Cell-Free DNA-Methylation-Based Methods and Applications in Oncology

 , ,
, , 
 and
and
Abstract
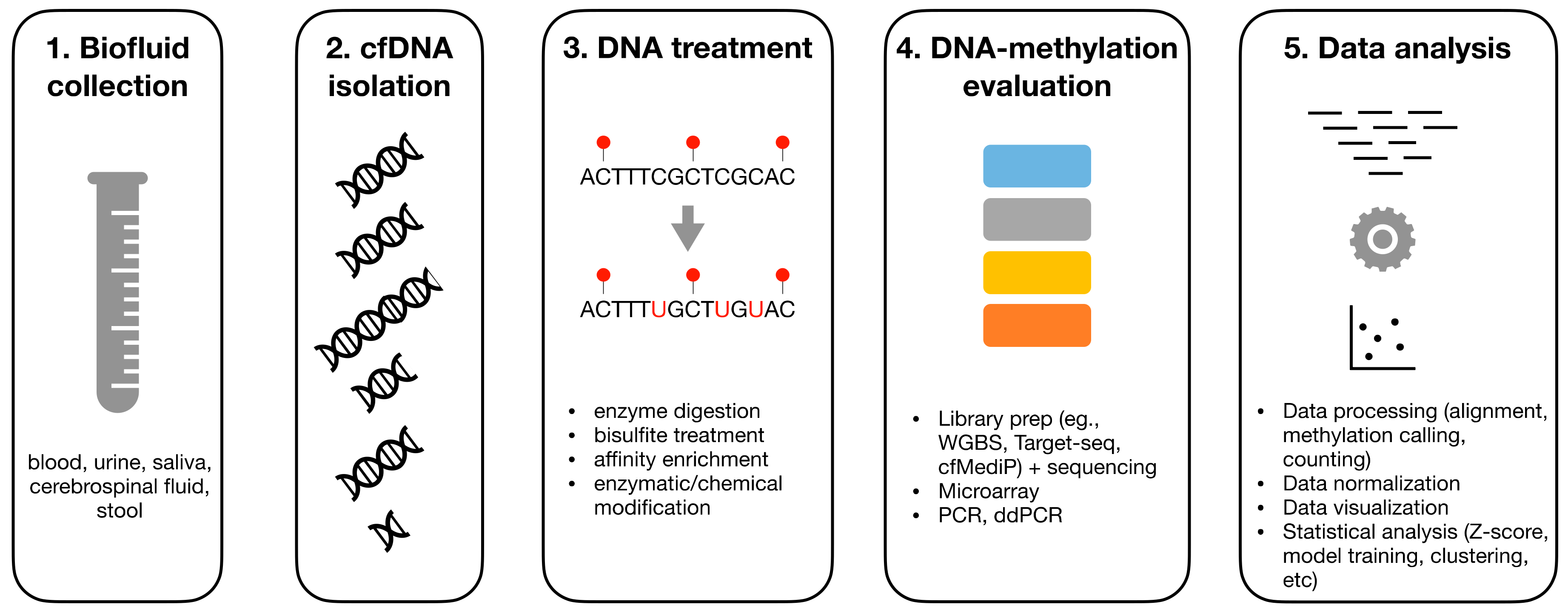
:1. Introduction
2. Overview on DNA-Methylation Analysis
3. Pre-Analytics of cfDNA
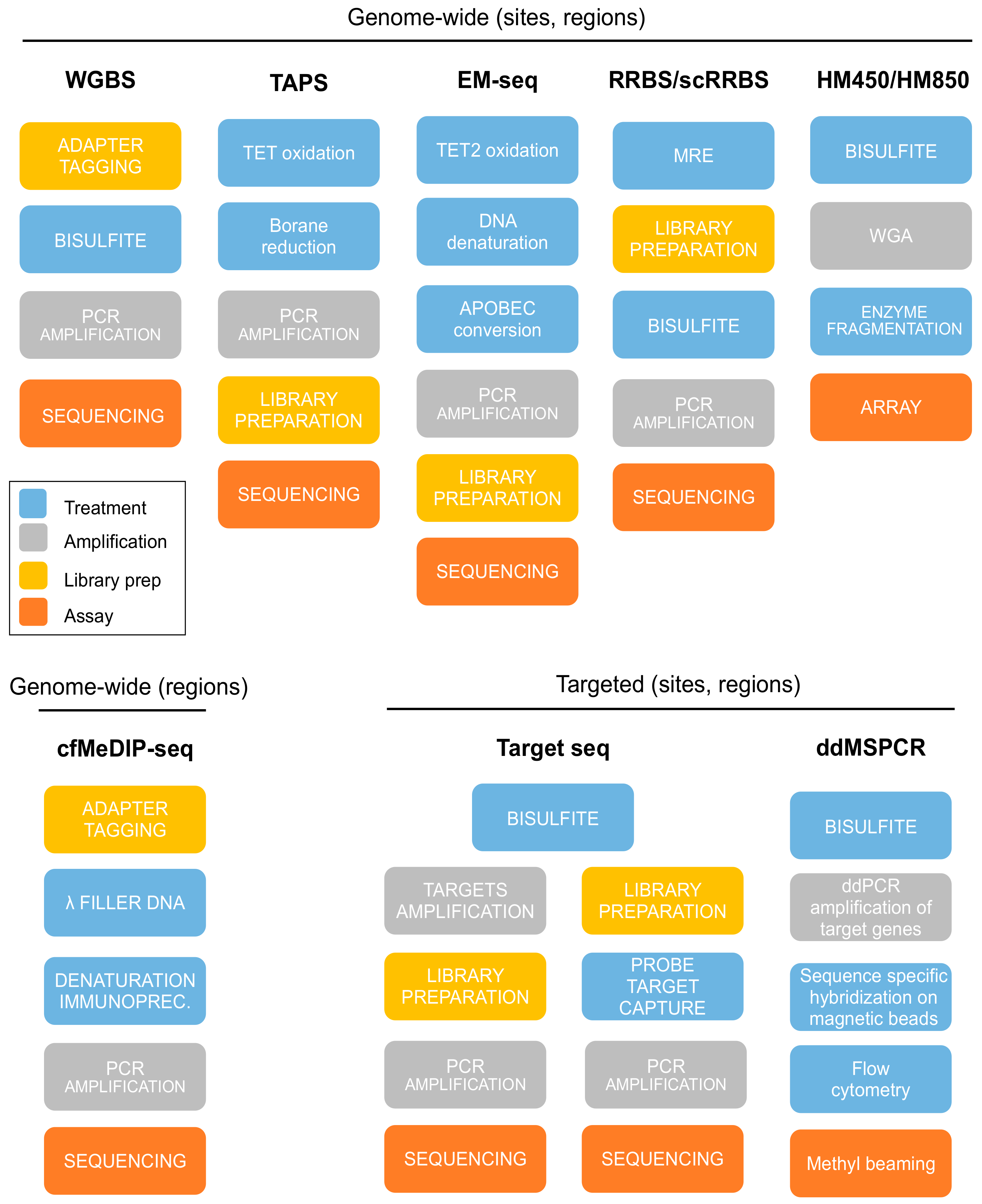
4. DNA Treatment
5. Experimental Assays for DNAm Evaluation
5.1. Whole-Genome Bisulfite Sequencing (WGBS)
5.2. Representative Genome-Wide Methods
5.3. Affinity Enrichment Sequencing
5.4. Microarrays
5.5. Targeted Sequencing
5.6. Methylation-Specific PCR and Droplet Digital PCR
6. Computational Analysis of cfDNA Data
6.1. Data Processing
6.1.1. Microarray
6.1.2. Bisulfite Sequencing Data
6.1.3. Affinity Enrichment Sequencing Data
6.2. Statistical Analysis of Cell-Free DNAm Data
7. Applications of Cell-Free DNA-Methylation Assays in Oncology
7.1. Tumor Detection and Monitoring
7.2. Drug Resistance
7.3. Cell-of-Origin Identification
8. Clinically Approved DNA-Methylation Assays
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diaz, L.A.; Bardelli, A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating Liquid Biopsies into the Management of Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and Future Perspectives of Liquid Biopsies in Genomics-Driven Oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, K.; Gupta, S. Cancer Epigenetics: An Introduction. Bioinform. MicroRNA Res. 2014, 1238, 3–25. [Google Scholar]
- Esteller, M.; Herman, J.G. Cancer as an Epigenetic Disease: DNA Methylation and Chromatin Alterations in Human Tumours. J. Pathol. 2002, 196, 1–7. [Google Scholar] [CrossRef]
- Esteller, M. Relevance of DNA Methylation in the Management of Cancer. Lancet Oncol. 2003, 4, 351–358. [Google Scholar] [CrossRef]
- Heyn, H.; Esteller, M. DNA Methylation Profiling in the Clinic: Applications and Challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef]
- Li, W.; Zhou, X.J. Methylation Extends the Reach of Liquid Biopsy in Cancer Detection. Nat. Rev. Clin. Oncol. 2020, 1–2. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Closing in on CfDNA-Based Detection and Diagnosis. Nat. Rev. Cancer 2020, 20, 481. [Google Scholar] [CrossRef]
- Song, C.-X.; Yin, S.; Amanda, W.; Wheeler, A.; Chen, Y.; Zhang, Y.; Liu, B.; Xiong, J.; Zhang, W.; Chun-Xiao, S.; et al. 5-Hydroxymethylcytosine Signatures in Cell-Free DNA Provide Information about Tumor Types and Stages. Cell Res. 2017, 27, 1231–1242. [Google Scholar] [CrossRef] [Green Version]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A.; Balasubramanian, S. 5-Hydroxymethylcytosine Is a Predominantly Stable DNA Modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Stroup, E.K.; Zhang, Z.; Chiu, B.C.; Zhang, W. Towards Precision Medicine: Advances in 5-Hydroxymethylcytosine Cancer Biomarker Discovery in Liquid Biopsy. Cancer Commun. 2019, 39, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.-X.; Yi, C. Mapping the Epigenetic Modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rodriguez, I.R.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and Death of Circulating Cell-Free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Chen, C.; Hulbert, A.; Brock, M.V.; Yu, F. Non-Blood Circulating Tumor DNA Detection in Cancer. Oncotarget 2017, 8, 69162–69173. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-Free Nucleic Acids as Biomarkers in Cancer Patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [Green Version]
- Johansson, G.; Andersson, D.; Filges, S.; Li, J.; Muth, A.; Godfrey, T.E.; Stahlberg, A. Considerations and Quality Controls When Analyzing Cell-Free Tumor DNA. Biomol. Detect. Quantif. 2019, 17, 100078. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Kefford, R.F.; Rizos, H. Evaluation of Commercial Kits for Purification of Circulating Free DNA. Cancer Genet. 2018, 229, 21–27. [Google Scholar] [CrossRef]
- Sozzi, G.; Roz, L.; Conte, D.; Mariani, L.; Andriani, F.; Verderio, P.; Pastorino, U. Effects of Prolonged Storage of Whole Plasma or Isolated Plasma DNA on the Results of Circulating DNA Quantification Assays. J. Natl. Cancer Inst. 2005, 97, 1848–1850. [Google Scholar] [CrossRef] [Green Version]
- Meddeb, R.; Pisareva, E.; Thierry, A.R. Guidelines for the Preanalytical Conditions for Analyzing Circulating Cell-Free DNA. Clin. Chem. 2019, 65, 623–633. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct Detection of DNA Methylation During Single-Molecule, Real-Time Sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Gouil, Q.; Keniry, A. Latest Techniques to Study DNA Methylation. Essays Biochem. 2019, 63, 639–648. [Google Scholar]
- Rand, A.C.; Jain, M.; Eizenga, J.M.; Musselman-Brown, A.; Olsen, H.E.; Akeson, M.; Paten, B. Mapping DNA Methylation with High-Throughput Nanopore Sequencing. Nat. Methods 2017, 14, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Khulan, B.; Thompson, R.F.; Ye, K.; Fazzari, M.J.; Suzuki, M.; Stasiek, E.; Figueroa, M.E.; Glass, J.L.; Chen, Q.; Montagna, C.; et al. Comparative Isoschizomer Profiling of Cytosine Methylation: The HELP Assay. Genome Res. 2006, 16, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Oda, M.; Glass, J.L.; Thompson, R.F.; Mo, Y.; Olivier, E.N.; Figueroa, M.E.; Selzer, R.R.; Richmond, T.A.; Zhang, X.; Dannenberg, L.; et al. High-Resolution Genome-Wide Cytosine Methylation Profiling with Simultaneous Copy Number Analysis and Optimization for Limited Cell Numbers. Nucleic Acids Res. 2009, 37, 3829–3839. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Bai, Y.; Cheng, Z.; Liu, F.; Wang, P.; Yang, D.; Li, G.; Jin, Q.; Mao, H.; Zhao, J. Absolute Quantification of DNA Methylation Using Microfluidic Chip-Based Digital PCR. Biosens. Bioelectron. 2017, 96, 339–344. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kokubun, S.; Itoi, E.; Roach, H.I. Improved Quantification of DNA Methylation Using Methylation-Sensitive Restriction Enzymes and Real-Time PCR. Epigenetics 2007, 2, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Zuo, T.; Tycko, B.; Liu, T.-M.; Lin, H.-J.L.; Huang, T.H.-M. Methods in DNA Methylation Profiling. Epigenomics 2009, 1, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, D.; Day, J. Faculty Opinions Recommendation of Conserved Role of Intragenic DNA Methylation in Regulating Alternative Promoters. Nature 2010, 466, 253–257. [Google Scholar]
- Park, P. Faculty Opinions Recommendation of Targeted and Genome-Scale Strategies Reveal Gene-Body Methylation Signatures in Human Cells. Nature 2009, 27, 361–368. [Google Scholar]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A Genomic Sequencing Protocol That Yields a Positive Display of 5-Methylcytosine Residues in Individual DNA Strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [Green Version]
- Grunau, C. Bisulfite Genomic Sequencing: Systematic Investigation of Critical Experimental Parameters. Nucleic Acids Res. 2001, 29, e65. [Google Scholar] [CrossRef]
- Warnecke, P.M.; Stirzaker, C.; Song, J.; Grunau, C.; Melki, J.R.; Clark, S. Identification and Resolution of Artifacts in Bisulfite Sequencing. Methods 2002, 27, 101–107. [Google Scholar] [CrossRef]
- Raizis, A.; Schmitt, F.; Jost, J. A Bisulfite Method of 5-Methylcytosine Mapping That Minimizes Template Degradation. Anal. Biochem. 1995, 226, 161–166. [Google Scholar] [CrossRef]
- Holmes, E.E.; Jung, M.; Meller, S.; Leisse, A.; Sailer, V.; Zech, J.; Mengdehl, M.; Garbe, L.-A.; Uhl, B.; Kristiansen, G.; et al. Performance Evaluation of Kits for Bisulfite-Conversion of DNA from Tissues, Cell Lines, FFPE Tissues, Aspirates, Lavages, Effusions, Plasma, Serum, and Urine. PLoS ONE 2014, 9, e93933. [Google Scholar] [CrossRef] [Green Version]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of Whole-Genome Bisulfite Sequencing Library Preparation Strategies Identifies Sources of Biases Affecting DNA Methylation Data. Genome Biol. 2018, 19, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, K.; Zotenko, E.; Luu, P.L.; Gould, C.M.; Nair, S.S.; Clark, S.; Stirzaker, C. Comprehensive Evaluation of Genome-Wide 5-Hydroxymethylcytosine Profiling Approaches in Human DNA. Epigenet. Chromatin 2017, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.J.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-Wide and Promoter-Specific Analyses Identify Sites of Differential DNA Methylation in Normal and Transformed Human Cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Down, T.A.; Rakyan, V.K.; Turner, D.J.; Flicek, P.; Li, H.; Kulesha, E.; Gräf, S.; Johnson, N.; Herrero, J.; Tomazou, E.M.; et al. A Bayesian Deconvolution Strategy for Immunoprecipitation-Based DNA Methylome Analysis. Nat. Biotechnol. 2008, 26, 779–785. [Google Scholar] [CrossRef]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome Analysis Using MeDIP-Seq with Low DNA Concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of CfMeDIP-Seq Libraries for Methylome Profiling of Plasma Cell-Free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef]
- Cross, S.H.; Charlton, J.A.; Nan, X.; Bird, A.P. Purification of CpG Islands Using a Methylated DNA Binding Column. Nat. Genet. 1994, 6, 236–244. [Google Scholar] [CrossRef]
- Brinkman, A.B.; Simmer, F.; Ma, K.; Kaan, A.; Zhu, J.; Stunnenberg, H.G. Whole-Genome DNA Methylation Profiling Using MethylCap-Seq. Methods 2010, 52, 232–236. [Google Scholar] [CrossRef]
- Jeltsch, A.; Broche, J.; Lungu, C.; Bashtrykov, P. Biotechnological Applications of MBD Domain Proteins for DNA Methylation Analysis. J. Mol. Biol. 2020, 432, 1816–1823. [Google Scholar] [CrossRef]
- Liu, Y.; Siejika-Zielinska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Bockler, B.; Song, C.-X. Bisulfite-Free Direct Detection of 5-Methylcytosine and 5-Hydroxymethylcytosine at Base Resolution. Nat. Biotech. 2019, 37, 424–429. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Romanel, A.; Conteduca, V.; Casiraghi, N.; Sigouros, M.; Franceschini, G.M.; Orlando, F.; Fedrizzi, T.; Ku, S.-Y.; Dann, E.; et al. Circulating Tumor DNA Profile Recognizes Transformation to Castration-Resistant Neuroendocrine Prostate Cancer. J. Clin. Investig. 2020, 130, 1653–1668. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA Methylation Loss in Late-Replicating Domains Is Linked to Mitotic Cell Division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Stirzaker, C.; Taberlay, P.C.; Statham, A.L.; Clark, S. Mining Cancer Methylomes: Prospects and Challenges. Trends Genet. 2014, 30, 75–84. [Google Scholar] [CrossRef]
- Plongthongkum, N.; Diep, D.H.; Zhang, K. Advances in the Profiling of DNA Modifications: Cytosine Methylation and Beyond. Nat. Rev. Genet. 2014, 15, 647–661. [Google Scholar] [CrossRef]
- Legendre, C.; Gooden, G.C.; Johnson, K.; Martinez, R.A.; Liang, W.S.; Salhia, B. Whole-Genome Bisulfite Sequencing of Cell-Free DNA Identifies Signature Associated with Metastatic Breast Cancer. Clin. Epigenet. 2015, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.C.A.; Jiang, P.; Chan, C.W.M.; Sun, K.; Wong, J.; Hui, E.P.; Chan, S.L.; Chan, W.C.; Hui, D.S.C.; Ng, S.S.M.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Cremaschi, P.; Wetterskog, D.; Conteduca, V.; Franceschini, G.M.; Kleftogiannis, D.; Jayaram, A.; Sandhu, S.; Wong, S.Q.; Benelli, M.; et al. Genome-Wide Plasma DNA Methylation Features of Metastatic Prostate Cancer. J. Clin. Investig. 2020, 130, 1991–2000. [Google Scholar] [CrossRef]
- Miura, F.; Ito, T. Post-Bisulfite Adaptor Tagging for PCR-Free Whole-Genome Bisulfite Sequencing. Adv. Struct. Saf. Stud. 2017, 1708, 123–136. [Google Scholar]
- Miura, F.; Shibata, Y.; Miura, M.; Sangatsuda, Y.; Hisano, O.; Araki, H.; Ito, T. Highly Efficient Single-Stranded DNA Ligation Technique Improves Low-Input Whole-Genome Bisulfite Sequencing by Post-Bisulfite Adaptor Tagging. Nucleic Acids Res. 2019, 47, e85. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-Cell Genome-Wide Bisulfite Sequencing for Assessing Epigenetic Heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Clark, S.J.; Smallwood, S.A.; Lee, H.J.; Krueger, F.; Reik, W.; Kelsey, G. Genome-Wide Base-Resolution Mapping of DNA Methylation in Single Cells Using Single-Cell Bisulfite Sequencing (scBS-seq). Nat. Protoc. 2017, 12, 534–547. [Google Scholar] [CrossRef]
- Erger, F.; Nörling, D.; Borchert, D.; Leenen, E.; Habbig, S.; Wiesener, M.S.; Bartram, M.P.; Wenzel, A.; Becker, C.; Toliat, M.R.; et al. CfNOMe—A Single Assay for Comprehensive Epigenetic Analyses of Cell-Free DNA. Genome Med. 2020, 12, 1–14. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, J.; Siejka-Zielińska, P.; Weldon, C.; Roberts, H.; Lopopolo, M.; Magri, A.; D’Arienzo, V.; Harris, J.M.; McKeating, J.A.; et al. Accurate Targeted Long-Read DNA Methylation and Hydroxymethylation Sequencing With TAPS. Genome Biol. 2020, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-Scale DNA Methylation Maps of Pluripotent and Differentiated Cells. Nat. Cell Biol. 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xia, Y.; Li, L.; Gong, D.; Yao, Y.; Luo, H.; Lu, H.; Yi, N.; Wu, H.; Zhang, X.; et al. Double Restriction-Enzyme Digestion Improves the Coverage and Accuracy of Genome-Wide CpG Methylation Profiling by Reduced Representation Bisulfite Sequencing. BMC Genom. 2013, 14, 11. [Google Scholar] [CrossRef] [Green Version]
- Schillebeeckx, M.; Schrade, A.; Löbs, A.-K.; Pihlajoki, M.; Wilson, D.B.; Mitra, R.D. Laser Capture microdissection–reduced Representation Bisulfite Sequencing (LCM-RRBS) Maps Changes in DNA Methylation Associated with Gonadectomy-Induced Adrenocortical Neoplasia in the Mouse. Nucleic Acids Res. 2013, 41, e116. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Wu, X.; Li, X.; Wen, L.; Tang, F. Single-Cell Methylome Landscapes of Mouse Embryonic Stem Cells and Early Embryos Analyzed Using Reduced Representation Bisulfite Sequencing. Genome Res. 2016, 23, 2126–2135. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.-L.; Zhang, K.Z.K.; Zhang, K. Identification of Methylation Haplotype Blocks Aids in Deconvolution of Heterogeneous Tissue Samples and Tumor Tissue-of-Origin Mapping from Plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Wen, L.; Li, J.; Guo, H.; Liu, X.; Zheng, S.; Zhang, D.; Zhu, W.; Qu, J.; Guo, L.; Du, D.; et al. Genome-Scale Detection of Hypermethylated CpG Islands in Circulating Cell-Free DNA of Hepatocellular Carcinoma Patients. Cell Res. 2015, 25, 1250–1264. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive Tumour Detection and Classification Using Plasma Cell-Free DNA Methylomes. Nat. Cell Biol. 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Aberg, K.A.; Chan, R.F.; Shabalin, A.A.; Zhao, M.; Turecki, G.; Staunstrup, N.H.; Starnawska, A.; Mors, O.; Xie, L.Y.; Oord, E.J.V.D. A MBD-Seq Protocol for Large-Scale Methylome-Wide Studies With (very) Low Amounts of DNA. Epigenetics 2017, 12, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-W.; Huang, T.H.-M.; Wang, L.-S. Profiling DNA Methylomes from Microarray to Genome-Scale Sequencing. Technol. Cancer Res. Treat. 2010, 9, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Ladd-Acosta, C.; Carvalho, B.; Wu, H.; Brandenburg, S.A.; Jeddeloh, J.A.; Wen, B.; Feinberg, A.P. Comprehensive High-Throughput Arrays for Relative Methylation (CHARM). Genome Res. 2008, 18, 780–790. [Google Scholar] [CrossRef] [Green Version]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Gordevičius, J.; Kriščiūnas, A.; Groot, D.E.; Yip, S.M.; Susic, M.; Kwan, A.; Kustra, R.; Joshua, A.M.; Chi, K.N.; Petronis, A.; et al. Cell-Free DNA Modification Dynamics in Abiraterone Acetate-Treated Prostate Cancer Patients. Clin. Cancer Res. 2018, 24, 3317–3324. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Mühlhäusler, B.S.; Stirzaker, C.; Clark, S. Critical Evaluation of the Illumina MethylationEPIC BeadChip Microarray for Whole-Genome DNA Methylation Profiling. Genome Biol. 2016, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Oliver, V.F.; Wan, J.; Agarwal, S.; Zack, D.J.; Qian, J.; Merbs, S.L. A Novel Methyl-Binding Domain Protein Enrichment Method for Identifying Genome-Wide Tissue-Specific DNA Methylation from Nanogram DNA Samples. Epigenet. Chromatin 2013, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Widschwendter, M.; Evans, I.; Jones, A.; Ghazali, S.; Reisel, D.; Ryan, A.; Gentry-Maharaj, A.; Zikan, M.; Cibula, D.; Eichner, J.; et al. Methylation Patterns in Serum DNA for Early Identification of Disseminated Breast Cancer. Genome Med. 2017, 9, 115. [Google Scholar] [CrossRef]
- Holmila, R.; Sklias, A.; Muller, D.C.; Degli Esposti, D.; Guilloreau, P.; McKay, J.; Sangrajrang, S.; Srivatanakul, P.; Hainaut, P.; Merle, P.; et al. Targeted Deep Sequencing of Plasma Circulating Cell-Free DNA Reveals Vimentin and Fibulin 1 As Potential Epigenetic Biomarkers for Hepatocellular Carcinoma. PLoS ONE 2017, 12, e0174265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V. Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-Free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Liu, L.; Toung, J.; Jassowicz, A.; Vijayaraghavan, R.; Kang, H.; Zhang, R.; Kruglyak, K.; Huang, H.; Hinoue, T.; Shen, H.; et al. Targeted Methylation Sequencing of Plasma Cell-Free DNA for Cancer Detection and Classification. Ann. Oncol. 2018, 29, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating Tumor DNA Methylation Profiles Enable Early Diagnosis, Prognosis Prediction, and Screening for Colorectal Cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef]
- Xu, R.-H.; Wei, W.; Krawczyk, M.; Wang, W.; Luo, H.; Flagg, K.; Yi, S.; Shi, W.; Quan, Q.; Li, K.; et al. Circulating Tumour DNA Methylation Markers for Diagnosis and Prognosis of Hepatocellular Carcinoma. Nat. Mater. 2017, 16, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Constâncio, V.; Nunes, S.P.; Henrique, R.; Jerónimo, C. DNA Methylation-Based Testing in Liquid Biopsies as Detection and Prognostic Biomarkers for the Four Major Cancer Types. Cells 2020, 9, 624. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.V.; Pulverer, W.; Kallmeyer, R.; Beikircher, G.; Pabinger, S.; Kriegner, A.; Weinhäusel, A. MSP-HTPrimer: A High-Throughput Primer Design Tool to Improve Assay Design for DNA Methylation Analysis in Epigenetics. Clin. Epigenet. 2016, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Marshall, O.J. PerlPrimer: Cross-Platform, Graphical Primer Design for Standard, Bisulphite and Real-Time PCR. Bioinformatics 2004, 20, 2471–2472. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-C.; Dahiya, R. MethPrimer: Designing Primers for Methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [Green Version]
- Hernández, H.G.; Tse, M.Y.; Pang, S.C.; Arboleda, H.; Forero, D.A. Optimizing Methodologies for PCR-Based DNA Methylation Analysis. Biotechniques 2013, 55, 181–197. [Google Scholar] [CrossRef]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A High-Throughput Assay to Measure DNA Methylation. Nucleic Acids Res. 2000, 28, 32e. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Gupta, S.; Badarukhiya, J.A.; Sachan, M. Detection of Aberrant Methylation of HOXA9 and HIC1 through Multiplex MethyLight Assay in Serum DNA for the Early Detection of Epithelial Ovarian Cancer. Int. J. Cancer 2020, 147, 1740–1752. [Google Scholar] [CrossRef]
- Bacolod, M.D.; Huang, J.; Giardina, S.F.; Feinberg, P.B.; Mirza, A.H.; Swistel, A.; Soper, S.A.; Barany, F. Prediction of Blood-Based Biomarkers and Subsequent Design of Bisulfite PCR-LDR-QPCR Assay for Breast Cancer Detection. BMC Cancer 2020, 20, 85. [Google Scholar] [CrossRef] [Green Version]
- Bacolod, M.D.; Mirza, A.H.; Huang, J.; Giardina, S.F.; Feinberg, P.B.; Soper, S.A.; Barany, F. Application of Multiplex Bisulfite PCR–Ligase Detection Reaction–Real-Time Quantitative PCR Assay in Interrogating Bioinformatically Identified, Blood-Based Methylation Markers for Colorectal Cancer. J. Mol. Diagn. 2020, 22, 885–900. [Google Scholar] [CrossRef]
- Hussmann, D.; Hansen, L.L. Methylation-Sensitive High Resolution Melting (MS-HRM). Bioinform. MicroRNA Res. 2018, 1708, 551–571. [Google Scholar]
- Wojdacz, T.K.; Dobrovic, A. Methylation-Sensitive High Resolution Melting (MS-HRM): A New Approach for Sensitive and High-Throughput Assessment of Methylation. Nucleic Acids Res. 2007, 35, e41. [Google Scholar] [CrossRef] [Green Version]
- Uehiro, N.; Sato, F.; Pu, F.; Tanaka, S.; Kawashima, M.; Kawaguchi, K.; Sugimoto, M.; Saji, S.; Toi, M. Circulating Cell-Free DNA-Based Epigenetic Assay Can Detect Early Breast Cancer. Breast Cancer Res. 2016, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Barault, L.; Amatu, A.; Siravegna, G.; Ponzetti, A.; Moran, S.; Cassingena, A.; Mussolin, B.; Falcomatà, C.; Binder, A.M.; Cristiano, C.; et al. Discovery of Methylated Circulating DNA Biomarkers for Comprehensive Non-Invasive Monitoring of Treatment Response in Metastatic Colorectal Cancer. Gut 2017, 67, 1995–2005. [Google Scholar] [CrossRef]
- Sato, H.; Soh, J.; Aoe, K.; Fujimoto, N.; Tanaka, S.; Namba, K.; Torigoe, H.; Shien, K.; Yamamoto, H.; Tomida, S.; et al. Droplet Digital PCR As a Novel System for the Detection of microRNA-34b/C Methylation in Circulating DNA in Malignant Pleural Mesothelioma. Int. J. Oncol. 2019, 54, 2139–2148. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Drabløs, F.; Rye, M.B. DNA Methylation Data by Sequencing: Experimental Approaches and Recommendations for Tools and Pipelines for Data Analysis. Clin. Epigenet. 2019, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Relton, C.L. Statistical and Integrative System-Level Analysis of DNA Methylation Data. Nat. Rev. Genet. 2018, 19, 129–147. [Google Scholar] [CrossRef]
- Bock, C. Analysing and Interpreting DNA Methylation Data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Grennan, K.; Badner, J.; Zhang, D.; Gershon, E.; Jin, L.; Liu, C. Removing Batch Effects in Analysis of Expression Microarray Data: An Evaluation of Six Batch Adjustment Methods. PLoS ONE 2011, 6, e17238. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Zhuang, J.; Widschwendter, M. Independent Surrogate Variable Analysis to Deconvolve Confounding Factors in Large-Scale Microarray Profiling Studies. Bioinformatics 2011, 27, 1496–1505. [Google Scholar] [CrossRef]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. CutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Siegmund, K.; Laird, P.W.; Berman, B.P. Bis-SNP: Combined DNA Methylation and SNP Calling for Bisulfite-Seq Data. Genome Biol. 2012, 13, R61. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.; Dhami, P.; Feber, A.; Cortázar, D.; Suzuki, Y.; Schulz, R.; Schär, P.; Beck, S. Resources for Methylome Analysis Suitable for Gene Knockout Studies of Potential Epigenome Modifiers. GigaScience 2012, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-Wide Differential Coverage Analysis of Sequencing Data Derived from DNA Enrichment Experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef]
- Robinson, M.D.; Kahraman, A.; Law, C.W.; Lindsay, H.; Nowicka, M.; Weber, L.M.; Zhou, X. Statistical Methods for Detecting Differentially Methylated Loci and Regions. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Benelli, M.; Romagnoli, D.; Demichelis, F. Tumor Purity Quantification by Clonal DNA Methylation Signatures. Bioinformatics 2018, 34, 1642–1649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wu, H.-J.; Zhang, W.; Wang, J.; Wu, H.; Zheng, X. Predicting Tumor Purity from Methylation Microarray Data. Bioinformatics 2015, 31, 3401–3405. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-Cancer Deconvolution of Tumour Composition Using DNA Methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.E.; Feinberg, A.P.; Irizarry, R.A.; Leek, J.T. Significance Analysis and Statistical Dissection of Variably Methylated Regions. Biostatics 2012, 13, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Prandi, D.; Baca, S.C.; Romanel, A.; Barbieri, C.E.; Mosquera, J.-M.M.; Fontugne, J.; Beltran, H.; Sboner, A.; Garraway, L.A.; Rubin, M.A.; et al. Unraveling the Clonal Hierarchy of Somatic Genomic Aberrations. Genome Biol. 2014, 15, 439. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-Invasive Early Detection of Cancer Four Years before Conventional Diagnosis Using a Blood Test. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and Discrimination of Intracranial Tumors Using Plasma Cell-Free DNA Methylomes. Nat. Med. 2020, 26, 1044–1047. [Google Scholar] [CrossRef]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Abou Alaiwi, S.; Chakravarthy, A.; Shen, S.Y.; Bakounty, Z.; Boccardo, F.; et al. Detection of Renal Cell Carcinoma Using Plasma and Urine Cell-Free DNA Methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef]
- McCartney, A.; Benelli, M.; Di Leo, A. Estimating the Magnitude of Clinical Benefit from (neo)adjuvant Chemotherapy in Patients With ER-positive/HER2-Negative Breast Cancer. Breast 2019, 48, S81–S84. [Google Scholar] [CrossRef]
- Fackler, M.J.; Lopez Bujanda, Z.; Umbricht, C.; Teo, W.W.; Cho, S.; Zhang, Z.; Visvanthan, K.; Jeter, S.; Arganni, P.; Wang, C.; et al. Novel Methylated Biomarkers and a Robust Assay to Detect Circulating Tumor DNA in Metastatic Breast Cancer. Cancer Res. 2014, 74, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Visvanathan, K.; Fackler, M.S.; Zhang, Z.; Lopez-Bujanda, Z.A.; Jeter, S.C.; Sokoll, L.J.; Garrett-Mayer, E.; Cope, L.M.; Umbricht, C.B.; Euhus, D.M.; et al. Monitoring of Serum DNA Methylation as an Early Independent Marker of Response and Survival in Metastatic Breast Cancer: TBCRC 005 Prospective Biomarker Study. J. Clin. Oncol. 2017, 35, 751–758. [Google Scholar] [CrossRef]
- Xu, W.; Lu, J.; Zhao, Q.; Wu, J.; Sun, J.; Han, B.; Zhao, X.-D.; Kang, Y. Genome-Wide Plasma Cell-Free DNA Methylation Profiling Identifies Potential Biomarkers for Lung Cancer. Dis. Markers 2019, 2019, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bs, K.L.; Nassar, A.H.; Hamieh, L.; Berchuck, J.E.; Nuzzo, P.V.; Korthauer, K.; Shinagare, A.B.; Ogorek, B.; McKay, R.; Thorner, A.R.; et al. Plasma Cell-Free DNA Variant Analysis Compared with Methylated DNA Analysis in Renal Cell Carcinoma. Genet. Med. 2020, 22, 1366–1373. [Google Scholar]
- Ruppin, E.; Robinson, W. Faculty Opinions Recommendation of SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science 2017, 355, 84–88. [Google Scholar]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-Like States, and Drug Resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Fiegl, H.; Millinger, S.; Mueller-Holzner, E.; Marth, C.; Ensinger, C.; Berger, A.; Klocker, H.; Goebel, G.; Widschwendter, M. Circulating Tumor-Specific DNA: A Marker for Monitoring Efficacy of Adjuvant Therapy in Cancer Patients. Cancer Res. 2005, 65, 1141–1145. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-Invasive Cancer Diagnosis and Tissue-of-Origin Prediction Using Methylation Profiles of Cell-Free DNA. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.-C.; Matsuoka, L.; Sher, L.; Wong, W.H.; et al. CancerDetector: Ultrasensitive and Non-Invasive Cancer Detection at the Resolution of Individual Reads Using Cell-Free DNA Methylation Sequencing Data. Nucleic Acids Res. 2018, 46, e89. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Joosten, S.C.; Feng, Z.; De Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA Methylation in Cancer: Location Revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N. Eng. J. Med. 2016, 370, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y.; Dhillon, S. Epi ProColon® 2.0 CE: A Blood-Based Screening Test for Colorectal Cancer. Mol. Diagn. Ther. 2017, 21, 225–232. [Google Scholar]
- Weiss, G.; Schlegel, A.; Kottwitz, D.; König, T.; Tetzner, R. Validation of the SHOX2 / PTGER4 DNA Methylation Marker Panel for Plasma-Based Discrimination Between Patients with Malignant and Nonmalignant Lung Disease. J. Thorac. Oncol. 2017, 12, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Liou, Y.-L.; Zhang, T.-L.; Yan, T.; Yeh, C.-T.; Kang, Y.-N.; Cao, L.; Wu, N.; Chang, C.-F.; Wang, H.-J.; Yen, C.; et al. Combined Clinical and Genetic Testing Algorithm for Cervical Cancer Diagnosis. Clin. Epigenet. 2016, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-J.; Chang, C.-F.; Lee, J.; Chen, H.-M.; Wang, H.-J.; Liou, Y.-L.; Yen, C.; Chiang, C.-P. Hypermethylated ZNF582 and PAX1 Are Effective Biomarkers for Detection of Oral Dysplasia and Oral Cancer. Oral Oncol. 2016, 62, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Van Kessel, K.E.M.; Van Neste, L.; Lurkin, I.; Zwarthoff, E.C.; Van Criekinge, W. Evaluation of an Epigenetic Profile for the Detection of Bladder Cancer in Patients with Hematuria. J. Urol. 2016, 195, 601–607. [Google Scholar] [CrossRef]
- Van Lanschot, M.C.J.; Carvalho, B.; Coupé, V.M.; Van Engeland, M.; Dekker, E.; Meijer, G.A. Molecular Stool Testing as an Alternative for Surveillance Colonoscopy: A Cross-Sectional Cohort Study. BMC Cancer 2017, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Beltrán-García, J.; Osca-Verdegal, R.; Mena, S.; García-Giménez, J.L. Epigenetic IVD Tests for Personalized Precision Medicine in Cancer. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical Epigenetics: Seizing Opportunities for Translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Gallardo-Gomez, M.; Moran, S.; De La Cadena, M.P.; Martínez-Zorzano, V.; Rodríguez-Berrocal, F.J.; Rodríguez-Girondo, M.; Esteller, M.; Cubiella, J.; Bujanda, L.; Castells, A. New Approach to Epigenome-Wide Discovery of Non-Invasive Methylation Biomarkers for Colorectal Cancer Screening in Circulating Cell-Free DNA Using Pooled Samples. Clin. Epigenet. 2018, 10, 53. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-Specific PCR: A Novel PCR Assay for Methylation Status of CpG Islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [Green Version]
- Watters, J.W.; Roberts, C.J. Developing Gene Expression Signatures of Pathway Deregulation in Tumors. Mol. Cancer Ther. 2006, 5, 2444–2449. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.J.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2006, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Name | Manufacturer | Biomarker(s) | Biosample | Application | Sensitivity (%) | Specificity (%) | Reference |
|---|---|---|---|---|---|---|---|
| Cologuard | Exact Sciences Corp. | NDRG4, BMP3 | Stool | CRC early detection | 92 | 87 | [138] |
| Epi proColon 2.0 | Epigenomics GA | SEPT9 | Plasma | CRC early detection | 81 | 97 | [139] |
| Epi proLung | Epigenomics GA | PTGER4, SHOX2 | Plasma | Lung cancer detection | 90 | 73 | [140] |
| Cervi-M | Epigene, iStat Biomedical Co | ZNF582, PAX1 | Cervical brush | Cervical cancer detection | 77 *, 70 ** | 87 *, 82 ** | [141] |
| Oral-M | Epigene, iStat Biomedical Co | ZNF582, PAX1 | Oral swab | Oral cancer detection | 85 *, 72 ** | 87 *, 86 ** | [142] |
| Assure MDx | MDxHealth | TWIST1, ONECUT2, OTX1 (+FGFR3, TERT, HRAS mutations) | Urine | Bladder cancer detection | 97 | 83 | [143] |
| Assay | Sample Requirement [Minimum] | DNA Treatment | Advantages | Disadvantages | Cost | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bisulfite Conversion | Restriction Enzyme | DNA Precipitation | Enzymatic Plus Chemical Modification | ||||||||
| Genome wide | Microarray | HM450 | 500 ng | ■ | pre-designed panel, easy to use, time efficient | DNA degradation; low coverage of intergenic regions | •• | [21,77,78,147] | |||
| HM850 | 250 ng | ■ | As above; includes enhancer regions; suitable for FFPE DNA; | As above | •• | ||||||
| MeKL-chip | 10–20 ng | ■ | low DNA input | Cross hybridization, PCR amplification, MBD binding ability | •• | [79] | |||||
| Whole genome sequencing | WGBS | 10 ng | ■ | full methylome | DNA degradation; requires high sequencing depth; low input DNA may induce PCR bias | ••••• | [51,53,54,56] | ||||
| PBAT | 125 pg–10 ng | ■ | full methylome; PCR free; suitable for single cell analysis | DNA degradation; requires high sequencing depth; low fraction of aligned reads | ••••• | [59,60,61,62] | |||||
| TAPS | 1 ng | ■ | no DNA degradation; low input DNA; suitable for third generation sequencing; detect 5mC and 5hmC | hyper-active TET1 preparation | •••• | [63,64] | |||||
| EM-seq | 100 pg | ■ | no bisulfite DNA degradation; very low DNA input; high mapping; uniform GC coverage; detect 5mC and 5hmC | low complexity sequencing library | ••• | [50,63] | |||||
| Representative genome wide methods | RRBS | 10–100 ng | ■ | ■ | high CpG coverage | DNA degradation, low coverage of intergenic regions | ••• | [66,67] | |||
| scRRBS | one cell | ■ | ■ | very low DNA input | ••• | [68,69] | |||||
| MCTA-seq | 7.5 pg | ■ | very low DNA input | DNA degradation, low coverage of intergenic regions | ••• | [70] | |||||
| cfMeDIP–seq | 1–10 ng | ■ | no bisulfite DNA degradation, no mutation introduced, genome wide CpG and no CpG, very low DNA input | Detect only regions, low GpG density bias, CNA bias, depending on antibody performance | ••• | [45,71,123,125] | |||||
| MBD-seq | 5 ng | ■ | no bisulfite DNA degradation, outperform meDIP-seq in regions with higher CpG density | hypermethylated regions bias, CNA bias, depending on antibody performance | ••• | [48,72] | |||||
| Targeted | Sequencing | Target bisulfite seq | 20-30 ng | ■ | high coverage on target loci | primer or probe design | ••/••• | [58,80,81,82,83,84,85,122] | |||
| PCR | MSPCR | Pg | ■ | ■ | low DNA in input; relative quantification of target loci | primer or probe design | • | [86,91,92,94,95,148] | |||
| ddMSPCR | pg | ■ | ■ | low DNA input; absolute quantification of target loci | primer or probe design; quantification depends on DNA input | • | [97,98,99] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galardi, F.; De Luca, F.; Romagnoli, D.; Biagioni, C.; Moretti, E.; Biganzoli, L.; Di Leo, A.; Migliaccio, I.; Malorni, L.; Benelli, M. Cell-Free DNA-Methylation-Based Methods and Applications in Oncology. Biomolecules 2020, 10, 1677. https://doi.org/10.3390/biom10121677
Galardi F, De Luca F, Romagnoli D, Biagioni C, Moretti E, Biganzoli L, Di Leo A, Migliaccio I, Malorni L, Benelli M. Cell-Free DNA-Methylation-Based Methods and Applications in Oncology. Biomolecules. 2020; 10(12):1677. https://doi.org/10.3390/biom10121677
Chicago/Turabian StyleGalardi, Francesca, Francesca De Luca, Dario Romagnoli, Chiara Biagioni, Erica Moretti, Laura Biganzoli, Angelo Di Leo, Ilenia Migliaccio, Luca Malorni, and Matteo Benelli. 2020. "Cell-Free DNA-Methylation-Based Methods and Applications in Oncology" Biomolecules 10, no. 12: 1677. https://doi.org/10.3390/biom10121677