
Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development

 , , ,
, , ,  and
and
Abstract
:1. Introduction
1.1. Retroviral Vectors
1.2. Lentiviral Vectors
1.3. Adenoviral Vectors
1.4. Adeno-Associated Viral Vectors
2. In Vitro Haematopoietic Stem Cell Gene Therapy
2.1. Metachromatic Leukodystrophy (MLD)
2.2. Mucopolysaccharidosis Type IIIA (MPS IIIA)
2.3. Mucopolysacchridosis Type I, Hurler Variant (MPS I-H)
2.4. Gaucher Disease
2.5. Cystinosis
2.6. Other Pre-Clinical Studies
2.7. Innovative Techniques
3. In Vivo Gene Therapy
3.1. Systemic Administration
3.2. Organs as Enzyme Factories for Systemic Expression
3.2.1. Liver
3.2.2. Lungs
3.2.3. Muscle
3.3. CNS-Directed Gene Therapy
3.4. Other Organ-Targeted Gene Therapy Approaches
3.4.1. Eye
3.4.2. Muscle
4. Gene Editing
4.1. Double-Strand Breaks (DSBs)
4.2. Zinc Finger Nucleases (ZFNs)
4.3. Transcription Activator-Like Effector Nucleases (TALENs)
4.4. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
4.5. Base Editing
4.6. Prime Editing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Martinoia, E.; Szabo, I. Organellar channels and transporters. Cell Calcium 2015, 58, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boustany, R.-M.N. Lysosomal storage diseases—the horizon expands. Nat. Rev. Neurol. 2013, 9, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Krivit, W. Microglia: The effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transplant. 1995, 4, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Katzmarski, N.; Haas, C.A.; Prinz, M. Bone Marrow Cell Recruitment to the Brain in the Absence of Irradiation or Parabiosis Bias. PLoS ONE 2013, 8, e58544. [Google Scholar] [CrossRef] [Green Version]
- De Ru, M.H.; Boelens, J.J.; Das, A.M.; Jones, S.A.; Van Der Lee, J.H.; Mahlaoui, N.; Mengel, E.; Offringa, M.; O’Meara, A.; Parini, R.; et al. Enzyme Replacement Therapy and/or Hematopoietic Stem Cell Transplantation at diagnosis in patients with Mucopolysaccharidosis type I: Results of a European consensus procedure. Orphanet J. Rare Dis. 2011, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.Y.; Boelens, J.J.; Jones, S.A.; Wynn, R.F. Hematopoietic Stem Cell Transplantation in Inborn Errors of Metabolism. Front. Pediatr. 2019, 7, 433. [Google Scholar] [CrossRef] [Green Version]
- Welling, L.; Marchal, J.P.; Van Hasselt, P.; Van Der Ploeg, A.T.; Wijburg, F.A.; Boelens, J.J.; Zschocke, J.; Baumgartner, M.; Gibson, K.M.; Patterson, M.; et al. Early Umbilical Cord Blood-Derived Stem Cell Transplantation Does Not Prevent Neurological Deterioration in Mucopolysaccharidosis Type III. JIMD Rep. 2014, 18, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Deduve, C. From Cytases to Lysosomes. Fed. Proc. 1964, 23, 1045–1049. [Google Scholar]
- Platt, F.M.; Lachmann, R.H. Treating lysosomal storage disorders: Current practice and future prospects. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1793, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.M.; Cachón-González, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2011, 226, 241–254. [Google Scholar] [CrossRef]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement Therapy for Inherited Enzyme Deficiency—Macrophage-Targeted Glucocerebrosidase for Gaucher’s Disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef]
- Aviezer, D.; Brill-Almon, E.; Shaaltiel, Y.; Hashmueli, S.; Bartfeld, D.; Mizrachi, S.; Liberman, Y.; Freeman, A.; Zimran, A.; Galun, E. A Plant-Derived Recombinant Human Glucocerebrosidase Enzyme—A Preclinical and Phase I Investigation. PLoS ONE 2009, 4, e4792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, G.A.; Golembo, M.; Shaaltiel, Y. Taliglucerase alfa: An enzyme replacement therapy using plant cell expression technology. Mol. Genet. Metab. 2014, 112, 1–8. [Google Scholar] [CrossRef]
- Zimran, A.; Altarescu, G.; Philips, M.; Attias, D.; Jmoudiak, M.; Deeb, M.; Wang, N.; Bhirangi, K.; Cohn, G.M.; Elstein, D. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood 2010, 115, 4651–4656. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Desnick, R.; Schuchman, E. Enzyme Replacement Therapy for Lysosomal Diseases: Lessons from 20 Years of Experience and Remaining Challenges. Annu. Rev. Genom. Hum. Genet. 2012, 13, 307–335. [Google Scholar] [CrossRef] [Green Version]
- Rombach, S.M.; Hollak, C.E.M.; Linthorst, G.E.; Dijkgraaf, M.G.W. Cost-effectiveness of enzyme replacement therapy for Fabry disease. Orphanet J. Rare Dis. 2013, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Baruteau, J.; Broomfield, A.; Crook, V.; Finnegan, N.; Harvey, K.; Burke, D.; Burch, M.; Shepherd, G.; Vellodi, A. Successful Desensitisation in a Patient with CRIM-Positive Infantile-Onset Pompe Disease. JIMD Rep. 2013, 12, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, A.; Jones, S.A.; Hughes, S.M.; Bigger, B.W. The impact of the immune system on the safety and efficiency of enzyme replacement therapy in lysosomal storage disorders. J. Inherit. Metab. Dis. 2016, 39, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Anderson, G.; Band, H.A.; Berkovic, S.F.; Cooper, J.D.; Holthaus, S.-M.K.; McKay, T.R.; Medina, D.L.; Rahim, A.A.; Schulz, A.; et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 2019, 18, 107–116. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Migalastat: First Global Approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, A. Heat shock response relieves ER stress. EMBO J. 2008, 27, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; Neises, G.R.; Reinkensmeier, G.; Townsend, M.J.; Perry, V.H.; Proia, R.L.; Winchester, B.; Dwek, R.A.; Butters, T.D. Prevention of Lysosomal Storage in Tay-Sachs Mice Treated with N-Butyldeoxynojirimycin. Science 1997, 276, 428–431. [Google Scholar] [CrossRef]
- Platt, F.M.; Jeyakumar, M. Substrate reduction therapy. Acta Paediatr. 2008, 97, 88–93. [Google Scholar] [CrossRef]
- Balwani, M.; Burrow, T.A.; Charrow, J.; Goker-Alpan, O.; Kaplan, P.; Kishnani, P.S.; Mistry, P.; Ruskin, J.; Weinreb, N. Recommendations for the use of eliglustat in the treatment of adults with Gaucher disease type 1 in the United States. Mol. Genet. Metab. 2016, 117, 95–103. [Google Scholar] [CrossRef]
- Belmatoug, N.; Di Rocco, M.; Fraga, C.; Giraldo, P.; Hughes, D.; Lukina, E.; Maison-Blanche, P.; Merkel, M.; Niederau, C.; Plöckinger, U.; et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur. J. Intern. Med. 2017, 37, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Elstein, D.; Dweck, A.; Attias, D.; Hadas-Halpern, I.; Zevin, S.; Altarescu, G.; Aerts, J.F.M.G.; Van Weely, S.; Zimran, A. Oral maintenance clinical trial with miglustat for type I Gaucher disease: Switch from or combination with intravenous enzyme replacement. Blood 2007, 110, 2296–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecarotta, S.; Romano, A.; Della Casa, R.; Del Giudice, E.; Bruschini, D.; Mansi, G.; Bembi, B.; Dardis, A.; Fiumara, A.; Di Rocco, M.; et al. Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann-Pick disease type C. Orphanet J. Rare Dis. 2015, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.H.; Smith, A.E. Gene therapy progress and prospects: Gene therapy of lysosomal storage disorders. Gene Ther. 2003, 10, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L. Ex vivo gene transfer and correction for cell-based therapies. Nat. Rev. Genet. 2011, 12, 301–315. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Neil, J.C. Safety of Retroviral Vectors in Clinical Applications: Lessons from Retroviral Biology and Pathogenesis. eLS 2017, 1–10. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, L.S.; Benedicenti, F.; Ambrosi, A.; Di Serio, C.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Mody, D.; DeRavin, S.S.; Hauer, J.; Lu, T.; Ma, Z.; Abina, S.H.-B.; Gray, J.T.; Greene, M.R.; Cavazzana-Calvo, M.; et al. A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells. Blood 2010, 116, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Vink, C.A.; Counsell, J.R.; Perocheau, D.P.; Karda, R.; Buckley, S.M.; Brugman, M.H.; Galla, M.; Schambach, A.; McKay, T.R.; Waddington, S.N.; et al. Eliminating HIV-1 Packaging Sequences from Lentiviral Vector Proviruses Enhances Safety and Expedites Gene Transfer for Gene Therapy. Mol. Ther. 2017, 25, 1790–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, M.V.; Fraietta, J.A.; Levine, B.L.; Kalos, M.; Zhao, Y.; June, C.H. Adoptive Immunotherapy for Cancer or Viruses. Annu. Rev. Immunol. 2014, 32, 189–225. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Michael, I.B. HIV-1 nuclear import: In search of a leader; update 1999. Front. Biosci. 1999, 4, d772-81. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Morsy, M.A.; Caskey, C. Expanded-capacity adenoviral vectors—the helper-dependent vectors. Mol. Med. Today 1999, 5, 18–24. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- Cunliffe, T.G.; Bates, E.A.; Parker, A.L. Hitting the Target but Missing the Point: Recent Progress towards Adenovirus-Based Precision Virotherapies. Cancers 2020, 12, 3327. [Google Scholar] [CrossRef] [PubMed]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Yakobson, B.; Koch, T.; Winocour, E. Replication of adeno-associated virus in synchronized cells without the addition of a helper virus. J. Virol. 1987, 61, 972–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, B.; Jayandharan, G.R. Basic Biology of Adeno-Associated Virus (AAV) Vectors Used in Gene Therapy. Curr. Gene Ther. 2014, 14, 86–100. [Google Scholar] [CrossRef]
- Pereira, D.J.; Mccarty, D.M.; Muzyczka, N. The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J. Virol. 1997, 71, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Calos, M.P.; Miller, J.H. Transposable elements. Cell 1980, 20, 579–595. [Google Scholar] [CrossRef]
- Rabinowitz, J.E.; Rolling, F.; Li, C.; Conrath, H.; Xiao, W.; Xiao, X.; Samulski, R.J. Cross-Packaging of a Single Adeno-Associated Virus (AAV) Type 2 Vector Genome into Multiple AAV Serotypes Enables Transduction with Broad Specificity. J. Virol. 2002, 76, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [Green Version]
- Somanathan, S.; Breous, E.; Bell, P.; Wilson, J.M. AAV Vectors Avoid Inflammatory Signals Necessary to Render Transduced Hepatocyte Targets for Destructive T Cells. Mol. Ther. 2010, 18, 977–982. [Google Scholar] [CrossRef]
- Buchlis, G.; Podsakoff, G.M.; Radu, A.; Hawk, S.M.; Flake, A.W.; Mingozzi, F.; High, K.A. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 2012, 119, 3038–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Dupaty, L.; Aillot, L.; Zhang, L.; Gallien, C.; Hallek, M.; Odenthal, M.; Adriouch, S.; Salvetti, A.; Büning, H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci. Rep. 2019, 9, 3631. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-Associated Viruses Are Widely Disseminated in Human Tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Hasilik, A.; Klein, U.; Waheed, A.; Strecker, G.; Von Figura, K. Phosphorylated oligosaccharides in lysosomal enzymes: Identification of alpha-N-acetylglucosamine(1)phospho(6)mannose diester groups. Proc. Natl. Acad. Sci. USA 1980, 77, 7074–7078. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, J. Reversal of Clinical Features of Hurler’s Disease and Biochemical Improvement after Treatment by Bone-Marrow Transplantation. Lancet 1981, 318, 709–712. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Boelens, J.J.; De Koning, T.J. The Clinical Outcome of Hurler Syndrome after Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2008, 14, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.A.; Nmdp, I.O.B.O.T.; Steward, C.G. Hematopoietic cell transplantation for inherited metabolic diseases: An overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003, 31, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steward, C.G. Haemopoietic stem cell transplantation for genetic disorders. Arch. Dis. Child. 2005, 90, 1259–1263. [Google Scholar] [CrossRef]
- Matzner, U.; Harzer, K.; Learish, R.; Barranger, J.; Gieselmann, V. Long-term expression and transfer of arylsulfatase A into brain of arylsulfatase A-deficient mice transplanted with bone marrow expressing the arylsulfatase A cDNA from a retroviral vector. Gene Ther. 2000, 7, 1250–1257. [Google Scholar] [CrossRef]
- Matzner, U.; Schestag, F.; Hartmann, D.; Lüllmann-Rauch, R.; D’Hooge, R.; De Deyn, P.P.; Gieselmann, V. Bone Marrow Stem Cell Gene Therapy of Arylsulfatase A-Deficient Mice, Using an Arylsulfatase A Mutant That Is Hypersecreted from Retrovirally Transduced Donor-Type Cells. Hum. Gene Ther. 2001, 12, 1021–1033. [Google Scholar] [CrossRef]
- Matzner, U.; Hartmann, D.; Lüllmann-Rauch, R.; Coenen, R.; Rothert, F.; Månsson, J.-E.; Fredman, P.; Hooge, R.D.; De Deyn, P.; Gieselmann, V. Bone marrow stem cell-based gene transfer in a mouse model for metachromatic leukodystrophy: Effects on visceral and nervous system disease manifestations. Gene Ther. 2002, 9, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; De Palma, M.; Quattrini, A.; Del Carro, U.; Amadio, S.; Visigalli, I.; Sessa, M.; Fasano, S.; Brambilla, R.; Marchesini, S.; et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Investig. 2004, 113, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Capotondo, A.; Fasano, S.; Del Carro, U.; Marchesini, S.; Azuma, H.; Malaguti, M.C.; Amadio, S.; Brambilla, R.; Grompe, M.; et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Investig. 2006, 116, 3070–3082. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [Green Version]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Calbi, V.; Fumagalli, F.; Consiglieri, G.; Penati, R.; Acquati, S.; Redaelli, D.; Attanasio, V.; Marcella, F.; Cicalese, M.P.; Migliavacca, M.; et al. Use of Defibrotide to help prevent post-transplant endothelial injury in a genetically predisposed infant with metachromatic leukodystrophy undergoing hematopoietic stem cell gene therapy. Bone Marrow Transplant. 2018, 53, 913–917. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, B.S.; Davison, J.; Jones, S.A.; Baruteau, J. Novel therapies for mucopolysaccharidosis type III. J. Inherit. Metab. Dis. 2021, 44, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, A.; Wilkinson, F.L.; Langford-Smith, K.J.; Holley, R.J.; Sergijenko, A.; Howe, S.J.; Bennett, W.R.; Jones, S.A.; Wraith, J.; Merry, C.L.; et al. Hematopoietic Stem Cell and Gene Therapy Corrects Primary Neuropathology and Behavior in Mucopolysaccharidosis IIIA Mice. Mol. Ther. 2012, 20, 1610–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergijenko, A.; Langford-Smith, A.; Liao, A.Y.; Pickford, C.E.; McDermott, J.C.; Nowinski, G.; Langford-Smith, K.J.; Merry, C.L.R.; Jones, S.A.; Wraith, J.E.; et al. Myeloid/Microglial Driven Autologous Hematopoietic Stem Cell Gene Therapy Corrects a Neuronopathic Lysosomal Disease. Mol. Ther. 2013, 21, 1938–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, S.M.; Liao, A.; Wood, S.; Taylor, J.; Youshani, A.S.; Rowlston, S.; Parker, H.; Armant, M.; Biffi, A.; Chan, L.; et al. Pre-clinical Safety and Efficacy of Lentiviral Vector-Mediated Ex Vivo Stem Cell Gene Therapy for the Treatment of Mucopolysaccharidosis IIIA. Mol. Ther. Methods Clin. Dev. 2019, 13, 399–413. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Rozengurt, N.; Ryazantsev, S.; Kohn, D.B.; Satake, N.; Neufeld, E.F. Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol. Genet. Metab. 2003, 79, 233–244. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; Von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef]
- De Palma, M.; Montini, E.; De Sio, F.R.S.; Benedicenti, F.; Gentile, A.; Medico, E.; Naldini, L. Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood 2005, 105, 2307–2315. [Google Scholar] [CrossRef]
- Orchard Therapeutics Outlines Comprehensive Presence at 2021 WORLDSymposiumTM. Orchard Therapeutics. Available online: https://ir.orchard-tx.com/news-releases/news-release-details/orchard-therapeutics-outlines-comprehensive-presence-2021 (accessed on 8 March 2021).
- Enquist, I.B.; Nilsson, E.C.; Ooka, A.; Månsson, J.-E.; Olsson, K.; Ehinger, M.; Brady, R.O.; Richter, J.; Karlsson, S. Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13819–13824. [Google Scholar] [CrossRef] [Green Version]
- Enquist, I.B.; Nilsson, E.; Månsson, J.-E.; Ehinger, M.; Richter, J.; Karlsson, S. Successful Low-Risk Hematopoietic Cell Therapy in a Mouse Model of Type 1 Gaucher Disease. STEM CELLS 2009, 27, 744–752. [Google Scholar] [CrossRef]
- Dahl, M.; Smith, E.M.; Warsi, S.; Rothe, M.; Ferraz, M.J.; Aerts, J.M.; Golipour, A.; Harper, C.; Pfeifer, R.; Pizzurro, D.; et al. Correction of pathology in mice displaying Gaucher disease type 1 by a clinically-applicable lentiviral vector. Mol. Ther. Methods Clin. Dev. 2021, 20, 312–323. [Google Scholar] [CrossRef]
- Jacobsen, L. The GuardOne Clinical Trial: A First-in-Human, Open-Label, Multinational Phase 1/2 Study of AVR-RD-02 Ex Vivo Lentiviral Vector, Autologous Gene Therapy for Gaucher Disease. In Proceedings of the WORLDSymposium, Manchester Grand Hyatt, San Diego, CA, USA, 8–12 February 2021. [Google Scholar]
- Harrison, F.; Yeagy, B.A.; Rocca, C.J.; Kohn, D.B.; Salomon, D.R.; Cherqui, S. Hematopoietic Stem Cell Gene Therapy for the Multisystemic Lysosomal Storage Disorder Cystinosis. Mol. Ther. 2013, 21, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef]
- Miwa, S.; Watabe, A.M.; Shimada, Y.; Higuchi, T.; Kobayashi, H.; Fukuda, T.; Kato, F.; Ida, H.; Ohashi, T. Efficient engraftment of genetically modified cells is necessary to ameliorate central nervous system involvement of murine model of mucopolysaccharidosis type II by hematopoietic stem cell targeted gene therapy. Mol. Genet. Metab. 2020, 130, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Shimada, Y.; Akiyama, K.; Higuchi, T.; Fukuda, T.; Kobayashi, H.; Eto, Y.; Ida, H.; Ohashi, T. Hematopoietic Stem Cell Gene Therapy Corrects Neuropathic Phenotype in Murine Model of Mucopolysaccharidosis Type II. Hum. Gene Ther. 2015, 26, 357–366. [Google Scholar] [CrossRef]
- Holley, R.J.; Ellison, S.M.; Fil, D.; O’Leary, C.; McDermott, J.; Senthivel, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; D’Souza, Z.; Parker, H.; et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain 2017, 141, 99–116. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Ryazantsev, S.; Ohmi, K.; Zhao, H.-Z.; Rozengurt, N.; Kohn, D.B.; Neufeld, E.F. Retrovirally transduced bone marrow has a therapeutic effect on brain in the mouse model of mucopolysaccharidosis IIIB. Mol. Genet. Metab. 2004, 82, 286–295. [Google Scholar] [CrossRef]
- Hofling, A.A.; Devine, S.; Vogler, C.; Sands, M.S. Human CD34+ hematopoietic progenitor cell-directed lentiviral-mediated gene therapy in a xenotransplantation model of lysosomal storage disease. Mol. Ther. 2004, 9, 856–865. [Google Scholar] [CrossRef]
- Sakurai, K.; Iizuka, S.; Shen, J.-S.; Meng, X.-L.; Mori, T.; Umezawa, A.; Ohashi, T.; Eto, Y. Brain transplantation of genetically modified bone marrow stromal cells corrects CNS pathology and cognitive function in MPS VII mice. Gene Ther. 2004, 11, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Capotondo, A.; Milazzo, R.; Politi, L.S.; Quattrini, A.; Palini, A.; Plati, T.; Merella, S.; Nonis, A.; Di Serio, C.; Montini, E.; et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. USA 2012, 109, 15018–15023. [Google Scholar] [CrossRef] [Green Version]
- Douillard-Guilloux, G.; Richard, E.; Batista, L.; Caillaud, C. Partial phenotypic correction and immune tolerance induction to enzyme replacement therapy after hematopoietic stem cell gene transfer of α-glucosidase in Pompe disease. J. Gene Med. 2009, 11, 279–287. [Google Scholar] [CrossRef]
- Piras, G.; Montiel-Equihua, C.; Chan, Y.-K.A.; Wantuch, S.; Stuckey, D.; Burke, D.; Prunty, H.; Phadke, R.; Chambers, D.; Partida-Gaytan, A.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Rescues Clinical Phenotypes in a Murine Model of Pompe Disease. Mol. Ther. Methods Clin. Dev. 2020, 18, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Stok, M.; De Boer, H.; Huston, M.W.; Jacobs, E.H.; Roovers, O.; Visser, T.P.; Jahr, H.; Duncker, D.J.; Van Deel, E.D.; Reuser, A.J.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Corrects Murine Pompe Disease. Mol. Ther. Methods Clin. Dev. 2020, 17, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Van Til, N.P.; Stok, M.; Kaya, F.S.F.A.; De Waard, M.C.; Farahbakhshian, E.; Visser, T.P.; Kroos, M.A.; Jacobs, E.H.; Willart, M.A.; Van Der Wegen, P.; et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 2010, 115, 5329–5337. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nat. Cell Biol. 2016, 539, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017, 9, eaan0820. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Scharenberg, S.G.; Poletto, E.; Lucot, K.L.; Colella, P.; Sheikali, A.; Montine, T.J.; Porteus, M.H.; Gomez-Ospina, N. Engineering monocyte/macrophage−specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat. Commun. 2020, 11, 11. [Google Scholar] [CrossRef]
- Chung, S.; Ma, X.; Liu, Y.; Lee, D.; Tittiger, M.; Ponder, K.P. Effect of neonatal administration of a retroviral vector expressing α-l-iduronidase upon lysosomal storage in brain and other organs in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2007, 90, 181–192. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, L.; Hennig, A.K.; Kovács, A.; Fu, A.; Chung, S.; Lee, D.; Wang, B.; Herati, R.S.; Ogilvie, J.M.; et al. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Mol. Ther. 2005, 11, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Mango, R.L. Neonatal retroviral vector-mediated hepatic gene therapy reduces bone, joint, and cartilage disease in mucopolysaccharidosis VII mice and dogs. Mol. Genet. Metab. 2004, 82, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Mango, R.L.; Sands, M.S.; Haskins, M.E.; Ellinwood, N.M.; Ponder, K.P. Evaluation of Pathological Manifestations of Disease in Mucopolysaccharidosis VII Mice after Neonatal Hepatic Gene Therapy. Mol. Ther. 2002, 6, 745–758. [Google Scholar] [CrossRef]
- Ponder, K.P.; Melniczek, J.R.; Xu, L.; Weil, M.A.; O’Malley, T.M.; O’Donnell, P.A.; Knox, V.W.; Aguirre, G.D.; Mazrier, H.; Ellinwood, N.M.; et al. Therapeutic neonatal hepatic gene therapy in mucopolysaccharidosis VII dogs. Proc. Natl. Acad. Sci. USA 2002, 99, 13102–13107. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Liu, Y.; Tittiger, M.; Hennig, A.; Kovacs, A.; Popelka, S.; Wang, B.; Herati, R.S.; Bigg, M.; Ponder, K.P.; et al. Improvements in Mucopolysaccharidosis I Mice After Adult Retroviral Vector–mediated Gene Therapy with Immunomodulation. Mol. Ther. 2007, 15, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.S.; Kang, Y.; Sauter, S.L.; Townsend, K.; Staber, P.; Derksen, T.A.; Martins, I.; Qian, J.; Davidson, B.L.; McCray, P.B.; et al. In Vivo Treatment of Hemophilia A and Mucopolysaccharidosis Type VII Using Nonprimate Lentiviral Vectors. Mol. Ther. 2001, 3, 850–856. [Google Scholar] [CrossRef]
- Kyosen, S.; Iizuka, S.; Kobayashi, H.; Kimura, T.; Fukuda, T.; Shen, J.; Shimada, Y.; Ida, H.; Eto, Y.; Ohashi, T. Neonatal gene transfer using lentiviral vector for murine Pompe disease: Long-term expression and glycogen reduction. Gene Ther. 2009, 17, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Carbonaro, D.; Pepper, K.; Petersen, D.; Ge, S.; Jackson, H.; Shimada, H.; Moats, R.; Kohn, D.B. Neonatal Gene Therapy of MPS I Mice by Intravenous Injection of a Lentiviral Vector. Mol. Ther. 2005, 11, 776–789. [Google Scholar] [CrossRef]
- Di Domenico, C.; Villani, G.R.; Di Napoli, D.; Reyero, E.G.Y.; Lombardo, A.; Naldini, L.; Di Natale, P. Gene Therapy for a Mucopolysaccharidosis Type I Murine Model with Lentiviral-IDUA Vector. Hum. Gene Ther. 2005, 16, 81–90. [Google Scholar] [CrossRef]
- Di Natale, P.; Di Domenico, C.; Gargiulo, N.; Castaldo, S.; Reyero, E.G.Y.; Mithbaokar, P.; De Felice, M.; Follenzi, A.; Naldini, L.; Villani, G.R.D. Treatment of the mouse model of mucopolysaccharidosis type IIIB with lentiviral-NAGLU vector. Biochem. J. 2005, 388, 639–646. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, C.; Roberts, A.L.D.; Ranieri, E.; Clements, P.R.; Byers, S.; Anson, D.S. Lentiviral-mediated gene therapy for murine mucopolysaccharidosis type IIIA. Mol. Genet. Metab. 2008, 93, 411–418. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.; Byers, S.; Anson, D.S. Correction of mucopolysaccharidosis type IIIA somatic and central nervous system pathology by lentiviral-mediated gene transfer. J. Gene Med. 2010, 12, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Bielicki, J.; McIntyre, C.; Anson, D.S. Comparison of ventricular and intravenous lentiviral-mediated gene therapy for murine MPS VII. Mol. Genet. Metab. 2010, 101, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.S.; Ghodsi, A.; Derksen, T.; Davidson, B.L. Systemic and Central Nervous System Correction of Lysosomal Storage in Mucopolysaccharidosis Type VII Mice. J. Virol. 1999, 73, 3424–3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, T.; Watabe, K.; Uehara, K.; Sly, W.S.; Vogler, C.; Eto, Y. Adenovirus-mediated gene transfer and expression of human -glucuronidase gene in the liver, spleen, and central nervous system in mucopolysaccharidosis type VII mice. Proc. Natl. Acad. Sci. USA 1997, 94, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, J.E.; Mignon, A.; Haase, G.; Caillaud, C.; McDonell, N.; Kahn, A.; Poenaru, L. Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. Hum. Mol. Genet. 1999, 8, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Heur, M.; Witte, D.P.; Ameis, D.; Grabowski, G.A. Lysosomal Acid Lipase Deficiency: Correction of Lipid Storage by Adenovirus-Mediated Gene Transfer in Mice. Hum. Gene Ther. 2002, 13, 1361–1372. [Google Scholar] [CrossRef]
- McVie-Wylie, A.J.; Ding, E.Y.; Lawson, T.; Serra, D.; Migone, F.K.; Pressley, D.; Mizutani, M.; Kikuchi, T.; Chen, Y.T.; Amalfitano, A. Multiple muscles in the AMD quail can be “cross-corrected” of pathologic glycogen accumulation after intravenous injection of an [E1-, polymerase-] adenovirus vector encoding human acid-α-glucosidase. J. Gene Med. 2002, 5, 399–406. [Google Scholar] [CrossRef]
- Amalfitano, A.; McVie-Wylie, A.J.; Hu, H.; Dawson, T.L.; Raben, N.; Plotz, P.; Chen, Y.T. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid- -glucosidase. Proc. Natl. Acad. Sci. USA 1999, 96, 8861–8866. [Google Scholar] [CrossRef] [Green Version]
- Pauly, D.F.; Fraites, T.J.; Toma, C.; Bayes, H.S.; Huie, M.L.; Hirschhorn, R.; Plotz, P.H.; Raben, N.; Kessler, P.D.; Byrne, B.J. Intercellular Transfer of the Virally Derived Precursor Form of Acid α-Glucosidase Corrects the Enzyme Deficiency in Inherited Cardioskeletal Myopathy Pompe Disease. Hum. Gene Ther. 2001, 12, 527–538. [Google Scholar] [CrossRef]
- Asokan, A.; Schaffer, D.V.; Samulski, R.J. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Ou, H.; Cui, M.R.; Jiang, M.N.; Zhang, M.M.; Li, M.X.; Ma, J.; Zhang, J.; Ma, D. Delivery of Glucosylceramidase Beta Gene Using AAV9 Vector Therapy as a Treatment Strategy in Mouse Models of Gaucher Disease. Hum. Gene Ther. 2019, 30, 155–167. [Google Scholar] [CrossRef]
- Weismann, C.M.; Ferreira, J.; Keeler, A.M.; Su, Q.; Qui, L.; Shaffer, S.A.; Xu, Z.; Gao, G.; Sena-Esteves, M. Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum. Mol. Genet. 2015, 24, 4353–4364. [Google Scholar] [CrossRef] [Green Version]
- Walia, J.S.; Altaleb, N.; Bello, A.; Kruck, C.; LaFave, M.C.; Varshney, G.K.; Burgess, S.M.; Chowdhury, B.; Hurlbut, D.; Hemming, R.; et al. Long-Term Correction of Sandhoff Disease Following Intravenous Delivery of rAAV9 to Mouse Neonates. Mol. Ther. 2015, 23, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Lahey, H.G.; Webber, C.J.; Golebiowski, D.; Izzo, C.M.; Horn, E.; Taghian, T.; Rodriguez, P.; Batista, A.R.; Ellis, L.E.; Hwang, M.; et al. Pronounced Therapeutic Benefit of a Single Bidirectional AAV Vector Administered Systemically in Sandhoff Mice. Mol. Ther. 2020, 28, 2150–2160. [Google Scholar] [CrossRef]
- Rafi, M.A.; Rao, H.Z.; Luzi, P.; Luddi, A.; Curtis, M.T.; Wenger, D.A. Intravenous injection of AAVrh10-GALC after the neonatal period in twitcher mice results in significant expression in the central and peripheral nervous systems and improvement of clinical features. Mol. Genet. Metab. 2015, 114, 459–466. [Google Scholar] [CrossRef]
- Belur, L.R.; Podetz-Pedersen, K.M.; Tran, T.A.; Mesick, J.A.; Singh, N.M.; Riedl, M.; Vulchanova, L.; Kozarsky, K.F.; McIvor, R.S. Intravenous delivery for treatment of mucopolysaccharidosis type I: A comparison of AAV serotypes 9 and rh10. Mol. Genet. Metab. Rep. 2020, 24, 100604. [Google Scholar] [CrossRef]
- Jung, S.-C.; Park, E.-S.; Choi, E.N.; Kim, C.H.; Kim, S.J.; Jin, D.-K. Characterization of a novel mucopolysaccharidosis type II mouse model and recombinant AAV2/8 vector-mediated gene therapy. Mol. Cells 2010, 30, 13–18. [Google Scholar] [CrossRef]
- Duncan, F.J.; Naughton, B.J.; Zaraspe, K.; Murrey, D.A.; Meadows, A.S.; Clark, K.R.; Newsome, D.E.; White, P.; Fu, H.; Mccarty, D.M. Broad Functional Correction of Molecular Impairments by Systemic Delivery of scAAVrh74-hSGSH Gene Delivery in MPS IIIA Mice. Mol. Ther. 2015, 23, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Ruzo, A.; Marcó, S.; Garcia, M.; Villacampa, P.; Ribera, A.; Ayuso, E.; Maggioni, L.; Mingozzi, F.; Haurigot, V.A.; Bosch, F. Correction of Pathological Accumulation of Glycosaminoglycans in Central Nervous System and Peripheral Tissues of MPSIIIA Mice Through Systemic AAV9 Gene Transfer. Hum. Gene Ther. 2012, 23, 1237–1246. [Google Scholar] [CrossRef]
- Naughton, B.J.; Duncan, F.J.; Murrey, D.; Ware, T.; Meadows, A.; Mccarty, D.M.; Fu, H. Amyloidosis, Synucleinopathy, and Prion Encephalopathy in a Neuropathic Lysosomal Storage Disease: The CNS-Biomarker Potential of Peripheral Blood. PLoS ONE 2013, 8, e80142. [Google Scholar] [CrossRef]
- Mccarty, D.M.; DiRosario, J.; Gulaid, K.; Muenzer, J.; Fu, H. Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 2009, 16, 1340–1352. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Claflin, K.; Geoghegan, J.C.; Davidson, B.L. Sialic Acid Deposition Impairs the Utility of AAV9, but Not Peptide-modified AAVs for Brain Gene Therapy in a Mouse Model of Lysosomal Storage Disease. Mol. Ther. 2012, 20, 1393–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaro, G.; Mattar, C.N.Z.; Wong, A.M.S.; Sirka, E.; Buckley, S.M.K.; Herbert, B.R.; Karlsson, S.; Perocheau, D.P.; Burke, D.; Heales, S.; et al. Fetal gene therapy for neurodegenerative disease of infants. Nat. Med. 2018, 24, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Massaro, G.; Hughes, M.P.; Whaler, S.M.; Wallom, K.-L.; Priestman, D.A.; Platt, F.M.; Waddington, S.N.; Rahim, A.A. Systemic AAV9 gene therapy using the synapsin I promoter rescues a mouse model of neuronopathic Gaucher disease but with limited cross-correction potential to astrocytes. Hum. Mol. Genet. 2020, 29, 1933–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartung, S.D.; Frandsen, J.L.; Pan, D.; Koniar, B.L.; Graupman, P.; Gunther, R.; Low, W.C.; Whitley, C.B.; McIvor, R.S. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human α-l-iduronidase gene. Mol. Ther. 2004, 9, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Polito, V.A.; Cosma, M.P. IDS Crossing of the Blood-Brain Barrier Corrects CNS Defects in MPSII Mice. Expanding the Spectrum of BAF-Related Disorders: De Novo Variants in SMARCC2 Cause a Syndrome with Intellectual Disability and Developmental Delay. AJHG 2009, 85, 296–301. [Google Scholar] [CrossRef] [Green Version]
- Daly, T.M.; Vogler, C.; Levy, B.; Haskins, M.E.; Sands, M.S. Neonatal gene transfer leads to widespread correction of pathology in a murine model of lysosomal storage disease. Proc. Natl. Acad. Sci. USA 1999, 96, 2296–2300. [Google Scholar] [CrossRef] [Green Version]
- Daly, T.M.; Ohlemiller, K.K.; Roberts, M.S.; Vogler, C.A.; Sands, M.S. Prevention of systemic clinical disease in MPS VII mice following AAV-mediated neonatal gene transfer. Gene Ther. 2001, 8, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Spampanato, C.; De Leonibus, E.; Dama, P.; Gargiulo, A.; Fraldi, A.; Sorrentino, N.C.; Russo, F.; Nusco, E.; Auricchio, A.; Surace, E.M.; et al. Efficacy of a Combined Intracerebral and Systemic Gene Delivery Approach for the Treatment of a Severe Lysosomal Storage Disorder. Mol. Ther. 2011, 19, 860–869. [Google Scholar] [CrossRef]
- Miyake, N.; Miyake, K.; Asakawa, N.; Yamamoto, M.; Shimada, T. Long-term correction of biochemical and neurological abnormalities in MLD mice model by neonatal systemic injection of an AAV serotype 9 vector. Gene Ther. 2014, 21, 427–433. [Google Scholar] [CrossRef]
- Lim, J.-A.; Yi, H.; Gao, F.; Raben, N.; Kishnani, P.S.; Sun, B. Intravenous Injection of an AAV-PHP.B Vector Encoding Human Acid α-Glucosidase Rescues Both Muscle and CNS Defects in Murine Pompe Disease. Mol. Ther. Methods Clin. Dev. 2019, 12, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliger, S.S.; Elliger, C.A.; Lang, C.; Watson, G.L. Enhanced Secretion and Uptake of β-Glucuronidase Improves Adeno-associated Viral-Mediated Gene Therapy of Mucopolysaccharidosis Type VII Mice. Mol. Ther. 2002, 5, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Kang, L.; Jennings, J.S.; Moy, S.S.; Perez, A.; DiRosario, J.; Mccarty, D.M.; Muenzer, J. Significantly increased lifespan and improved behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther. 2007, 14, 1065–1077. [Google Scholar] [CrossRef]
- Gray, S.J.; Matagne, V.; Bachaboina, L.; Yadav, S.; Ojeda, S.R.; Samulski, R.J. Preclinical Differences of Intravascular AAV9 Delivery to Neurons and Glia: A Comparative Study of Adult Mice and Nonhuman Primates. Mol. Ther. 2011, 19, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.M.; Flotte, T.R. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Chandler, R.J.; Williams, I.M.; Gibson, A.L.; Davidson, C.D.; Incao, A.A.; Hubbard, B.T.; Porter, F.D.; Pavan, W.J.; Venditti, C.P. Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Hum. Mol. Genet. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Gray, J.T.; McIntosh, J.; Ng, C.Y.C.; Zhou, J.; Spence, Y.; Cochrane, M.; Gray, E.; Tuddenham, E.G.D.; Davidoff, A.M. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood 2006, 109, 1414–1421. [Google Scholar] [CrossRef] [Green Version]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Baruteau, J.; Waddington, S.N.; Alexander, I.E.; Gissen, P. Gene therapy for monogenic liver diseases: Clinical successes, current challenges and future prospects. J. Inherit. Metab. Dis. 2017, 40, 497–517. [Google Scholar] [CrossRef]
- Kattenhorn, L.M.; Tipper, C.H.; Stoica, L.; Geraghty, D.S.; Wright, T.L.; Clark, K.R.; Wadsworth, S.C. Adeno-Associated Virus Gene Therapy for Liver Disease. Hum. Gene Ther. 2016, 27, 947–961. [Google Scholar] [CrossRef]
- Paulk, N.K.; Pekrun, K.; Zhu, E.; Nygaard, S.; Li, B.; Xu, J.; Chu, K.; Leborgne, C.; Dane, A.P.; Haft, A.; et al. Bioengineered AAV Capsids with Combined High Human Liver Transduction In Vivo and Unique Humoral Seroreactivity. Mol. Ther. 2018, 26, 289–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzzo, F.; Colella, P.; Biferi, M.G.; Bali, D.; Paulk, N.K.; Vidal, P.; Collaud, F.; Simon-Sola, M.; Charles, S.; Hardet, R.; et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid α-glucosidase. Sci. Transl. Med. 2017, 9, eaam6375. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.J.; Canavese, M.; Chisari, E.; Pandya, J.; Cocita, C.; Portillo, M.; McIntosh, J.; Kia, A.; Foley, J.H.; Dane, A.; et al. Liver-Directed AAV Gene Therapy for Gaucher Disease. Blood 2019, 134, 3354. [Google Scholar] [CrossRef]
- Jung, S.-C.; Han, I.P.; Limaye, A.; Xu, R.; Gelderman, M.P.; Zerfas, P.; Tirumalai, K.; Murray, G.J.; During, M.J.; Brady, R.O.; et al. Adeno-associated viral vector-mediated gene transfer results in long-term enzymatic and functional correction in multiple organs of Fabry mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2676–2681. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, R.J.; Lonning, S.M.; Armentano, D.; Li, C.; Souza, D.W.; Cherry, M.; Ford, C.; Barbon, C.M.; Desnick, R.J.; Gao, G.; et al. AAV2 Vector Harboring a Liver-Restricted Promoter Facilitates Sustained Expression of Therapeutic Levels of α-Galactosidase A and the Induction of Immune Tolerance in Fabry Mice. Mol. Ther. 2004, 9, 231–240. [Google Scholar] [CrossRef]
- McEachern, K.A.; Nietupski, J.B.; Chuang, W.-L.; Armentano, D.; Johnson, J.; Hutto, E.; Grabowski, G.A.; Cheng, S.H.; Marshall, J. AAV8-mediated expression of glucocerebrosidase ameliorates the storage pathology in the visceral organs of a mouse model of Gaucher disease. J. Gene Med. 2006, 8, 719–729. [Google Scholar] [CrossRef]
- Franco, L.M.; Sun, B.; Yang, X.; Bird, A.; Zhang, H.; Schneider, A.; Brown, T.; Young, S.P.; Clay, T.M.; Amalfitano, A.; et al. Evasion of Immune Responses to Introduced Human Acid α-Glucosidase by Liver-Restricted Expression in Glycogen Storage Disease Type II. Mol. Ther. 2005, 12, 876–884. [Google Scholar] [CrossRef]
- Wang, G.; Young, S.P.; Bali, D.; Hutt, J.; Li, S.; Benson, J.; Koeberl, D.D. Assessment of toxicity and biodistribution of recombinant AAV8 vector–mediated immunomodulatory gene therapy in mice with Pompe disease. Mol. Ther. Methods Clin. Dev. 2014, 1, 14018. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-O.; Ronzitti, G.; Arnson, B.; Leborgne, C.; Li, S.; Mingozzi, F.; Koeberl, D. Low-Dose Liver-Targeted Gene Therapy for Pompe Disease Enhances Therapeutic Efficacy of ERT via Immune Tolerance Induction. Mol. Ther. Methods Clin. Dev. 2017, 4, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Bell, P.; Gurda, B.L.; Wang, Q.; Louboutin, J.-P.; Zhu, Y.; Bagel, J.; O’Donnell, P.; Sikora, T.; Ruane, T.; et al. Liver-directed gene therapy corrects cardiovascular lesions in feline mucopolysaccharidosis type I. Proc. Natl. Acad. Sci. USA 2014, 111, 14894–14899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferla, R.; Alliegro, M.; Marteau, J.-B.; Dell’Anno, M.; Nusco, E.; Pouillot, S.; Galimberti, S.; Valsecchi, M.G.; Zuliani, V.; Auricchio, A. Non-clinical Safety and Efficacy of an AAV2/8 Vector Administered Intravenously for Treatment of Mucopolysaccharidosis Type VI. Mol. Ther. Methods Clin. Dev. 2017, 6, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Cotugno, G.; Annunziata, P.; Tessitore, A.; O’Malley, T.; Capalbo, A.; Faella, A.; Bartolomeo, R.; O’Donnell, P.; Wang, P.; Russo, F.; et al. Long-term Amelioration of Feline Mucopolysaccharidosis VI After AAV-mediated Liver Gene Transfer. Mol. Ther. 2011, 19, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Sferra, T.J.; Backstrom, K.; Wang, C.; Rennard, R.; Miller, M.; Hu, Y. Widespread Correction of Lysosomal Storage Following Intrahepatic Injection of a Recombinant Adeno-associated Virus in the Adult MPS VII Mouse. Mol. Ther. 2004, 10, 478–491. [Google Scholar] [CrossRef]
- Perocheau, D.P.; Cunningham, S.C.; Lee, J.; Diaz, J.A.; Waddington, S.N.; Gilmour, K.; Eaglestone, S.; Lisowski, L.; Thrasher, A.J.; Alexander, I.E.; et al. Age-Related Seroprevalence of Antibodies Against AAV-LK03 in a UK Population Cohort. Hum. Gene Ther. 2019, 30, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Rucker, M.; Fraites, T.J.; Porvasnik, S.L.; Lewis, M.A.; Zolotukhin, I.; Cloutier, D.A.; Byrne, B.J. Rescue of enzyme deficiency in embryonic diaphragm in a mouse model of metabolic myopathy: Pompe disease. Development 2004, 131, 3007–3019. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, S.C.; Dane, A.P.; Spinoulas, A.; Alexander, I.E. Gene Delivery to the Juvenile Mouse Liver Using AAV2/8 Vectors. Mol. Ther. 2008, 16, 1081–1088. [Google Scholar] [CrossRef]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef] [Green Version]
- La Bella, T.; Imbeaud, S.; Peneau, C.; Mami, I.; Datta, S.; Bayard, Q.; Caruso, S.; Hirsch, T.Z.; Calderaro, J.; Morcrette, G.; et al. Adeno-associated virus in the liver: Natural history and consequences in tumour development. Gut 2020, 69, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Auricchio, A.; O’Connor, E.; Weiner, D.; Gao, G.-P.; Hildinger, M.; Wang, L.; Calcedo, R.; Wilson, J.M. Noninvasive gene transfer to the lung for systemic delivery of therapeutic proteins. J. Clin. Investig. 2002, 110, 499–504. [Google Scholar] [CrossRef]
- Li, C.; Ziegler, R.J.; Cherry, M.; Lukason, M.; Desnick, R.J.; Yew, N.S.; Cheng, S.H. Adenovirus-Transduced Lung as a Portal for Delivering α-Galactosidase A into Systemic Circulation for Fabry Disease. Mol. Ther. 2002, 5, 745–754. [Google Scholar] [CrossRef]
- Muraine, M.L.; Bensalah, M.; Dhiab, M.J.; Cordova, G.; Arandel, L.; Marhic, M.A.; Chapart, M.M.; Vasseur, S.; Benkhelifa-Ziyyat, S.; Bigot, A.; et al. Transduction Efficiency of Adeno-Associated Virus Serotypes After Local Injection in Mouse and Human Skeletal Muscle. Hum. Gene Ther. 2020, 31, 233–240. [Google Scholar] [CrossRef]
- Daly, T.M.; Okuyama, T.; Vogler, C.; Haskins, M.E.; Muzyczka, N.; Sands, M.S. Neonatal Intramuscular Injection with Recombinant Adeno-Associated Virus Results in Prolonged beta-Glucuronidase Expression in Situ and Correction of Liver Pathology in Mucopolysaccharidosis Type VII Mice. Hum. Gene Ther. 1999, 10, 85–94. [Google Scholar] [CrossRef]
- Fraites, T.J.; Schleissing, M.R.; Shanely, R.; Walter, G.A.; Cloutier, D.A.; Zolotukhin, I.; Pauly, D.F.; Raben, N.; Plotz, P.H.; Powers, S.K.; et al. Correction of the Enzymatic and Functional Deficits in a Model of Pompe Disease Using Adeno-associated Virus Vectors. Mol. Ther. 2002, 5, 571–578. [Google Scholar] [CrossRef]
- Takahashi, H.; Hirai, Y.; Migita, M.; Seino, Y.; Fukuda, Y.; Sakuraba, H.; Kase, R.; Kobayashi, T.; Hashimoto, Y.; Shimada, T. Long-term systemic therapy of Fabry disease in a knockout mouse by adeno-associated virus-mediated muscle-directed gene transfer. Proc. Natl. Acad. Sci. USA 2002, 99, 13777–13782. [Google Scholar] [CrossRef] [Green Version]
- Shyng, C.; Nelvagal, H.R.; Dearborn, J.T.; Tyynelä, J.; Schmidt, R.E.; Sands, M.S.; Cooper, J.D. Synergistic effects of treating the spinal cord and brain in CLN1 disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5920–E5929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passini, M.A.; Dodge, J.C.; Bu, J.; Yang, W.; Zhao, Q.; Sondhi, L.; Hackett, N.R.; Kaminsky, S.M.; Mao, Q.; Shihabuddin, L.S.; et al. Intracranial Delivery of CLN2 Reduces Brain Pathology in a Mouse Model of Classical Late Infantile Neuronal Ceroid Lipofuscinosis. J. Neurosci. 2006, 26, 1334–1342. [Google Scholar] [CrossRef] [Green Version]
- Sondhi, L.; Scott, E.C.; Chen, A.; Hackett, N.R.; Wong, A.M.; Kubiak, A.; Nelvagal, H.R.; Pearse, Y.; Cotman, S.L.; Cooper, J.D.; et al. Partial Correction of the CNS Lysosomal Storage Defect in a Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis by Neonatal CNS Administration of an Adeno-Associated Virus Serotype rh.10 Vector Expressing the Human CLN3 Gene. Hum. Gene Ther. 2014, 25, 223–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, S.; Kam, T.-I.; Lee, S.; Ham, S.; et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Baek, R.C.; Broekman, M.L.D.; Leroy, S.G.; Tierney, L.A.; Sandberg, M.A.; D’Azzo, A.; Seyfried, T.N.; Sena-Esteves, M. AAV-Mediated Gene Delivery in Adult GM1-Gangliosidosis Mice Corrects Lysosomal Storage in CNS and Improves Survival. PLoS ONE 2010, 5, e13468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargeant, T.J.; Wang, S.; Bradley, J.; Smith, N.J.; Raha, A.A.; McNair, R.; Ziegler, R.J.; Cheng, S.H.; Cox, T.M.; Cachón-González, M.B. Adeno-associated virus-mediated expression of β-hexosaminidase prevents neuronal loss in the Sandhoff mouse brain. Hum. Mol. Genet. 2011, 20, 4371–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafi, M.A.; Rao, H.Z.; Passini, M.A.; Curtis, M.; Vanier, M.T.; Zaka, M.; Luzi, P.; Wolfe, J.H.; Wenger, D.A. AAV-Mediated expression of galactocerebrosidase in brain results in attenuated symptoms and extended life span in murine models of globoid cell leukodystrophy. Mol. Ther. 2005, 11, 734–744. [Google Scholar] [CrossRef]
- Lin, D.; Fantz, C.R.; Levy, B.; Rafi, M.A.; Vogler, C.; Wenger, D.A.; Sands, M.S. AAV2/5 vector expressing galactocerebrosidase ameliorates CNS disease in the murine model of globoid-cell leukodystrophy more efficiently than AAV2. Mol. Ther. 2005, 12, 422–430. [Google Scholar] [CrossRef]
- Piguet, F.; Sondhi, D.; Piraud, M.; Fouquet, F.; Hackett, N.R.; Ahouansou, O.; Vanier, M.-T.; Bieche, I.; Aubourg, P.; Crystal, R.G.; et al. Correction of Brain Oligodendrocytes by AAVrh.10 Intracerebral Gene Therapy in Metachromatic Leukodystrophy Mice. Hum. Gene Ther. 2012, 23, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Desmaris, N.; Verot, L.; Puech, J.P.; Caillaud, C.; Heard, J.M. Prevention of neuropathology in the mouse model of hurler syndrome. Ann. Neurol. 2004, 56, 68–76. [Google Scholar] [CrossRef]
- Winner, L.K.; Beard, H.; Hassiotis, S.; Lau, A.A.; Luck, A.J.; Hopwood, J.J.; Hemsley, K.M. A Preclinical Study Evaluating AAVrh10-Based Gene Therapy for Sanfilippo Syndrome. Hum. Gene Ther. 2016, 27, 363–375. [Google Scholar] [CrossRef]
- Hocquemiller, M.; Hemsley, K.M.; Douglass, M.L.; Tamang, S.J.; Neumann, D.; King, B.M.; Beard, H.; Trim, P.J.; Winner, L.K.; Lau, A.A.; et al. AAVrh10 Vector Corrects Disease Pathology in MPS IIIA Mice and Achieves Widespread Distribution of SGSH in Large Animal Brains. Mol. Ther. Methods Clin. Dev. 2020, 17, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Cressant, A.; Desmaris, N.; Verot, L.; Bréjot, T.; Froissart, R.; Vanier, M.-T.; Maire, I.; Heard, J.M. Improved Behavior and Neuropathology in the Mouse Model of Sanfilippo Type IIIB Disease after Adeno-Associated Virus-Mediated Gene Transfer in the Striatum. J. Neurosci. 2004, 24, 10229–10239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldermon, C.D.; Ohlemiller, K.K.; Herzog, E.D.; Vogler, C.; Qin, E.; Wozniak, D.F.; Tan, Y.; Orrock, J.L.; Sands, M.S. Therapeutic Efficacy of Bone Marrow Transplant, Intracranial AAV-mediated Gene Therapy, or Both in the Mouse Model of MPS IIIB. Mol. Ther. 2010, 18, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Tordo, J.; O’Leary, C.; Antunes, A.S.L.M.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain 2018, 141, 2014–2031. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chen, Y.H.; He, X.; Martins, I.; Heth, J.A.; Chiorini, J.A.; Davidson, B.L. Adeno-associated Virus Type 5 Reduces Learning Deficits and Restores Glutamate Receptor Subunit Levels in MPS VII Mice CNS. Mol. Ther. 2007, 15, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Cearley, C.N.; Wolfe, J.H. A Single Injection of an Adeno-Associated Virus Vector into Nuclei with Divergent Connections Results in Widespread Vector Distribution in the Brain and Global Correction of a Neurogenetic Disease. J. Neurosci. 2007, 27, 9928–9940. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Macauley, S.L.; Huff, M.R.; Taksir, A.T.V.; Bu, J.; Wu, I.-H.; Piepenhagen, P.A.; Dodge, J.C.; Shihabuddin, L.S.; O’Riordan, C.R.; et al. AAV Vector-Mediated Correction of Brain Pathology in a Mouse Model of Niemann–Pick A Disease. Mol. Ther. 2005, 11, 754–762. [Google Scholar] [CrossRef]
- McCurdy, V.J.; Johnson, A.K.; Gray-Edwards, H.L.; Randle, A.N.; Brunson, B.L.; Morrison, N.E.; Salibi, N.; Johnson, J.A.; Hwang, M.; Beyers, R.J.; et al. Sustained Normalization of Neurological Disease after Intracranial Gene Therapy in a Feline Model. Sci. Transl. Med. 2014, 6, 231ra48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, A.M.; Cochran, J.N.; McCurdy, V.J.; Johnson, A.K.; Brunson, B.L.; Gray-Edwards, H.; Leroy, S.G.; Hwang, M.; Randle, A.N.; Jackson, L.S.; et al. Therapeutic Response in Feline Sandhoff Disease Despite Immunity to Intracranial Gene Therapy. Mol. Ther. 2013, 21, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Gray-Edwards, H.L.; Brunson, B.L.; Holland, M.; Hespel, A.-M.; Bradbury, A.M.; McCurdy, V.J.; Beadlescomb, P.M.; Randle, A.N.; Salibi, N.; Denney, T.S.; et al. Mucopolysaccharidosis-like phenotype in feline Sandhoff disease and partial correction after AAV gene therapy. Mol. Genet. Metab. 2015, 116, 80–87. [Google Scholar] [CrossRef]
- Ellinwood, N.M.; Ausseil, J.; Desmaris, N.; Bigou, S.; Liu, S.; Jens, J.K.; Snella, E.M.; Mohammed, E.E.A.; Thomson, C.B.; Raoul, S.; et al. Safe, Efficient, and Reproducible Gene Therapy of the Brain in the Dog Models of Sanfilippo and Hurler Syndromes. Mol. Ther. 2011, 19, 251–259. [Google Scholar] [CrossRef]
- Sondhi, D.; Peterson, D.A.; Giannaris, E.L.; Sanders, C.T.; Mendez, B.S.; De, B.; Rostkowski, A.B.; Blanchard, B.; Bjugstad, K.; Sladek, J.R.; et al. AAV2-mediated CLN2 gene transfer to rodent and non-human primate brain results in long-term TPP-I expression compatible with therapy for LINCL. Gene Ther. 2005, 12, 1618–1632. [Google Scholar] [CrossRef] [Green Version]
- Karumuthil-Melethil, S.; Marshall, M.S.; Heindel, C.; Jakubauskas, B.; Bongarzone, E.R.; Gray, S.J. Intrathecal administration of AAV/GALC vectors in 10-11-day-old twitcher mice improves survival and is enhanced by bone marrow transplant. J. Neurosci. Res. 2016, 94, 1138–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, G.; Bastacky, J.; Belichenko, P.; Buddhikot, M.; Jungles, S.; Vellard, M.; Mobley, W.C.; Kakkis, E. Intrathecal administration of AAV vectors for the treatment of lysosomal storage in the brains of MPS I mice. Gene Ther. 2006, 13, 917–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliger, S.S.; Elliger, C.A.; Aguilar, C.P.; Raju, N.R.; Watson, G.L. Elimination of lysosomal storage in brains of MPS VII mice treated by intrathecal administration of an adeno-associated virus vector. Gene Ther. 1999, 6, 1175–1178. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.L.; Gray-Edwards, H.; Murdock, B.; Taylor, A.; Brunson, B.; Randle, A.N.; Stocia, L.; Todessa, S.; Lata, J.; Sena-Esteves, M.; et al. 605. Cerebrospinal Fluid for Delivery of AAV Gene Therapy in GM1 Gangliosidosis. Mol. Ther. 2016, 24, S240. [Google Scholar] [CrossRef]
- 604. Intrathecal AAV9-Mediated Gene Delivery Corrects Lysosomal Storage Throughout the CNS in a Large Animal Model of Mucopolysaccharidosis Type I. Mol. Ther. 2014, 22, S233–S234. [CrossRef] [Green Version]
- Haurigot, V.; Marcó, S.; Ribera, A.; Garcia, M.; Ruzo, A.; Villacampa, P.; Ayuso, E.; Añor, S.; Andaluz, A.; Pineda, M.; et al. Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J. Clin. Investig. 2013, 123, 3254–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribera, A.; Haurigot, V.; Garcia, M.; Marcó, S.; Motas, S.; Villacampa, P.; Maggioni, L.; León, X.; Molas, M.; Sánchez, V.; et al. Biochemical, histological and functional correction of mucopolysaccharidosis Type IIIB by intra-cerebrospinal fluid gene therapy. Hum. Mol. Genet. 2015, 24, 2078–2095. [Google Scholar] [CrossRef] [Green Version]
- Roca, C.; Motas, S.; Marcó, S.; Ribera, A.; Sánchez, V.; Sánchez, X.; Bertolin, J.; León, X.; Pérez, J.; Garcia, M.; et al. Disease correction by AAV-mediated gene therapy in a new mouse model of mucopolysaccharidosis type IIID. Hum. Mol. Genet. 2017, 26, 1535–1551. [Google Scholar] [CrossRef]
- Hordeaux, J.; Dubreil, L.; Robveille, C.; Deniaud, J.; Pascal, Q.; Dequéant, B.; Pailloux, J.; Lagalice, L.; Ledevin, M.; Babarit, C.; et al. Long-term neurologic and cardiac correction by intrathecal gene therapy in Pompe disease. Acta Neuropathol. Commun. 2017, 5, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.Y.; Bagel, J.H.; O’Donnell, P.A.; Vite, C.H.; Wolfe, J.H. Clinical Improvement of Alpha-mannosidosis Cat Following a Single Cisterna Magna Infusion of AAV1. Mol. Ther. 2016, 24, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, C.; Bell, P.; Gurda, B.L.; Wang, Q.; Louboutin, J.-P.; Zhu, Y.; Bagel, J.; O’Donnell, P.; Sikora, T.; Ruane, T.; et al. Intrathecal Gene Therapy Corrects CNS Pathology in a Feline Model of Mucopolysaccharidosis I. Mol. Ther. 2014, 22, 2018–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, C.; Bell, P.; Louboutin, J.-P.; Zhu, Y.; Yu, H.; Lin, G.; Choa, R.; Gurda, B.L.; Bagel, J.; O’Donnell, P.; et al. Neonatal Systemic AAV Induces Tolerance to CNS Gene Therapy in MPS I Dogs and Nonhuman Primates. Mol. Ther. 2015, 23, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, C.; Bell, P.; Louboutin, J.-P.; Katz, N.; Zhu, Y.; Lin, G.; Choa, R.; Bagel, J.; O’Donnell, P.; Fitzgerald, C.A.; et al. Neonatal tolerance induction enables accurate evaluation of gene therapy for MPS I in a canine model. Mol. Genet. Metab. 2016, 119, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Gurda, B.L.; Lataillade, A.D.G.D.; Bell, P.; Zhu, Y.; Yu, H.; Wang, P.; Bagel, J.; Vite, C.H.; Sikora, T.; Hinderer, C.; et al. Evaluation of AAV-mediated Gene Therapy for Central Nervous System Disease in Canine Mucopolysaccharidosis VII. Mol. Ther. 2016, 24, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Holthaus, S.-M.K.; Herranz-Martin, S.; Massaro, G.; Aristorena, M.; Hoke, J.; Hughes, M.P.; Maswood, R.; Semenyuk, O.; Basche, M.; Shah, A.Z.; et al. Neonatal brain-directed gene therapy rescues a mouse model of neurodegenerative CLN6 Batten disease. Hum. Mol. Genet. 2019, 28, 3867–3879. [Google Scholar] [CrossRef]
- Johnson, T.B.; White, K.A.; Brudvig, J.J.; Cain, J.T.; Langin, L.; Pratt, M.A.; Booth, C.D.; Timm, D.J.; Davis, S.S.; Meyerink, B.; et al. AAV9 Gene Therapy Increases Lifespan and Treats Pathological and Behavioral Abnormalities in a Mouse Model of CLN8-Batten Disease. Mol. Ther. 2021, 29, 162–175. [Google Scholar] [CrossRef]
- Broekman, M.L.D.; Baek, R.C.; Comer, L.A.; Fernandez, J.L.; Seyfried, T.N.; Sena-Esteves, M. Complete Correction of Enzymatic Deficiency and Neurochemistry in the GM1-gangliosidosis Mouse Brain by Neonatal Adeno-associated Virus–mediated Gene Delivery. Mol. Ther. 2007, 15, 30–37. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Hirai, Y.; Miyake, K.; Shimada, T. Targeted gene transfer into ependymal cells through intraventricular injection of AAV1 vector and long-term enzyme replacement via the CSF. Sci. Rep. 2014, 4, 5506. [Google Scholar] [CrossRef] [Green Version]
- Hironaka, K.; Yamazaki, Y.; Hirai, Y.; Yamamoto, M.; Miyake, N.; Miyake, K.; Okada, T.; Morita, A.; Shimada, T. Enzyme replacement in the CSF to treat metachromatic leukodystrophy in mouse model using single intracerebroventricular injection of self-complementary AAV1 vector. Sci. Rep. 2015, 5, 13104. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.A.; Lenander, A.W.; Nan, Z.; Belur, L.R.; Whitley, C.B.; Gupta, P.; Low, W.C.; McIvor, R.S. Direct gene transfer to the CNS prevents emergence of neurologic disease in a murine model of mucopolysaccharidosis type I. Neurobiol. Dis. 2011, 43, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Janson, C.G.; Romanova, L.G.; Leone, P.; Nan, Z.; Belur, L.; McIvor, R.S.; Low, W.C. Comparison of Endovascular and Intraventricular Gene Therapy With Adeno-Associated Virus–α-L-Iduronidase for Hurler Disease. Neurosurg. 2013, 74, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Fraldi, A.; Hemsley, K.; Crawley, A.; Lombardi, A.; Lau, A.; Sutherland, L.; Auricchio, A.; Ballabio, A.; Hopwood, J.J. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 2007, 16, 2693–2702. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, N.C.; Cacace, V.; De Risi, M.; Maffia, V.; Strollo, S.; Tedesco, N.; Nusco, E.; Romagnoli, N.; Ventrella, D.; Huang, Y.; et al. Enhancing the Therapeutic Potential of Sulfamidase for the Treatment of Mucopolysaccharidosis IIIA. Mol. Ther. Methods Clin. Dev. 2019, 15, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Passini, M.A.; Watson, D.J.; Vite, C.H.; Landsburg, D.J.; Feigenbaum, A.L.; Wolfe, J.H. Intraventricular Brain Injection of Adeno-Associated Virus Type 1 (AAV1) in Neonatal Mice Results in Complementary Patterns of Neuronal Transduction to AAV2 and Total Long-Term Correction of Storage Lesions in the Brains of β-Glucuronidase-Deficient Mice. J. Virol. 2003, 77, 7034–7040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Martins, I.; Wemmie, J.A.; Chiorini, J.A.; Davidson, B.L. Functional Correction of CNS Phenotypes in a Lysosomal Storage Disease Model Using Adeno-Associated Virus Type 4 Vectors. J. Neurosci. 2005, 25, 9321–9327. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.P.; Smith, D.A.; Morris, L.; Fletcher, C.; Colaço, A.; Huebecker, M.; Tordo, J.; Palomar, N.; Massaro, G.; Henckaerts, E.; et al. AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann–Pick type C1 disease. Hum. Mol. Genet. 2018, 27, 3079–3098. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Tecedor, L.; Chen, Y.; Williamson, B.G.; Lysenko, E.; Wininger, F.A.; Young, W.M.; Johnson, G.C.; Whiting, R.E.H.; Coates, J.R.; et al. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci. Transl. Med. 2015, 7, 313ra180. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, N.L.; Russell, K.N.; Wellby, M.P.; Wicky, H.E.; Schoderboeck, L.; Barrell, G.K.; Melzer, T.R.; Gray, S.J.; Hughes, S.M.; Palmer, D.N. Longitudinal In Vivo Monitoring of the CNS Demonstrates the Efficacy of Gene Therapy in a Sheep Model of CLN5 Batten Disease. Mol. Ther. 2018, 26, 2366–2378. [Google Scholar] [CrossRef] [Green Version]
- Rockwell, H.E.; McCurdy, V.J.; Eaton, S.C.; Wilson, D.U.; Johnson, A.K.; Randle, A.N.; Bradbury, A.M.; Gray-Edwards, H.L.; Baker, H.J.; Hudson, J.A.; et al. AAV-Mediated Gene Delivery in a Feline Model of Sandhoff Disease Corrects Lysosomal Storage in the Central Nervous System. ASN Neuro 2015, 7. [Google Scholar] [CrossRef]
- Gray-Edwards, H.L.; Randle, A.N.; Maitland, S.A.; Benatti, H.R.; Hubbard, S.M.; Canning, P.F.; Vogel, M.B.; Brunson, B.L.; Hwang, M.; Ellis, L.E.; et al. Adeno-Associated Virus Gene Therapy in a Sheep Model of Tay–Sachs Disease. Hum. Gene Ther. 2018, 29, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.R.; Gray, S.J.; Quach, E.T.; Huang, J.W.; Leung, C.H.; Samulski, R.J.; Boulis, N.M.; Federici, T. Comparison of Adeno-Associated Viral Vector Serotypes for Spinal Cord and Motor Neuron Gene Delivery. Hum. Gene Ther. 2011, 22, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Federici, T.; Taub, J.S.; Baum, G.R.; Gray, S.J.; Grieger, J.C.; Matthews, K.A.; Handy, C.R.; Passini, M.A.; Samulski, R.J.; Boulis, N.M. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2011, 19, 852–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, S.J.; Kalburgi, S.N.; McCown, T.J.; Samulski, R.J. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013, 20, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, C.; Bell, P.; Vite, C.H.; Louboutin, J.-P.; Grant, R.; Bote, E.; Yu, H.; Pukenas, B.; Hurst, R.; Wilson, J.M. Widespread gene transfer in the central nervous system of cynomolgus macaques following delivery of AAV9 into the cisterna magna. Mol. Ther. Methods Clin. Dev. 2014, 1, 14051. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Dyer, C.; Goode, T.; Johansson, J.; Bell, P.; Richman, L.; Buza, E.; Wilson, J.M. Translational Feasibility of Lumbar Puncture for Intrathecal AAV Administration. Mol. Ther. Methods Clin. Dev. 2020, 17, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Bell, P.; Katz, N.; Vite, C.H.; Louboutin, J.-P.; Bote, E.; Yu, H.; Zhu, Y.; Casal, M.L.; Bagel, J.; et al. Evaluation of Intrathecal Routes of Administration for Adeno-Associated Viral Vectors in Large Animals. Hum. Gene Ther. 2018, 29, 15–24. [Google Scholar] [CrossRef]
- Cain, J.T.; Likhite, S.; White, K.A.; Timm, D.J.; Davis, S.S.; Johnson, T.B.; Dennys-Rivers, C.N.; Rinaldi, F.; Motti, D.; Corcoran, S.; et al. Gene Therapy Corrects Brain and Behavioral Pathologies in CLN6-Batten Disease. Mol. Ther. 2019, 27, 1836–1847. [Google Scholar] [CrossRef] [Green Version]
- Ohno, K.; Samaranch, L.; Hadaczek, P.; Bringas, J.R.; Allen, P.C.; Sudhakar, V.; Stockinger, D.E.; Snieckus, C.; Campagna, M.V.; Sebastian, W.S.; et al. Kinetics and MR-Based Monitoring of AAV9 Vector Delivery into Cerebrospinal Fluid of Nonhuman Primates. Mol. Ther. Methods Clin. Dev. 2019, 13, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Hordeaux, J.; Hinderer, C.; Goode, T.; Katz, N.; Buza, E.L.; Bell, P.; Calcedo, R.; Richman, L.K.; Wilson, J.M. Toxicology Study of Intra-Cisterna Magna Adeno-Associated Virus 9 Expressing Human Alpha-L-Iduronidase in Rhesus Macaques. Mol. Ther. Methods Clin. Dev. 2018, 10, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Katz, N.; Goode, T.; Hinderer, C.; Hordeaux, J.; Wilson, J.M. Standardized Method for Intra-Cisterna Magna Delivery Under Fluoroscopic Guidance in Nonhuman Primates. Hum. Gene Ther. Methods 2018, 29, 212–219. [Google Scholar] [CrossRef]
- Samaranch, L.; Salegio, E.A.; Sebastian, W.S.; Kells, A.P.; Foust, K.D.; Bringas, J.R.; Lamarre, C.; Forsayeth, J.; Kaspar, B.K.; Bankiewicz, K.S. Adeno-Associated Virus Serotype 9 Transduction in the Central Nervous System of Nonhuman Primates. Hum. Gene Ther. 2012, 23, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Samaranch, L.; Bringas, J.; Pivirotto, P.; Sebastian, W.S.; Forsayeth, J.; Bankiewicz, K.S. Cerebellomedullary Cistern Delivery for AAV-Based Gene Therapy: A Technical Note for Nonhuman Primates. Hum. Gene Ther. Methods 2016, 27, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Hordeaux, J.; Buza, E.L.; Jeffrey, B.; Song, C.; Jahan, T.; Yuan, Y.; Zhu, Y.; Bell, P.; Li, M.; Chichester, J.A.; et al. MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates. Sci. Transl. Med. 2020, 12, eaba9188. [Google Scholar] [CrossRef]
- Taghian, T.; Marosfoi, M.G.; Puri, A.S.; Cataltepe, O.; King, R.M.; Diffie, E.B.; Maguire, A.S.; Martin, D.R.; Fernau, D.; Batista, A.R.; et al. A Safe and Reliable Technique for CNS Delivery of AAV Vectors in the Cisterna Magna. Mol. Ther. 2020, 28, 411–421. [Google Scholar] [CrossRef]
- Nevoret, M.-L. TRGX-121 Gene Therapy for Severe Mucopolysaccharidosis Type II (MPS II): Interim Results of an Ongoing First in Human Trialitle. In Proceedings of the WORLDSymposium, Manchester Grand Hyatt, San Diego, CA, USA, 8–12 February 2021. [Google Scholar]
- Consiglio, A.; Quattrini, A.; Martino, S.; Bensadoun, J.C.; Dolcetta, D.; Trojani, A.; Benaglia, G.; Marchesini, S.; Cestari, V.; Oliverio, A.; et al. In vivo gene therapy of metachromatic leukodystrophy by lentiviral vectors: Correction of neuropathology and protection against learning impairments in affected mice. Nat. Med. 2001, 7, 310–316. [Google Scholar] [CrossRef]
- Penzien, J.M.; Kappler, J.; Herschkowitz, N.; Schuknecht, B.; Leinekugel, P.; Propping, P.; Tønnesen, T.; Lou, H.; Moser, H.; Zierz, S.; et al. Compound heterozygosity for metachromatic leukodystrophy and arylsulfatase A pseudodeficiency alleles is not associated with progressive neurological disease. Am. J. Hum. Genet. 1993, 52, 557–564. [Google Scholar]
- Ghodsi, A.; Stein, C.; Derksen, T.; Yang, G.; Anderson, R.D.; Davidson, B.L. Extensiveβ-Glucuronidase Activity in Murine Central Nervous System after Adenovirus-Mediated Gene Transfer to Brain. Hum. Gene Ther. 1998, 9, 2331–2340. [Google Scholar] [CrossRef]
- Bru, T.; Salinas, S.; Kremer, E.J. An Update on Canine Adenovirus Type 2 and Its Vectors. Viruses 2010, 2, 2134–2153. [Google Scholar] [CrossRef] [Green Version]
- Ariza, L.; Giménez-Llort, L.; Cubizolle, A.; Pagès, G.; García-Lareu, B.; Serratrice, N.; Cots, D.; Thwaite, R.; Chillón, M.; Kremer, E.J.; et al. Central Nervous System Delivery of Helper-Dependent Canine Adenovirus Corrects Neuropathology and Behavior in Mucopolysaccharidosis Type VII Mice. Hum. Gene Ther. 2014, 25, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Cubizolle, A.; Serratrice, N.; Skander, N.; Colle, M.-A.; Ibanes, S.; Gennetier, A.; Bayo-Puxan, N.; Mazouni, K.; Mennechet, F.; Joussemet, B.; et al. Corrective GUSB Transfer to the Canine Mucopolysaccharidosis VII Brain. Mol. Ther. 2014, 22, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.A.; Hopwood, J.J.; Kremer, E.J.; Hemsley, K.M. SGSH gene transfer in mucopolysaccharidosis type IIIA mice using canine adenovirus vectors. Mol. Genet. Metab. 2010, 100, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Beucher, B.; Lavigne, M.; Wehbi, A.; Dopeso-Reyes, I.G.; Saggio, I.; Kremer, E.J. CAV-2 Vector Development and Gene Transfer in the Central and Peripheral Nervous Systems. Front. Mol. Neurosci. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Zussy, C.; Loustalot, F.; Junyent, F.; Gardoni, F.; Bories, C.; Valero, J.; Desarménien, M.G.; Bernex, F.; Henaff, D.; Bayo-Puxan, N.; et al. Coxsackievirus Adenovirus Receptor Loss Impairs Adult Neurogenesis, Synapse Content, and Hippocampus Plasticity. J. Neurosci. 2016, 36, 9558–9571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.B.; Cain, J.T.; White, K.A.; Ramirez-Montealegre, D.; Pearce, D.A.; Weimer, J.M. Therapeutic landscape for Batten disease: Current treatments and future prospects. Nat. Rev. Neurol. 2019, 15, 161–178. [Google Scholar] [CrossRef]
- Holthaus, S.-M.K.; Ribeiro, J.; Abelleira-Hervas, L.; Pearson, R.A.; Duran, Y.; Georgiadis, A.; Sampson, R.D.; Rizzi, M.; Hoke, J.; Maswood, R.; et al. Prevention of Photoreceptor Cell Loss in a Cln6 Mouse Model of Batten Disease Requires CLN6 Gene Transfer to Bipolar Cells. Mol. Ther. 2018, 26, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Davidson, B.L. Phenotype correction in retinal pigment epithelium in murine mucopolysaccharidosis VII by adenovirus-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1995, 92, 7700–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennig, A.K.; Ogilvie, J.M.; Ohlemiller, K.K.; Timmers, A.M.; Hauswirth, W.W.; Sands, M.S. AAV-mediated intravitreal gene therapy reduces lysosomal storage in the retinal pigmented epithelium and improves retinal function in adult MPS VII mice. Mol. Ther. 2004, 10, 106–116. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, H.; Franco, L.M.; Brown, T.; Bird, A.; Schneider, A.; Koeberl, D.D. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol. Ther. 2005, 11, 889–898. [Google Scholar] [CrossRef]
- Todd, A.G.; Bsc, J.A.M.; Grange, R.W.; Fuller, D.D.; Walter, G.A.; Byrne, B.J.; Falk, D.J. Correcting Neuromuscular Deficits With Gene Therapy in Pompe Disease. Ann. Neurol. 2015, 78, 222–234. [Google Scholar] [CrossRef] [Green Version]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef] [Green Version]
- Mah, C.S.; Falk, D.J.; Germain, S.A.; Kelley, J.S.; Lewis, M.A.; Cloutier, D.A.; DeRuisseau, L.R.; Conlon, T.J.; Cresawn, K.O.; Fraites, T.J.F., Jr.; et al. Gel-mediated Delivery of AAV1 Vectors Corrects Ventilatory Function in Pompe Mice With Established Disease. Mol. Ther. 2010, 18, 502–510. [Google Scholar] [CrossRef] [Green Version]
- McCall, A.L.; Elmallah, M.K. Macroglossia, Motor Neuron Pathology, and Airway Malacia Contribute to Respiratory Insufficiency in Pompe Disease: A Commentary on Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases. Int. J. Mol. Sci. 2019, 20, 751. [Google Scholar] [CrossRef] [Green Version]
- ElMallah, M.K.; Falk, D.J.; Nayak, S.; Federico, R.A.; Sandhu, M.S.; Poirier, A.; Byrne, B.J.; Fuller, D.D. Sustained Correction of Motoneuron Histopathology Following Intramuscular Delivery of AAV in Pompe Mice. Mol. Ther. 2014, 22, 702–712. [Google Scholar] [CrossRef]
- Falk, D.J.; Mah, C.S.; Soustek, M.S.; Lee, K.-Z.; Elmallah, M.K.; Cloutier, D.A.; Fuller, D.D.; Byrne, B.J. Intrapleural Administration of AAV9 Improves Neural and Cardiorespiratory Function in Pompe Disease. Mol. Ther. 2013, 21, 1661–1667. [Google Scholar] [CrossRef] [Green Version]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, H.H.Y.C.N.R.P.M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Leal, A.F.; Espejo-Mojica, A.J.; Sánchez, O.F.; Ramírez, C.M.; Reyes, L.H.; Cruz, J.C.; Alméciga-Díaz, C.J. Lysosomal storage diseases: Current therapies and future alternatives. J. Mol. Med. 2020, 98, 931–946. [Google Scholar] [CrossRef]
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In Vivo Genome Editing as a Therapeutic Approach. Int. J. Mol. Sci. 2018, 19, 2721. [Google Scholar] [CrossRef] [Green Version]
- Rouet, P.; Smih, F.; Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. USA 1994, 91, 6064–6068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, P.; Klein, H.L. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durai, S. Zinc finger nucleases: Custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005, 33, 5978–5990. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Pabo, C.O. Zinc finger-DNA recognition: Crystal structure of a Zif268-DNA complex at 2.1 A. Science 1991, 252, 809–817. [Google Scholar] [CrossRef]
- Liu, Q.; Segal, D.J.; Ghiara, J.B.; Barbas, C.F. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 5525–5530. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.A.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015, 126, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; DeKelver, R.C.; Rohde, M.; Tom, S.; Radeke, R.; Martin, S.J.S.; Santiago, Y.; Sproul, S.; Przybilla, M.J.; Koniar, B.L.; et al. ZFN-Mediated In Vivo Genome Editing Corrects Murine Hurler Syndrome. Mol. Ther. 2019, 27, 178–187. [Google Scholar] [CrossRef]
- Laoharawee, K.; DeKelver, R.C.; Podetz-Pedersen, K.M.; Rohde, M.; Sproul, S.; Nguyen, H.-O.; Nguyen, T.; Martin, S.J.S.; Ou, L.; Tom, S.; et al. Dose-Dependent Prevention of Metabolic and Neurologic Disease in Murine MPS II by ZFN-Mediated In Vivo Genome Editing. Mol. Ther. 2018, 26, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
- Scholze, H.; Boch, J. TAL effectors are remote controls for gene activation. Curr. Opin. Microbiol. 2011, 14, 47–53. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xiangdong, M.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2010, 29, 143–148. [Google Scholar] [CrossRef]
- Ramalingam, S.; Annaluru, N.; Kandavelou, K.; Chandrasegaran, S. TALEN-mediated generation and genetic correction of disease-specific human induced pluripotent stem cells. Curr. Gene Ther. 2014, 14, 461–472. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Broeders, M.; Herrero-Hernandez, P.; Ernst, M.P.; van der Ploeg, A.T.; Pijnappel, W.P. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience 2020, 23, 100789. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; Przybilla, M.J.; Ahlat, O.; Kim, S.; Overn, P.; Jarnes, J.; O’Sullivan, M.G.; Whitley, C.B. A Highly Efficacious PS Gene Editing System Corrects Metabolic and Neurological Complications of Mucopolysaccharidosis Type I. Mol. Ther. 2020, 28, 1442–1454. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Tăbăran, A.-F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay–Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236. [Google Scholar] [CrossRef]
- Karumuthil-Melethil, S.; Kalburgi, S.N.; Thompson, P.; Tropak, M.; Kaytor, M.D.; Keimel, J.G.; Mark, B.L.; Mahuran, D.; Walia, J.S.; Gray, S.J. Novel Vector Design and Hexosaminidase Variant Enabling Self-Complementary Adeno-Associated Virus for the Treatment of Tay-Sachs Disease. Hum. Gene Ther. 2016, 27, 509–521. [Google Scholar] [CrossRef]
- Osmon, K.J.; Woodley, E.; Thompson, P.; Ong, K.; Karumuthil-Melethil, S.; Keimel, J.G.; Mark, B.L.; Mahuran, D.; Gray, S.J.; Walia, J.S. Systemic Gene Transfer of a Hexosaminidase Variant Using an scAAV9.47 Vector Corrects GM2Gangliosidosis in Sandhoff Mice. Hum. Gene Ther. 2016, 27, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Schuh, R.S.; de Carvalho, T.G.; Giugliani, R.; Matte, U.; Baldo, G.; Teixeira, H.F. Gene editing of MPS I human fibroblasts by co-delivery of a CRISPR/Cas9 plasmid and a donor oligonucleotide using nanoemulsions as nonviral carriers. Eur. J. Pharm. Biopharm. 2018, 122, 158–166. [Google Scholar] [CrossRef]
- Miki, T.; Vazquez, L.; Yanuaria, L.; Lopez, O.; Garcia, I.M.; Ohashi, K.; Rodriguez, N.S. Induced Pluripotent Stem Cell Derivation and Ex Vivo Gene Correction Using a Mucopolysaccharidosis Type 1 Disease Mouse Model. Stem Cells Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.; Yeh, W.-H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020, 4, 97–110. [Google Scholar] [CrossRef]
- Truong, D.-J.J.; Kühner, K.; Kühn, R.; Werfel, S.; Engelhardt, S.; Wurst, W.; Ortiz, O. Development of an intein-mediated split–Cas9 system for gene therapy. Nucleic Acids Res. 2015, 43, 6450–6458. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nat. Cell Biol. 2019, 576, 149–157. [Google Scholar] [CrossRef]



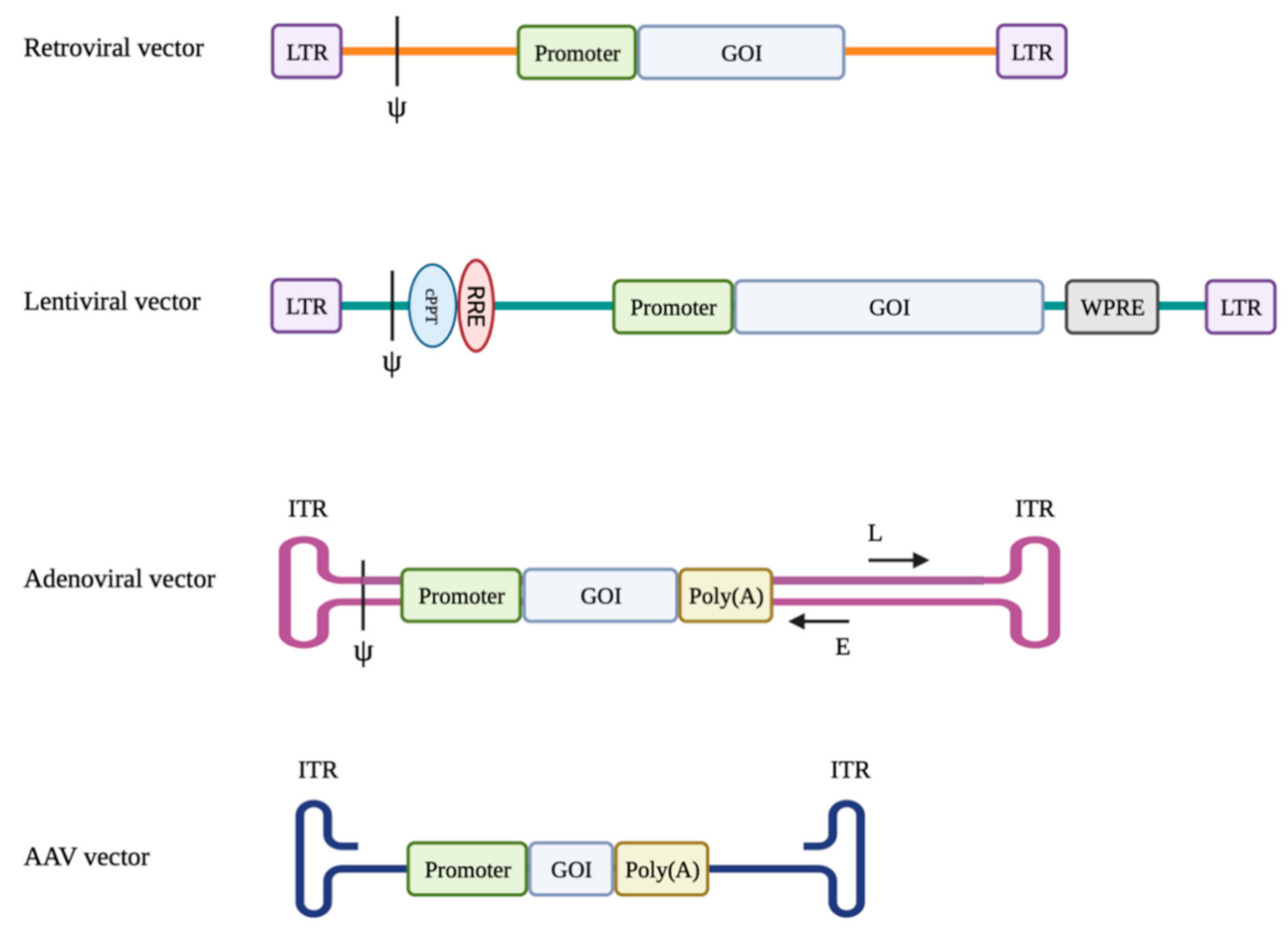{kind=link}
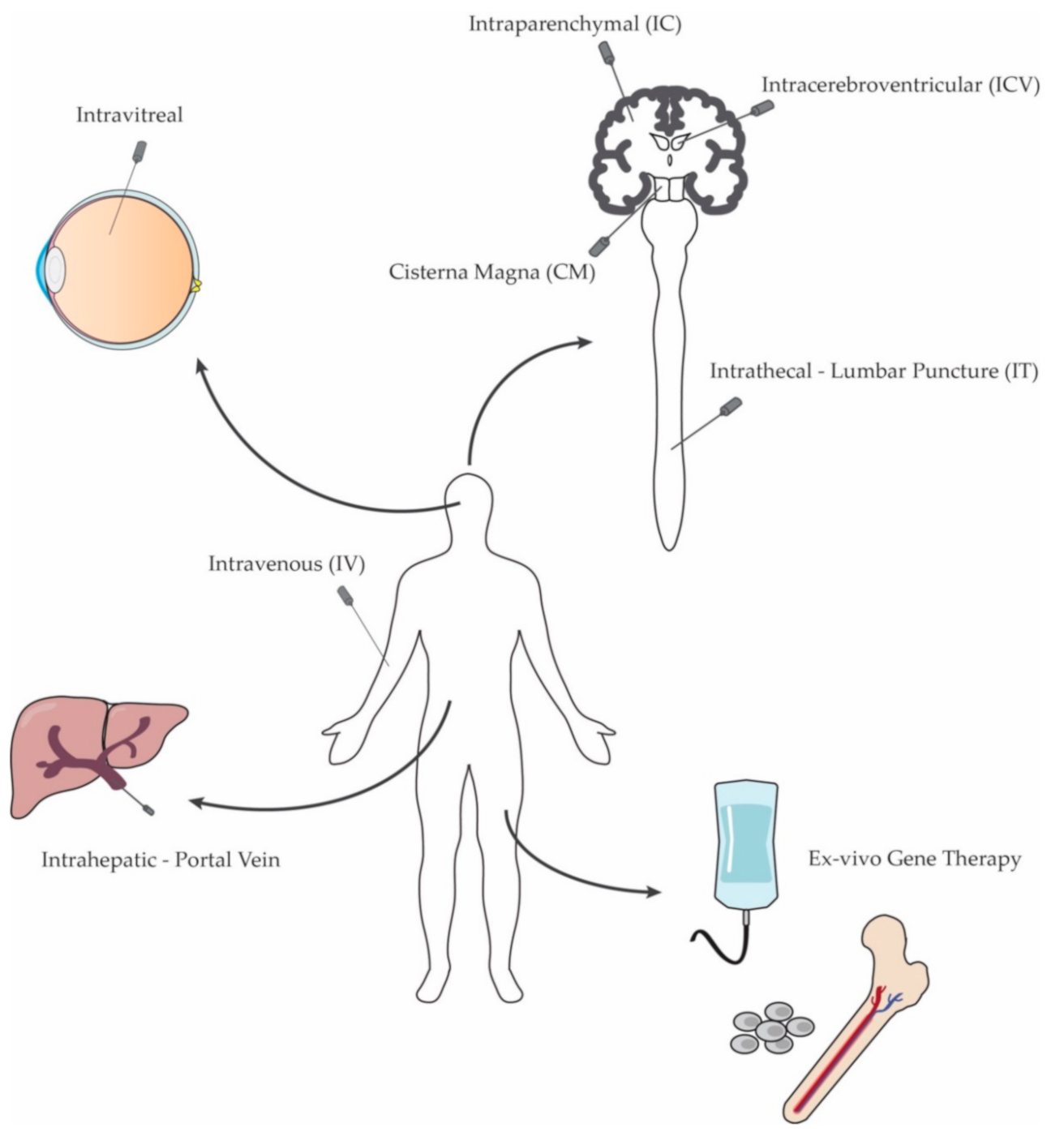{kind=link}
| Vector | Size (nm) | Genome | Packaging Capacity (Kb) | Cell Type | Integration Profile |
|---|---|---|---|---|---|
| Retroviral | 100 | RNA | 8 | Dividing | Integrating |
| Lentiviral | 100 | RNA | 14 | Dividing and non-dividing | Integrating |
| Adenoviral | 70–100 | dsDNA | 8–10 | Dividing and non-dividing | Non-integrating |
| AAV | 25–20 | ssDNA | 4.7 | Dividing and non-dividing | Non-integrating |
| Disease | NCT Ref. | Intervention | Status | Est. Participants |
|---|---|---|---|---|
| MLD | NCT01560182 | OTL-200 (autologous CD34+ enriched cell fraction transduced with human ARSA LV) | Phase 1/2; Active, not recruiting | 20 |
| MLD | NCT03392987 | OTL-200 | Phase 2; Active, not recruiting | 10 |
| MLD | NCT04283227 | OTL-200 | Phase 3; Recruiting | 6 |
| MLD | NCT02559830 | Transduced CD34+ autologous HSCs | Phase 1/2; Recruiting | 50 |
| MPS IIIA | NCT04201405 | Autologous CD34+ cells transduced with the human SGSH LV | Phase 1/2; Recruiting | 5 |
| MPS I-H | NCT03488394 | Autologous CD34+ cells transduced with human IDUA LV | Phase 1/2; Recruiting | 8 |
| Cystinosis | NCT03897361 | CTNS-RD-04 (autologous CD34+ enriched cells transduced with human CTNS LV) | Phase 1/2; Recruiting | 6 |
| Fabry | NCT03454893 | AVR-RD-01 (autologous CD34+ cell-enriched fraction transduced with human AGA LV) | Phase 1/2; Recruiting | 12 |
| Gaucher | NCT04145037 | AVR-RD-02 (autologous CD34+ enriched HSCs transduced with human GBA gene LV) | Phase 1/2; Recruiting | 16 |
| Delivery Route | Disease | Species | Model | Vector | Dose | Time of injection | Ref. |
|---|---|---|---|---|---|---|---|
| Tail Vein | |||||||
| Gaucher | Mouse | UBC-creERT2-Gbaflox/flox | AAV9-CMV-Gba or AAV9-SYN-Gba | 3–5 × 1011 vg | 4 weeks | [134] | |
| GM1 | Mouse | C57BL/6J-βgal−/− | AAV9-CAG-βgal | 1 × 1011 or 3 × 1011 vg | 6 weeks | [135] | |
| GM2 (Sandhoff) | Mouse | C57BL/6:C129-Hexb−/− | AAV9-CMV-HEXB | 2.5 × 1014 or 3.5 × 1013 vg/kg | P1 or 6 weeks | [136] | |
| GM2 (Sandhoff) | Mouse | Hexb−/− | AAV9 or PHP.B-BiCBA-mHEXA/mHEXB, AAV9 or PHP.B-P2-HEX | AAV9: 1 × 1012–4 × 1012 vg; PHP.B: 3 × 1011–1 × 1012 vg | 4 weeks | [137] | |
| Krabbe | Mouse | C57BL-twi/twi | AAVhr10-CAG-mGALC | 2 × 1011 vg | P10–12 | [138] | |
| MPS I | Mouse | C57BL/6-Idua−/− | AAV9-CAG-IDUA or AAVrh10-CAG-IDUA | 1 × 1012 vg | 8–21 weeks | [139] | |
| MPS II | Mouse | idsy/- | AAV8-EF1α-IDS | 1 × 1011 vg | NA | [140] | |
| MPS IIIA | Mouse | C57BL/6-Sgsh−/− | scAAVrh74-U1a-hSGSH | 5 × 1012 vg/kg | 4–6 weeks | [141] | |
| MPS IIIA | Mouse | C57BL/6-SgshG91A | AAV9-CAG-mSGSH | 1 × 1012 vg | 2 months | [142] | |
| MPS IIIB | Mouse | C57BL/6-Naglu−/− | AAV9-CAG-hNAGLU | 1 × 1013 vg/kg | 4–6 weeks | [143] | |
| MPS IIIB | Mouse | C57BL/6-Naglu−/− | AAV2-CMV-hNAGLU | 4 × 1011 vg | 4–6 weeks | [144] | |
| MPS VII | Mouse | b6.C-H-abm1/byBir-gusmps/mps | AAV9-βGluc | 1 × 1012 vg | 6 weeks | [145] | |
| Temporal/Cephalic Vein | |||||||
| Gaucher | Mouse | CD1-K14-ln/ln | AAV9-GUSB-hGBA1 | 4 × 1011 gc | P0–P1 | [146] | |
| Gaucher | Mouse | CD1-K14-ln/ln | AAV9-SYN-hGBA1 | 2.4 × 1012 vg | P0–P1 | [147] | |
| MPS I | Mouse | Idua−/− | AAV2-CAG-hIDUA | 1010 vp | P1–P2 | [148] | |
| MPS II | Mouse | idsy/- | AAV5-CMV-hIDS | 1 × 1011 vg | P2 | [149] | |
| MPS VII | Mouse | B6.C-H-2bm1/ByBir-gusmps/mps | AAV-CAG-hGUSB | 5 × 109 iu/kg | P2 | [150] | |
| MPS VII | Mouse | B6.C-H-2bm1/ByBir-gusmps/mps | AAV-CAG-hGUSB | 8 × 1010 iu/kg | P2 | [151] | |
| MSD | Mouse | C57B6/S129J-Sgsh−/− | AAV9-CMV-hSUMF1 | 2 × 1011 vg | P1 | [152] | |
| Jugular Vein | |||||||
| MLD | Mouse | C57BL/6-ASA−/− | AAV9-CAG-hARSA | 2 × 1012 vg | P1–2 | [153] | |
| Retro-orbital Sinus | |||||||
| Pompe | Mouse | C57BL/6J-GAA−/− | AAV-PHP.B-CAG-hGAA | 5 × 1012 vg/kg | 2 weeks | [154] | |
| Combination | |||||||
| IV + ICV | MSD | Mouse | C57B6/S129J-Sgsh−/− | AAV9-CMV-hSUMF1 | IV: 2 × 1011 vg; ICV: 6 × 109 vg | P1 | [152] |
| IV + IT | MPS VII | Mouse | C57BL6/J-Gusb/b | AAV2-CMV-GUSB | IV: 4.1 × 1011 vg; IT: 3 × 1010 vg | P3–P4 | [155] |
| IV + CM | MPS IIIB | Mouse | C57BL/6-Naglu−/− | AAV2-CMV-hNAGLU | IV: 4 × 1011 vg; CM: 5 × 1010 vg | 4–8 weeks | [156] |
| Delivery Route | Disease | Species | Model | Vector | Dose | Time of injection | Ref. |
|---|---|---|---|---|---|---|---|
| Intraparenchymal (IC) | |||||||
| CLN1 Batten | Mouse | C57BL/6-Ptt1−/− | AAV9-CAG-hPTT1 | 1 × 1012 vg/mL | P1 | [191] | |
| CLN2 Batten | Mouse | C57BL/6-CLN2−/− | AAV2 or AAV5-CAG-hCLN2 | 3.6 × 109 gc | 6 or 10 weeks | [192] | |
| CLN2 Batten | Mouse | C57BL/6:129Sv-TPPI−/− | AAV2 or AAV5-CAG-hCLN2 | 1.2–3.6 × 109 gc | 6 or 10 weeks | [192] | |
| CLN3 Batten | Mouse | C57BL/6N-Cln3Δex7/8 | AAVrh10-CAG-hCLN3 | 3 × 1010 gc | P2 | [193] | |
| Gaucher | Mouse | GBAL444P/L444P | AVV5-CAG-hGBA | 3.5 × 1013 vg/mL | 8 months | [194] | |
| GM1 | Mouse | C57BL/6-βgal−/− | AAV1-CAG-mβgal | 4 × 1010 gc | 6–8 weeks | [195] | |
| GM2 (Sandhoff) | Mouse | C57BL/6-Hexb−/− | AAV1-HEXA and AAV1-HEXB | 9.9 × 109 and 1.4 × 1010 gc | 1 month | [196] | |
| Krabbe | Mouse | C57BL/6-twiW339X or C57BL/129SVJ/FVB/N-twi-trs | AAV1-CMV-mGALC | 3 × 1010 vp | P0–P1 | [197] | |
| Krabbe | Mouse | B6.CE-Galctwi/J | AAV2 or AAV5-CAG-GALC | 5.5 × 106 iu | P3 | [198] | |
| MLD | Mouse | C57BL/6J-ASA−/− | AAVrh10-CAG-hARSA or AAV5-PGK-hARSA | 2.3 × 109 vg | 8 and 16 months | [199] | |
| MPS I | Mouse | C57BL/6-Idua−/− | AAV2 or AAV5-PGK-hIDUA | 109 vg | 6–7 weeks | [200] | |
| MPS I | Mouse | C57BL/6-Idua−/− | AAV2 or AVA5-mPGK-hIDUA | 109 vg | 6–7 weeks | [200] | |
| MPS IIIA | Mouse | C57BL/6-Sgsh−/− | AAVrh10-PGK-SGSH-IRES-SUMF1 | 7.5 × 109 gc | 5 weeks | [201] | |
| MPS IIIA | Mouse | C57BL/6-Sgsh−/− | AAVrh10-CMV-SGSH-IRES-SUMF1 | 7.5 × 109 gc | 5 weeks | [201] | |
| MPS IIIA | Mouse | C57BL/6-Sgsh−/− | AAVrh10-CAG-hSGSH | 8.4 × 108, 4.1 × 1010, 9 × 1010 vg | 5–6 weeks | [202] | |
| MPS IIIB | Mouse | C57BL/6-Naglu−/− | AAV2 or AAV5-mPGK-hNAGLU | 109 vg | 6 weeks | [203] | |
| MPS IIIB | Mouse | C57BL/6-Naglu−/− | AAV5-CAG-hNAGLU | 1.8 × 1010 vp | P2–4 | [204] | |
| MPS IIIC | Mouse | Hgsnat−/− | AAV2TT-CAG-hcoHGSNAT | 5.2 × 109 vg | 8 weeks | [205] | |
| MPS VII | Mouse | B6.C-H-2bm1/byBirgusmps/+ | AAV5-RSV-GUSB | 3 × 109 vg | 6–8 weeks | [206] | |
| MPS VII | Mouse | C3H/HeOuJ-Gusbmps-2J | AAV1, AAV9 or AAVrh10-GUSB-hGUSB | 1.2–1.3 × 1010 vg | >2 months | [207] | |
| NP-A | Mouse | C57BL/6-ASMKO−/− | AVV2-CMV-hASM | 1.86 × 1010 gc | 10 weeks | [208] | |
| GM1 | Cat | fGM1 | AAV1 or AAVrh8-CAG-fβgal | 3 × 1011, 4 × 1012 or 1.2 × 1013 vg | 1.3–3 months | [209] | |
| GM2 (Sandhoff) | Cat | fGM2 | AAV1-CAG-fHEXA/B or AAV8-DC172-HEXA/B | 3 × 1011–3.2 × 1012 vg | 0.9–2.6 months | [210] | |
| GM2 (Sandhoff) | Cat | fGM2 | AAVrh8-CAG-HEXA, AAVrh8-CAG-HEXB | 1.6–4.5 × 1011 vg | 1 month | [211] | |
| MPS I | Dog | IDUA−/− | AAV5-PGK-hIDUA | 5 × 1011–2.1 × 1012 vg | 18–30 months | [212] | |
| MPS IIIB | Dog | Schipperke Naglu−/− | AAV5-PGK-hNAGLU | 5 × 1011–2.1 × 1012 vg | 18–30 months | [212] | |
| CLN2 Batten | Rat | Fisher 344 | AAV2-CAG-TPP1 | 109–1010 pu | NA | [213] | |
| CLN2 Batten | NHP | C. sabaeus | AAV2-CAG-TPP1 | 3.6 × 1011 pu | 5–10 years | [213] | |
| Intrathecal (IT) | |||||||
| Krabbe | Mouse | B6.CE-Galctwi/J | AAV9, AAVrh10 or AAVOlig001-CAGGS-mβgal | 2 × 1011 vg | P10–11 | [214] | |
| MPS I | Mouse | C57BL/6-IDUA−/− | AAV2-CMV-hIDUA | 2 × 109–4 × 1010 vp | 2–4 months | [215] | |
| MPS VII | Mouse | Gusb−/− | AAV2-CMV-mGUSB | 1 × 1011 and 5 × 1011 vp | P3 and 7–13 weeks | [216] | |
| GM1 | Cat | βgal−/− | AAVrh10-βgal | 1 × 1012 vg/kg | NA | [217] | |
| MPS I | Cat | IDUA−/− | AVV9-IDUA | 1 × 1012 gc/kg | NA | [218] | |
| Cisterna Magna (CM) | |||||||
| MPS IIIA | Mouse | C57BL/6-SGSHD31N | AAV9-CAG-mSgsh | 5 × 109–5 × 1010 vg | 2 months | [219] | |
| MPS IIIB | Mouse | C57BL/6-Naglu−/− | AVV9-CAG-mNaglu | 3 × 1010 vg | 2 months | [220] | |
| MPS IIID | Mouse | C57BL/6NTac-Gnstm1e(EUCOMM)Hmgu | AAV9-CAG-mGns | 5 × 1010 vg | 2 months | [221] | |
| Pompe | Mouse | 6neo/6neo | AAV9 or AAVrh10-CAG-hGAA | 1011 vg | 1 month | [222] | |
| AMD | Cat | MANB−/− | AAV1-GUSB-fMANB | 1 × 1013 gc | 4–6 weeks | [223] | |
| GM1 | Cat | βgal−/− | AAVrh10-βgal | 1 × 1012 vg/kg | NA | [217] | |
| MPS I | Cat | IDUA−/− | AAV9-CB-fIDUA, AAV9-CMV-fIDUA | 1012 gc/kg | 4–7 months | [224] | |
| MPS I | Dog | IDUA−/− | AAV9-CAG-IDUA | 1012 gc/kg | 1 month | [225] | |
| MPS I | Dog | IDUA−/− | AAV9-CAG-IDUA | 1011–1012 gc/kg | 1 month | [226] | |
| MPS VII | Dog | GUSB−/− | AAV9 or AAVrh10-CAG-caGUSB | 5 × 1012 gc/kg | 3 weeks | [227] | |
| Intracerebroventricular (ICV) | |||||||
| CLN6 Batten | Mouse | Cln6nclf | AAV9-CMV-hCLN6 | 5 × 1010–5 × 1011 vg | P0 | [228] | |
| CLN8 Batten | Mouse | C57BL/6J-Cln8mnd | AAV9.pT-MecP2.CLN8 | 5 × 1010 vg | P0–P1 | [229] | |
| Gaucher | Mouse | CD1-K14-ln/ln | AAV9-GUSB-hGBA1 | 5 × 1010 gc | E16 and P0 | [146] | |
| GM1 | Mouse | βgal−/− | AAV1-CAG-mβgal | 3.2 × 109 vg | P0 | [230] | |
| Krabbe | Mouse | C57BL/6-twiW339X or C57BL/129SVJ/FVB/N-twi-trs | AAV1-CMV-mGALC | 3 × 1010 vp | P0–P1 | [197] | |
| MLD | Mouse | C57BL/6J-Asratm1Gie | AAV1-CAG-ARSA | 2 × 1011 vg | 8–12 weeks | [231] | |
| MLD | Mouse | C57BL/6J-Asratm1Gie | AAV1, 9-CAG-ARSA | 1.1 × 1011–2.3 × 1011 vg | 8 and 18 weeks | [232] | |
| MPS I | Mouse | C57BL/6-IDUA−/− | AAV8-CAG-hIDUA | 2 × 1010 vg | P4-P6 | [233] | |
| MPS I | Mouse | C57BL/6-IDUAnull | AAV5-CBA-hIDUA | 1 × 1011 vp | 8–11 weeks | [234] | |
| MPS IIIA | Mouse | B6Cg-Sgsh(mps3/PstJ) | AAV5-CMV-SGSH-IRES-SUMF1 | 6 × 109–3 × 1010 vp | P0 | [235] | |
| MPS III A | Mouse | B6Cg-Sgsh(mps3a/PstJ) | AAV9-CMV-IDSspSGSH-IRES-SUMF1 | 5.4 × 1012 gc/kg | P60 | [236] | |
| MPS VII | Mouse | C3H/HeOuJ-Gusbmps-2J | AVV1, 2, 5-GUSB-GUSB | 1.8 × 1010 vg | P0 | [237] | |
| MPS VII | Mouse | B6.C-H-2bm1/byBirgusmps/+ | AAV4-RSV-hGUSB | 1 × 1010 vg | 6–8 weeks | [238] | |
| MSD | Mouse | C57B6/S129J-Sumf−/− | AAV4, 9-CMV-SUMF1 | 1.2 × 1010 vg | P1 | [152] | |
| NP-C | Mouse | BALB/cNctr-Npc1m1N/J | AAV9-hSYN-hNPC1 | 4.6 × 109 vg | P0 | [239] | |
| GM1 | Cat | βgal−/− | AAVrh10-βgal | 1 × 1012 vg/kg | NA | [217] | |
| CLN2 Batten | Dog | Dachshunds TPP1null | AAV2-CAG-caTPP1 | 1.6 × 107 vg | 3 months | [240] | |
| CLN5 Batten | Sheep | CLN5−/− | AAV9-CBh-CLN5 | 4 × 1012 vg | 2–3 months | [241] | |
| Combination | |||||||
| IC + IT | CLN1 Batten | Mouse | C57BL/6-Ptt1−/− | AAV9-CAG-hPTT1 | 1 × 1012 vg/ml | P1 | [191] |
| IC + ICV | GM2 (Sandhoff) | Cat | fGM2 | AAVrh8-CAG-HEXA/B | 1.1 × 1012 vg | 1.1–1.6 months | [242] |
| IC + ICV | GM2 (Sandhoff) | Cat | fGM2 | AAVrh8-CAG-HEXA, AAVrh8-CAG-HEXB | IC: 1.6–4.5 × 1011 vg; ICV: 6.4 × 1011 vg | 1 month | [211] |
| CM + ICV | CLN2 Batten | Dog | Dachshunds TPP1null | AAV2-CAG-caTPP1 | 1.6 × 107 vg | 3 months | [240] |
| IC + ICV | CLN5 Batten | Sheep | CLN5−/− | AAV9-CBh-CLN5 | 3.1 × 1012 vg | 2–3 months | [241] |
| IC + ICV | GM2 (Tay-Sachs) | Sheep | TSD | AAVrh8-CAG-HEXA, AAVrh8-CAG-HEXA/B | 6.3 × 1012 vg, 4.2 × 1012–1.3 × 1013 vg | 2–4 months | [243] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massaro, G.; Geard, A.F.; Liu, W.; Coombe-Tennant, O.; Waddington, S.N.; Baruteau, J.; Gissen, P.; Rahim, A.A. Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development. Biomolecules 2021, 11, 611. https://doi.org/10.3390/biom11040611
Massaro G, Geard AF, Liu W, Coombe-Tennant O, Waddington SN, Baruteau J, Gissen P, Rahim AA. Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development. Biomolecules. 2021; 11(4):611. https://doi.org/10.3390/biom11040611
Chicago/Turabian StyleMassaro, Giulia, Amy F. Geard, Wenfei Liu, Oliver Coombe-Tennant, Simon N. Waddington, Julien Baruteau, Paul Gissen, and Ahad A. Rahim. 2021. "Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development" Biomolecules 11, no. 4: 611. https://doi.org/10.3390/biom11040611