Structure-Based Virtual Screening of Ultra-Large Library Yields Potent Antagonists for a Lipid GPCR

 , , ,
, , ,  , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Receptor Model Preparation and Optimization
2.2. Screening Libraries
2.3. Virtual Ligand Screening
2.4. IP1 Production Assay
3. Results
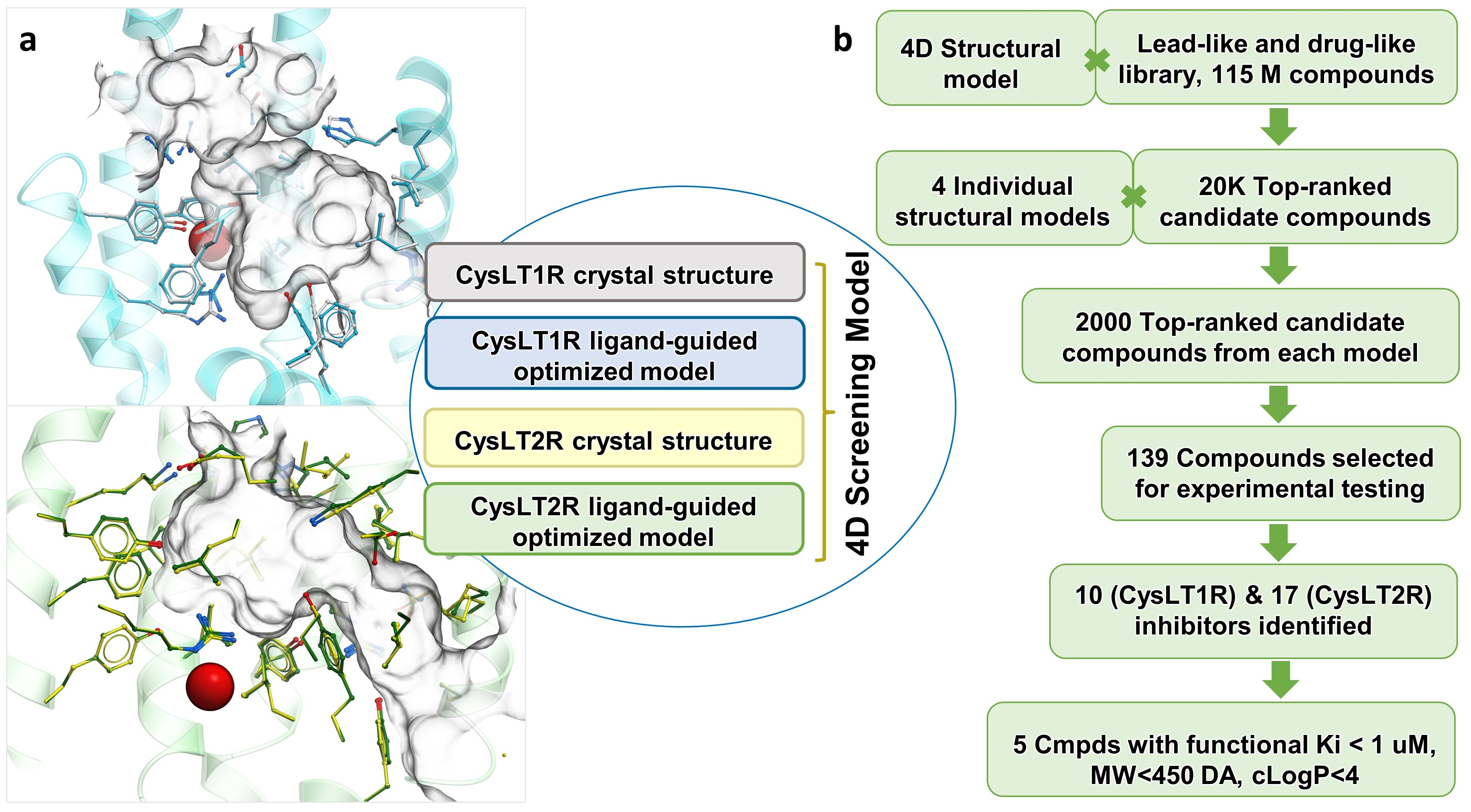
3.1. Optimization of Receptor Models
3.2. Prospective VLS and Hits Selections
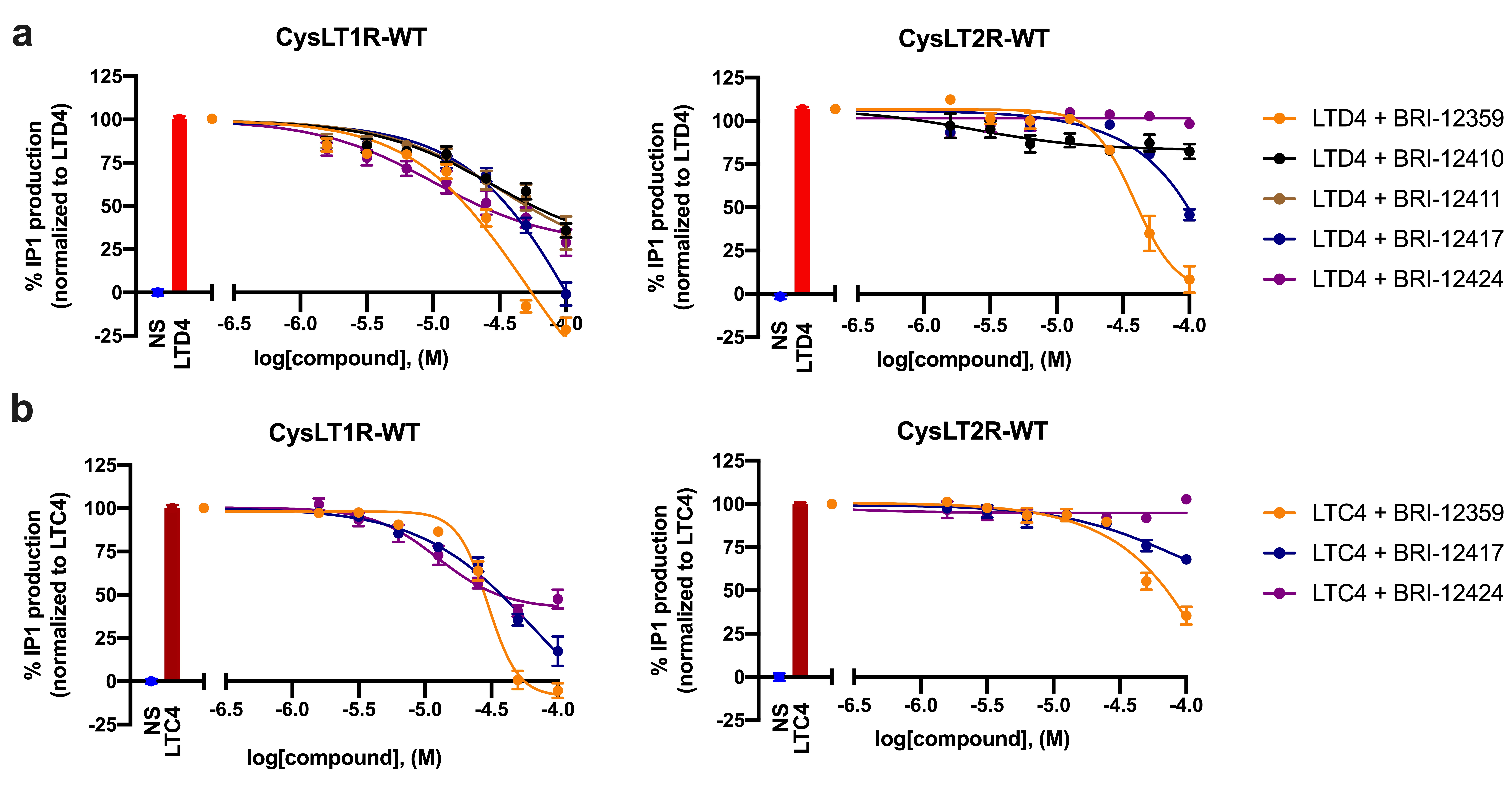
3.3. Functional Activity of Selected Compounds in IP1 Production Assay
3.4. Characterization of Hits for Their Ability to Inhibit LTC4-Induced IP1 Production
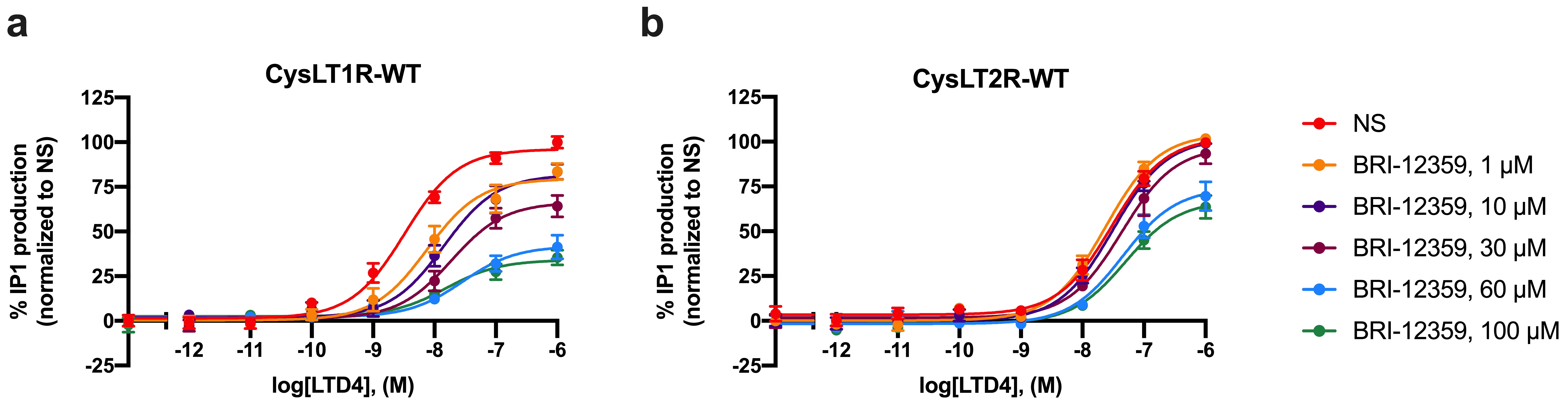
3.5. Further Functional Characterization of Best Hits in IP1 Production Assay
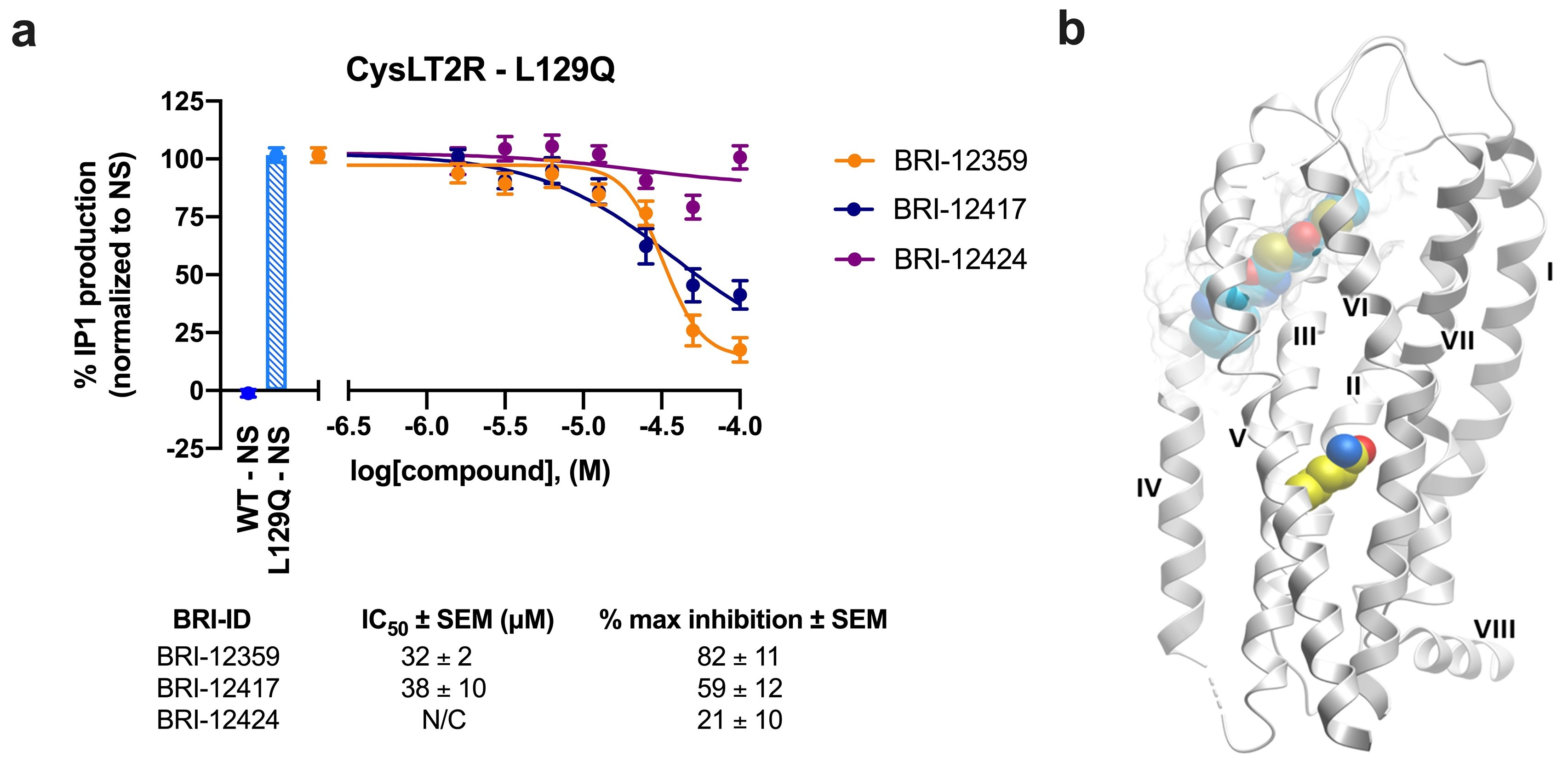
3.6. Inhibition of Constitutively Active CysLT2R Mutant L129Q Involved in Uveal Melanoma
3.7. Chemical and Conformational Diversity of Hit Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bäck, M.; Powell, W.S.; Dahlén, S.-E.; Drazen, J.M.; Evans, J.F.; Serhan, C.N.; Shimizu, T.; Yokomizo, T.; Rovati, G.E. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br. J. Pharmacol. 2014, 171, 3551–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovati, G.E.; Capra, V. Cysteinyl-leukotriene receptors and cellular signals. Sci. World J. Hindawi Ltd. 2007, 7, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, T.M.; Boyce, J.A. Cysteinyl leukotriene receptors, old and new; implications for asthma. Clin. Exp. Allergy 2012, 42, 1313–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelsson, E.; Yin, L.; Bäck, M. Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J. Allergy Clin. Immunol. 2012, 129, 702–707.e2. [Google Scholar] [CrossRef] [PubMed]
- Burke, L.; Butler, C.T.; Murphy, A.; Moran, B.; Gallagher, W.M.; O’Sullivan, J.; Kennedy, B.N. Evaluation of Cysteinyl leukotriene signaling as a therapeutic target for colorectal cancer. Front. Cell Dev. Biol. 2016, 4, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäck, M.; Dahlén, S.-E.; Drazen, J.M.; Evans, J.F.; Serhan, C.N.; Shimizu, T.; Yokomizo, T.; Rovati, G.E. International Union of Basic and Clinical Pharmacology. LXXXIV: Leukotriene receptor nomenclature, distribution, and pathophysiological functions. Pharmacol. Rev. 2011, 63, 539–584. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; A Kazmi, M.; Taylor, M.T.C.B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Küsters-Vandevelde, H.V.N.; Germans, M.R.; Rabbie, R.; Rashid, M.; Broek, R.T.; Blokx, W.A.M.; Prinsen, C.F.M.; Adams, D.J.; Ter Laan, M. Whole-exome sequencing of a meningeal melanocytic tumour reveals activating CYSLTR2 and EIF1AX hotspot mutations and similarities to uveal melanoma. Brain Tumor Pathol. 2018, 35, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Van De Nes, J.A.; Koelsche, C.; Gessi, M.; Möller, I.; Sucker, A.; Scolyer, R.A.; Buckland, M.E.; Pietsch, T.; Murali, R.; Schadendorf, D.; et al. Activating CYSLTR2 and PLCB4 mutations in primary leptomeningeal melanocytic tumors. J. Investig. Dermatol. 2017, 137, 2033–2035. [Google Scholar] [CrossRef] [Green Version]
- Möller, I.; Murali, R.; Müller, H.; Wiesner, T.; A Jackett, L.; Scholz, S.L.; Cosgarea, I.; Ap Van De Nes, J.; Sucker, A.; Hillen, U.; et al. Activating cysteinyl leukotriene receptor 2 (CYSLTR2) mutations in blue nevi. Mod. Pathol. 2017, 30, 350–356. [Google Scholar] [CrossRef]
- Ceraudo, E.; Horioka, M.; Mattheisen, J.M.; Hitchman, T.D.; Moore, A.R.; Kazmi, M.A.; Chi, P.; Chen, Y.; Sakmar, T.P.; Huber, T. Uveal melanoma oncogene CYSLTR2 encodes a constitutively active GPCR highly biased toward Gq signaling. bioRxiv 2019, 1–60. [Google Scholar] [CrossRef] [Green Version]
- Miligkos, M.; Bannuru, R.R.; Alkofide, H.; Kher, S.R.; Schmid, C.H.; Balk, E.M. Leukotriene receptor antagonists versus placebo in the treatment of asthma in adults and adolescents: A systematic review and meta-analysis. Ann. Intern. Med. 2015, 163, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, S.W.Y.; Wong, A.Y.S.; Anand, S.; Wong, I.C.K.; Chan, E.W. Neuropsychiatric events associated with leukotriene-modifying agents: A systematic review. Drug Saf. 2018, 41, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimbo, J.; Onodera, O.; Tanaka, K.; Tsuji, S. Churg-Strauss syndrome and the leukotriene receptor antagonist pranlukast. Clin. Rheumatol. 2005, 24, 661–662. [Google Scholar] [CrossRef]
- Yonetomi, Y.; Sekioka, T.; Kadode, M.; Kitamine, T.; Kamiya, A.; Inoue, A.; Nakao, T.; Nomura, H.; Murata, M.; Nakao, S.; et al. Effects of ONO-6950, a novel dual cysteinyl leukotriene 1 and 2 receptors antagonist, in a guinea pig model of asthma. Eur. J. Pharmacol. 2015, 765, 242–248. [Google Scholar] [CrossRef]
- Reynolds, A.L.; Alvarez, Y.; Sasore, T.; Waghorne, N.; Butler, C.T.; Kilty, C.; Smith, A.J.; McVicar, C.; Wong, V.H.Y.; Galvin, O.; et al. Phenotype-Based discovery of 2-[(E)-2-(Quinolin-2-yl)vinyl]phenol as a novel regulator of ocular angiogenesis. J. Biol. Chem. 2016, 291, 7242–7255. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.G.; Casey, R.; Maguire, A.; Tosetto, M.; Butler, C.T.; Conroy, E.; Reynolds, A.L.; Sheahan, K.; O’Donoghue, D.; Gallagher, W.M.; et al. Preclinical validation of the small molecule drug quininib as a novel therapeutic for colorectal cancer. Sci. Rep. 2016, 6, 34523. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Huang, X.-P.; Grandner, J.M.; Johansson, L.C.; Stauch, B.; McCorvy, J.D.; Liu, Y.; Roth, B.; Katritch, V. Structure-Based discovery of potent and selective melatonin receptor agonists. eLife 2020, 9. [Google Scholar] [CrossRef]
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.C.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.-P.; Savych, O.; Moroz, Y.S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-Large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, X.-P.; Mangano, T.J.; Zou, R.; Chen, X.; Zaidi, S.A.; Roth, B.L.; Stevens, R.C.; Katritch, V. Structure-Based discovery of new antagonist and biased agonist chemotypes for the Kappa opioid receptor. J. Med. Chem. 2017, 60, 3070–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, M.A.; Roth, C.B.; Jo, E.; Griffith, M.T.; Scott, F.L.; Reinhart, G.; Desale, H.; Clemons, B.; Cahalan, S.M.; Schuerer, S.C.; et al. Crystal structure of a lipid G protein-coupled receptor. Science 2012, 335, 851–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audet, M.; Stevens, R.C. Emerging structural biology of lipid G protein-coupled receptors. Protein Sci. 2019, 28, 292–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- REAL Database—Enamine. Available online: https://www.enamine.net/library-synthesis/real-compounds/real-database (accessed on 21 September 2020).
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiriaeva, A.; Besserer-Offroy, É.; Longpré, J.-M.; Lyapina, E.; et al. Structure-Based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 2019, 5, eaax2518. [Google Scholar] [CrossRef] [Green Version]
- Gusach, A.; Luginina, A.; Marin, E.; Brouillette, R.L.; Besserer-Offroy, É.; Longpré, J.-M.; Ishchenko, A.; Popov, P.; Patel, N.; Fujimoto, T.; et al. Structural basis of ligand selectivity and disease mutations in cysteinyl leukotriene receptors. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Bottegoni, G.; Kufareva, I.; Totrov, M.; Abagyan, R. Four-Dimensional docking: A fast and accurate account of discrete receptor flexibility in ligand docking. J. Med. Chem. 2009, 52, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Katritch, V.; Rueda, M.; Abagyan, R. Ligand-Guided receptor optimization. Methods Mol. Biol. 2012, 189–205. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Gatica, E.A.; Cavasotto, C.N. Ligand and decoy sets for docking to G protein-coupled receptors. J. Chem. Inf. Model. 2012, 52, 1–6. [Google Scholar] [CrossRef]
- ICM v.3.9 User’s Guide. Available online: http://www.molsoft.com/icmpro/ (accessed on 21 September 2020).
- Totrov, M.; Abagyan, R. Derivation of Sensitive Discrimination Potential for Virtual Ligand Screening; (RECOMB 99); ACM Press: Lyon, France, 1999; pp. 312–317. [Google Scholar]
- Lane, R.; Chubukov, P.; Liu, W.; Canals, M.; Cherezov, V.; Abagyan, R.; Stevens, R.C.; Katritch, V. Structure-Based Ligand Discovery Targeting Orthosteric and Allosteric Pockets of Dopamine Receptors. Mol. Pharmacol. 2013, 84, 794–807. [Google Scholar] [CrossRef] [Green Version]
- Besserer-Offroy, E.; Brouillette, R.; Longpré, J.-M.; Sarret, P. Assessing Gαq/15-signaling with IP-One: Single plate transfection and assay protocol for cell-based high-throughput assay. BIO PROTOC 2020, 10, e3715. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Goto, K.; Pissaloux, D.; Paindavoine, S.; Tirode, F.; de la Fouchardière, A. CYSLTR2-mutant cutaneous melanocytic neoplasms frequently simulate “Pigmented Epithelioid Melanocytoma”, expanding the morphologic spectrum of blue tumors: A clinicopathologic study of 7 cases. Am. J. Surg. Pathol. 2019, 43, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Knappe, U.J.; Tischoff, I.; Tannapfel, A.; Möller, I.; Sucker, A.; Schadendorf, D.; Griewank, K.G.; Van De Nes, J.A.; Reinbold, W.-D. Intraventricular melanocytoma diagnosis confirmed by gene mutation profile. Neuropathology 2018, 38, 288–292. [Google Scholar] [CrossRef]
- Jiang, Y.; Borrelli, L.A.; Kanaoka, Y.; Bacskai, B.J.; Boyce, J.A. CysLT2 receptors interact with CysLT1 receptors and down-modulate cysteinyl leukotriene–dependent mitogenic responses of mast cells. Blood 2007, 110, 3263–3270. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.-J.; Xiao, L.; Zhao, B.; Zhang, X.-Y.; Wang, X.-R.; Xu, D.-M.; Yu, S.-Y.; Fang, S.-H.; Lu, Y.-B.; Zhang, W.-P.; et al. Intracerebroventricular injection of HAMI 3379, a selective cysteinyl leukotriene receptor 2 antagonist, protects against acute brain injury after focal cerebral ischemia in rats. Brain Res. 2012, 1484, 57–67. [Google Scholar] [CrossRef]
- Shi, Q.; Wang, H.; Liu, Z.; Fang, S.; Song, X.; Lu, Y.; Zhang, W.; Sa, X.; Ying, H.; Wei, E. HAMI 3379, a CysLT2R antagonist, dose- and time-dependently attenuates brain injury and inhibits microglial inflammation after focal cerebral ischemia in rats. Neuroscience 2015, 291, 53–69. [Google Scholar] [CrossRef]
- Hoxha, M.; Rovati, G.E.; Cavanillas, A.B. The leukotriene receptor antagonist montelukast and its possible role in the cardiovascular field. Eur. J. Clin. Pharmacol. 2017, 73, 799–809. [Google Scholar] [CrossRef]
- Ni, N.C.; Ballantyne, L.L.; Mewburn, J.D.; Funk, C.D. Multiple-Site activation of the cysteinyl leukotriene receptor 2 is required for exacerbation of ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Slater, K.; Hoo, P.S.; Buckley, A.M.; Piulats, J.M.; Villanueva, A.; Portela, A.; Kennedy, B.N. Evaluation of oncogenic cysteinyl leukotriene receptor 2 as a therapeutic target for uveal melanoma. Cancer Metastasis Rev. 2018, 37, 335–345. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
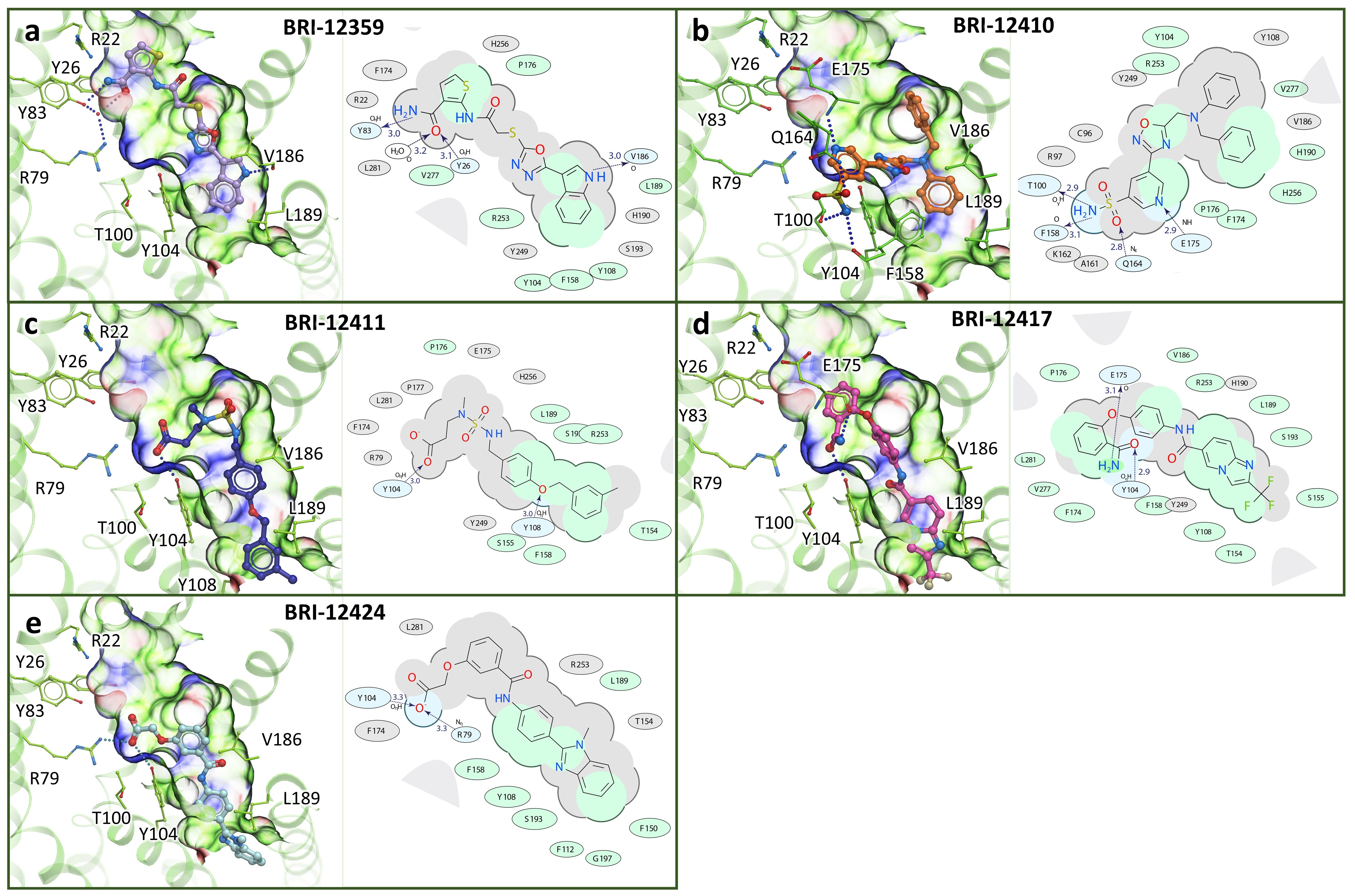{kind=link}
| Number of Water Molecules | ROC | NSA | Score Average | Best Score | |
|---|---|---|---|---|---|
| CyLT1, crystal structure | 0 | 80 | 60 | −37 | −41 |
| CyLT1, LiBERO structure | 1 | 86 | 74 | −46 | −52 |
| CyLT2, crystal structure | 1 | 87 | 77 | −34 | −37 |
| CyLT2, LiBERO structure | 1 | 89 | 82 | −39 | −43 |
| 4D screening model (combined) | as above | 90 | 82 | −45 | −50 |
| CysLT1R (LTD4) | CysLT2R (LTD4) | CysLT1R (LTC4) | CysLT2R (LTC4) | MW, Da | cLogP | cLogS | Tanimoto Distance | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BRI-ID | Ki ± SEM μM | % Max Inhibition ± SEM | Ki ± SEM μM | % Max Inhibition ± SEM | Ki ± SEM μM | % Max Inhibition ± SEM | Ki ± SEM μM | % Max Inhibition ± SEM | ||||
| BRI-12359 | 1.03 ± 0.28 | 121 ± 13 | 6.46 ± 0.43 | 92 ± 15 | 0.72 ± 0.26 | 105 ± 7 | N/C | 65 ± 11 | 399 | 1.59 | −2.25 | 0.57 |
| BRI-12410 | 0.59 ± 0.12 | 64 ± 7 | N/C | 18 ± 9 | 421 | 3.29 | −3.43 | 0.62 | ||||
| BRI-12411 | 0.72 ± 0.16 | 66 ± 18 | 392 | 3.48 | −3.64 | 0.54 | ||||||
| BRI-12417 | 3.1 ± 0.63 | 100 ± 17 | N/C | 54 ± 6 | 0.68 ± 0.12 | 83 ± 14 | N/C | 32 ± 5 | 440 | 3.69 | −4.29 | 0.54 |
| BRI-12424 | 0.22 ± 0.03 | 71 ± 19 | N/C | 7 ± 5 | 0.18 ± 0.03 | 52 ± 9 | N/C | 9 ± 5 | 400 | 3.32 | −3.71 | 0.43 |
| CysLT1R-WT | CysLT2R-WT | |||||
|---|---|---|---|---|---|---|
| [BRI-12359] μM | LTD4 EC50 ± SEM nM | LTD4 Emax ± SEM% | pA2 ± SEM −log | LTD4 EC50 ± SEM nM | LTD4 Emax ± SEM % | pA2 ± SEM −log |
| 0 | 3.3 ± 0.3 | 100 ± 6 | 6.5 ± 0.1 | 30 ± 3 | 100 ± 3 | 4.0 ± 0.2 |
| 1 | 8 ± 1 | 84 ± 9 | 24 ± 2 | 101 ± 3 | ||
| 10 | 14 ± 2 | 83 ± 9 | 31 ± 3 | 99 ± 3 | ||
| 30 | 19 ± 4 | 64 ± 12 | 43 ± 6 | 93 ± 10 | ||
| 60 | 29 ± 8 | 41 ± 13 | 45 ± 8 | 70 ± 16 | ||
| 100 | 15 ± 4 | 35 ± 8 | 50 ± 8 | 64 ± 13 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadybekov, A.A.; Brouillette, R.L.; Marin, E.; Sadybekov, A.V.; Luginina, A.; Gusach, A.; Mishin, A.; Besserer-Offroy, É.; Longpré, J.-M.; Borshchevskiy, V.; et al. Structure-Based Virtual Screening of Ultra-Large Library Yields Potent Antagonists for a Lipid GPCR. Biomolecules 2020, 10, 1634. https://doi.org/10.3390/biom10121634
Sadybekov AA, Brouillette RL, Marin E, Sadybekov AV, Luginina A, Gusach A, Mishin A, Besserer-Offroy É, Longpré J-M, Borshchevskiy V, et al. Structure-Based Virtual Screening of Ultra-Large Library Yields Potent Antagonists for a Lipid GPCR. Biomolecules. 2020; 10(12):1634. https://doi.org/10.3390/biom10121634
Chicago/Turabian StyleSadybekov, Arman A., Rebecca L. Brouillette, Egor Marin, Anastasiia V. Sadybekov, Aleksandra Luginina, Anastasiia Gusach, Alexey Mishin, Élie Besserer-Offroy, Jean-Michel Longpré, Valentin Borshchevskiy, and et al. 2020. "Structure-Based Virtual Screening of Ultra-Large Library Yields Potent Antagonists for a Lipid GPCR" Biomolecules 10, no. 12: 1634. https://doi.org/10.3390/biom10121634