Can We Assume the Gene Expression Profile as a Proxy for Signaling Network Activity?

 , , , and
, , , and
Abstract
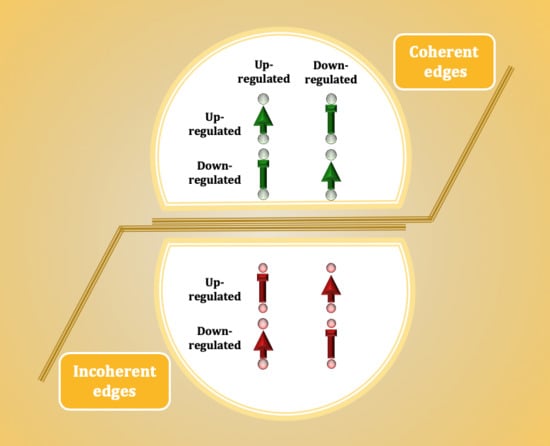
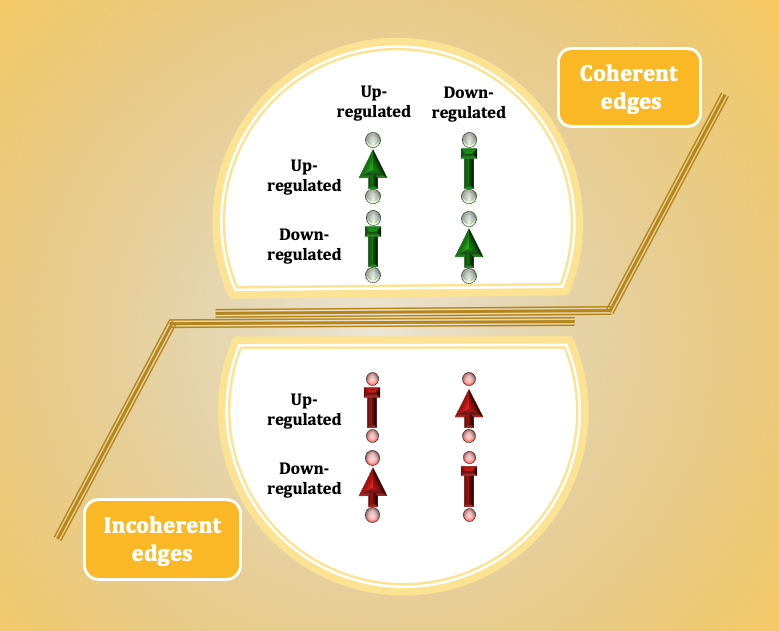
:
1. Introduction
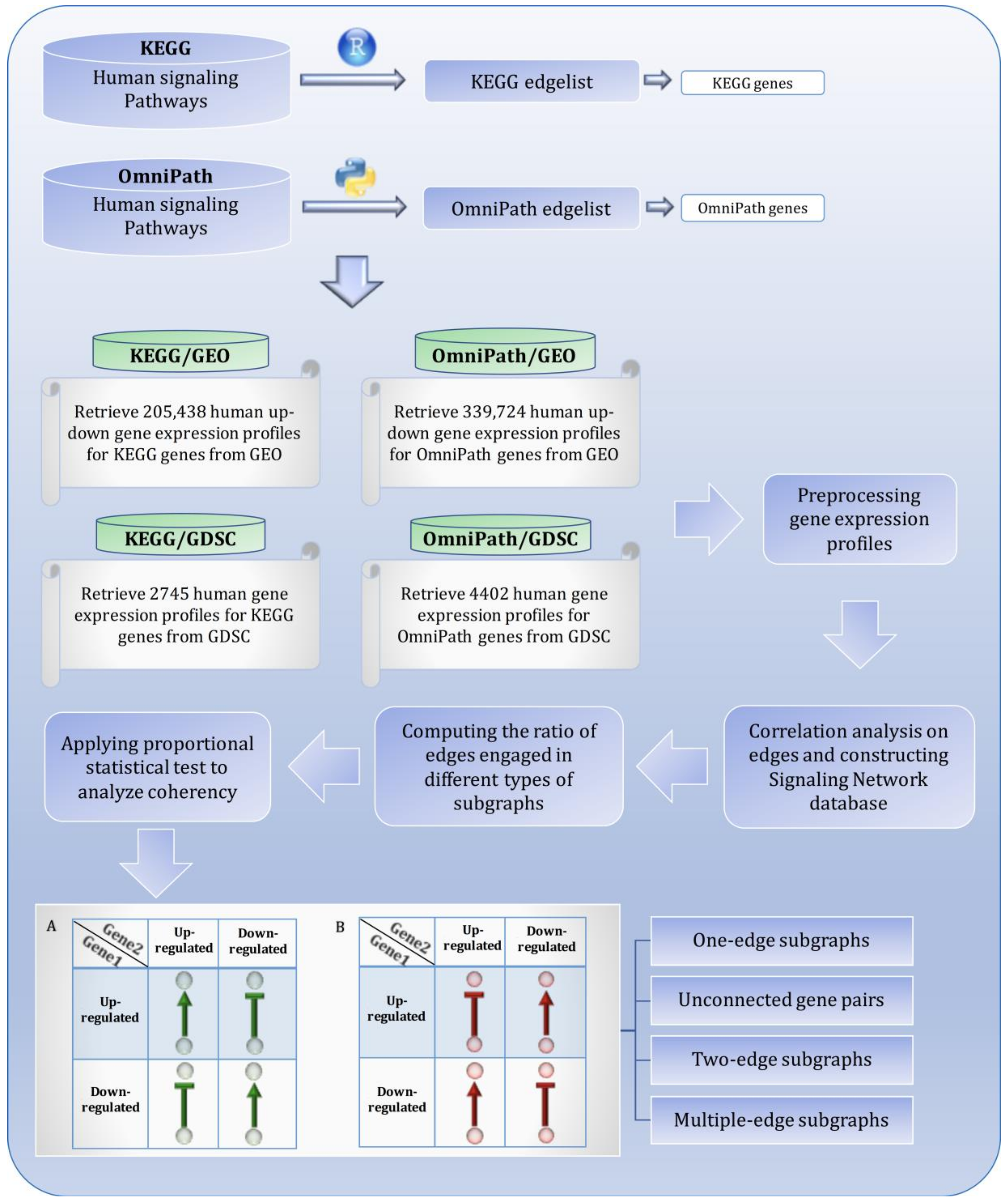
2. Materials and Methods
2.1. Signaling Network Reconstruction
2.2. Gene Expression Profiles Extraction
2.3. Mutual Association Analysis
2.4. Randomly Selected Unconnected Gene Pairs
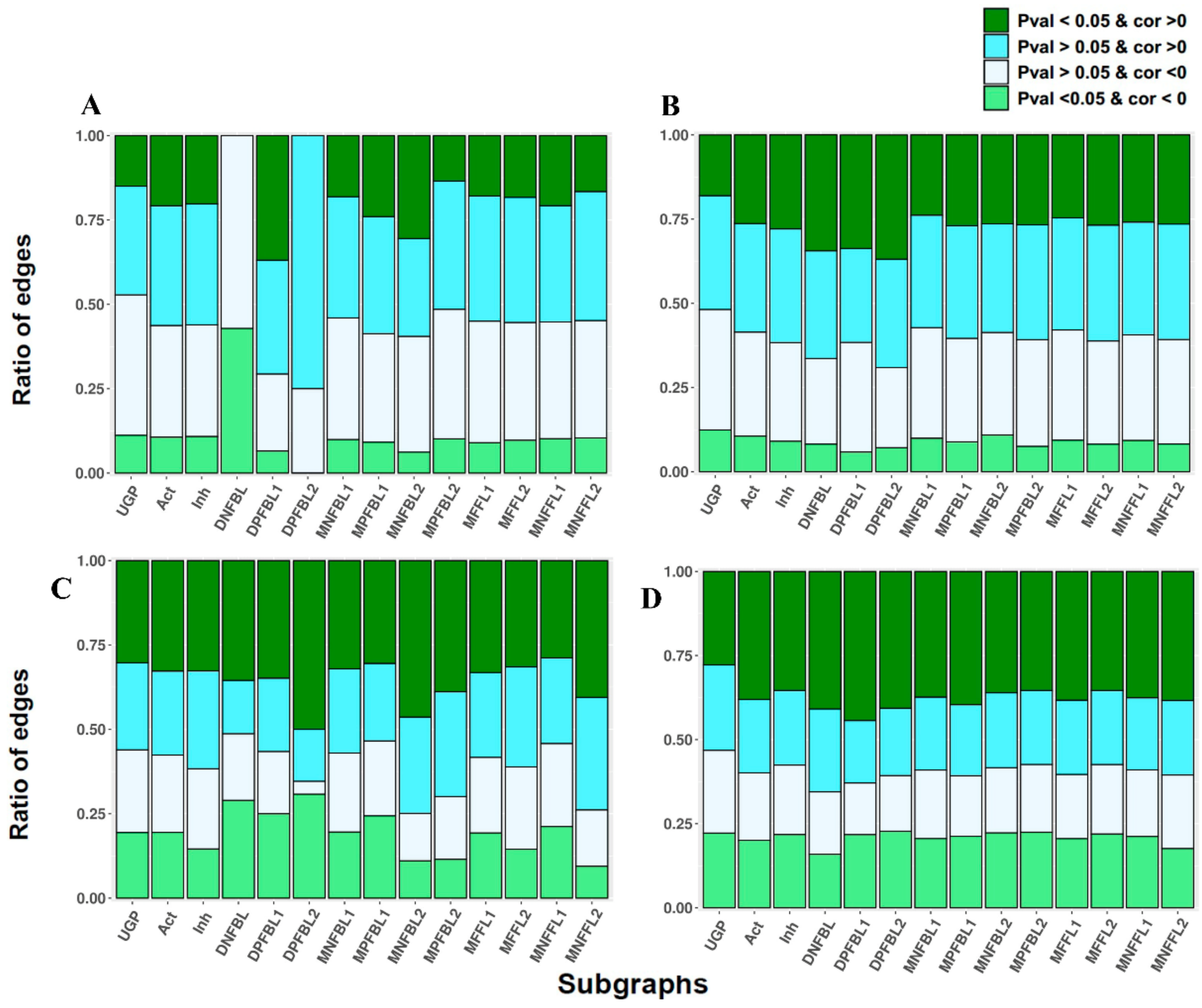
2.5. Complex Subgraphs
3. Results
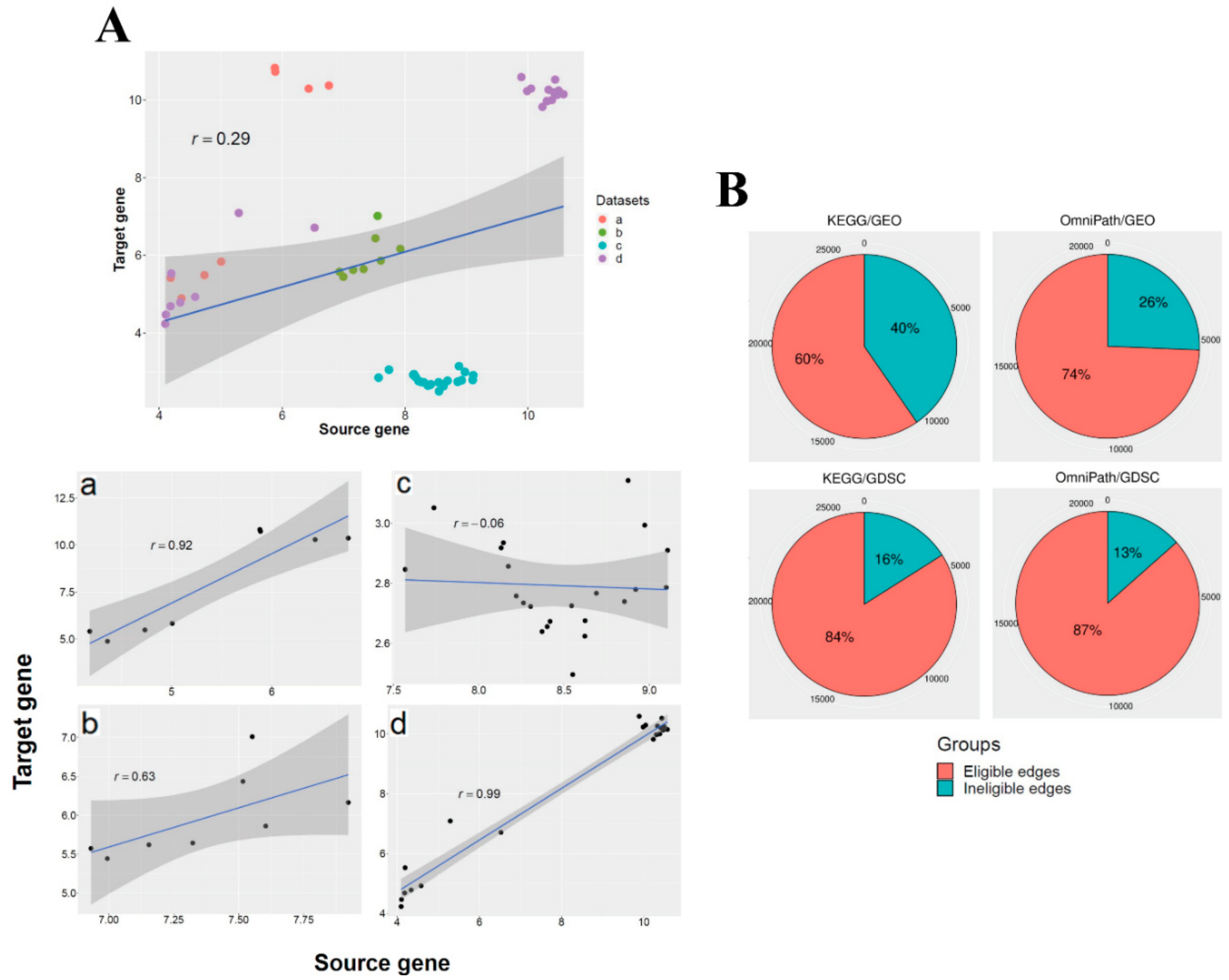
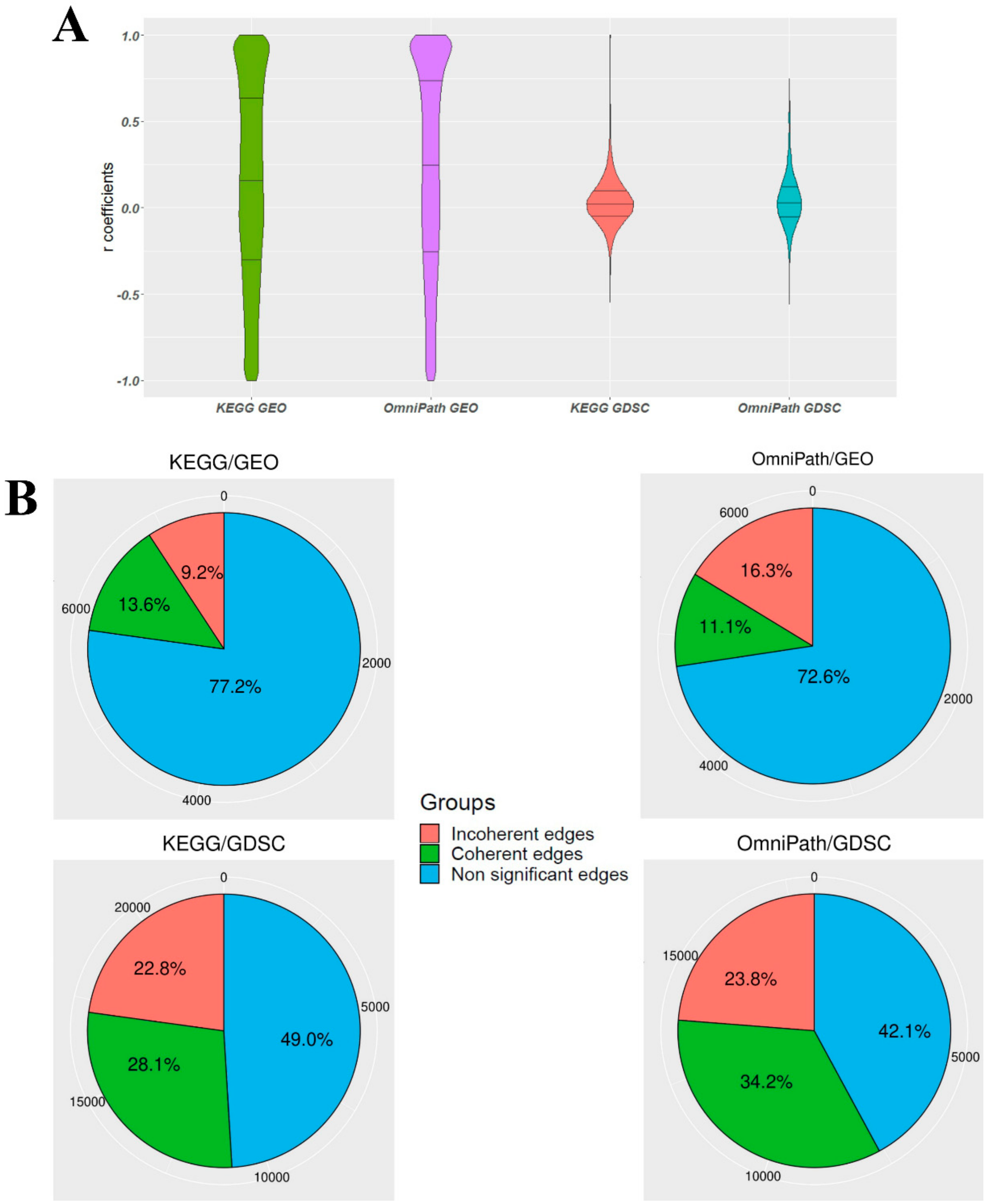
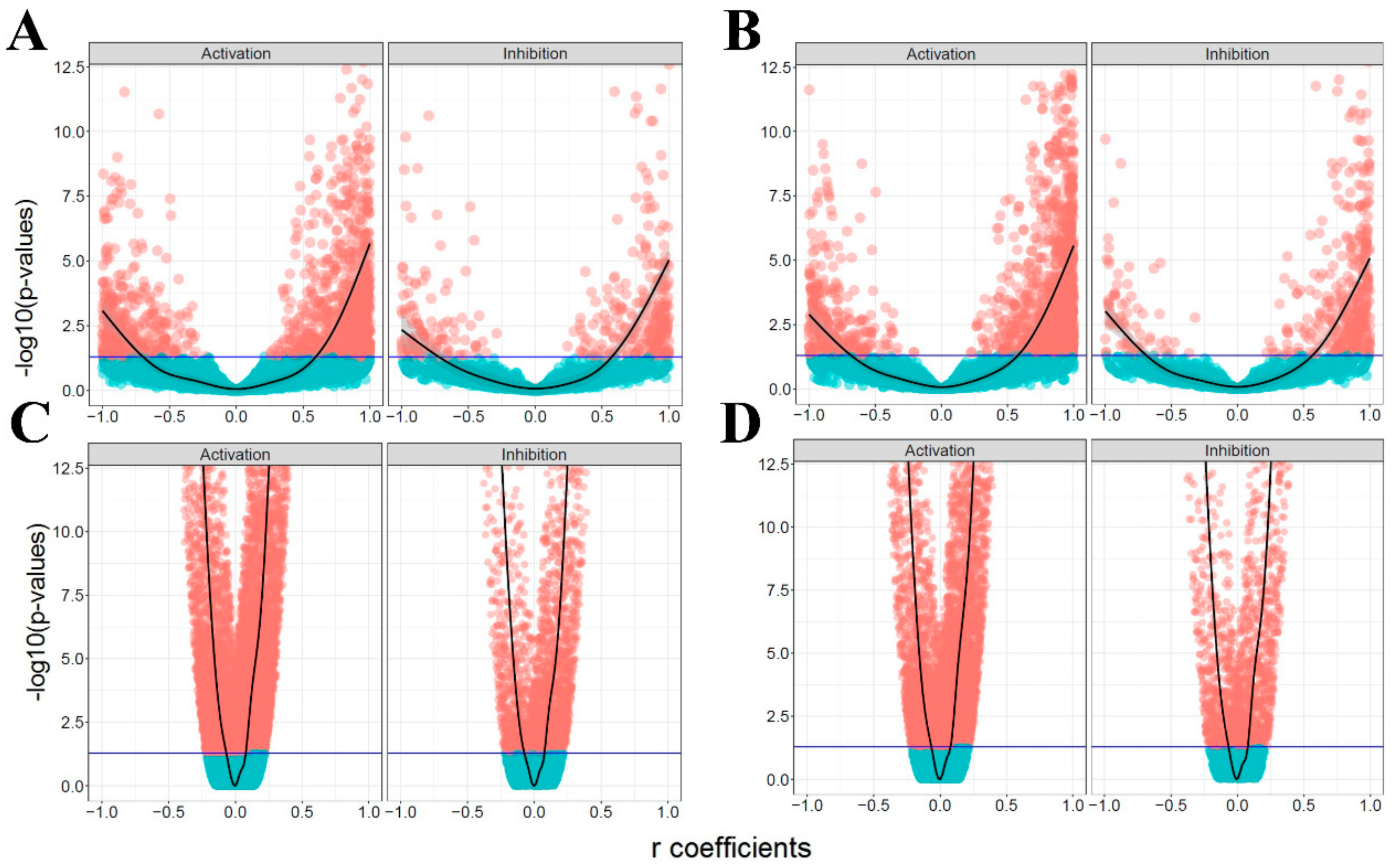
3.1. The Ratio of Coherency for Gene Pairs
3.2. The Ratio of Coherency on Subgraphs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, A.; Trairatphisan, P.; Gjerga, E.; Didangelos, A.; Barratt, J.; Saez-Rodriguez, J. From expression footprints to causal pathways: Contextualizing large signaling networks with CARNIVAL. NPJ Syst. Biol. Appl. 2019, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Ma’ayan, A. Introduction to network analysis in systems biology. Sci. Signal. 2011, 4, tr5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, M.; Lambeck, S.; Toepfer, S.; Van Someren, E.; Guthke, R. Gene regulatory network inference: Data integration in dynamic models—A review. Biosystems 2009, 96, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Shipley, B. Cause and Correlation in Biology: A User’s Guide to Path Analysis, Structural Equations and Causal Inference with R; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Bansal, M.; Belcastro, V.; Ambesi-Impiombato, A.; Di Bernardo, D. How to infer gene networks from expression profiles. Mol. Syst. Biol. 2007, 3, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [Green Version]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Guo, H.; Cao, Y.; Zohrabian, S.; Zhou, P.; Ma, Q. Acetylation of VGLL4 regulates Hippo-YAP signaling and postnatal cardiac growth. Dev. Cell 2016, 39, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Li, Z.-X.; Liu, X.; Wang, R.; Li, L.-W.; Zhang, Q. The Wnt/β-catenin and PI3K/Akt signaling pathways promote EMT in gastric cancer by epigenetic regulation via H3 lysine 27 acetylation. Tumor Biol. 2017, 39, 1010428317712617. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.M.; Heiser, L.M.; Cokelaer, T.; Unger, M.; Nesser, N.K.; Carlin, D.E. Inferring causal molecular networks: Empirical assessment through a community-based effort. Nat. Methods 2016, 13, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Jafari, M.; Ansari-Pour, N.; Azimzadeh, S.; Mirzaie, M. A logic-based dynamic modeling approach to explicate the evolution of the central dogma of molecular biology. PLoS ONE 2017, 12, e0189922. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Shi, X.; Zhang, Y.; Xu, Y.; Jiang, Y.; Zhang, C. ESEA: Discovering the dysregulated pathways based on edge set enrichment analysis. Sci. Rep. 2015, 5, 13044. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966. [Google Scholar] [CrossRef]
- Zhang, J.D.; Wiemann, S. KEGGgraph: A graph approach to KEGG PATHWAY in R and bioconductor. Bioinformatics 2009, 25, 1470–1471. [Google Scholar] [CrossRef]
- Luke, D.A. A User’s Guide to Network Analysis in R; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Csardi, G.; Nepusz, T.J.I. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. BioSyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.; de Sousa Abreu, R.; Ko, D.; Le, S.Y.; Shapiro, B.A.; Burns, S.C. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 2010, 6, 400. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Vogel, C.; Wang, R.; Yao, X.; Marcotte, E.M. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007, 25, 117–124. [Google Scholar] [CrossRef] [PubMed]
- MacKay, V.L.; Li, X.; Flory, M.R.; Turcott, E.; Law, G.L.; Serikawa, K.A. Gene expression analyzed by high-resolution state array analysis and quantitative proteomics response of yeast to mating pheromone. Mol. Cell. Proteom. 2004, 3, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Mata, J.; Marguerat, S.; Bähler, J. Post-transcriptional control of gene expression: A genome-wide perspective. Trends Biochem. Sci. 2005, 30, 506–514. [Google Scholar] [CrossRef]
- Jafari, M.; Mirzaie, M.; Sadeghi, M.; Marashi, S.-A.; Rezaei-Tavirani, M. Exploring biological processes involved in embryonic stem cell differentiation by analyzing proteomic data. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2013, 1834, 1063–1069. [Google Scholar] [CrossRef]
- Ansari-Pour, N.; Razaghi-Moghadam, Z.; Barneh, F.; Jafari, M. Testis-Specific Y-Centric Protein–Protein Interaction Network Provides Clues to the Etiology of Severe Spermatogenic Failure. J. Proteome Res. 2016, 15, 1011–1022. [Google Scholar] [CrossRef]
- Inui, M.; Martello, G.; Piccolo, S. MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 2010, 11, 252–263. [Google Scholar] [CrossRef]
- Mørk, S.; Pletscher-Frankild, S.; Palleja Caro, A.; Gorodkin, J.; Jensen, L.J. Protein-driven inference of miRNA–disease associations. Bioinformatics 2014, 30, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Song, Y.-X.; Ma, B.; Wang, J.-J.; Sun, J.-X.; Chen, X.-W. Regulatory roles of non-coding RNAs in colorectal cancer. Int. J. Mol. Sci. 2015, 16, 19886–19919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Zhou, S.; Yi, S.; Gu, X. The regulatory roles of non-coding RNAs in nerve injury and regeneration. Prog. Neurobiol. 2015, 134, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Biggar, K.K.; Li, S.S.-C. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef]
- Larsen, S.J.; Röttger, R.; Schmidt, H.H.H.W.; Baumbach, J.E. coli gene regulatory networks are inconsistent with gene expression data. Nucleic Acids Res. 2019, 47, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Gautam, P.; Gupta, A.; He, L.; Timonen, S.; Akimov, Y. Network pharmacology modeling identifies synergistic Aurora B and ZAK interaction in triple-negative breast cancer. NPJ Syst. Biol. Appl. 2019, 5, 20. [Google Scholar] [CrossRef]
- D’Souza, R.C.; Knittle, A.M.; Nagaraj, N.; van Dinther, M.; Choudhary, C.; Ten Dijke, P. Time-resolved dissection of early phosphoproteome and ensuing proteome changes in response to TGF-β. Sci. Signal. 2014, 7, rs5. [Google Scholar]
- Azimzadeh, S.; Mirzaie, M.; Jafari, M.; Mehrani, H.; Shariati, P.; Khodabandeh, M. Signaling network of lipids as a comprehensive scaffold for omics data integration in sputum of COPD patients. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 1383–1393. [Google Scholar] [CrossRef]
- Azimzadeh Jamalkandi, S.; Mozhgani, S.-H.; Gholami Pourbadie, H.; Mirzaie, M.; Noorbakhsh, F.; Vaziri, B. Systems Biomedicine of Rabies Delineates the Affected Signaling Pathways. Front. Microbiol. 2016, 7, 1688. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simple Subgraphs | ||||
|---|---|---|---|---|
| Structures | Names | Abbrev. | KEGG | OmniPath |
 | Unconnected Gene Pairs | UGP | __ | __ |
 | Activation | Act | 19,170 | 15,841 |
 | Inhibition | Inh | 7320 | 5012 |
| Complex Subgraphs | ||||
 | Dual Negative Feedback Loop | DNFBL | 37 | 279 |
 | Dual Positive Feedback Loop1 | DPFBL1 | 186 | 912 |
 | Dual Positive Feedback Loop2 | DPFBL2 | 14 | 173 |
 | Multiple Negative Feedback Loop1 | MNFBL1 | 17,712 | 14,913 |
 | Multiple Positive Feedback Loop1 | MPFBL1 | 3731 | 4104 |
 | Multiple Negative Feedback Loop2 | MNFBL2 | 2417 | 1005 |
 | Multiple Positive Feedback Loop2 | MPFBL2 | 3232 | 3279 |
 | Multiple Feed-Forward Loop1 | MFFL1 | 12,869 | 6729 |
 | Multiple Feed-Forward Loop2 | MFFL2 | 6618 | 4718 |
 | Multiple Negative Feed-Forward Loop1 | MNFFL1 | 8918 | 9663 |
 | Multiple Negative Feed-Forward Loop2 | MNFFL2 | 2925 | 842 |
| KEGG | OmniPath | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Date Retrieved | DEGs | Samples | Giant Component | Diameter | Ratio | Date Retrieved | DEGs | Samples | Giant Component | Diameter | Ratio | |
| GEO | 2017.08 | 3047 | 40,903 | 2549 | 17 | 0.95 | 201,905 | 4724 | 40,774 | 3848 | 17 | 0.95 |
| GDSC | 2017.10 | 2745 | 1018 | 2583 | 17 | 0.16 | 201,905 | 4402 | 1018 | 4045 | 15 | 0.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piran, M.; Karbalaei, R.; Piran, M.; Aldahdooh, J.; Mirzaie, M.; Ansari-Pour, N.; Tang, J.; Jafari, M. Can We Assume the Gene Expression Profile as a Proxy for Signaling Network Activity? Biomolecules 2020, 10, 850. https://doi.org/10.3390/biom10060850
Piran M, Karbalaei R, Piran M, Aldahdooh J, Mirzaie M, Ansari-Pour N, Tang J, Jafari M. Can We Assume the Gene Expression Profile as a Proxy for Signaling Network Activity? Biomolecules. 2020; 10(6):850. https://doi.org/10.3390/biom10060850
Chicago/Turabian StylePiran, Mehran, Reza Karbalaei, Mehrdad Piran, Jehad Aldahdooh, Mehdi Mirzaie, Naser Ansari-Pour, Jing Tang, and Mohieddin Jafari. 2020. "Can We Assume the Gene Expression Profile as a Proxy for Signaling Network Activity?" Biomolecules 10, no. 6: 850. https://doi.org/10.3390/biom10060850