Antiproliferative and Antimigration Activities of Fluoro-Neplanocin A via Inhibition of Histone H3 Methylation in Triple-Negative Breast Cancer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemicals
2.2. Western Blotting Analysis
2.3. Cell Proliferation Assay (SRB assay)
2.4. DOT1L Enzyme Activity Assay
2.5. Cell Viability (MTT Assay)
2.6. Wound Healing Assay
2.7. Transwell Cell Invasion Assay
2.8. RNA Isolation and Real-Time Polymerase Chain Reaction (Real-Time PCR) Analysis
2.9. Statistical Analysis
3. Results
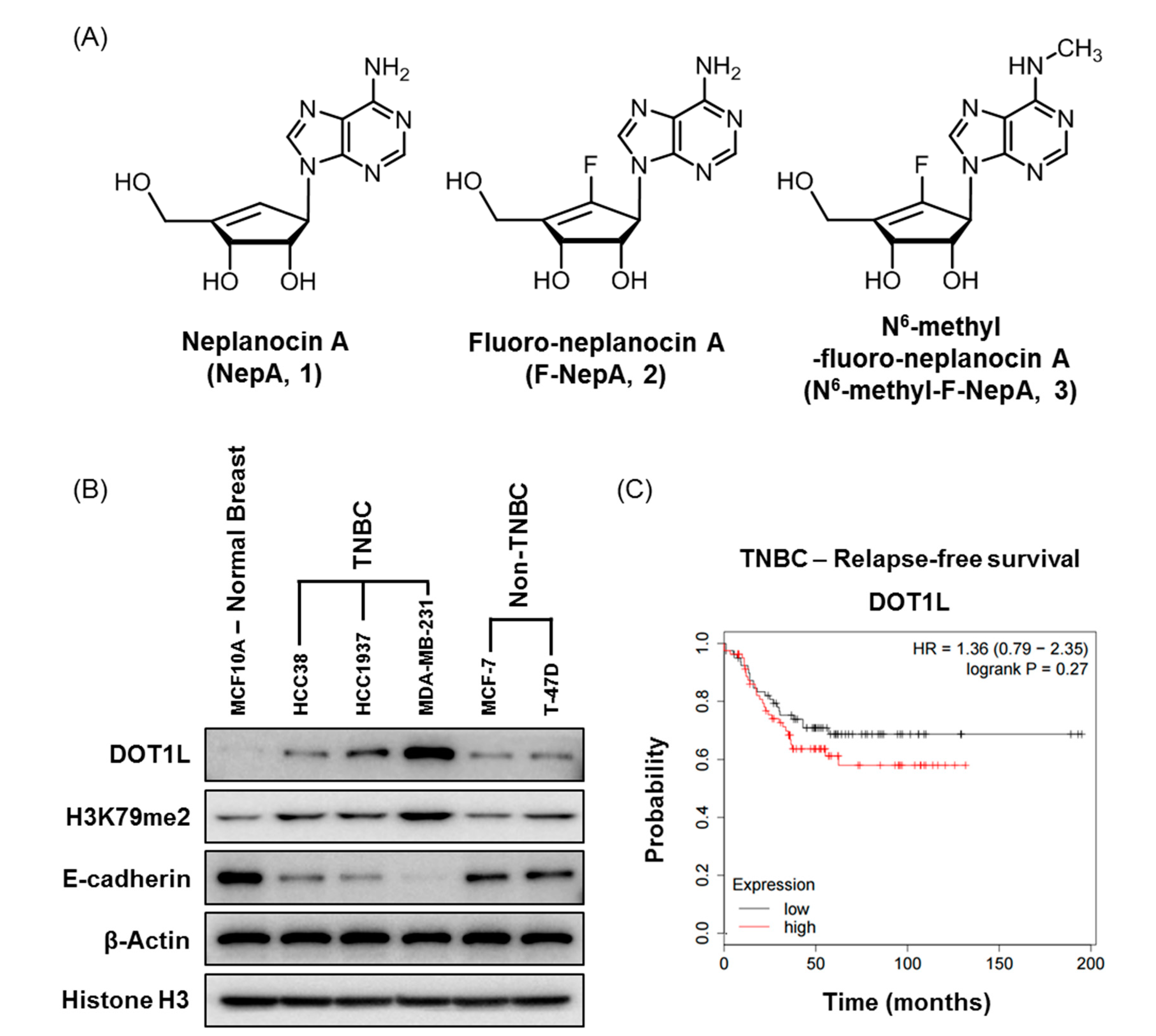
3.1. Expression Levels of DOT1L and Antiproliferative Activities of NepA Analogs in Human BC Cells
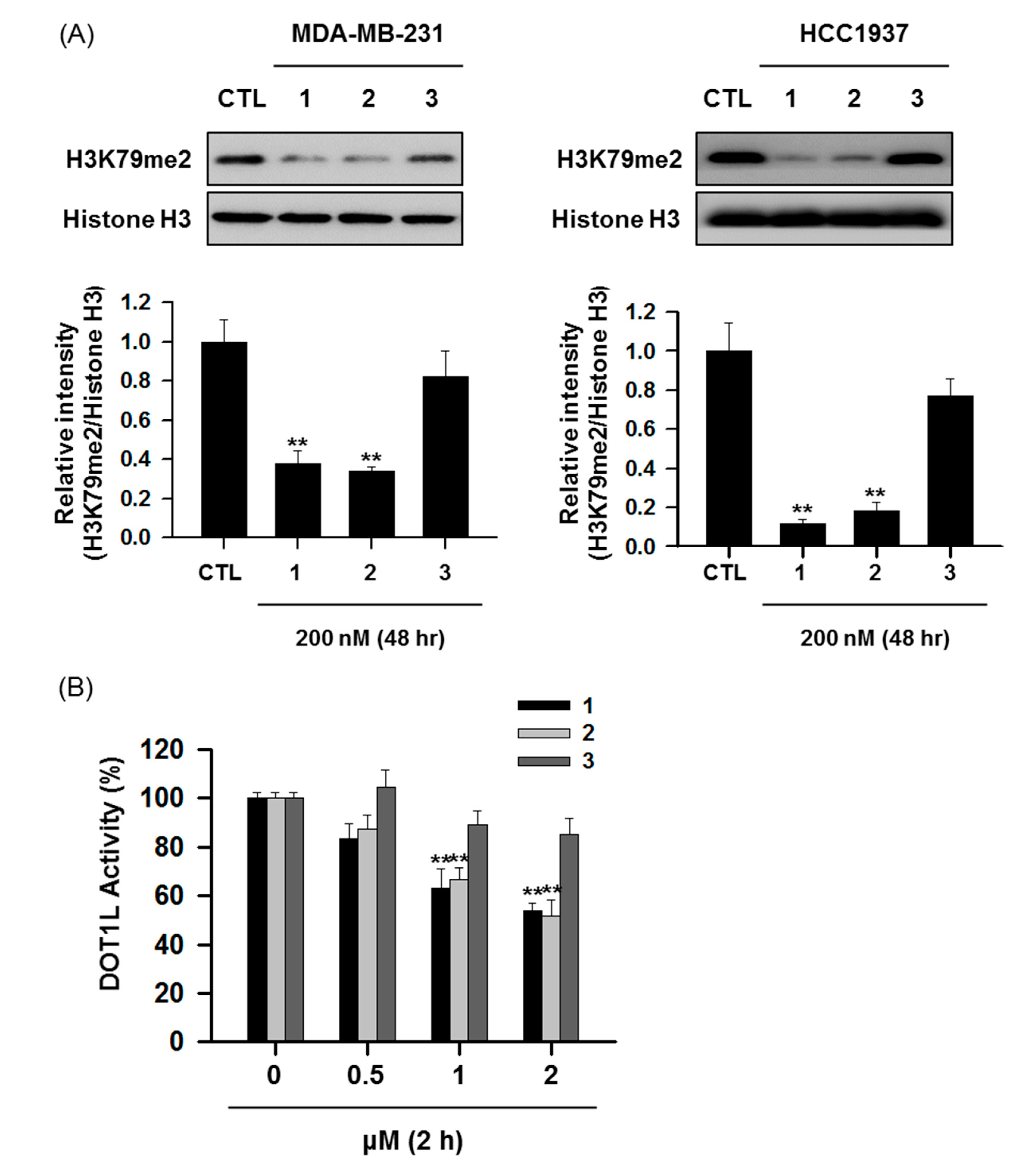
3.2. Effects of NepA Analogs on DOT1L Activity and H3K79me2 Expression
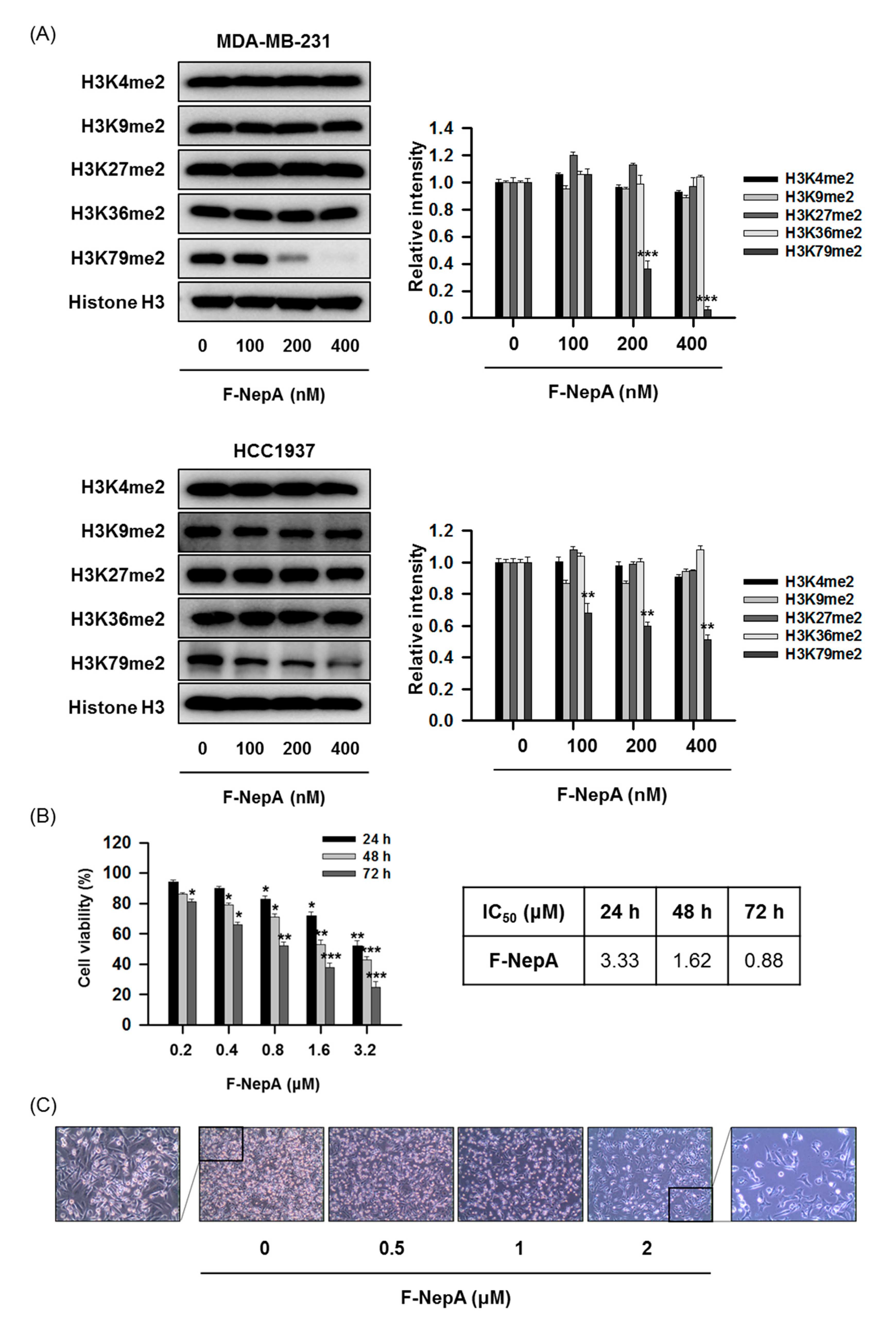
3.3. F-NepA Selectively Suppresses DOT1L-Mediated H3K79 Methylation in Human TNBC Cells
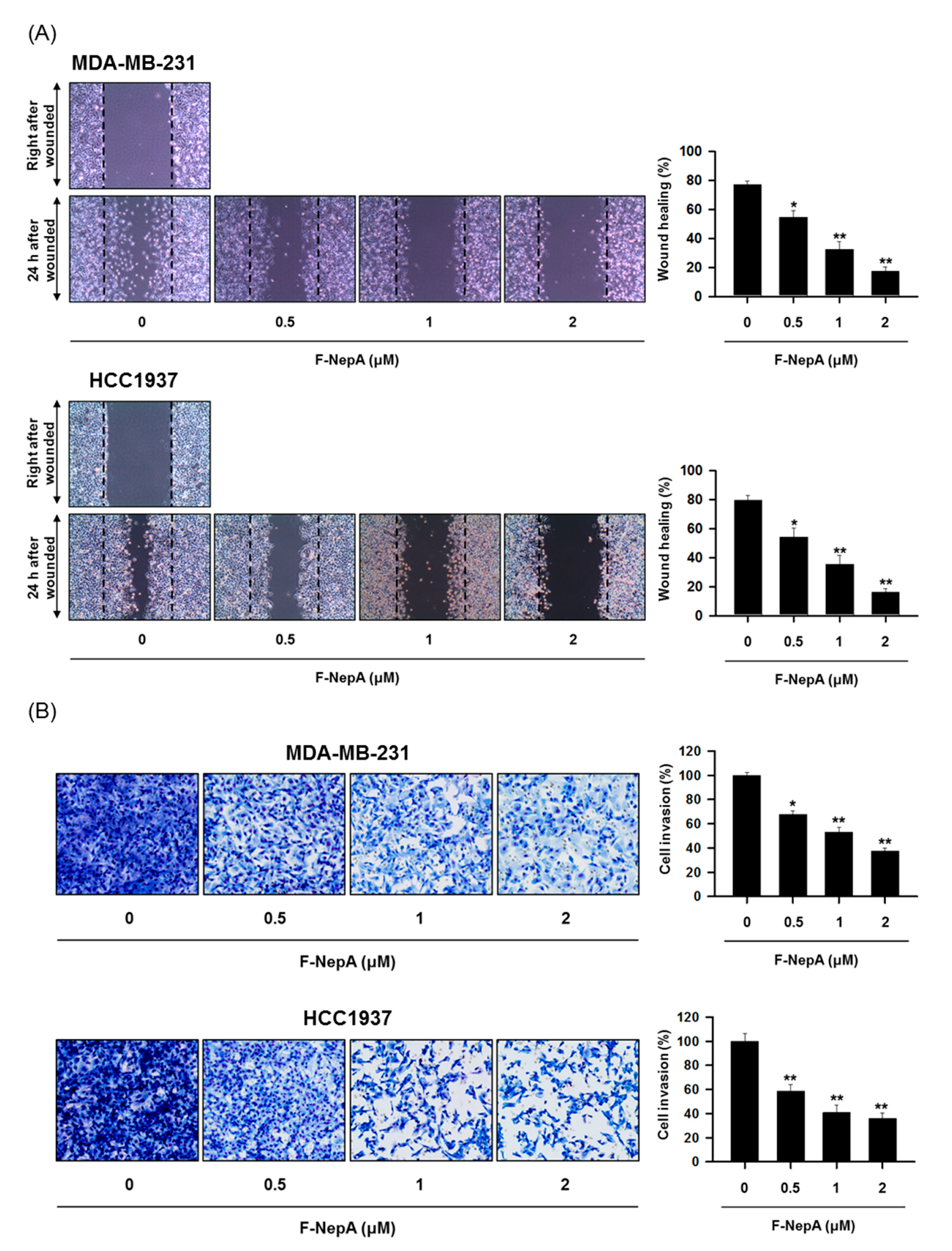
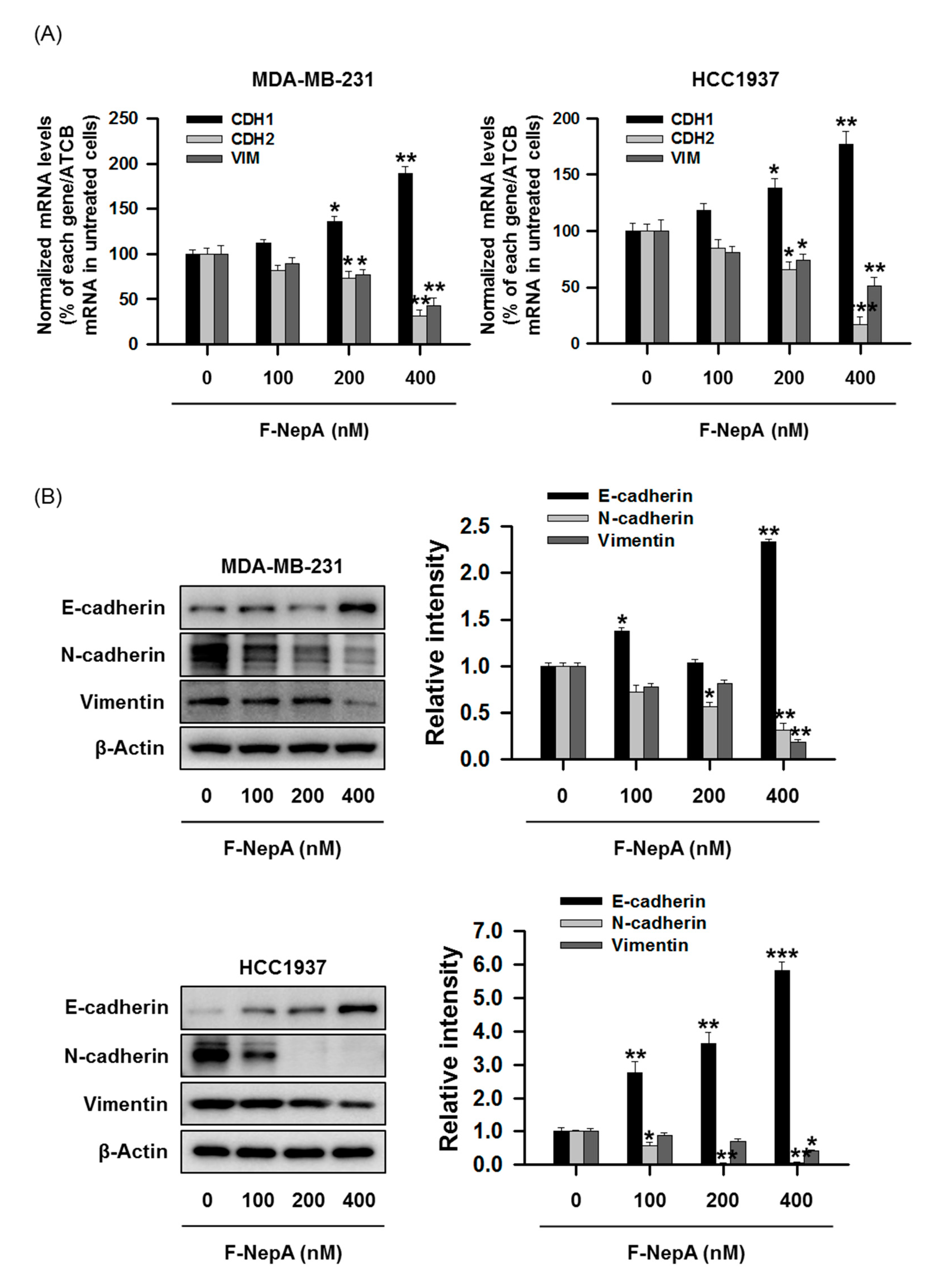
3.4. F-NepA Modulates EMT-Associated Gene Expressions in Human TNBC Cells
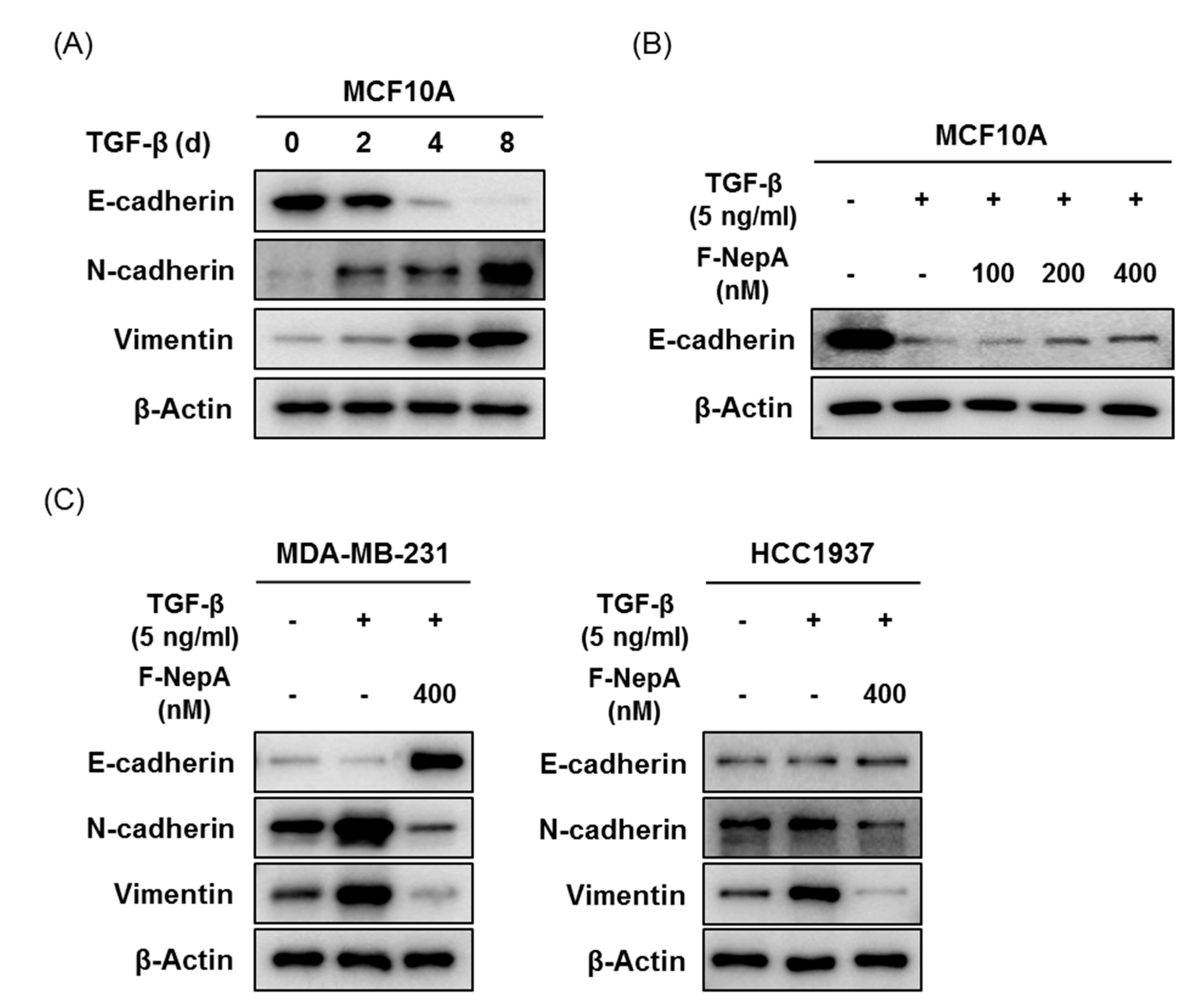
3.5. F-NepA Reverses TGF-β Induced EMT Biomarker Expressions
4. Discussion
5. Conclusion
6. Patents
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, W.S.; Kim, W.K.; Han, H.J.; Chung, H.-J.; Jang, K.; Kim, H.S.; Kim, S.; Kim, D.; Bae, E.S.; Park, S.; et al. Targeting histone methyltransferase DOT1L by a novel psammaplin A analog inhibits growth and metastasis of triple-negative breast cancer. Mol. Ther. Oncolytics 2019, 15, 140–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colleoni, M.; Sun, Z.; Price, K.N.; Karlsson, P.; Forbes, J.F.; Thürlimann, B.; Gianni, L.; Castiglione, M.; Gelber, R.D.; Coates, A.S.; et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: Results from the international breast cancer study group trials I to V. J. Clin. Oncol. 2016, 34, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, F.; Shi, L.; Cai, H.; Zheng, Y.; Zheng, W.; Bao, P.; Shu, X.O. Accelerated aging in breast cancer survivors and its association with mortality and cancer recurrence. Breast Cancer Res. Treat. 2020, 180, 449–459. [Google Scholar] [CrossRef]
- Itani, N.; Grogan, N.; Mott, S.; Phadke, S. Metastatic presentations of previously treated early-stage breast cancer patients and association with survival. Clin. Breast Cancer 2019. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Valentini, A.; Hanna, W.; Rawlinson, E.; Rakovitch, E.; Sun, P.; Narod, S.A. Factors associated with breast cancer mortality after local recurrence. Curr. Oncol. 2014, 21, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Melvin, J.C.; Purushotham, A.D.; Garmo, H.; Pinder, S.E.; Fentiman, I.S.; Gillett, C.; Mera, A.; Lüctehnborg, M.; Holmberg, L.; Van Hemelrijck, M. Progression of breast cancer following locoregional ipsilateral recurrence: Importance of interval time. Br. J. Cancer 2016, 114, 88–95. [Google Scholar] [CrossRef]
- Park, J.H.; Ahn, J.-H.; Kim, S.-B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3, 352–372. [Google Scholar] [CrossRef] [Green Version]
- McArthur, H.L. Innovations and challenges in the treatment of metastatic triple-negative breast cancer. J. Oncol. Pract. 2018, 14, 290–291. [Google Scholar] [CrossRef]
- Jang, M.H.; Kim, H.J.; Kim, E.J.; Chung, Y.R.; Park, S.Y. Expression of epithelial-mesenchymal transition–related markers in triple-negative breast cancer: ZEB1 as a potential biomarker for poor clinical outcome. Hum. Pathol. 2015, 46, 1267–1274. [Google Scholar] [CrossRef]
- Rogers, T.J.; Christenson, J.L.; Greene, L.I.; O’Neill, K.I.; Williams, M.M.; Gordon, M.A.; Nemkov, T.; D’Alessandro, A.; Degala, G.D.; Shin, J.; et al. Reversal of triple-negative breast cancer EMT by miR-200c decreases tryptophan catabolism and a program of immunosuppression. Mol. Cancer Res. 2019, 17, 30–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, A.H.; van Lohuizen, M. Epigenetics and cancer. Genes Dev. 2004, 18, 2315–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29–48. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Tsai, C.T.; So, C.W. Epigenetic therapies by targeting aberrant histone methylome in AML: Molecular mechanisms, current preclinical and clinical development. Oncogene 2017, 36, 1753–1759. [Google Scholar] [CrossRef]
- Zhang, L.; Deng, L.; Chen, F.; Yao, Y.; Wu, B.; Wei, L.; Mo, Q.; Song, Y. Inhibition of histone H3K79 methylation selectively inhibits proliferation, self-renewal and metastatic potential of breast cancer. Oncotarget 2014, 5, 10665–10677. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.H.; Park, J.H.; Choi, H.J.; Park, M.K.; Won, H.Y.; Park, Y.J.; Lee, C.H.; Oh, S.H.; Song, Y.S.; Kim, H.S.; et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 2015, 6, 7821–7834. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Kong, G. DOT1L: A new therapeutic target for aggressive breast cancer. Oncotarget 2015, 6, 30451–30452. [Google Scholar] [CrossRef]
- Chen, J.-F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets--what are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saenger, W. Structure and function of nucleosides and nucleotides. Angew. Chem. Int. Ed. Engl. 1973, 12, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Eyer, L.; Nencka, R.; de Clercq, E.; Seley-Radtke, K.; Růžek, D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir. Chem. Chemother. 2018, 26, 1–28. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Byun, W.S.; Jin, M.; Yu, J.; Kim, W.K.; Song, J.; Chung, H.J.; Jeong, L.S.; Lee, S.K. A novel selenonucleoside suppresses tumor growth by targeting Skp2 degradation in paclitaxel-resistant prostate cancer. Biochem. Pharmacol. 2018, 158, 84–94. [Google Scholar] [CrossRef]
- Shin, Y.S.; Jarhad, D.B.; Jang, M.H.; Kovacikova, K.; Kim, G.; Yoon, J.S.; Kim, H.R.; Hyun, Y.E.; Tipnis, A.S.; Chang, T.S.; et al. Identification of 6’-beta-fluoro-homoaristeromycin as a potent inhibitor of chikungunya virus replication. Eur. J. Med. Chem. 2020, 187, 111956–111965. [Google Scholar] [CrossRef]
- Borchardt, R.T.; Keller, B.T.; Patel-Thombre, U. Neplanocin A. A potent inhibitor of S-adenosylhomocysteine hydrolase and of vaccinia virus multiplication in mouse L929 cells. J. Biol. Chem. 1984, 259, 4353–4358. [Google Scholar]
- Obara, T.; Shuto, S.; Saito, Y.; Snoeck, R.; Andrei, G.; Balzarini, J.; De Clercq, E.; Matsuda, A. New neplanocin analogues. 7. Synthesis and antiviral activity of 2-halo derivatives of neplanocin A. J. Med. Chem. 1996, 39, 3847–3852. [Google Scholar] [CrossRef]
- Chandra, G.; Moon, Y.W.; Lee, Y.; Jang, J.Y.; Song, J.; Nayak, A.; Oh, K.; Mulamoottil, V.A.; Sahu, P.K.; Kim, G.; et al. Structure-activity relationships of neplanocin A analogues as S-adenosylhomocysteine hydrolase inhibitors and their antiviral and antitumor activities. J. Med. Chem. 2015, 58, 5108–5120. [Google Scholar] [CrossRef]
- Moon, H.R.; Kim, H.O.; Lee, K.M.; Chun, M.W.; Kim, J.H.; Jeong, L.S. Stereoselective synthesis of a novel apio analogue of neplanocin A as potential S-adenosylhomocysteine hydrolase inhibitor. Org. Lett. 2002, 4, 3501–3503. [Google Scholar] [CrossRef] [PubMed]
- Jeong, L.S.; Yoo, S.J.; Lee, K.M.; Koo, M.J.; Choi, W.J.; Kim, H.O.; Moon, H.R.; Lee, M.Y.; Park, J.G.; Lee, S.K.; et al. Design, synthesis, and biological evaluation of fluoroneplanocin A as the novel mechanism-based inhibitor of S-adenosylhomocysteine hydrolase. J. Med. Chem. 2003, 46, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Jeong, L.S.; Moon, H.R.; Park, J.G.; Shin, D.H.; Choi, W.J.; Lee, K.M.; Kim, H.O.; Chun, M.W.; Kim, H.D.; Kim, J.H. Synthesis and biological evaluation of halo-neplanocin A as novel mechanism-based inhibitors of S-adenosylhomocysteine hydrolase. Nucleosides Nucleotides Nucleic Acids 2003, 22, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.O.; Yoo, S.J.; Ahn, H.S.; Choi, W.J.; Moon, H.R.; Lee, K.M.; Chun, M.W.; Jeong, L.S. Synthesis of fluorinated cyclopentenyladenine as potent inhibitor of S-adenosylhomocysteine hydrolase. Bioorg. Med. Chem. Lett. 2004, 14, 2091–2093. [Google Scholar] [CrossRef] [PubMed]
- Barve, A.; Vega, A.; Shah, P.P.; Ghare, S.; Casson, L.; Wunderlich, M.; Siskind, L.J.; Beverly, L.J. Perturbation of methionine/S-adenosylmethionine metabolism as a novel vulnerability in MLL rearranged leukemia. Cells 2019, 8, 1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Jarhad, D.B.; Kim, G.; Nayak, A.; Zhao, L.X.; Yu, J.; Kim, H.R.; Lee, J.Y.; Mulamoottil, V.A.; Chandra, G.; et al. Design, synthesis and anticancer activity of fluorocyclopentenyl-purines and - pyrimidines. Eur. J. Med. Chem. 2018, 155, 406–417. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Kwon, Y.; Byun, W.S.; Kim, B.Y.; Song, M.C.; Bae, M.; Yoon, Y.J.; Shin, J.; Lee, S.K.; Oh, D.C. Depsidomycins B and C: New cyclic peptides from a ginseng farm soil-derived Actinomycete. Molecules 2018, 23, 1266. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.K.; Byun, W.S.; Chung, H.J.; Oh, J.; Park, H.J.; Choi, J.S.; Lee, S.K. Esculetin suppresses tumor growth and metastasis by targeting Axin2/E-cadherin axis in colorectal cancer. Biochem. Pharmacol. 2018, 152, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Byun, W.S.; Kim, S.; Shin, Y.H.; Kim, W.K.; Oh, D.C.; Lee, S.K. Antitumor activity of ohmyungsamycin A through the regulation of the Skp2-p27 axis and MCM4 in human colorectal cancer cells. J. Nat. Prod. 2020, 83, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Borchardt, R.T. S-adenosyl-L-homocysteine hydrolase as a target for antiviral chemotherapy. J. Med. Chem. 1991, 34, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Tamaru, H.; Selker, E.U. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 2001, 414, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.R.; Bahirvani, A.G.; Rao, V.K.; Bharathy, N.; Ow, J.R.; Taneja, R. G9a, a multipotent regulator of gene expression. Epigenetics 2013, 8, 16–22. [Google Scholar] [CrossRef]
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERα gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, 5590–5603. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Cha, J.; Kim, S.K.; Park, J.H.; Song, K.H.; Kim, P.; Kim, M.-Y. c-MYC drives breast cancer metastasis to the brain, but promotes synthetic lethality with TRAIL. Mol. Cancer Res. 2018, 17, 544–554. [Google Scholar] [CrossRef] [Green Version]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef] [Green Version]
- Tsesmetzis, N.; Paulin, C.B.J.; Rudd, S.G.; Herold, N. Nucleobase and nucleoside analogues: Resistance and re-sensitisation at the level of pharmacokinetics, pharmacodynamics and metabolism. Cancers 2018, 10, 240. [Google Scholar] [CrossRef] [Green Version]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Klanova, M.; Lorkova, L.; Vit, O.; Maswabi, B.; Molinsky, J.; Pospisilova, J.; Vockova, P.; Mavis, C.; Lateckova, L.; Kulvait, V.; et al. Downregulation of deoxycytidine kinase in cytarabine-resistant mantle cell lymphoma cells confers cross-resistance to nucleoside analogs gemcitabine, fludarabine and cladribine, but not to other classes of anti-lymphoma agents. Mol. Cancer 2014, 13, 159–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.M.; Lamont, I.L. Nucleoside analogues as antibacterial agents. Front. Microbiol. 2019, 10, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Wang, M.-W. Histone lysine methyltransferases as anti-cancer targets for drug discovery. Acta Pharmacol. Sin. 2016, 37, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagni, C.; Chiacchio, U.; Rescifina, A. Histone methyltransferase inhibitors: Novel epigenetic agents for cancer treatment. Curr. Med. Chem. 2013, 20, 167–185. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.; Wong, G.; Shiina, M. Up-regulation of histone methyltransferase, DOT1L, by matrix hyaluronan promotes microRNA-10 expression leading to tumor cell invasion and chemoresistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem. 2016, 291, 10571–10585. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Chen, P.; Diao, J.; Cheng, G.; Deng, L.; Anglin, J.L.; Prasad, B.V.V.; Song, Y. Selective inhibitors of Histone methyltransferase DOT1L: Design, synthesis, and crystallographic studies. J. Am. Chem. Soc. 2011, 133, 16746–16749. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of neplanocin A and its analogs are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (μM)a | |||
|---|---|---|---|---|
| 1 | 2 | 3 | Etoposideb | |
| MCF10A | 22.28 ± 1.98 | 38.97 ± 1.77 | >50 | 31.86 ± 3.45 |
| HCC38 | 1.22 ± 0.26 | 0.87 ± 0.17 | 16.25 ± 2.10 | 1.36 ± 0.33 |
| HCC1937 | 0.98 ± 0.14 | 1.12 ± 0.28 | 14.36 ± 0.82 | 6.24 ± 0.39 |
| MDA-MB-231 | 0.42 ± 0.11 | 0.47 ± 0.16 | 9.23 ± 0.69 | 4.42 ± 0.47 |
| MCF-7 | 4.94 ± 0.31 | 5.24 ± 0.09 | 34.01 ± 1.78 | 1.11 ± 0.05 |
| T-47D | 6.22 ± 0.29 | 6.17 ± 0.88 | 29.63 ± 2.33 | 3.52 ± 0.12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byun, W.S.; Kim, W.K.; Yoon, J.-s.; Jarhad, D.B.; Jeong, L.S.; Lee, S.K. Antiproliferative and Antimigration Activities of Fluoro-Neplanocin A via Inhibition of Histone H3 Methylation in Triple-Negative Breast Cancer. Biomolecules 2020, 10, 530. https://doi.org/10.3390/biom10040530
Byun WS, Kim WK, Yoon J-s, Jarhad DB, Jeong LS, Lee SK. Antiproliferative and Antimigration Activities of Fluoro-Neplanocin A via Inhibition of Histone H3 Methylation in Triple-Negative Breast Cancer. Biomolecules. 2020; 10(4):530. https://doi.org/10.3390/biom10040530
Chicago/Turabian StyleByun, Woong Sub, Won Kyung Kim, Ji-seong Yoon, Dnyandev B. Jarhad, Lak Shin Jeong, and Sang Kook Lee. 2020. "Antiproliferative and Antimigration Activities of Fluoro-Neplanocin A via Inhibition of Histone H3 Methylation in Triple-Negative Breast Cancer" Biomolecules 10, no. 4: 530. https://doi.org/10.3390/biom10040530