and mediated by apical membrane megalin-cubilin complex and transferrin receptor 1. J. Biol. Chem. 2019, 294, 7025–7036. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.K.; Haldar, S.; Qian, J.; Beserra, A.; Suda, S.; Singh, A.; Hopfer, U.; Chen, S.G.; Garrick, M.D.; Turner, J.R.; et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic Biol. Med. 2015, 84, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Asthana, A.; Baksi, S.; Desai, V.; Haldar, S.; Hari, S.; Tripathi, A.K. The prion-ZIP connection: From cousins to partners in iron uptake. Prion 2015, 9, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevenod, F.; Wolff, N.A. Iron transport in the kidney: Implications for physiology and cadmium nephrotoxicity. Metallomics 2016, 8, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Van Raaij, S.E.G.; Rennings, A.J.; Biemond, B.J.; Schols, S.E.M.; Wiegerinck, E.T.G.; Roelofs, H.M.J.; Hoorn, E.J.; Walsh, S.B.; Nijenhuis, T.; Swinkels, D.W.; et al. Iron handling by the human kidney: Glomerular filtration and tubular reabsorption both contribute to urinary iron excretion. Am. J. Physiol. Renal Physiol. 2019, 316, F606–F614. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Cellular iron metabolism. Kidney Int. 1999, 55 (Suppl. 69), S2–S11. [Google Scholar] [CrossRef] [Green Version]
- Martines, A.M.; Masereeuw, R.; Tjalsma, H.; Hoenderop, J.G.; Wetzels, J.F.; Swinkels, D.W. Iron metabolism in the pathogenesis of iron-induced kidney injury. Nat. Rev. Nephrol. 2013, 9, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Bahrainwala, J.; Berns, J.S. Diagnosis of Iron-Deficiency Anemia in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S. Iron Homeostasis Pathways as Therapeutic Targets in Acute Kidney Injury. Nephron 2018, 140, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Van Swelm, R.P.L.; Wetzels, J.F.M.; Swinkels, D.W. The multifaceted role of iron in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 77–98. [Google Scholar] [CrossRef]
- Liu, H.; Liu, D.; Liu, Y.; Xia, M.; Li, Y.; Li, M.; Liu, H. Comprehensive analysis of circRNA expression profiles and circRNA-associated competing endogenous RNA networks in IgA nephropathy. PeerJ 2020, 8, e10395. [Google Scholar] [CrossRef]
- Doshi, S.M.; Friedman, A.N. Diagnosis and Management of Type 2 Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1366–1373. [Google Scholar] [CrossRef]
- Chatauret, N.; Badet, L.; Barrou, B.; Hauet, T. Ischemia-reperfusion: From cell biology to acute kidney injury. Prog Urol 2014, 24 (Suppl. 1), S4–S12. [Google Scholar] [CrossRef]
- Bindroo, S.; Quintanilla Rodriguez, B.S.; Challa, H.J. Renal Failure; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Winocour, P.H. Diabetes and chronic kidney disease: An increasingly common multi-morbid disease in need of a paradigm shift in care. Diabet. Med. 2018, 35, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cooper, M.E.; Zimmet, P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.H.; Say, Y.H. Resistance against tumour necrosis factor alpha apoptosis by the cellular prion protein is cell-specific for oral, colon and kidney cancer cell lines. Cell Biol. Int. 2012, 36, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Kusuma, G.D.; Carthew, J.; Lim, R.; Frith, J.E. Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem Cells Dev. 2017, 26, 617–631. [Google Scholar] [CrossRef]
- Mashima, T.; Lee, J.H.; Kamatari, Y.O.; Hayashi, T.; Nagata, T.; Nishikawa, F.; Nishikawa, S.; Kinoshita, M.; Kuwata, K.; Katahira, M. Development and structural determination of an anti-PrP(C) aptamer that blocks pathological conformational conversion of prion protein. Sci. Rep. 2020, 10, 4934. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Black, R.A. Tumor necrosis factor-alpha converting enzyme. Int. J. Biochem. Cell Biol. 2002, 34, 1–5. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Xu, Y.; Zhang, Y.; Lee, M.H. Translocating a High-Affinity Designer TIMP-1 to the Cell Membrane for Total Renal Carcinoma Inhibition: Putting the Prion Protein to Good Use. Mol. Cell. Biol. 2019, 39, e00128-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Yu, S.M.; Bonventre, J.V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Kishi, S.; Brooks, C.R.; Taguchi, K.; Ichimura, T.; Mori, Y.; Akinfolarin, A.; Gupta, N.; Galichon, P.; Elias, B.C.; Suzuki, T.; et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Investig. 2019, 129, 4797–4816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Mitu, G.; Hirschberg, R. Bone morphogenetic protein-7 (BMP7) in chronic kidney disease. Front. Biosci. 2008, 13, 4726–4739. [Google Scholar] [CrossRef] [PubMed]
- Li, R.X.; Yiu, W.H.; Tang, S.C. Role of bone morphogenetic protein-7 in renal fibrosis. Front. Physiol. 2015, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.M.; Go, G.; Yun, C.W.; Lim, J.H.; Lee, J.H.; Lee, S.H. Melatonin Suppresses Renal Cortical Fibrosis by Inhibiting Cytoskeleton Reorganization and Mitochondrial Dysfunction through Regulation of miR-4516. Int. J. Mol. Sci. 2020, 21, 5323. [Google Scholar] [CrossRef]
- Chen, D.Q.; Cao, G.; Zhao, H.; Chen, L.; Yang, T.; Wang, M.; Vaziri, N.D.; Guo, Y.; Zhao, Y.Y. Combined melatonin and poricoic acid A inhibits renal fibrosis through modulating the interaction of Smad3 and beta-catenin pathway in AKI-to-CKD continuum. Ther. Adv. Chronic Dis. 2019, 10, 2040622319869116. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Qi, X.; Yang, W.; Xia, L.; Wu, Y. Melatonin Ameliorates Renal Fibrosis Through the Inhibition of NF-kappaB and TGF-beta1/Smad3 Pathways in db/db Diabetic Mice. Arch. Med. Res. 2020, 51, 524–534. [Google Scholar] [CrossRef]
- Watts, J.C.; Bourkas, M.E.C.; Arshad, H. The function of the cellular prion protein in health and disease. Acta Neuropathol. 2018, 135, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Acute kidney injury and chronic kidney disease: From the laboratory to the clinic. Nephrol. Ther. 2016, 12 (Suppl. 1), S41–S48. [Google Scholar] [CrossRef] [Green Version]
- Drawz, P.; Rahman, M. Chronic kidney disease. Ann. Intern. Med. 2015, 162, ITC1-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, H.; Strauer, B.E. [Renal failure]. Internist (Berl.) 2012, 53, 789–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuelo, J.G. Renal failure caused by chemicals, foods, plants, animal venoms, and misuse of drugs. An overview. Arch. Intern. Med. 1990, 150, 505–510. [Google Scholar] [CrossRef]
- Kemmner, S.; Verbeek, M.; Heemann, U. Renal dysfunction following bone marrow transplantation. J. Nephrol. 2017, 30, 201–209. [Google Scholar] [CrossRef]
- Shen, Y.; Cai, R.; Sun, J.; Dong, X.; Huang, R.; Tian, S.; Wang, S. Diabetes mellitus as a risk factor for incident chronic kidney disease and end-stage renal disease in women compared with men: A systematic review and meta-analysis. Endocrine 2017, 55, 66–76. [Google Scholar] [CrossRef]
- Scolari, F.; Ravani, P. Atheroembolic renal disease. Lancet 2010, 375, 1650–1660. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Negi, S.; Koreeda, D.; Kobayashi, S.; Yano, T.; Tatsuta, K.; Mima, T.; Shigematsu, T.; Ohya, M. Acute kidney injury: Epidemiology, outcomes, complications, and therapeutic strategies. Semin. Dial. 2018, 31, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Levin, A.; for the Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: Synopsis of the kidney disease: Improving global outcomes 2012 clinical practice guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Pannu, N. Bidirectional relationships between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2013, 22, 351–356. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Renal Inj. Prev. 2015, 4, 20–27. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Liu, J.; Kumar, S.; Dolzhenko, E.; Alvarado, G.F.; Guo, J.; Lu, C.; Chen, Y.; Li, M.; Dessing, M.C.; Parvez, R.K.; et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017, 2, e94716. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.L.; Zhang, Y.; Li, X.; Fu, Q.L. Mechanisms underlying the protective effects of mesenchymal stem cell-based therapy. Cell Mol. Life Sci. 2020, 77, 2771–2794. [Google Scholar] [CrossRef] [Green Version]
- Morigi, M.; Rota, C.; Remuzzi, G. Mesenchymal Stem Cells in Kidney Repair. Methods Mol. Biol. 2016, 1416, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Papazova, D.A.; Oosterhuis, N.R.; Gremmels, H.; van Koppen, A.; Joles, J.A.; Verhaar, M.C. Cell-based therapies for experimental chronic kidney disease: A systematic review and meta-analysis. Dis. Model. Mech. 2015, 8, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, S.; Gonzalez, F.; Lorca, E.; Tapia, A.; Lopez, V.G.; Strodthoff, R.; Fajre, F.; Carreno, J.E.; Valjalo, R.; Vergara, C.; et al. Adipose tissue-derived mesenchymal stromal cells for treating chronic kidney disease: A pilot study assessing safety and clinical feasibility. Kidney Res. Clin. Pract 2019, 38, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.H. Use of mesenchymal stem cells for chronic kidney disease. Kidney Res. Clin. Pract. 2019, 38, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, S.H. Potential and Therapeutic Efficacy of Cell-Based Therapy Using Mesenchymal Stem Cells for Acute/Chronic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1619. [Google Scholar] [CrossRef] [Green Version]
- Urt-Filho, A.; Oliveira, R.J.; Hermeto, L.C.; Pesarini, J.R.; David, N.; Cantero Wde, B.; Falcao, G.; Marks, G.; Antoniolli-Silva, A.C. Mesenchymal stem cell therapy promotes the improvement and recovery of renal function in a preclinical model. Genet. Mol. Biol. 2016, 39, 290–299. [Google Scholar] [CrossRef]
- Huang, Y.C.; Leung, V.Y.; Lu, W.W.; Luk, K.D. The effects of microenvironment in mesenchymal stem cell-based regeneration of intervertebral disc. Spine J. 2013, 13, 352–362. [Google Scholar] [CrossRef]
- Teng, X.; Chen, L.; Chen, W.; Yang, J.; Yang, Z.; Shen, Z. Mesenchymal Stem Cell-Derived Exosomes Improve the Microenvironment of Infarcted Myocardium Contributing to Angiogenesis and Anti-Inflammation. Cell Physiol. Biochem. 2015, 37, 2415–2424. [Google Scholar] [CrossRef]
- Han, Y.S.; Lee, J.H.; Yoon, Y.M.; Yun, C.W.; Noh, H.; Lee, S.H. Hypoxia-induced expression of cellular prion protein improves the therapeutic potential of mesenchymal stem cells. Cell Death Dis. 2016, 7, e2395. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. TUDCA-Treated Mesenchymal Stem Cells Protect against ER Stress in the Hippocampus of a Murine Chronic Kidney Disease Model. Int. J. Mol. Sci. 2019, 20, 613. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.P.; Yoon, Y.M.; Lee, J.H.; Kook, M.; Han, Y.S.; Jung, S.K.; Lee, S.H. Tauroursodeoxycholic Acid Protects against the Effects of P-Cresol-Induced Reactive Oxygen Species via the Expression of Cellular Prion Protein. Int. J. Mol. Sci. 2018, 19, 352. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; Ordonez, R.; Reiter, R.J.; Gonzalez-Gallego, J.; Mauriz, J.L. Melatonin and endoplasmic reticulum stress: Relation to autophagy and apoptosis. J. Pineal. Res. 2015, 59, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Mias, C.; Trouche, E.; Seguelas, M.H.; Calcagno, F.; Dignat-George, F.; Sabatier, F.; Piercecchi-Marti, M.D.; Daniel, L.; Bianchi, P.; Calise, D.; et al. Ex vivo pretreatment with melatonin improves survival, proangiogenic/mitogenic activity, and efficiency of mesenchymal stem cells injected into ischemic kidney. Stem Cells 2008, 26, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Kim, S.M.; Lee, J.H.; Jung, S.K.; Noh, H.; Lee, S.H. Melatonin protects chronic kidney disease mesenchymal stem cells against senescence via PrPC-dependent enhancement of the mitochondrial function. J. Pineal Res. 2019, 66, e12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Porta, S.; Bussolati, B. Extracellular vesicles in renal tissue damage and regeneration. Eur. J. Pharmacol. 2016, 790, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, F.T.; Reis, L.A.; Schor, N. Extracellular vesicles: Structure, function, and potential clinical uses in renal diseases. Braz. J. Med. Biol. Res. 2013, 46, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhou, X.; Zhang, H.; Yao, Q.; Liu, Y.; Dong, Z. Extracellular vesicles in diagnosis and therapy of kidney diseases. Am. J. Physiol. Renal Physiol. 2016, 311, F844–F851. [Google Scholar] [CrossRef] [Green Version]
- Jing, H.; Tang, S.; Lin, S.; Liao, M.; Chen, H.; Zhou, J. The role of extracellular vesicles in renal fibrosis. Cell Death Dis. 2019, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.L.; Wu, W.J.; Feng, Y.; Li, Z.L.; Tang, T.T.; Liu, B.C. Therapeutic application of extracellular vesicles in kidney disease: Promises and challenges. J. Cell Mol. Med. 2018, 22, 728–737. [Google Scholar] [CrossRef]
- Shen, B.; Liu, J.; Zhang, F.; Wang, Y.; Qin, Y.; Zhou, Z.; Qiu, J.; Fan, Y. CCR2 Positive Exosome Released by Mesenchymal Stem Cells Suppresses Macrophage Functions and Alleviates Ischemia/Reperfusion-Induced Renal Injury. Stem Cells Int. 2016, 2016, 1240301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Wu, J.; Wang, J.; Li, Y.; Hu, X.; Luo, S.; Xiang, D. Extracellular vesicles derived from different sources of mesenchymal stem cells: Therapeutic effects and translational potential. Cell Biosci. 2020, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Cantaluppi, V.; Gatti, S.; Medica, D.; Figliolini, F.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012, 82, 412–427. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.L.; Feng, Y.; Wu, M.; Wang, B.; Li, Z.L.; Zhong, X.; Wu, W.J.; Chen, J.; Ni, H.F.; Tang, T.T.; et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. 2020, 27, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Grange, C.; Collino, F.; Deregibus, M.C.; Cantaluppi, V.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS ONE 2012, 7, e33115. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Zhu, X.Y.; Puranik, A.S.; Tang, H.; McGurren, K.A.; van Wijnen, A.J.; Lerman, A.; Lerman, L.O. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. 2017, 92, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Bruno, S.; Incarnato, D.; Dettori, D.; Neri, F.; Provero, P.; Pomatto, M.; Oliviero, S.; Tetta, C.; Quesenberry, P.J.; et al. AKI Recovery Induced by Mesenchymal Stromal Cell-Derived Extracellular Vesicles Carrying MicroRNAs. J. Am. Soc. Nephrol. 2015, 26, 2349–2360. [Google Scholar] [CrossRef]
- Vinas, J.L.; Burger, D.; Zimpelmann, J.; Haneef, R.; Knoll, W.; Campbell, P.; Gutsol, A.; Carter, A.; Allan, D.S.; Burns, K.D. Transfer of microRNA-486-5p from human endothelial colony forming cell-derived exosomes reduces ischemic kidney injury. Kidney Int. 2016, 90, 1238–1250. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.M.; Han, Y.S.; Yun, C.W.; Lee, J.H.; Kim, R.; Lee, S.H. Pioglitazone Protects Mesenchymal Stem Cells against P-Cresol-Induced Mitochondrial Dysfunction via Up-Regulation of PINK-1. Int. J. Mol. Sci. 2018, 19, 2898. [Google Scholar] [CrossRef] [Green Version]
- Starke, R.; Mackie, I.; Drummond, O.; MacGregor, I.; Harrison, P.; Machin, S. Prion protein in patients with renal failure. Transfus. Med. 2006, 16, 165–168. [Google Scholar] [CrossRef]
- Van der Vorm, L.N.; Visser, R.; Huskens, D.; Veninga, A.; Adams, D.L.; Remijn, J.A.; Hemker, H.C.; Rensma, P.L.; van Horssen, R.; de Laat, B. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-stage renal disease. Clin. Kidney J. 2020, 13, 72–74. [Google Scholar] [CrossRef] [PubMed]
- McEwan, J.F.; Windsor, M.L.; Cullis-Hill, S.D. Antibodies to prion protein inhibit human colon cancer cell growth. Tumour Biol. 2009, 30, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, H.; Sidana, S. Antibody-Based Treatment Approaches in Multiple Myeloma. Curr. Hematol. Malig Rep. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bodyak, N.D.; Mosher, R.; Yurkovetskiy, A.V.; Yin, M.; Bu, C.; Conlon, P.R.; Demady, D.R.; DeVit, M.J.; Gumerov, D.R.; Gurijala, V.R.; et al. The Dolaflexin-based antibody-drug conjugate XMT-1536 targets the solid tumor lineage antigen SLC34A2/NaPi2b. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef]
- Sutherland, M.S.; Sanderson, R.J.; Gordon, K.A.; Andreyka, J.; Cerveny, C.G.; Yu, C.; Lewis, T.S.; Meyer, D.L.; Zabinski, R.F.; Doronina, S.O.; et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J. Biol. Chem. 2006, 281, 10540–10547. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Xuan, W.; Peng, Y.; Deng, Z.; Peng, T.; Kuai, H.; Li, Y.; He, J.; Jin, C.; Liu, Y.; Wang, R.; et al. A basic insight into aptamer-drug conjugates (ApDCs). Biomaterials 2018, 182, 216–226. [Google Scholar] [CrossRef]
- Zhu, G.; Niu, G.; Chen, X. Aptamer-Drug Conjugates. Bioconjug. Chem. 2015, 26, 2186–2197. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Liu, B.; Yu, B.; Zhong, W.; Lu, Y.; Zhang, J.; Liao, J.; Liu, J.; Pu, Y.; Qiu, L.; et al. Advances in the development of aptamer drug conjugates for targeted drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1438. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, Z.; Gao, F.; Lei, Y.; Li, Z. Melatonin ameliorates renal fibroblast-myofibroblast transdifferentiation and renal fibrosis through miR-21-5p regulation. J. Cell Mol. Med. 2020, 24, 5615–5628. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, N.; Yan, S.; Lu, Y.; Miao, X.; Gu, Z.; Shao, Y. Melatonin attenuates renal fibrosis in diabetic mice by activating the AMPK/PGC1alpha signaling pathway and rescuing mitochondrial function. Mol. Med. Rep. 2019, 19, 1318–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramljak, S.; Herlyn, H.; Zerr, I. Cellular Prion Protein (PrPc) and Hypoxia: True to Each Other in Good Times and in Bad, in Sickness, and in Health. Front. Cell. Neurosci. 2016, 10, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Kidney Disease | Potential PrPc-Based Therapeutic Strategy | Proposed Mechanism for the Roles/Effects of PrPc | Validated in Renal Cells | Related References |
|---|---|---|---|---|
| AKI/CKD | Expressing PrPc in renal tissues to ameliorate IRI renal damage via gene delivery with the adenovirus vector | PrPc suppresses ERK1/2-dependent tissue damage via apoptosis and mitochondrial regulation | Yes | [35,120,121] |
| Suppressing PrPc in the kidney to treat the dysregulation of iron homeostasis | PrPc ferrireductase activity promotes DMT1 and ZIP14-mediated iron uptake in kidney tissues | No | [23,119,122] | |
| Upregulating PrPc in MSC with melatonin, TUDCA (bile acid), or pioglitazone treatment to improve MSC or EV-based cell therapies | PrPc protects MSCs against the adverse microenvironment in damaged kidneys, enhancing the efficacy of the treatment | Yes | [24,123,124,125,126] | |
| Renal Fibrosis | Targeting PrPc downstream of BMP-7 to stop the progression of TGF-β-induced renal fibrosis | PrPc precursor gene is regulated by BMP-7, which disappears in fibrosis. Interventions at the PrPc level could help ameliorate the fibrotic effects | No | [127] |
| Renal Carcinoma | Cotreatment of the PrPc antibody and chemotherapeutic agents | Reduction of PrPc-related drug resistance and metastasis leads to a better outcome | No | [38,128,129] |
| Using a PrPc aptamer-conjugated drug delivery system or PrPc antibody–drug conjugates for cancer treatment | Targeting differentially expressed PrPc in cancer cells achieves targeted drug delivery | No | [38,130,131,132,133,134,135,136,137,138] | |
| Fusion of TIMPs with the GPI anchor domain for cell surface expression | Increased colocalization of PrPc-fused TIMP with its inhibitory target (MMP) increases the anticancer effect | No | [139,140,141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.; Go, G.; Yoon, Y.; Lim, J.; Lee, G.; Lee, S. Harnessing the Physiological Functions of Cellular Prion Protein in the Kidneys: Applications for Treating Renal Diseases. Biomolecules 2021, 11, 784. https://doi.org/10.3390/biom11060784
Yoon S, Go G, Yoon Y, Lim J, Lee G, Lee S. Harnessing the Physiological Functions of Cellular Prion Protein in the Kidneys: Applications for Treating Renal Diseases. Biomolecules. 2021; 11(6):784. https://doi.org/10.3390/biom11060784
Chicago/Turabian StyleYoon, Sungtae, Gyeongyun Go, Yeomin Yoon, Jiho Lim, Gaeun Lee, and Sanghun Lee. 2021. "Harnessing the Physiological Functions of Cellular Prion Protein in the Kidneys: Applications for Treating Renal Diseases" Biomolecules 11, no. 6: 784. https://doi.org/10.3390/biom11060784