Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Model
2.2. Liver Histological Analysis
2.3. Analysis of the Liver and Plasma Lipid Contents
2.4. Immunoblotting
2.5. Determination of the Oxidative and Antioxidative Parameters
2.6. Data Analysis
3. Results
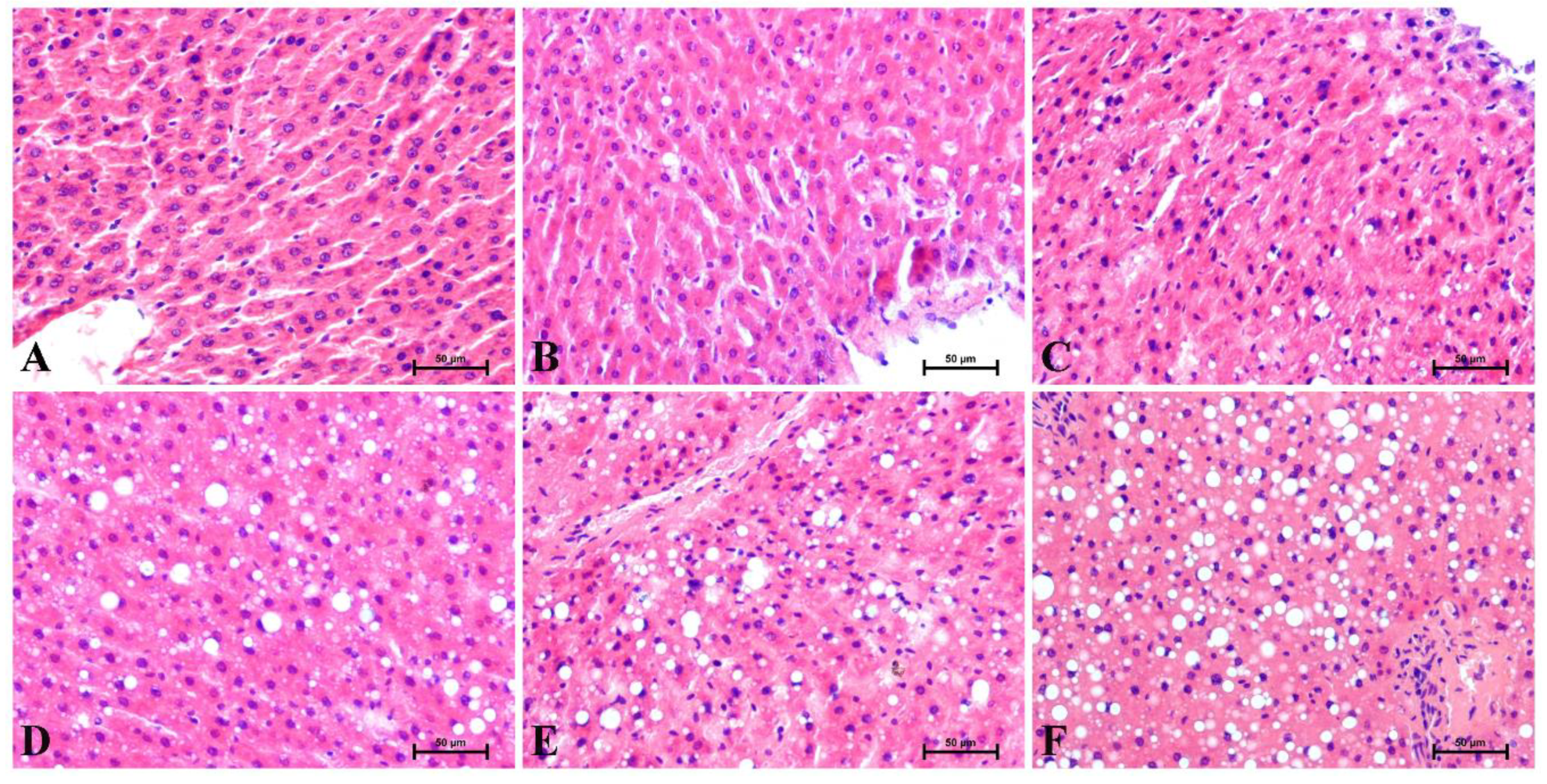
3.1. Changes in the Liver Histology
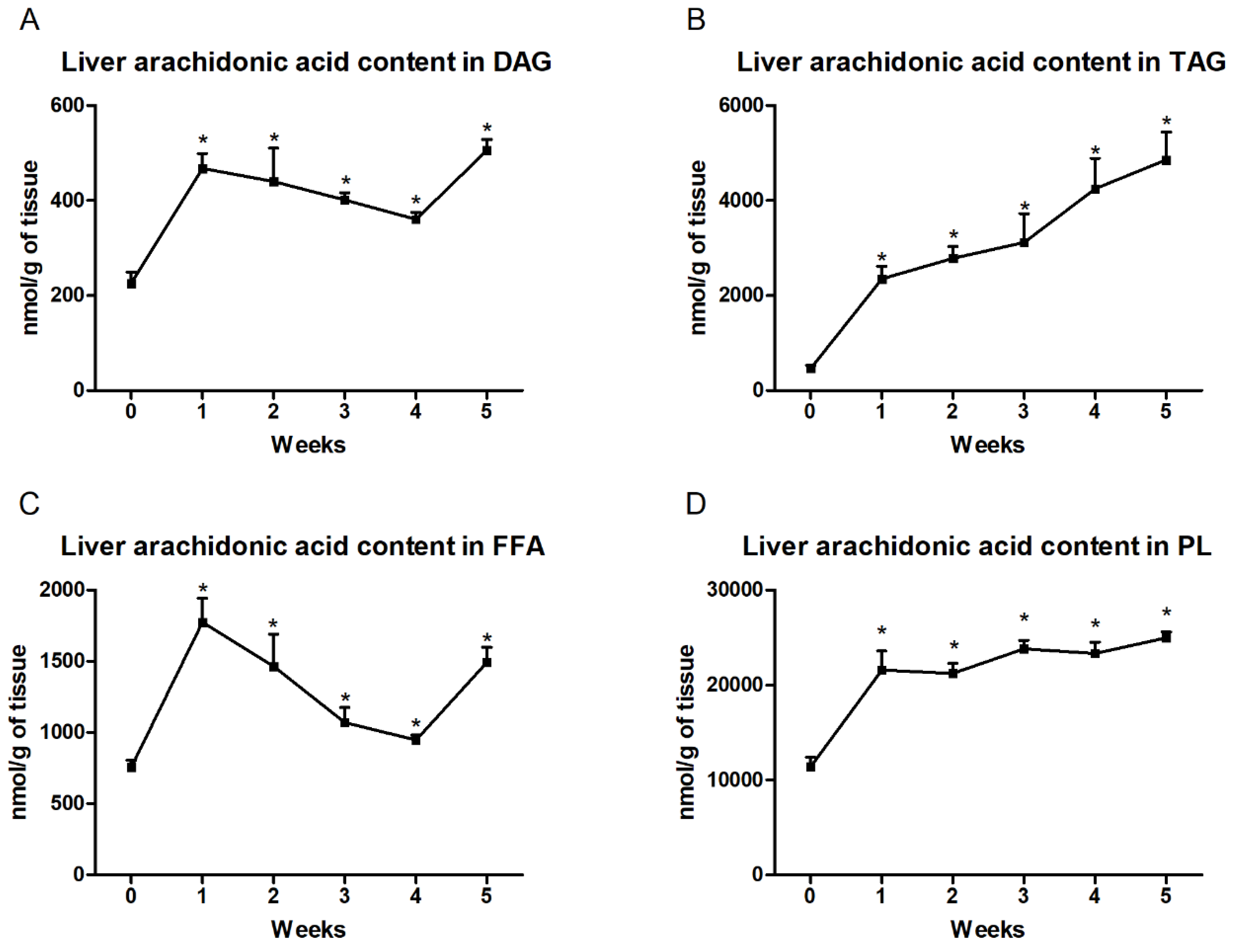
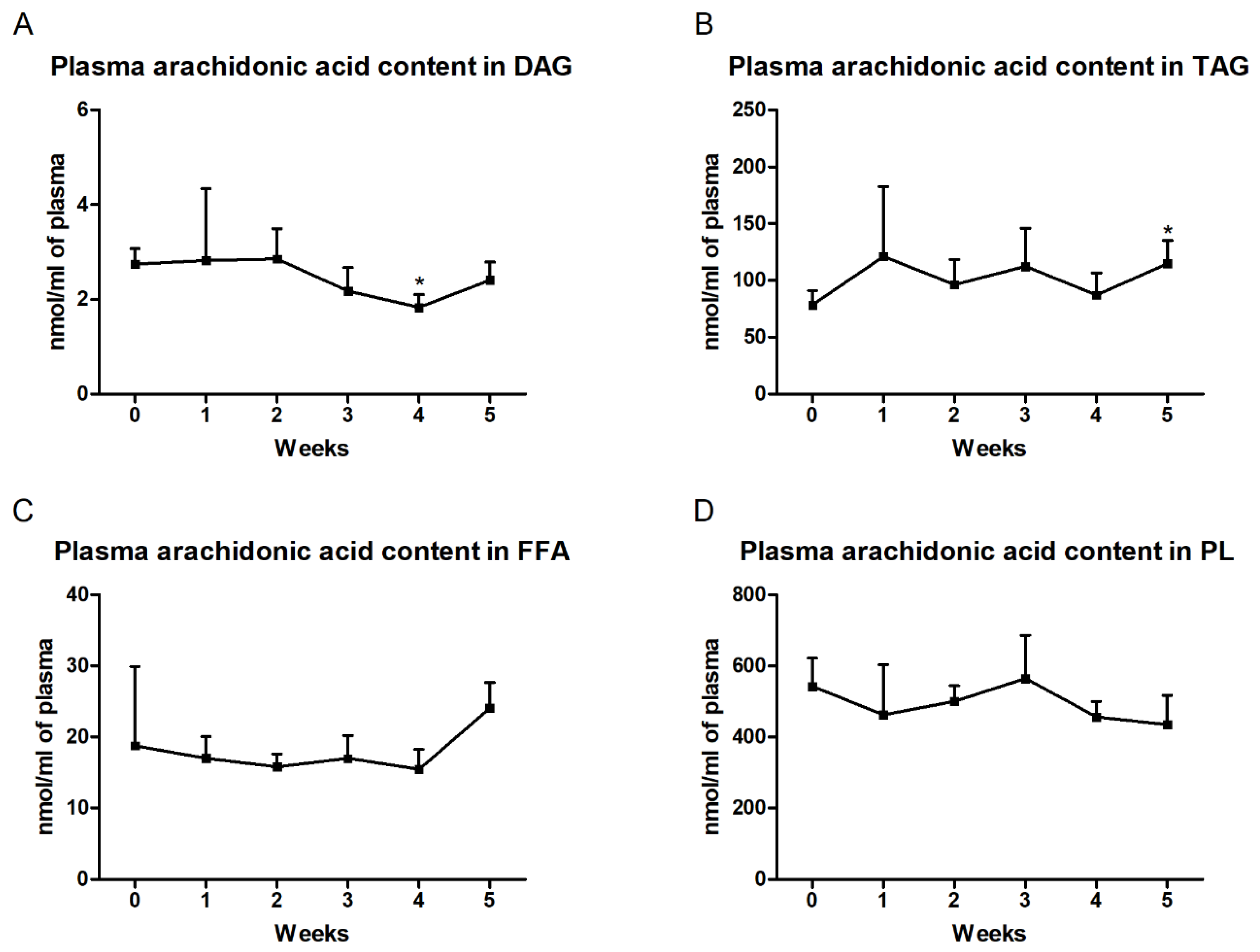
3.2. Changes in the Arachidonic Acid Content in the Liver and Plasma
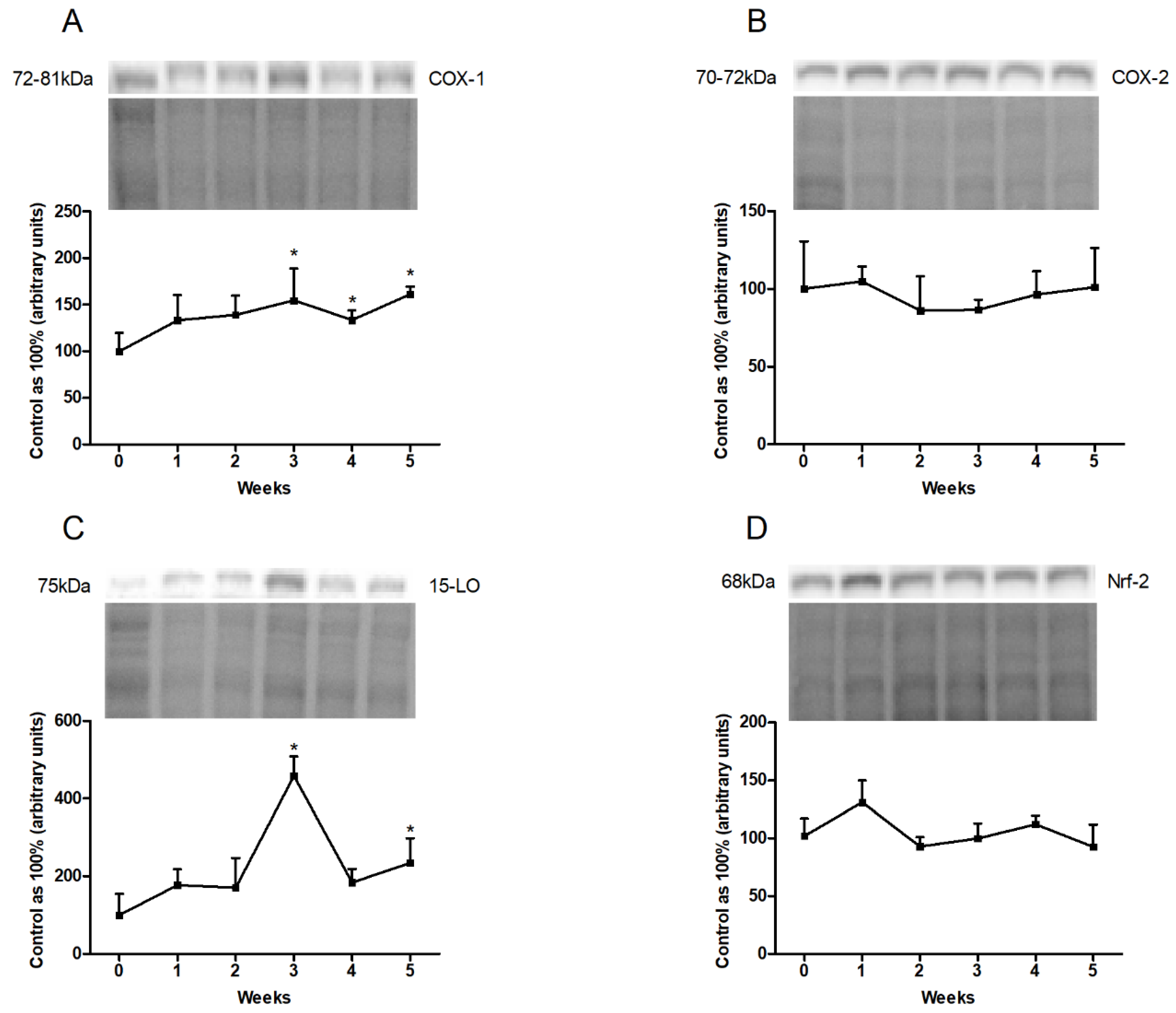
3.3. Changes in the Expression of Proteins Involved in Eicosanoid and Prostanoid Synthesis Pathways
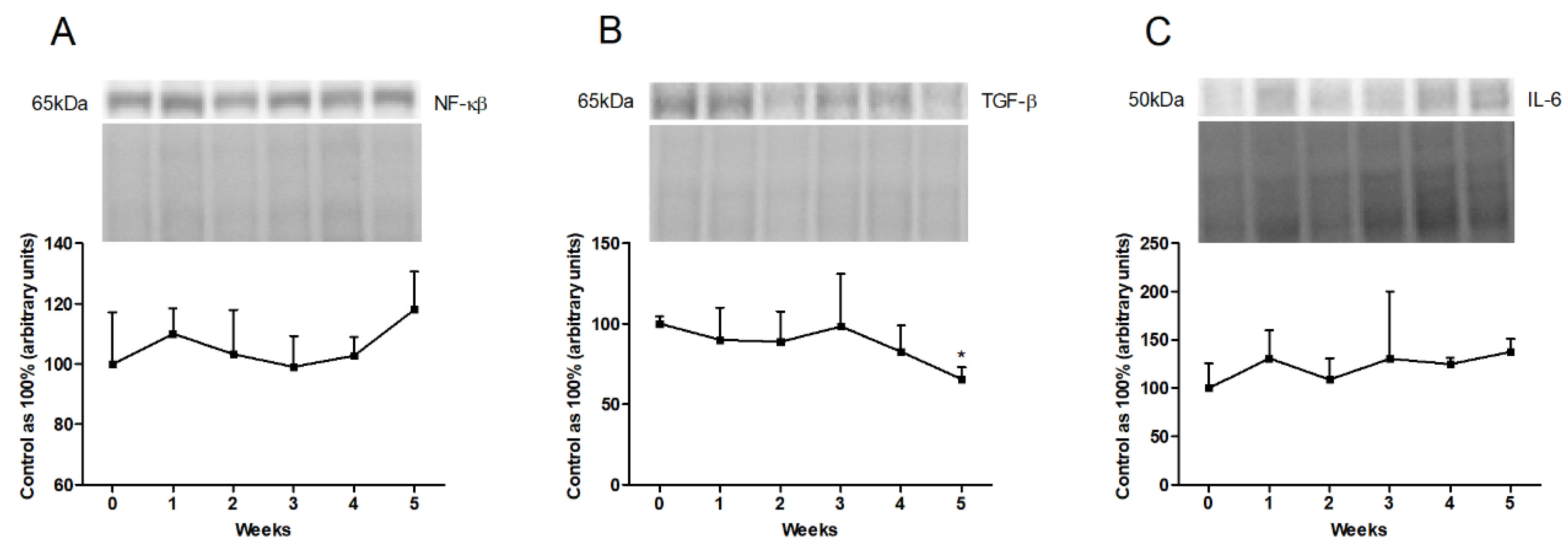
3.4. Changes in the Expression of Inflammatory Pathway Proteins
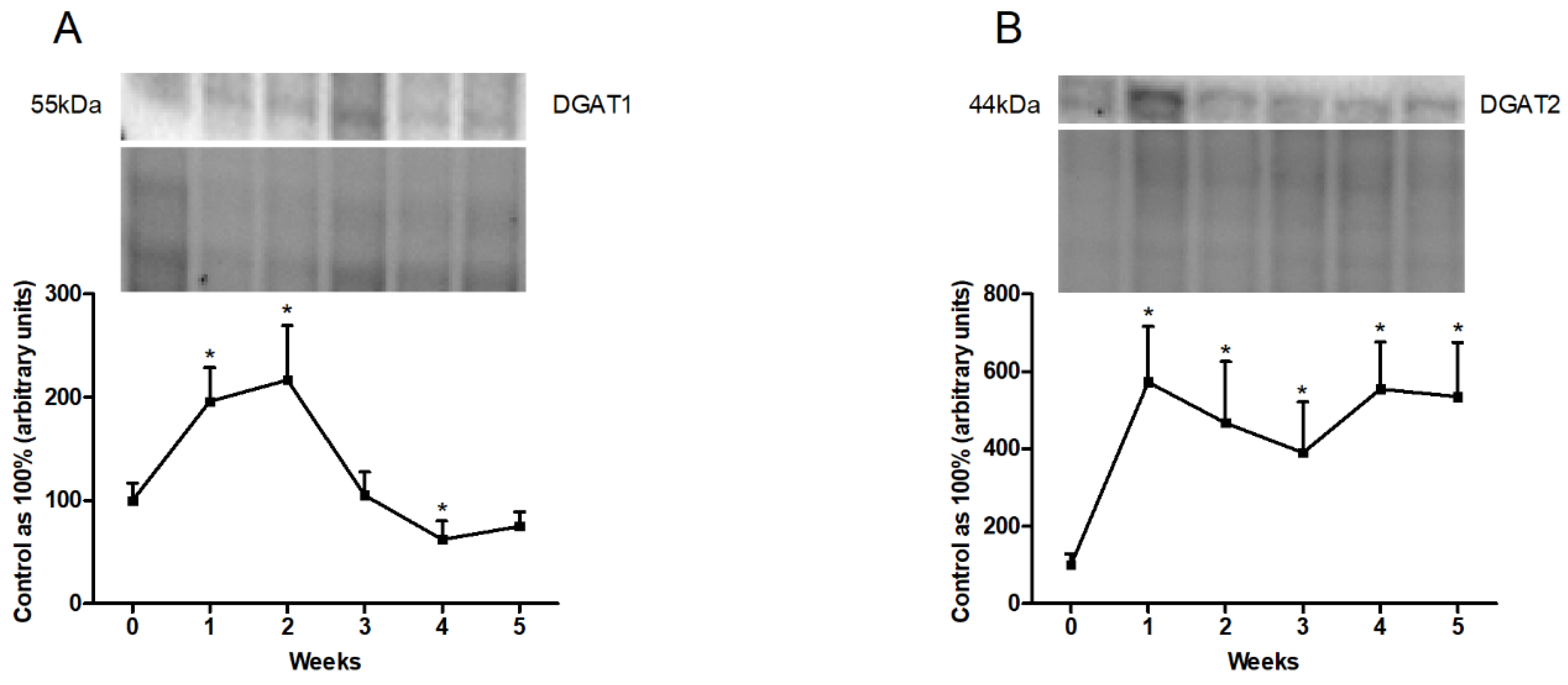
3.5. Changes in the Expression of Proteins Directly Involved in Lipogenesis Pathway
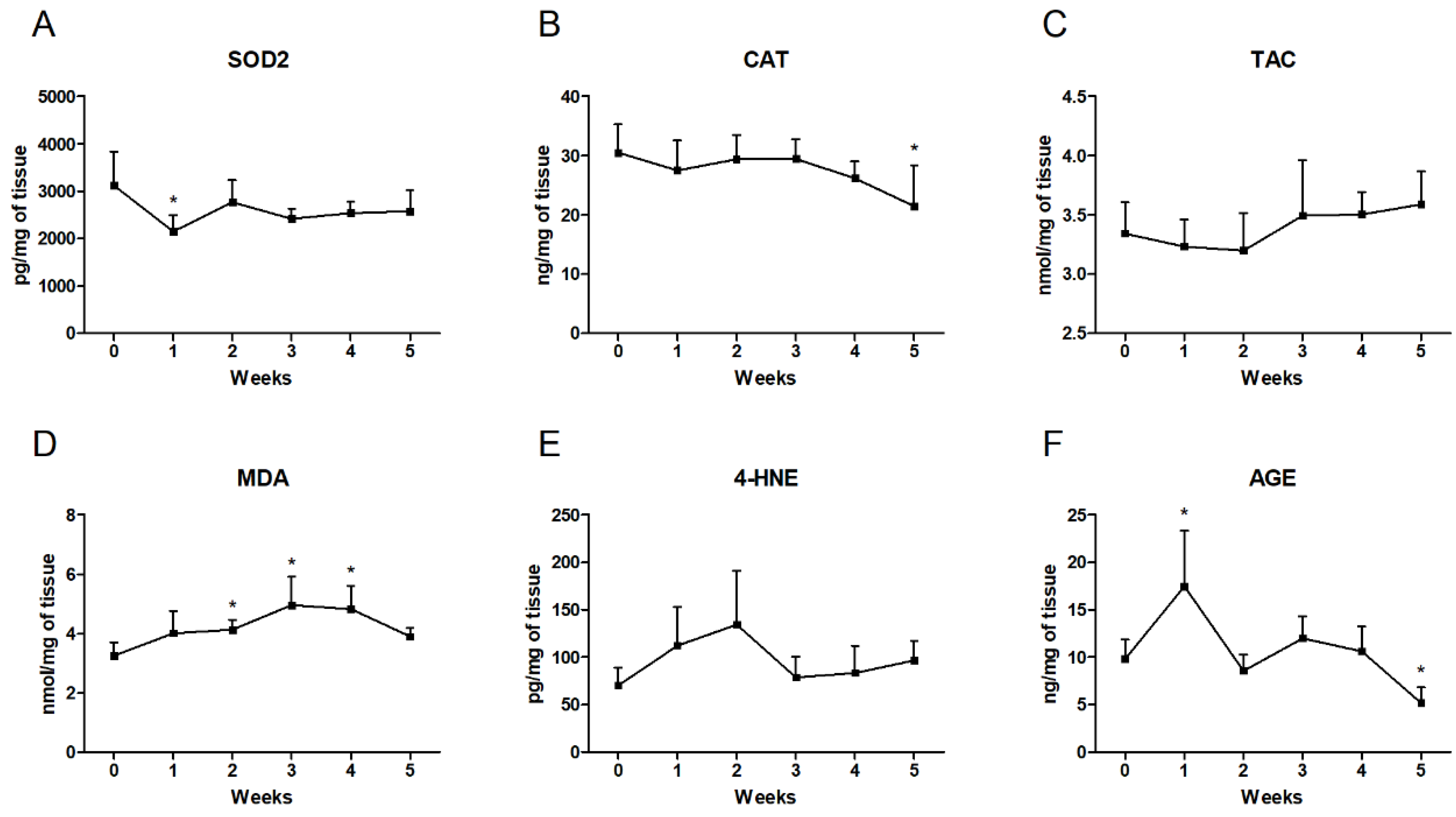
3.6. Changes in the Oxidative and Antioxidative Parameters
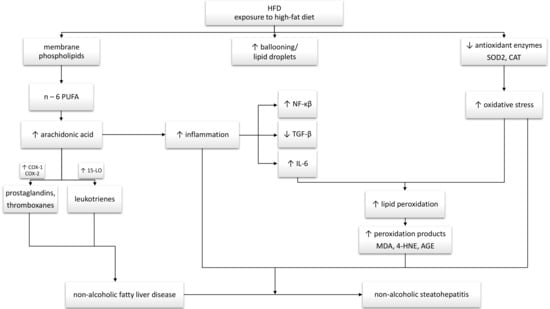
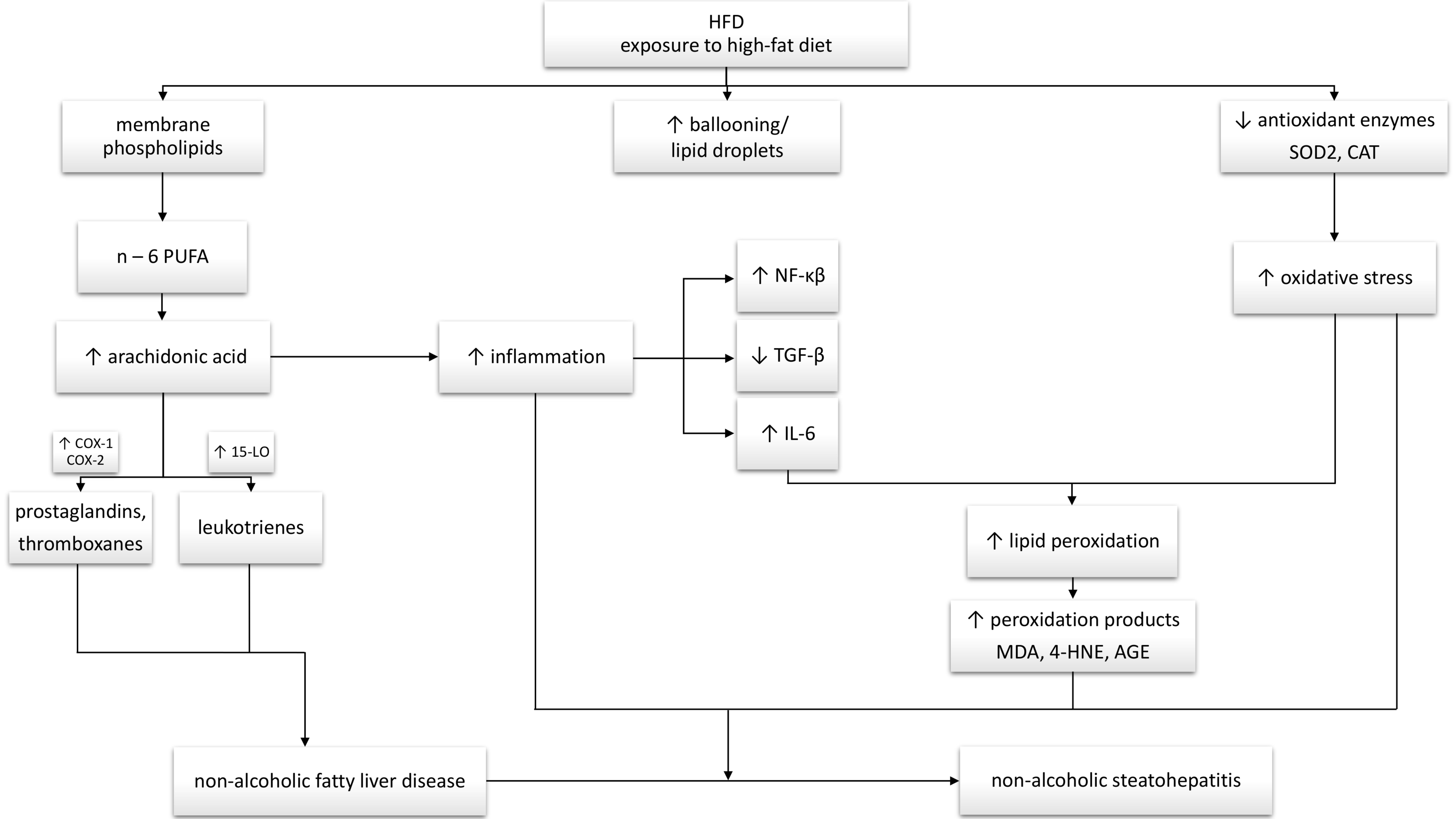
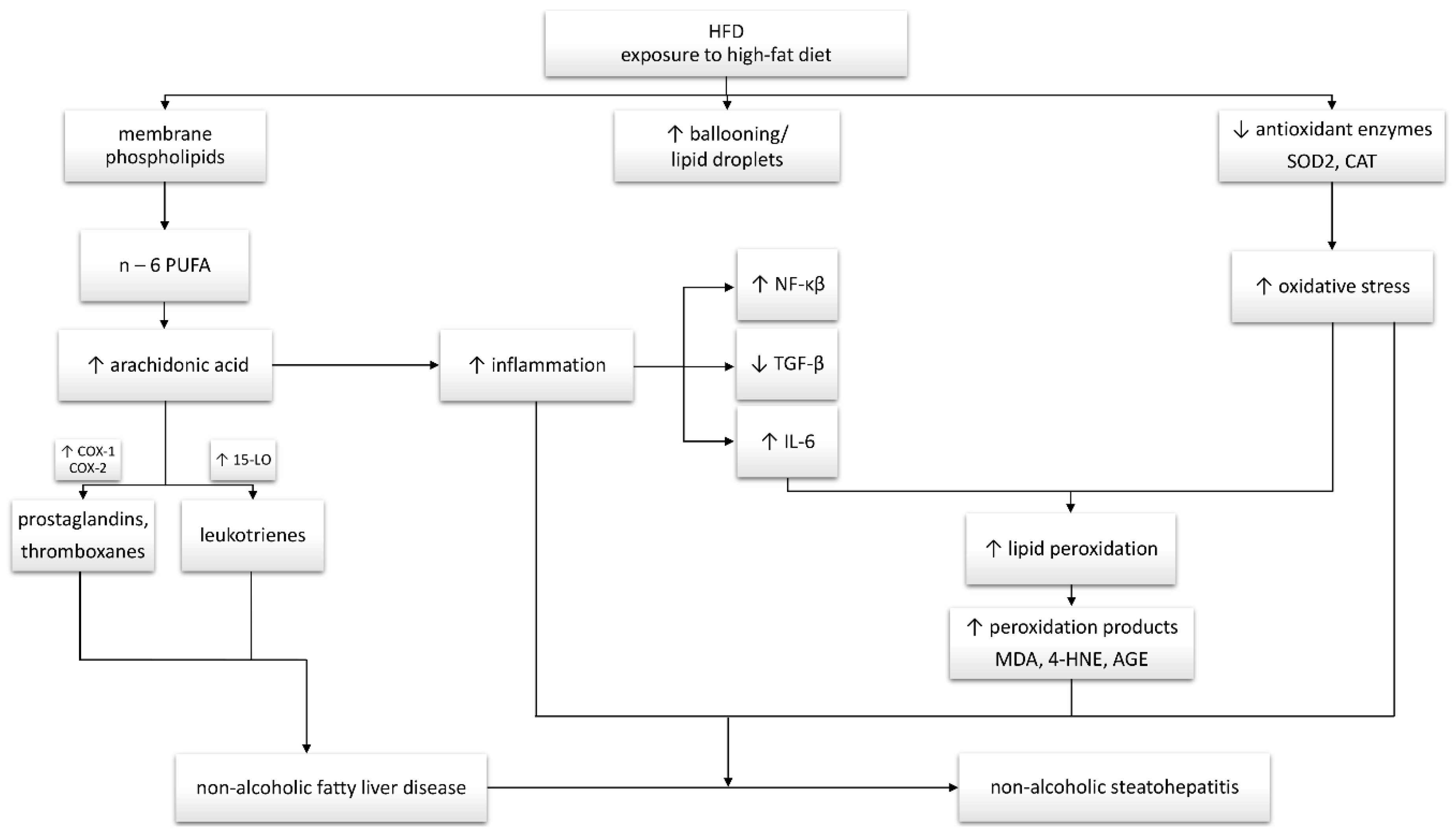
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jensen, V.S.; Tveden-Nyborg, P.; Zacho-Rasmussen, C.; Quaade, M.L.; Ipsen, D.H.; Hvid, H.; Fledelius, C.; Wulff, E.M.; Lykkesfeldt, J. Variation in diagnostic NAFLD/NASH read-outs in paired liver samples from rodent models. J. Pharm. Toxicol. Methods 2020, 101, 106651. [Google Scholar] [CrossRef] [PubMed]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svegliati-Baroni, G.; Pierantonelli, I.; Torquato, P.; Marinelli, R.; Ferreri, C.; Chatgilialoglu, C.; Bartolini, D.; Galli, F. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019, 144, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Wu, L.; Ma, W.; Ju, L.; Bai, M.; Chen, X.; Liu, S.; Yang, X.; Shi, J. Nonalcoholic Fatty Liver Disease: Pathogenesis and Treatment in Traditional Chinese Medicine and Western Medicine. Evid. Based Complement. Altern. Med. 2020, 2020, 8749564. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Kim, S.U.; Song, K.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Han, K.H. Sarcopenia is associated with significant liver fibrosis independently of obesity and insulin resistance in nonalcoholic fatty liver disease: Nationwide surveys (KNHANES 2008-2011). Hepatology 2016, 63, 776–786. [Google Scholar] [CrossRef] [Green Version]
- Ore, A.; Akinloye, O.A. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [Green Version]
- Abenavoli, L.; Luigiano, C.; Guzzi, P.H.; Milic, N.; Morace, C.; Stelitano, L.; Consolo, P.; Miraglia, S.; Fagoonee, S.; Virgilio, C.; et al. Serum adipokine levels in overweight patients and their relationship with non-alcoholic fatty liver disease. Panminerva Med. 2014, 56, 189–193. [Google Scholar]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e6. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Stanković, M.N.; Mladenović, D.; Ninković, M.; Duričić, I.; Šobajić, S.; Jorgačević, B.; De Luka, S.; Vukicevic, R.J.; Radosavljević, T.S. The effects of α-lipoic acid on liver oxidative stress and free fatty acid composition in methionine-choline deficient diet-induced NAFLD. J. Med. Food 2014, 17, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated fatty acids. J. Nutr. Metab. 2012, 2012, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Konstantynowicz-Nowicka, K.; Berk, K.; Chabowski, A.; Kasacka, I.; Bielawiec, P.; Łukaszuk, B.; Harasim-Symbor, E. High-fat feeding in time-dependent manner affects metabolic routes leading to nervonic acid synthesis in NAFLD. Int. J. Mol. Sci. 2019, 20, 3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G. A simple method for the isolation and purfication of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Chabowski, A.; Żendzian-Piotrowska, M.; Konstantynowicz, K.; Pankiewicz, W.; Mikłosz, A.; Łukaszuk, B.; Górski, J. Fatty acid transporters involved in the palmitate and oleate induced insulin resistance in primary rat hepatocytes. Acta Physiol. 2013, 207, 346–357. [Google Scholar] [CrossRef]
- Konstantynowicz-Nowicka, K.; Harasim, E.; Baranowski, M.; Chabowski, A. New evidence for the role of ceramide in the development of hepatic insulin resistance. PLoS ONE 2015, 10, e0116858. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Zhao, Y.; Shi, X.; Zhang, N.; Zu, G.; Li, Z.; Zhou, J.; Gao, D.; Lv, L.; Tian, X.; et al. New insights into salvianolic acid A action: Regulation of the TXNIP/NLRP3 and TXNIP/ChREBP pathways ameliorates HFD-induced NAFLD in rats. Sci. Rep. 2016, 6, 28734. [Google Scholar] [CrossRef]
- Banaszczak, M.; Stachowska, E. Patogeneza NAFLD w świetle wyników najnowszych badań. Postepy Biochem. 2017, 63, 190–197. [Google Scholar]
- Meli, R.; Mattace Raso, G.; Irace, C.; Simeoli, R.; Di Pascale, A.; Paciello, O.; Pagano, T.B.; Calignano, A.; Colonna, A.; Santamaria, R. High Fat Diet Induces Liver Steatosis and Early Dysregulation of Iron Metabolism in Rats. PLoS ONE 2013, 8, e66570. [Google Scholar] [CrossRef]
- Heden, T.D.; Morris, E.M.; Kearney, M.L.; Liu, T.W.; Park, Y.M.; Kanaley, J.A.; Thyfault, J.P. Differential effects of low-fat and high-fat diets on fed-state hepatic triacylglycerol secretion, hepatic fatty acid profiles, and DGAT-1 protein expression in obese-prone Sprague–Dawley rats. Appl. Physiol. Nutr. Metab. 2014, 39, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Asrih, M.; Jornayvaz, F.R. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J. Endocrinol. 2013, 218, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, N.; Hu, G.; Ma, M. Comparison of long-term effects of egg yolk consumption under normal and high fat diet on lipid metabolism and fatty acids profile in mice. Food Sci. Biotechnol. 2019, 28, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, L.; Zhu, L.; Yu, Y. Free fatty acids, not triglycerides, are associated with non-alcoholic liver injury progression in high fat diet induced obese rats. Lipids Health Dis. 2016, 15, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.W.; Heden, T.D.; Morris, E.M.; Fritsche, K.L.; Vieira-Potter, V.J.; Thyfault, J.P. High-fat diet alters serum fatty acid profiles in obesity prone rats: Implications for in-vitro studies. Lipids 2015, 50, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, R.; Espinosa, A.; González-Mañán, D.; D’Espessailles, A.; Fernández, V.; Videla, L.A.; Tapia, G. N-3 long-chain polyunsaturated fatty acid supplementation significantly reduces liver oxidative stress in high fat induced steatosis. PLoS ONE 2012, 7, e46400. [Google Scholar] [CrossRef] [Green Version]
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachnik, J. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin. Sci. 2004, 106, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Chen, Y.; Liang, X.; Miao, J.; Zhao, Q. Inhibition of 5-lipoxygenase inhibitor zileuton in high-fat diet-induced nonalcoholic fatty liver disease progression model. Iran. J. Basic Med. Sci. 2017, 20, 1207–1212. [Google Scholar]
- Xu, Y.; Han, J.; Dong, J.; Fan, X.; Cai, Y.; Li, J.; Wang, T.; Zhou, J.; Shang, J. Metabolomics characterizes the effects and mechanisms of quercetin in nonalcoholic fatty liver disease development. Int. J. Mol. Sci. 2019, 20, 1220. [Google Scholar] [CrossRef] [Green Version]
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180. [Google Scholar] [CrossRef]
- Scheja, L.; Toedter, K.; Mohr, R.; Niederfellner, G.; Michael, M.D.; Meissner, A.; Schoettler, A.; Pospisil, H.; Beisiegel, U.; Heeren, J. Liver TAG transiently decreases while PL n-3 and n-6 Fatty acids are persistently elevated in insulin resistant mice. Lipids 2008, 43, 1039–1051. [Google Scholar] [CrossRef]
- Bozza, P.T.; Bakker-Abreu, I.; Navarro-Xavier, R.A.; Bandeira-Melo, C. Lipid body function in eicosanoid synthesis: An update. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Rius, B.; López-Vicario, C.; González-Périz, A.; Morán-Salvador, E.; García-Alonso, V.; Clària, J.; Titos, E. Resolution of inflammation in obesity-induced liver disease. Front. Immunol. 2012, 3, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujimoto, S.; Kishina, M.; Koda, M.; Yamamoto, Y.; Tanaka, K.; Harada, Y.; Yoshida, A.; Hisatome, I. Nimesulide, a cyclooxygenase-2 selective inhibitor, suppresses obesity-related non-alcoholic fatty liver disease and hepatic insulin resistance through the regulation of peroxisome proliferator-activated receptor γ. Int. J. Mol. Med. 2016, 38, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Tuzcu, Z.; Orhan, C.; Sahin, N.; Juturu, V.; Sahin, K. Cinnamon Polyphenol Extract Inhibits Hyperlipidemia and Inflammation by Modulation of Transcription Factors in High-Fat Diet-Fed Rats. Oxid. Med. Cell. Longev. 2017, 2017, 1583098. [Google Scholar] [CrossRef] [PubMed]
- Tuzcu, M.; Sahin, N.; Orhan, C.; Agca, C.A.; Akdemir, F.; Tuzcu, Z.; Komorowski, J.; Sahin, K. Impact of chromium histidinate on high fat diet induced obesity in rats. Nutr. Metab. 2011, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Itoh, Y.; Yokomizo, C.; Nishimura, T.; Niimi, T.; Fujii, H.; Okanoue, T.; Yoshikawa, T. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab. Investig. 2010, 90, 1169–1178. [Google Scholar] [CrossRef]
- Tao, W.; Sun, W.; Liu, L.; Wang, G.; Xiao, Z.; Pei, X.; Wang, M. Chitosan oligosaccharide attenuates nonalcoholic fatty liver disease induced by high fat diet through reducing lipid accumulation, inflammation and oxidative stress in C57BL/6 mice. Mar. Drugs 2019, 17, 645. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.J.; Fan, J.G.; Ding, X.D.; Qiao, L.; Wang, G.L. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Dig. Dis. Sci. 2010, 55, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- da Pereira, E.N.G.S.; Silvares, R.R.; Flores, E.E.I.; Rodrigues, K.L.; Ramos, I.P.; da Silva, I.J.; Machado, M.P.; Miranda, R.A.; Pazos-Moura, C.C.; Gonçalves-De-Albuquerque, C.F.; et al. Hepatic microvascular dysfunction and increased advanced glycation end products are components of non-alcoholic fatty liver disease. PLoS ONE 2017, 12, e0179654. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sztolsztener, K.; Chabowski, A.; Harasim-Symbor, E.; Bielawiec, P.; Konstantynowicz-Nowicka, K. Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development. Biomolecules 2020, 10, 1133. https://doi.org/10.3390/biom10081133
Sztolsztener K, Chabowski A, Harasim-Symbor E, Bielawiec P, Konstantynowicz-Nowicka K. Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development. Biomolecules. 2020; 10(8):1133. https://doi.org/10.3390/biom10081133
Chicago/Turabian StyleSztolsztener, Klaudia, Adrian Chabowski, Ewa Harasim-Symbor, Patrycja Bielawiec, and Karolina Konstantynowicz-Nowicka. 2020. "Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development" Biomolecules 10, no. 8: 1133. https://doi.org/10.3390/biom10081133