Deoxycholic Acid Upregulates Serum Golgi Protein 73 through Activating NF-κB Pathway and Destroying Golgi Structure in Liver Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Animals
2.3. Cell Lines
2.4. Quantification of GP73 in Cell Culture Supernatant
2.5. Quantification of Serum GP73, TBA, Alanine Transaminase (ALT), and Aspartate Transaminase (AST)
2.6. RNA Extraction and Quantitative Reverse-Transcriptase PCR (qRT-PCR)
2.7. Western Blot (WB) Analysis
2.8. Luciferase Reporter Assay
2.9. Cytoimmunofluorescence
2.10. Chromatin Immunoprecipitation (ChIP) Assay
2.11. Statistical Analysis
3. Results
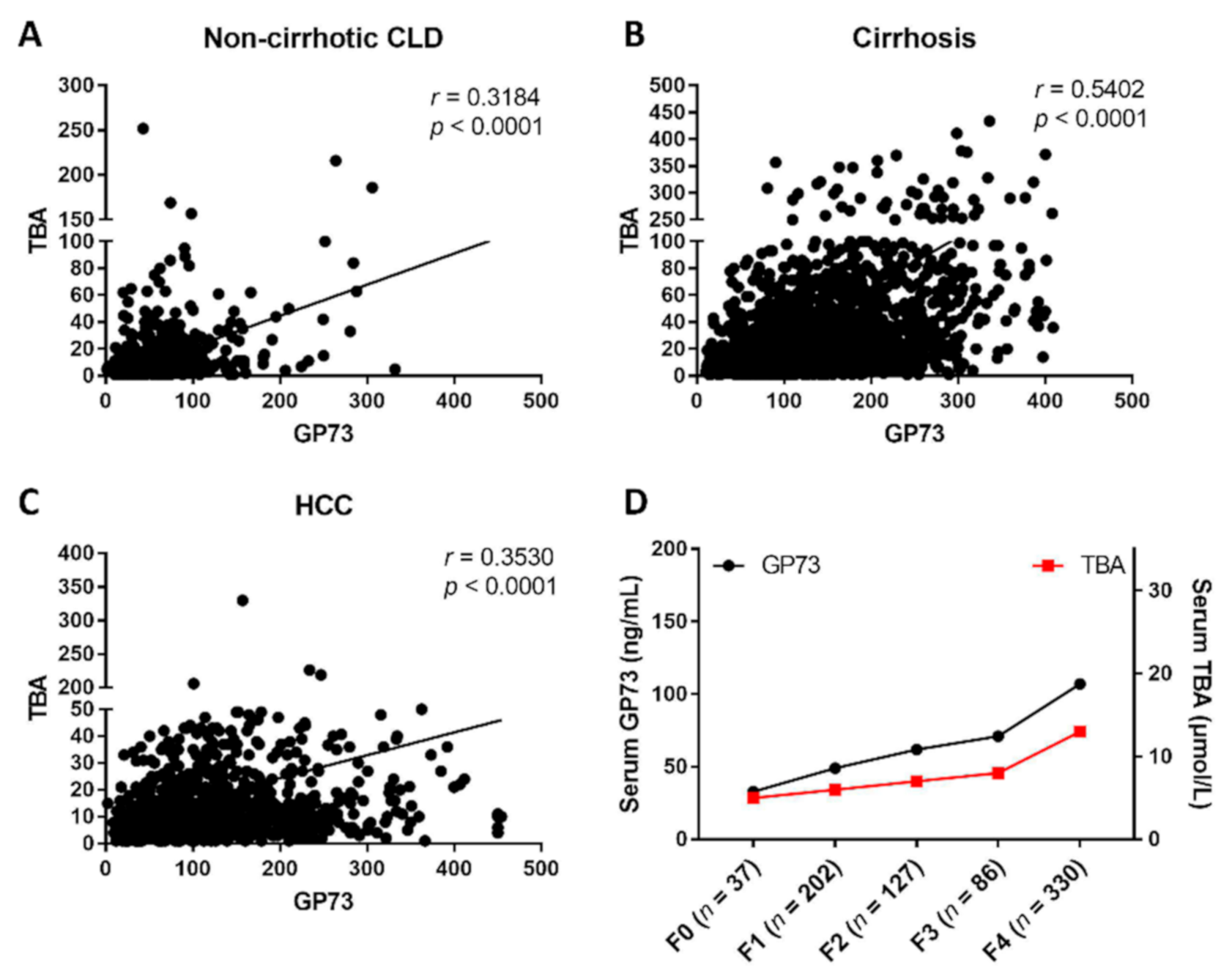
3.1. Serum GP73 and TBA Are Significantly Increased in Patients with CLD or HCC
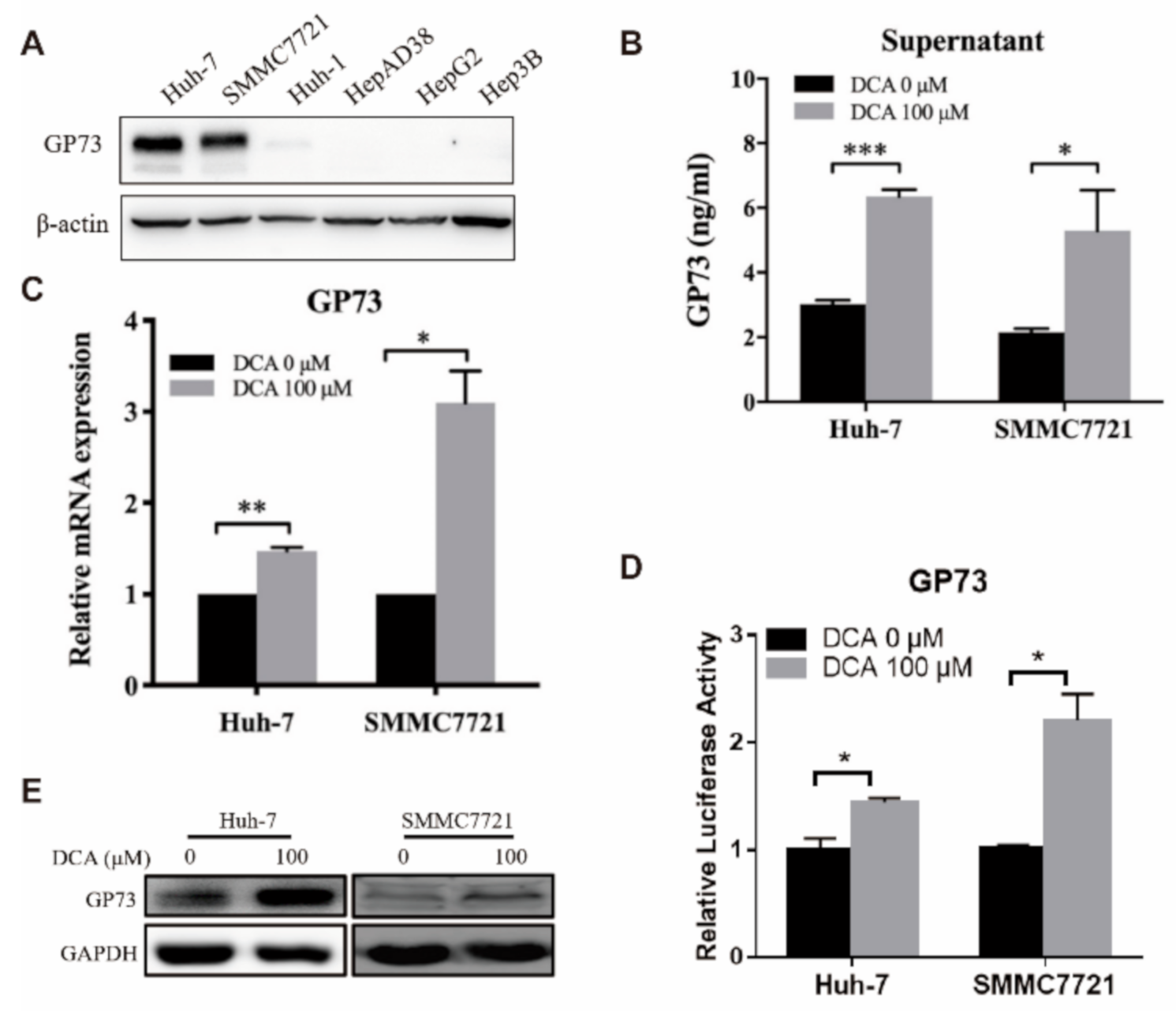
3.2. DCA Upregulates the Expression of Endogenous GP73
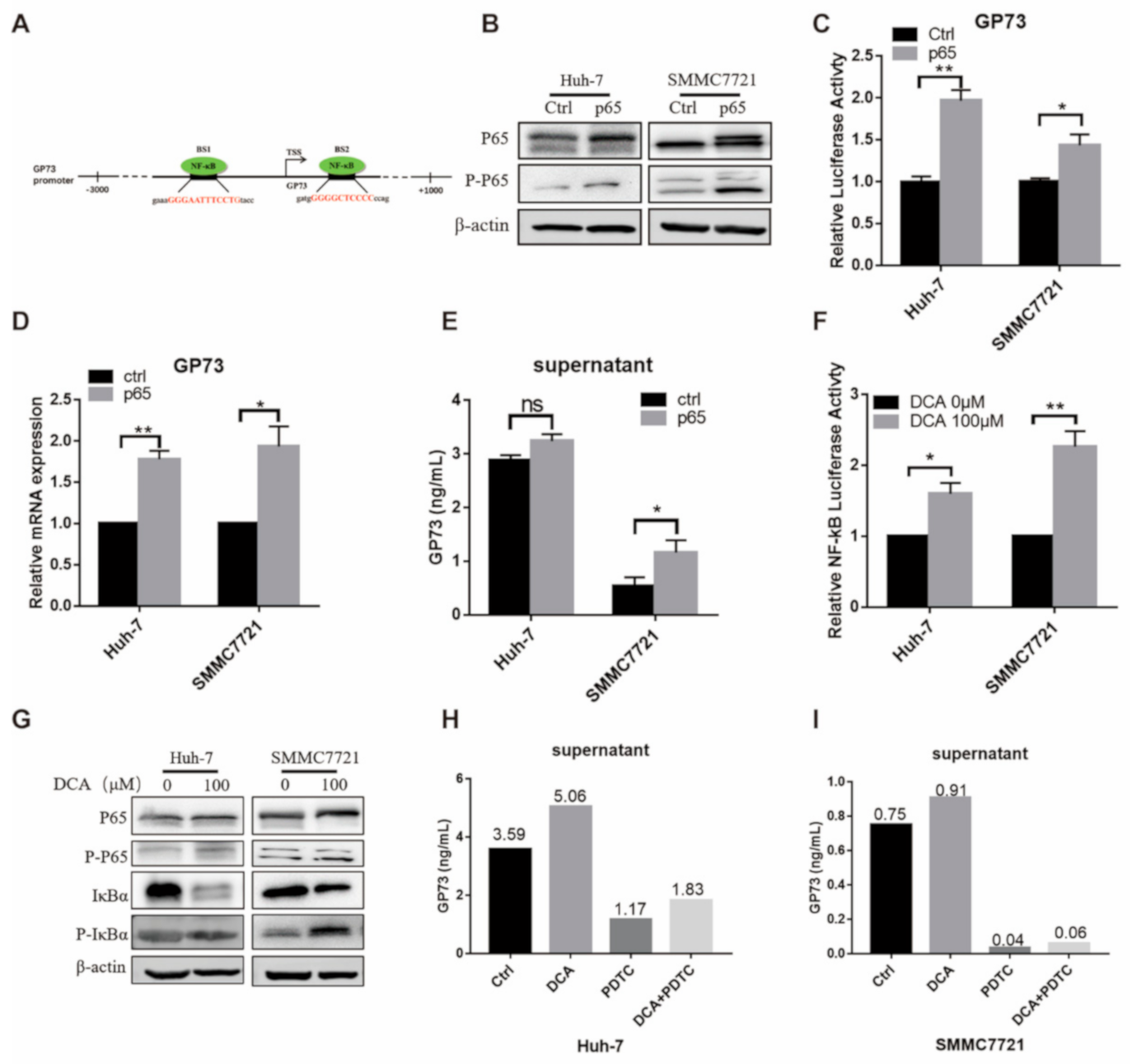
3.3. DCA Upregulates the Expression of Endogenous GP73 through NF-κB Pathway Activation
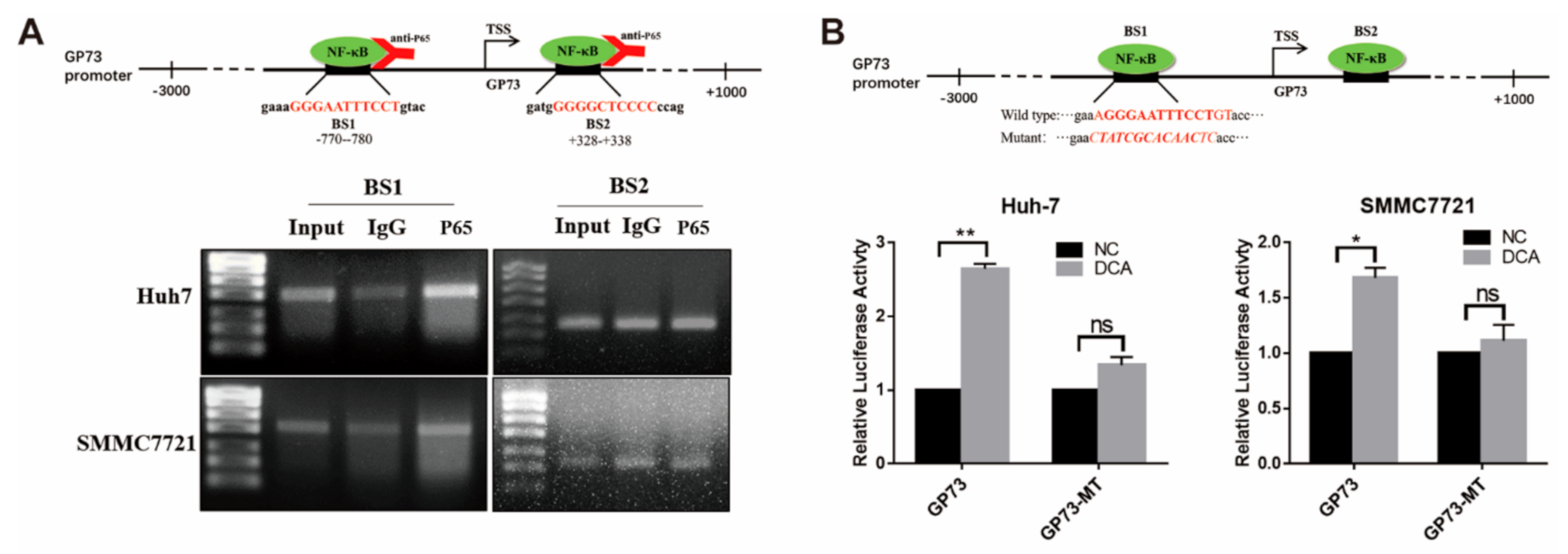
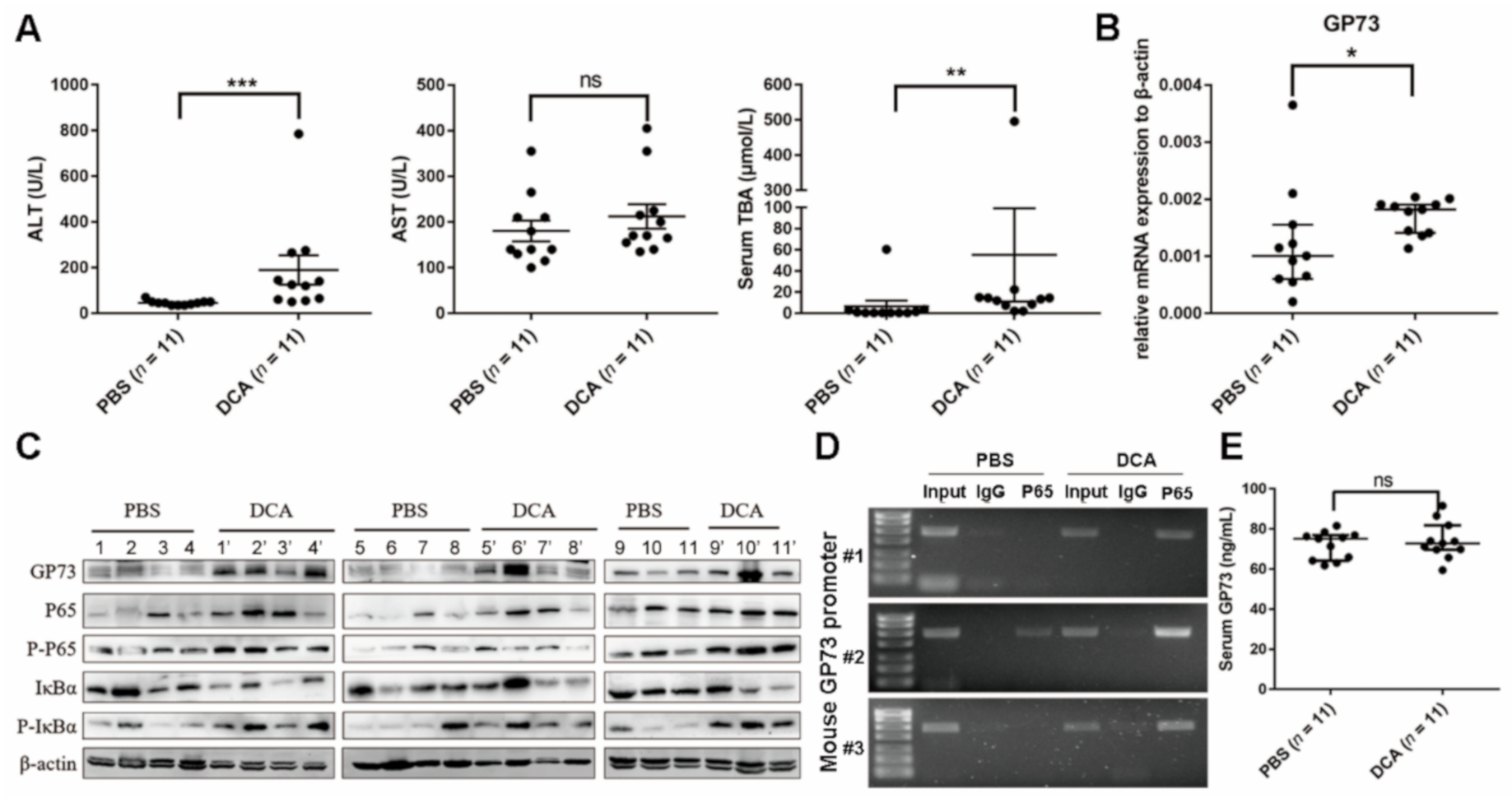
3.4. NF-κB P65 Directly Binds to the Promoter of GP73 to Upregulate Its Expression
3.5. DCA Upregulates NF-κB-Mediated GP73 Expression In Vivo
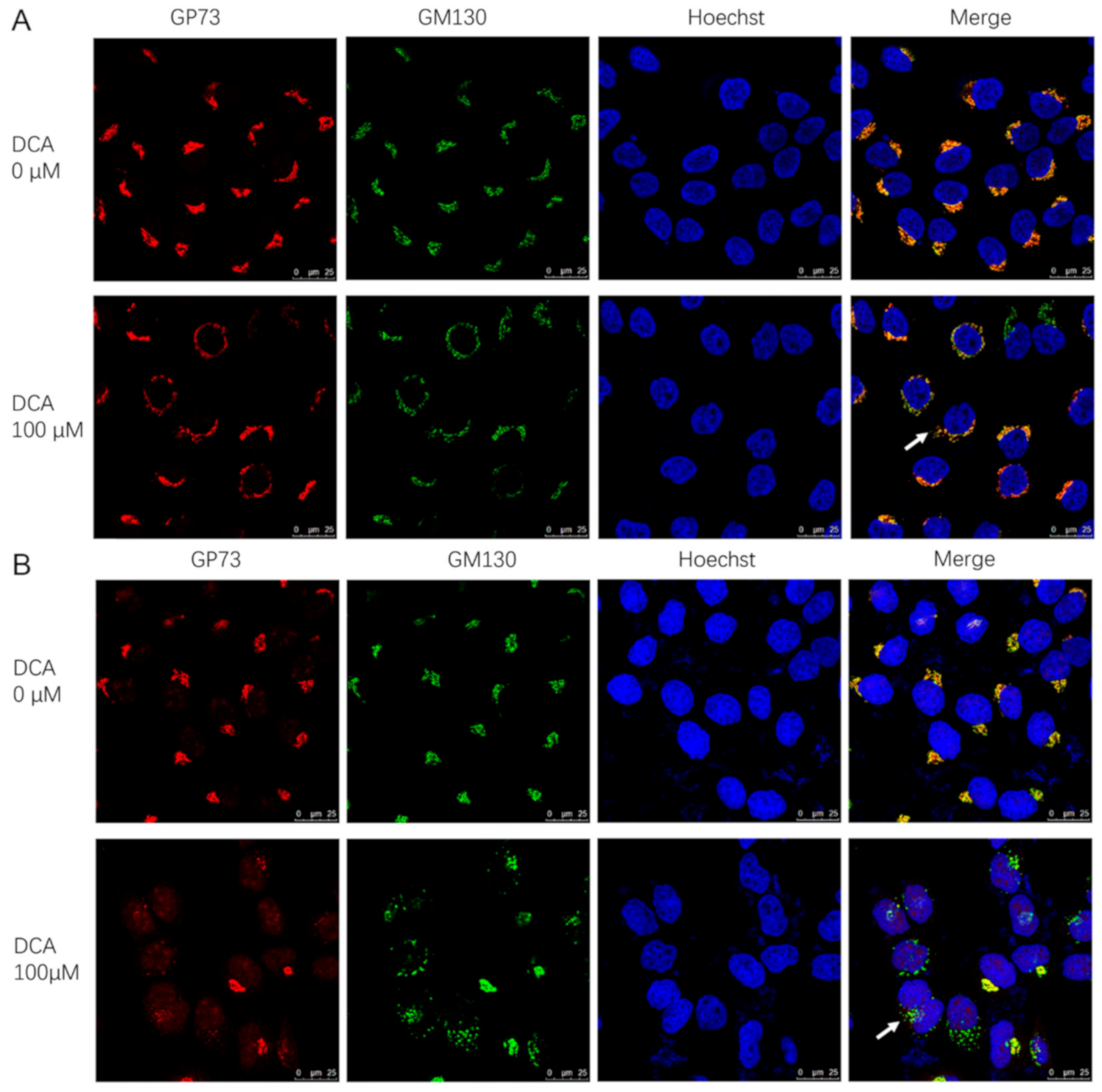
3.6. DCA May Promote GP73 Release by Causing Golgi Structure Disassembly
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ganem, D.; Prince, A.M. Hepatitis B Virus Infection—Natural History and Clinical Consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thein, H.H.; Yi, Q.; Dore, G.J.; Krahn, M.D. Estimation of stage-specific fibrosis progression rates in chronic hepatitis C virus infec-tion: A meta-analysis and meta-regression. Hepatology 2008, 48, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schawkat, K.; Reiner, C.S. Diffuse Liver Disease: Cirrhosis, Focal Lesions in Cirrhosis, and Vascular Liver Disease. In Diseases of the Abdomen and Pelvis 2018–2021: Diagnostic Imaging—IDKD Book. Cham (CH); Ho-dler, J., Kubik-Huch, R.A., von Schulthess, G.K., Eds.; Springer: Berlin, Germany, 2018; pp. 229–236. [Google Scholar]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Kladney, R.D.; Bulla, G.A.; Guo, L.; Mason, A.L.; Tollefson, A.E.; Simon, D.J.; Koutoubi, Z.; Fimmel, C.J. GP73, a novel Golgi-localized protein upregulated by viral infection. Gene 2000, 249, 53–65. [Google Scholar] [CrossRef]
- Munro, S. Localization of proteins to the Golgi apparatus. Trends Cell Biol. 1998, 8, 11–15. [Google Scholar] [CrossRef]
- Kladney, R.D.; Cui, X.; Bulla, G.A.; Brunt, E.M.; Fimmel, C.J. Expression of GP73, a resident Golgi membrane protein, in viral and nonviral liver disease. Hepatology 2002, 35, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.-S.; Ye, W.; Jiang, Y.-K.; Ma, J.; Zhu, M.-Q.; Ma, J.-M.; Zhou, H.; Yu, L.-Q.; Yang, Y.-F.; Wang, S.-C. The Value of GPC3 and GP73 in Clinical Diagnosis of Hepatocellular Carcinoma. Clin. Lab. 2017, 63, 1903–1909. [Google Scholar] [CrossRef]
- Liu, T.; Yao, M.; Liu, S.; Wang, L.; Wang, L.; Hou, J.; Ma, X.; Jia, J.; Zhao, J.; Zhuang, H.; et al. Serum Golgi protein 73 is not a suitable diagnostic marker for hepatocellular carcinoma. Oncotarget 2017, 8, 16498–16506. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Wang, L.; Leung, P.S.; Li, Y.; Liu, S.; Wang, L.; Guo, X.; Zhou, G.; Yan, Y.; Guan, G.; et al. The Clinical Significance of GP73 in Immunologically Mediated Chronic Liver Dis-eases: Experimental Data and Literature Review. Clin. Rev. Allergy Immunol. 2018, 54, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Quilty, F.; Gilmer, J.F.; Long, A.; Byrne, A.M. Unconjugated secondary bile acids activate the unfolded protein response and in-duce golgi fragmentation via a src-kinase-dependant mechanism. Oncotarget 2017, 8, 967–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, A.-M.; Bekiaris, S.; Duggan, G.; Prichard, D.; Kirca, M.; Finn, S.; Reynolds, J.V.; Kelleher, D.; Long, A. Golgi phosphoprotein 2 (GOLPH2) is a novel bile acid-responsive modulator of oesophageal cell migration and invasion. Br. J. Cancer 2015, 113, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Xie, G.; Wang, X.; Fan, J.; Qiu, Y.; Zheng, X.; Qi, X.; Cao, Y.; Su, M.; Wang, X.; et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellu-lar carcinoma. Mol. Cell. Proteom. 2011, 10, M110.004945. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Xie, G.; Wang, X.; Fan, J.; Qiu, Y.; Zheng, X.; Qi, X.; Cao, Y.; Su, M.; Wang, X.; et al. Biomarkers for the early diagnosis of hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 10573–10583. [Google Scholar]
- Wang, J.; Ding, S.; Duan, Z.; Xie, Q.; Zhang, T.; Zhang, X.; Wang, Y.; Chen, X.; Zhuang, H.; Lu, F. Role of p14ARF-HDM2-p53 axis in SOX6-mediated tumor suppression. Oncogene 2015, 35, 1692–1702. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Wang, J.; Cheng, J.; Cao, Z.; Chen, R.; Li, H.; Liu, S.; Chen, X.; Sui, J.; Lu, F. Down-regulation of NTCP expression by cyclin D1 in hepatitis B virus-related hepatocel-lular carcinoma has clinical significance. Oncotarget 2017, 8, 56041–56050. [Google Scholar] [CrossRef] [Green Version]
- Bachert, C.; Fimmel, C.; Linstedt, A.D. Endosomal Trafficking and Proprotein Convertase Cleavage of cis Golgi Protein GP73 Produces Marker for Hepatocellular Carcinoma. Traffic 2007, 8, 1415–1423. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, T.; Huang, P.; Zhao, H.; Zhang, R.; Ma, B.; Chen, K.; Huang, F.; Zhou, X.; Cui, C.; et al. A novel Golgi protein (GOLPH2)-regulated oncolytic adenovirus exhibits potent anti-tumor efficacy in hepatocellular carcinoma. Oncotarget 2015, 6, 13564–13578. [Google Scholar] [CrossRef] [Green Version]
- Wright, L.M.; Yong, S.; Picken, M.M.; Rockey, D.; Fimmel, C.J. Decreased survival and hepato-renal pathology in mice with C-terminally trun-cated GP73 (GOLPH2). Int. J. Clin. Exp. Pathol. 2009, 2, 34–47. [Google Scholar] [PubMed]
- Liu, X.; Wan, X.; Li, Z.; Lin, C.; Zhan, Y.; Lu, X. Golgi protein 73(GP73), a useful serum marker in liver diseases. Clin. Chem. Lab. Med. 2011, 49, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Small, D.M. Detergent Properties of Bile Salts: Correlation with Physiological Function. Annu. Rev. Med. 1967, 18, 333–376. [Google Scholar] [CrossRef] [PubMed]
- Meadows, V.; Kennedy, L.; Kundu, D.; Alpini, G.; Francis, H. Bile Acid Receptor Therapeutics Effects on Chronic Liver Diseases. Front. Med. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Veysey, M.; French, G.L.; Hylemon, P.B.; Murphy, G.M.; Dowling, R.H. Bile acid metabolism by fresh human colonic contents: A comparison of caecal versus faecal samples. Gut 2001, 49, 835–842. [Google Scholar] [CrossRef]
- Liu, L.; Dong, W.; Wang, S.; Zhang, Y.; Liu, T.; Xie, R.; Wang, B.; Cao, H. Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumor-igenesis. Food Funct. 2018, 9, 5588–5597. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Xu, M.; Dong, W.; Deng, B.; Wang, S.; Zhang, Y.; Wang, S.; Luo, S.; Wang, W.; Qi, Y.; et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int. J. Cancer 2017, 140, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Gong, Z.; Du, X.; Tian, C.; Wang, L.; Zhou, J.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Deoxycholic Acid-Mediated Sphingosine-1-Phosphate Receptor 2 Signaling Exacerbates DSS-Induced Colitis through Promoting Cathepsin B Release. J. Immunol. Res. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Surman, D.R.; Diggs, L.; Xi, S.; Gao, S.; Gurusamy, D.; McLoughlin, K.; Drake, J.; Feingold, P.; Brown, K.; et al. Bile acid-induced “Minority MOMP” promotes esophageal carcinogenesis while main-taining apoptotic resistance via Mcl-1. Oncogene 2020, 39, 877–890. [Google Scholar] [CrossRef]
- Prichard, D.O.; Byrne, A.M.; Murphy, J.O.; Reynolds, J.V.; O’Sullivan, J.; Feighery, R.; Doyle, B.; Eldin, O.S.; Finn, S.P.; Maguire, A.; et al. Deoxycholic acid promotes development of gastroesophageal reflux disease and Barrett’s oesophagus by modulating integrin-alphav trafficking. J. Cell. Mol. Med. 2017, 21, 3612–3625. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through se-nescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, V.I. Physiological and molecular biochemical mechanisms of bile formation. World J. Gastroenterol. 2013, 19, 7341–7360. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Y.; Tao, J.; Shi, Y.; Gai, X.; Huang, F.; Ma, Q.; Zhou, Z.; Chen, H.; Zhang, H.; et al. mTORC1 Up-Regulates GP73 to Promote Proliferation and Migration of Hepatocellular Carcinoma Cells and Growth of Xenograft Tumors in Mice. Gastroenterology 2015, 149, 741–752.e14. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Block, T.M.; Wang, M.; Nefsky, B.; Long, R.; Hafner, J.; Mehta, A.S.; Marrero, J.; Gish, R.; Norton, P.A. Interleukin-6 and oncostatin M are elevated in liver disease in conjunction with can-didate hepatocellular carcinoma biomarker GP73. Cancer Biomark. 2012, 11, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Longoni, N.; Sarti, M.; Albino, D.; Civenni, G.; Malek, A.; Ortelli, E.; Pinton, S.; Mello-Grand, M.; Ostano, P.; D’Ambrosio, G.; et al. ETS transcription factor ESE1/ELF3 orchestrates a positive feedback loop that con-stitutively activates NF-kappaB and drives prostate cancer progression. Cancer Res. 2013, 73, 4533–4547. [Google Scholar] [CrossRef] [Green Version]
- Youn, S.Y.; Ji, G.E.; Han, Y.R.; Park, M.S. Development of Strain-Specific Primers for Identification of Bifidobacterium bifidum BGN4. J. Microbiol. Biotechnol. 2017, 27, 909–915. [Google Scholar] [CrossRef]
- Reynolds, J.V.; Abdel-Latif, M.M.M.; Inoue, H.; Kelleher, D. Factors regulating nuclear factor-kappa B activation in esophageal cancer cells: Role of bile acids and acid. J. Cancer Res. Ther. 2016, 12, 364. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Cirrhotic CLD (n = 985) | Cirrhosis (n = 2141) | HCC (n = 1085) | p | |
|---|---|---|---|---|
| Sex (M/F) | 620/365 | 1338/803 | 926/159 | <0.0001 |
| Age (year) | 44.00 (35.00–51.00) | 50.00 (43.00–58.00) | 52.00 (45.00–59.00) | <0.0001 |
| BMI (kg/m2) | 24.26 (21.86–26.62) | 23.83 (21.50–26.37) | 23.88 (21.79–25.95) | 0.033 |
| ALT (U/L) | 26.00 (17.00–48.00) | 30.00 (20.00–55.00) | 36.00 (24.00–60.00) | <0.0001 |
| AST (U/L) | 25.00 (20.00–38.00) | 41.00 (27.00–71.00) | 40.00 (28.00–69.75) | <0.0001 |
| GGT (U/L) | 25.00 (16.00–54.00) | 44.00 (24.00–97.00) | 58.00 (33.00–127.00) | <0.0001 |
| ALP (U/L) | 77.00 (62.00–99.00) | 99.00 (75.00–138.50) | 98.00 (76.00–136.00) | <0.0001 |
| ALB (g/L) | 40.00 (38.00–43.00) | 35.00 (30.00–39.00) | 38.00 (35.00–41.00) | <0.0001 |
| PA (mg/L) | 198.00 (158.00–231.00) | 113.00 (72.75–162.00) | 153.00 (110.00–198.00) | <0.0001 |
| TBiL (μmol/L) | 11.70 (8.90–16.10) | 18.30 (12.05–31.80) | 14.60 (10.70–20.08) | <0.0001 |
| DBiL (μmol/L) | 4.20 (3.00–5.95) | 7.50 (4.50–16.20) | 6.20 (4.30–10.20) | <0.0001 |
| CHE (U/L) | 7216.00 (5945.50–8329.50) | 4525.00 (2925.50–6248.00) | 4375.00 (3027.50–6093.50) | <0.0001 |
| GP73 (ng/mL) | 46.42 (30.60–66.90) | 127.90 (82.56–192.40) | 100.50 (60.62–161.50) | <0.0001 |
| PLT (× 109/L) | 176.00 (139.00–215.00) | 92.00 (57.00–142.00) | 134.00 (87.25–181.00) | <0.0001 |
| TBA (μmol/L) | 5.00 (3.00–10.00) | 21.00 (7.00–53.00) | 9.00 (5.00–18.00) | <0.0001 |
| PT (s) | 11.00 (10.40–11.70) | 12.50 (11.40–14.30) | 11.90 (11.10–12.90) | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, D.; Yao, M.; Yan, Y.; Liu, Y.; Wen, X.; Chen, X.; Lu, F. Deoxycholic Acid Upregulates Serum Golgi Protein 73 through Activating NF-κB Pathway and Destroying Golgi Structure in Liver Disease. Biomolecules 2021, 11, 205. https://doi.org/10.3390/biom11020205
Yang D, Yao M, Yan Y, Liu Y, Wen X, Chen X, Lu F. Deoxycholic Acid Upregulates Serum Golgi Protein 73 through Activating NF-κB Pathway and Destroying Golgi Structure in Liver Disease. Biomolecules. 2021; 11(2):205. https://doi.org/10.3390/biom11020205
Chicago/Turabian StyleYang, Danli, Mingjie Yao, Ying Yan, Yanna Liu, Xiajie Wen, Xiangmei Chen, and Fengmin Lu. 2021. "Deoxycholic Acid Upregulates Serum Golgi Protein 73 through Activating NF-κB Pathway and Destroying Golgi Structure in Liver Disease" Biomolecules 11, no. 2: 205. https://doi.org/10.3390/biom11020205