
Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation
by
 , and
, and
Ghaith A. Bahader
1,
Antonisamy William James
1,
Daniyah A. Almarghalani
2 and
Zahoor A. Shah
1,* 1
Department of Medicinal and Biological Chemistry, The University of Toledo, 3000 Arlington Avenue, Toledo, OH 43614, USA
2
Department of Pharmacology and Experimental Therapeutics, College of Pharmacy and Pharmaceutical Sciences, The University of Toledo, Toledo, OH 43614, USA
*
Author to whom correspondence should be addressed.
Biology 2023, 12(4), 630; https://doi.org/10.3390/biology12040630
Submission received: 1 March 2023
/
Revised: 4 April 2023
/
Accepted: 15 April 2023
/
Published: 21 April 2023
(This article belongs to the Special Issue Advances in the Fields of Neurotrauma and Neuroregeneration)
Abstract
:Simple Summary
Traumatic brain injury (TBI) is a significant healthcare problem and a leading cause of death in the United States. There is a critical need to develop potential therapeutics to treat TBI-related injuries. Oxidative stress is considered a major mechanism that worsens the damage. Microglia, the first line of defense in the brain, is overactivated following injury causing the death of neuronal cells. Cofilin is a cytoskeleton protein that is activated during such brain injuries. As reported in previous in vivo studies, targeting cofilin has already been shown as a promising therapeutic strategy in other brain diseases. This study investigated the potential benefits of a new cofilin inhibitor in reducing microglial cell activation and the death of neurons in in vitro immortalized cells. We also explored this cofilin inhibitor’s effect in a mouse TBI model. Following brain injury, we measured the levels of different genes and proteins in mice brains. We found that the administration of cofilin inhibitor reduced various inflammatory and oxidative markers in in vitro and in vivo mice models.
Abstract
Microglial activation and failure of the antioxidant defense mechanisms are major hallmarks in different brain injuries, particularly traumatic brain injury (TBI). Cofilin is a cytoskeleton-associated protein involved in actin binding and severing. In our previous studies, we identified the putative role of cofilin in mediating microglial activation and apoptosis in ischemic and hemorrhagic conditions. Others have highlighted the involvement of cofilin in ROS production and the resultant neuronal death; however, more studies are needed to delineate the role of cofilin in oxidative stress conditions. The present study aims to investigate the cellular and molecular effects of cofilin in TBI using both in vitro and in vivo models as well as the first-in-class small-molecule cofilin inhibitor (CI). An in vitro H2O2-induced oxidative stress model was used in two different types of cells, human neuroblastoma (SH-SY5Y) and microglia (HMC3), along with an in vivo controlled cortical impact model of TBI. Our results show that treatment with H2O2 increases the expression of cofilin and slingshot-1 (SSH-1), an upstream regulator of cofilin, in microglial cells, which was significantly reduced in the CI-treated group. Cofilin inhibition significantly attenuated H2O2-induced microglial activation by reducing the release of proinflammatory mediators. Furthermore, we demonstrate that CI protects against H2O2-induced ROS accumulation and neuronal cytotoxicity, activates the AKT signaling pathway by increasing its phosphorylation, and modulates mitochondrial-related apoptogenic factors. The expression of NF-E2-related factor 2 (Nrf2) and its associated antioxidant enzymes were also increased in CI-treated SY-SY5Y. In the mice model of TBI, CI significantly activated the Nrf2 and reduced the expression of oxidative/nitrosative stress markers at the protein and gene levels. Together, our data suggest that cofilin inhibition provides a neuroprotective effect in in vitro and in vivo TBI mice models by inhibiting oxidative stress and inflammatory responses, the pivotal mechanisms involved in TBI-induced brain damage.
1. Introduction
Traumatic brain injury (TBI) is a significant cause of death and long-term disability in the United States. An estimated 1.5 million individuals are suffering from TBI each year, with (230,000 hospitalized and 50,000 death) [1], with the major cause being sports, vehicle accidents, and war [2,3]. Moreover, TBI has an economic burden on society, with an estimated annual cost of USD 48.6 billion in the United States alone [4]. Although no effective therapeutics can alleviate TBI-associated brain damage and neurological impairment, preclinical studies have shown that targeting key pathological mechanisms is an effective approach [5]. Microglial activation, neuroinflammation, and oxidative stress have been involved in the pathophysiology of several neurodegenerative diseases including TBI [6,7]. Oxidative stress is caused by the imbalance between the antioxidant defense and the production of oxygen-derived radicals such as hydrogen peroxide (H2O2), superoxide, and nitric oxide, leading to the excessive generation of reactive oxygen species (ROS) [8]. Excessive ROS production and accumulation represent a major hallmark in the pathogenesis of TBI that can damage the structure of cell membranes and other key cellular molecules such as lipids, proteins, and DNA, which eventually disrupt cellular integrity [9]. Among the various species, H2O2-induced ROS production and oxidative stress response can lead to mitochondrial dysfunction and initiate cellular apoptosis in different types of cells [10]. H2O2-induced mitochondrial dysfunction and cell apoptosis are partially regulated by the expression of pro-apoptotic BAX and anti-apoptotic Bcl-2 proteins [11]. Studies have shown that activating antioxidant cellular defense systems may prevent oxidative stress-induced neuronal apoptosis in different neurological diseases [12,13]. The nuclear factor erythroid 2–related factor 2 (Nrf2) is a major regulator of cellular resistance to oxidative stress. It regulates the cellular response to oxidative damage by mediating the expression of antioxidant enzyme genes such as heme oxygenase 1 (HO-1), sodium oxide dismutase 2 (SOD2), NAD(P)H quinone oxidoreductase 1 (NQO1), and glutathione peroxidase (GPx) [14].
Often referred to as “CNS resident macrophage”, microglia play an important role in the pathogenesis of several neurodegenerative diseases. Although it has some beneficial functions, such as clearance of cellular debris following brain injury, excessive microglia activation is deleterious and aggravates secondary brain injury [15]. Activated microglia mediates brain neuroinflammation and subsequent neurotoxicity by releasing proinflammatory cytokines and cytotoxic substances [16]. A major transcription factor controlling proinflammatory gene expression in microglial cells, nuclear factor-κB (NF-κB), is activated by exogenous H2O2 application or endogenous H2O2 release, such as through the activation of NADPH oxidase (NOX) enzymes. The activation of NF-κB induces an inflammatory phenotype of microglia, which is exhibited by increased expression of inflammatory mediators such as tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), and high mobility group box1 (HMGB1) [17,18].
Cofilin is an actin-associated protein that regulates the dynamics of the actin cytoskeleton through binding, depolarization, and severing of filamentous actin (F-actin) into globular actin (G-actin) [19]. The activity and dynamics of cofilin are regulated by its phosphorylation/dephosphorylation process by various kinases and phosphatases. LIM kinase mediates cofilin phosphorylation at ser3 and the formation of phospho-cofilin (p-cofilin), while slingshot phosphatase 1 (SSH1) mediates its dephosphorylation [20]. Evidence suggests that cofilin activation and the formation of cofilin-actin rods trigger the initiation of apoptosis [21] and are involved in the pathogenesis of neurodegenerative diseases such as Alzheimer’s and ischemic stroke [22,23]. Using gene-editing techniques such as siRNA, studies from our lab have demonstrated that inhibiting cofilin expression is effective in restoring neuronal viability and attenuating microglial activation in different stressful conditions [16,24,25]. However, using a small molecule that can deliver the same therapeutic outcomes is an integral approach to overcoming challenges associated with gene therapy. A recent report from our lab showed that treatment with a newly synthesized novel small molecule cofilin inhibitor (CI) reduced thrombin-induced microglial activation and neuronal apoptosis in vitro [26].
In the present study, we investigated the role of cofilin in TBI by using oxidative stress models of H2O2-induced microglial activation and neurotoxicity as well as controlled cortical impact representing both in vitro and in vivo TBI models. In addition, we explored the potential therapeutic effects of CI in reducing microglial-related inflammatory markers, neuronal apoptosis, and oxidative stress both in vitro and in vivo.
2. Material and Methods
2.1. Cell Culture
The human microglia clone 3 cell line (HMC3) (ATCC, Manassas, VA, USA) was cultured in DMEMF12 (Thermo Scientific, West Palm Beach, FL, USA) supplemented with 5% heat-inactivated fetal bovine serum (FBS), 5% horse serum, 2 mM L-glutamine, 1 mM sodium pyruvate, and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 95% air and 5% CO2. HMC3 were seeded at a density of 1 × 105 cells in 6-well plates with H2O2 and H2O2 + cofilin inhibitor (CI) cultured 24 h for protein expression studies.
SH-SY5Y (neuroblastoma cell line) cells (ATCC, Manassas, VA, USA) were cultured in DMEMF12 (Thermo Scientific) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 95% air/5% CO2. SH-SY5Y were seeded at a density of 1 × 105 cells in 6-well plates for protein expression studies and a density of 1 × 104 cells in 96-well plates for the viability studies.
2.2. Animals and TBI Model
In this study, we used male C57BL/6J (10–12 weeks old) mice obtained from The Jackson Laboratories (Bar Harbor, ME, USA). Mice were housed in the Department of Laboratory Animal Resources (DLAR), University of Toledo, Health Science Campus (HSC) under a 12 h light–dark cycle, with free food and water access. All animal procedures were carried out in accordance with protocols approved by the Institutional Animal Care and Utilization Committee (IACUC) of the University of Toledo and under the guidelines of the National Institute of Health (NIH).
A total of 30 mice were randomly subdivided into three groups (sham, TBI + vehicle, and TBI + CI) and subjected to moderate controlled cortical impact (CCI) or sham surgery. Briefly, mice were anesthetized with isoflurane (3% for induction and 1–2% for maintenance) and placed into the stereotaxic frame. A longitudinal midline incision was made, the skin retracted, and a 4 mm craniectomy was performed over the left hemisphere midway to the lambda and bregma sutures and laterally midway between the central suture and the temporalis muscle. The skull cap was removed carefully without damaging the underlying dura. CCI injury was induced in mice using an impactor with a 3 mm in diameter piston that was angled and placed perpendicular to the exposed cortex. The CCI injury was performed with a velocity of 3 m/s, a depth of 1.5 mm, and a dwelling time of 300 milliseconds. After the injury, the incision was sutured, and the mice were allowed to recover on a heating pad for 30 min. Sham mice underwent the same procedure but without the CCI impact.
2.3. MTT Assay
SH-SY5Y cells (1 × 104) were seeded in 96-well plates and after 36 h, cells were treated with different concentrations of H2O2 (10 to 250 μM) for 24 h; then, 10 μL of CCK-8 reagent was added into each well and incubated at 37 °C for 2 h according to manufactures instruction. The [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2, 4-disulfophenyl)-2H tetrazolium, monosodium salt] is reduced by dehydrogenase in cells to give an orange-colored product (formazan), which is soluble in the tissue culture medium. The amount of the formazan dye generated by dehydrogenases in cells is directly proportional to the number of living cells. The absorbance was measured at 450 nm using a microplate reader, normalized with control, and calculated as a percentage change of cell viability or death. For the CI experiments, SH-SY5Y cells (1 × 104) were seeded in 96-well plates, and after 36 h, cells were co-treated with vehicle or different concentrations of CI (3, 5, 7, 10, 20 μM) with H2O2 (200 μM) for 24 h followed by same steps mentioned above.
2.4. Western Blotting (WB)
Cells or brain tissue samples were harvested by homogenizing them in an ice-cold RIPA buffer (Thermofisher Scientific, Waltham, MA, USA) supplemented with a protease and phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) for 30 min. Supernatants containing the total protein fraction were collected after centrifugation at 14,000× g for 15 min. Protein concentration was determined using Bradford reagent (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer protocol. Equivalent amounts of proteins from each sample were loaded onto 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels, separated by electrophoresis, and then transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 3% BSA for 1 h to avoid non-specific binding. Next, membranes were incubated overnight at 4 °C with different primary antibodies, including cofilin, p-cofilin, TNFα, NF-κB, SOD2, NQO1, p-Akt, BAX, Bcl-2, caspase-3, cleaved caspase-3, GAPDH, β-actin (Cell Signaling Technology, Danvers, MA, USA), SSH-1, HMGB1 (Abcam, Cambridge, MA, USA), Nrf2, and HO-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The membranes were then washed and incubated with horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit secondary antibodies (Cell Signaling Technology) for 1 h at room temperature. GAPDH and β-actin were used as loading controls. Finally, images were captured using Syngene Imaging System (Frederick, MD, USA), and bands were analyzed using ImageJ software 1.53t (National Institutes of Health, Bethesda, Maryland, USA).
2.5. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Total mRNA was isolated from mice brain tissue samples from the perilesional cortex region using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Complementary DNA (cDNA) synthesis was conducted using an iScript cDNA synthesis kit (Bio-rad). The mRNA expression level was detected using Advanced Universal SYBR Green Supermix (Bio-Rad) in the Step OnePlus Real-Time PCR System (Thermofisher Scientific). GAPDH was used as a housekeeping control. Relative gene abundance levels were analyzed by double delta Ct analysis 2−ΔΔCT. The primer pairs that were used for RT-qPCR measurements are mentioned in Table 1.
2.6. DCF-DA ROS Staining
SH-SY5Y cells were seeded in 6-well plates, and after 36 h, cells were co-treated with 200 μM concentration of H2O2 with or without 10 μM concentration of CI for 24 h. Cells were washed with 1× buffer and then stained with 20 μM DCF-DA for 30 min at 37 °C, and cells were rewashed with 1× buffer to remove overstaining. Live cell imaging was performed using a fluorescence microscope at 20× magnification with fluorescein (FITC) and maintained at low light to avoid photobleaching and photo-oxidation.
2.7. Immunohistochemistry
Three days following TBI, mice were anesthetized with Ketamine/Xylazine (100/10 mg/kg, intraperitoneally) and then transcardially perfused with 1× phosphate buffer saline (PBS) followed by 4% paraformaldehyde (PFA). Mice brains were dissected, placed in 4% PFA for 24 h, and paraffin-embedded, and 8 µm sections were made using a microtome and used for the staining. The staining procedure started with deparaffinized brain sections in different gradients of xylene/ethanol, followed by antigen retrieval in a pressure cooker for 15 min, washing with 1× PBS, and blocking in 5% BSA for 2 h at room temperature. The following primary antibodies were added subsequently and incubated overnight at 4 °C: mouse anti-iNOS (1:200, Sigma), and rabbit anti-3NT (1:200, Cell Signaling). After washing with 1× PBS, slides were incubated with secondary antibodies for 1 h at room temperature (Taxes red labeled donkey anti-rabbit IgG and Alexa Fluor donkey anti-mouse IgG) (1:1000; Jackson, Immunoresearch, West Grove, PA, USA). Then, the slides were washed with 1× PBS, mounted with ProLong® Gold Antifade Mountant with DAPI (Molecular Probes), and images were captured using fluorescence microscopy. Fluorescence intensity was analyzed using ImageJ software (NIH, USA).
2.8. Drug Treatments/Administration
For in vitro studies, cofilin inhibitor (CI) was dissolved in DMSO to prepare 10 mM stock; then, 3, 5, 7, 10, and 15 μM/mL concentrations were used in corresponding experiments. After stimulating cells with 200 μM H2O2 and co-treating with 10 μM CI for 24 h, cells were harvested for protein expression studies. For in vivo studies, CI was dissolved in 4.9% DMSO, 4.9% Tween-20, 88.9% solubilizing agent, and the final concentration was 1.3%. The first intravenous injection of CI (25 mg/kg) was given 4 h after the TBI followed by 25 mg/kg intraperitoneal injections every 12 h for three days.
2.9. Statistical Analysis
Experimental data were presented as the mean ± SEM and were analyzed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA). Each experiment was repeated three times, and data were analyzed using one-way analysis of variance (ANOVA) followed by the Newman–Keuls multiple comparisons test. Additionally, Student’s unpaired t test was used to determine the significance between the two groups. A p < 0.05 value was considered significant.
3. Results
3.1. Cofilin and SSH1 Are Activated in Microglial Cells after H2O2 Treatment
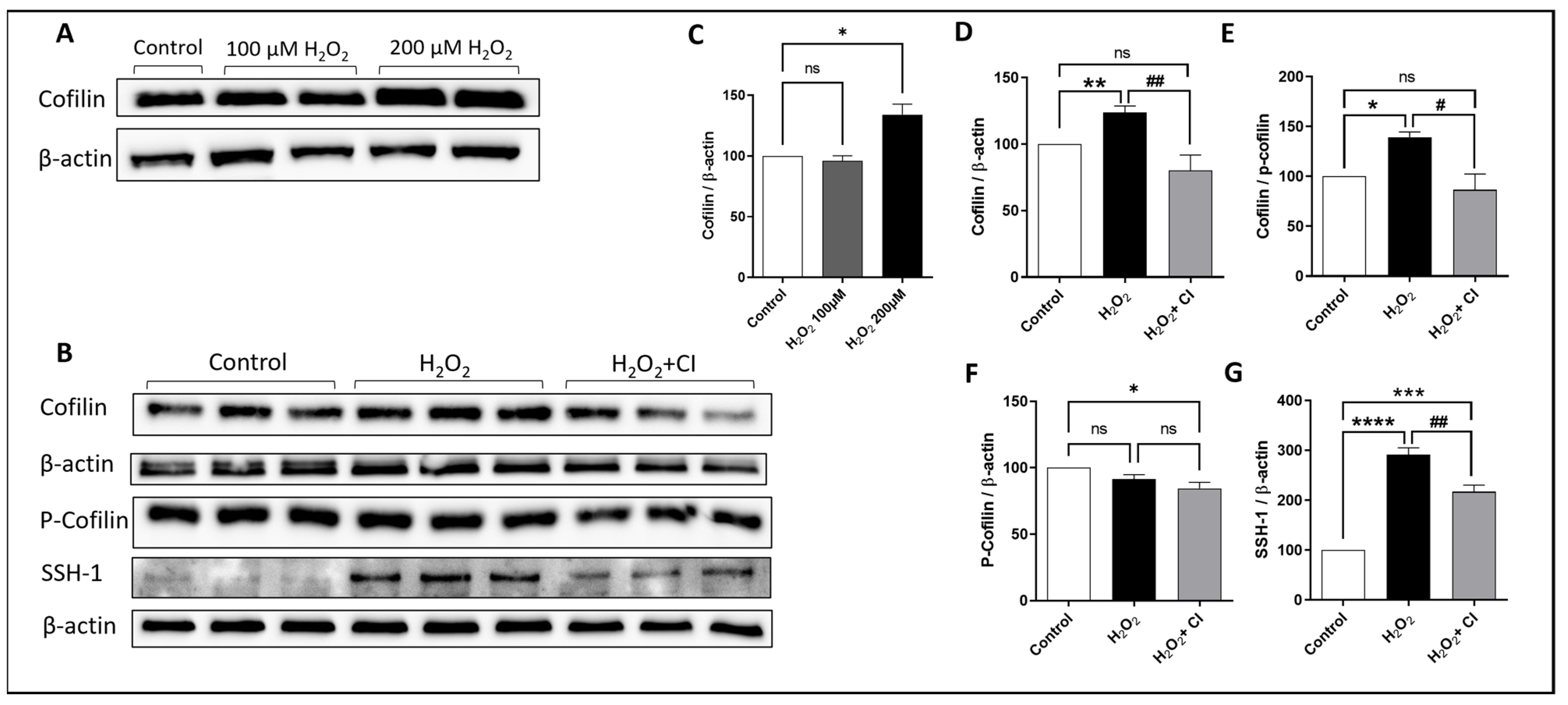
To investigate the activation of cofilin in microglia after H2O2 treatment, HMC3 human microglial cells were treated with two doses of H2O2 (100 μM and 200 μM) for 24 h, and the expression of cofilin was analyzed by WB. Although there was no difference with 100 μM H2O2 treatment, cofilin expression was significantly increased when treated with 200 μM H2O2 (Figure 1A,C). Accordingly, 200 μM H2O2 treatment was used in the subsequent experiments. Next, we tested the effect of CI on inhibiting cofilin activation in H2O2-treated microglial cells. HMC3 cells were challenged with 200 μM H2O2 in the presence of 10 μM CI or vehicle for 24 h. WB analysis confirmed the increase in cofilin expression in the H2O2-treated group; however, treatment with CI significantly reduced the cofilin expression compared to the H2O2 group (Figure 1D). H2O2 treatment did not change the expression of p-cofilin (Figure 1F) in the H2O2 group; however, the cofilin/p-cofilin ratio was significantly reduced in the CI-treated group as compared to the H2O2 (Figure 1E). Cofilin activity is modulated through phosphorylation/dephosphorylation processes. The activation of cofilin is mediated through dephosphorylation by multiple phosphatases, and SSH1 phosphatase is one of them [27]. Our results showed that SSH1 protein expression was significantly increased in microglial cells treated with H2O2 as compared to the control, and it was significantly reduced in the CI-treated cultures (Figure 1G). These results indicate that cofilin is activated during oxidative stress conditions induced by H2O2, which can be inhibited by CI treatment.
3.2. CI Treatment Reduced the H2O2-Induced Microglial Activation
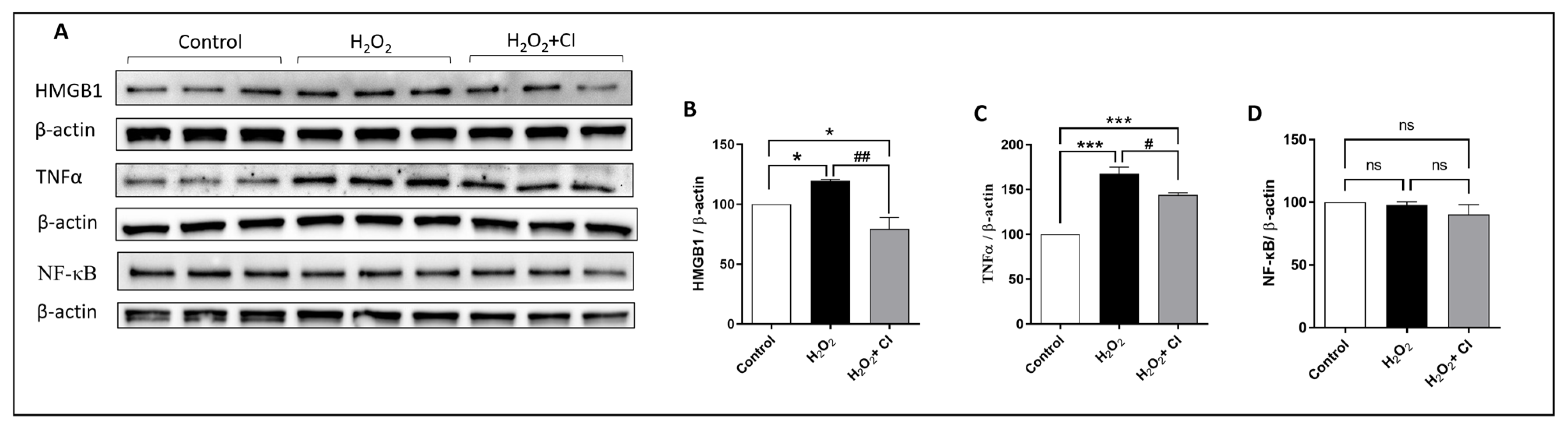
ROS, in particular H2O2, induces microglial proliferation and activation through different regulators, and the NF-κB signaling pathway is one of them. NF-κB is considered a master transcription factor that controls the proinflammatory gene expression in microglia [28]. Proinflammatory substances such as HMGB1 bind to cellular receptors (such as Toll-like receptor 4), which activates NF-κB and the production of inflammatory cytokines such as TNFα. Therefore, our next objective was to understand the impact of CI treatment on H2O2-induced microglial activation. WB analysis showed that the expression of HMGB1 and TNFα was significantly reduced in the CI-treated cultures compared to the vehicle-treated cultures (Figure 2B,C). Of note, there was no change in the expression level of total NF-κB (Figure 2D).
3.3. H2O2 Activates Cofilin Signaling, and CI Treatment Protects against H2O2-Induced Neurotoxicity in SH-SH5Y Cells
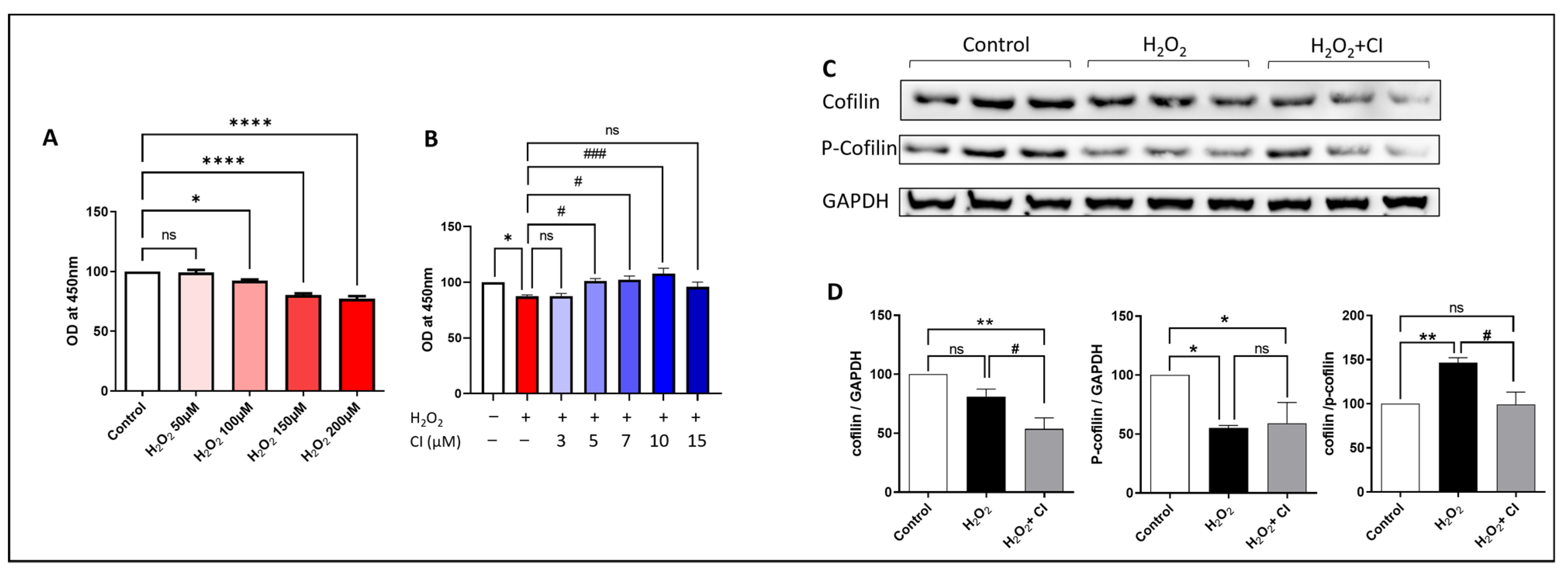
Next, we tested the neuroprotective potential of CI against H2O2-induced cell death using the SH-SY5Y neuroblastoma cell line. CCK8 assay was used to assess the viability of SH-SY5Y cells. First, we treated the cells with increasing concentrations of H2O2 for 24 h and then measured the cell viability. We found that SH-SY5Y cell viability was remarkably reduced starting from 100 µM H2O2 concentration, and the reduction is proportionally increased with increased H2O2 concentration compared to controls (Figure 3A). Accordingly, we decided to choose the 200 µM concentration, which was associated with around 20–25% reduction in cell viability. In the following experiment, we incubated the SH-SY5Y cells with 200 µM H2O2 plus vehicle or increasing concentration of CI and assessed the cell viability. Our results showed that the viability of SH-SY5Y cells was significantly increased starting from 5 µM concentration of CI with the highest increase with 10 µM concentration; however, there was no increase in the viability of the cells with 3 and 15 µM concentrations of CI as compared to the H2O2 vehicle-treated cultures (Figure 3B). Accordingly, a concentration of 10 µM CI was used for the followed experiments. Next, we investigated cofilin signaling in SH-SY5Y cells 24 h following stimulation with 200 µM H2O2 with or without CI. Although the cofilin expression was not significantly changed in the H2O2-stimulated cells, there was a significant reduction in the expression of p-cofilin (Figure 3C,D). Analyzing the ratio of cofilin to p-cofilin showed a significant increase in the cofilin dephosphorylation state. Moreover, CI treatment significantly reduced the total cofilin and the cofilin/p-cofilin ratio compared to vehicle-treated cells.
3.4. CI Treatment Reduces the H2O2-Induced ROS Production in SH-SY5Y Cells
ROS generation is an important indicator of oxidative stress and mitochondrial dysfunction in cells, resulting in cell apoptosis. Therefore, to determine the effect of CI in reducing ROS production, SH-SY5Y cells were treated with H2O2 in the presence of 10 µM CI or vehicle, and ROS level was measured using DCF-DA fluorescence staining. Treatment with H2O2 markedly increased the green fluorescence level in SH-SY5Y cells as an indicator of increased intracellular ROS levels. Co-treatment with 10 µM CI effectively attenuated this increase and reduced ROS production (Figure 4).
3.5. CI Treatment Enhances the Expression of Anti-Apoptotic and Survival Proteins after H2O2 Stimulation in SH-SY5Y Cells
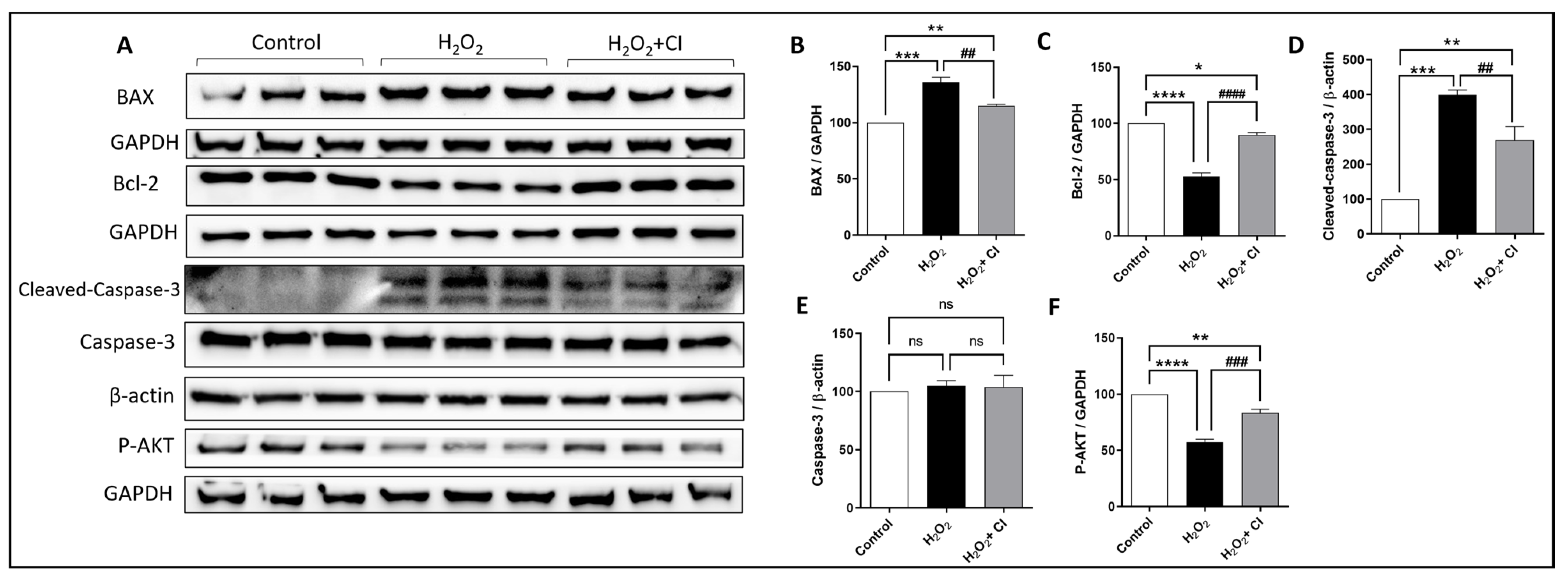
Cell survival, proliferation, and apoptosis are regulated by many factors, including the Bcl-2 family proteins, transcription factors, caspases, and Akt signaling pathway [29]. Thus, we used WB to investigate the protein expression changes of pro- and anti-apoptotic proteins in SH-SY5Y cells after H2O2 stimulation with or without CI treatments. As shown in Figure 5, CI treatment significantly attenuated the increased expression of BAX, a pro-apoptotic protein, and recovered the diminished expression of Bcl-2, an anti-apoptotic protein, in SH-SY5Y cells (Figure 5A–C). In addition, the expression of cleaved caspase-3, as a marker of cell apoptosis, was significantly suppressed in CI-treated cultures, while there was no difference in the protein expression of total caspase-3 (Figure 5A,D,E). Moreover, the reduced expression of p-Akt in the H2O2-stimulated cells was significantly rescued by CI treatment (Figure 5A,F). These observations demonstrate that cofilin activation mediated mitochondrial dysfunction and apoptotic cell death in models of oxidative stress in the SH-SY5Y cells.
3.6. CI Treatment Induces the Expression of Nrf2 and Antioxidant Enzymes in SY-SY5Y Cells
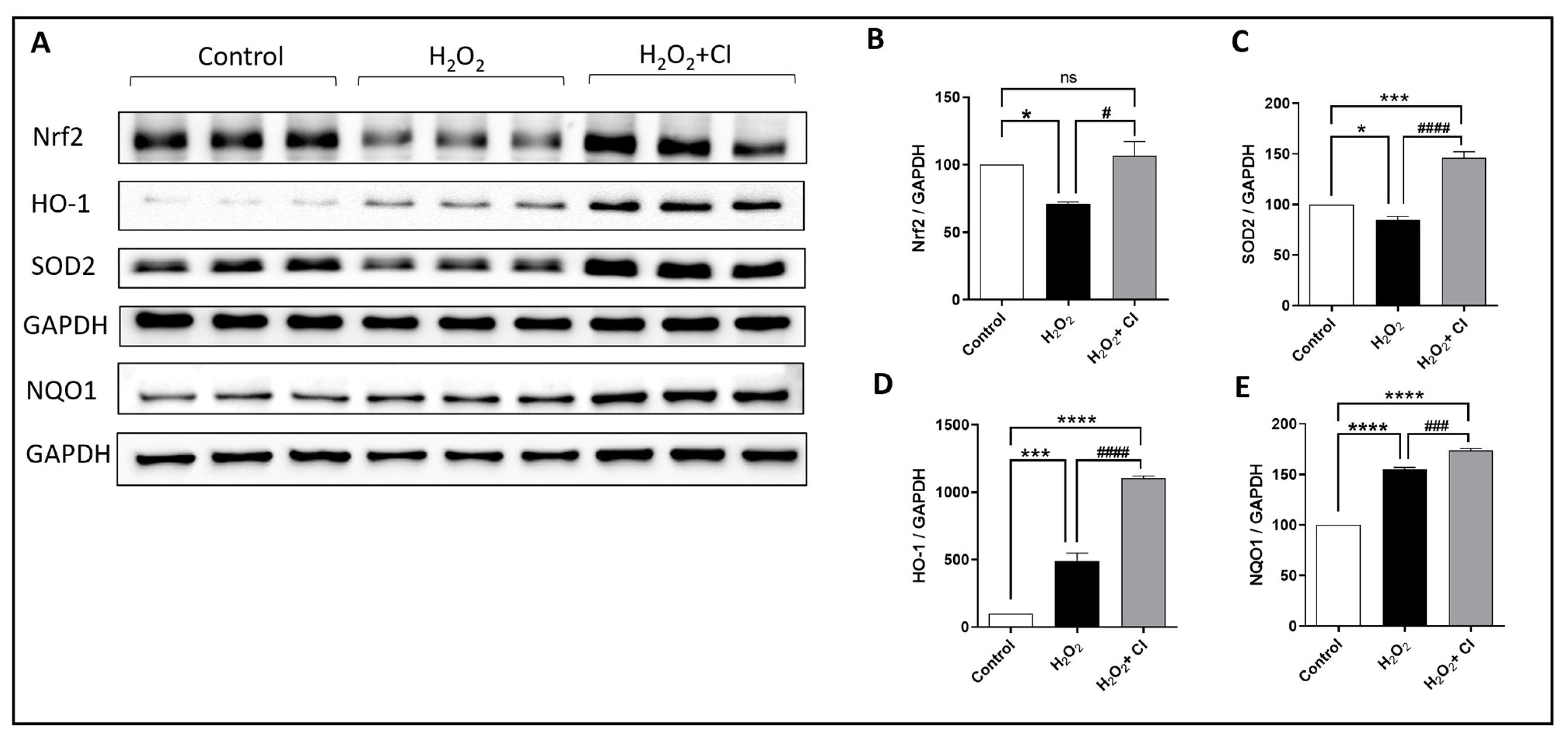
Nrf2 is a major transcription factor that protects against oxidative stress damage due to various diseases by regulating the expression of several cytoprotective enzymes [30]. Therefore, we tested the effect of CI on the expression of Nrf2 and relevant antioxidant enzymes, including SOD2, HO-1, and NQO1. In brief, SH-SY5Y cells were incubated with 200 μm H2O2 with or without 10 μM CI for 24 h; then, protein expression levels were measured (Figure 6A). Results from this experiment revealed that the reduced expression of Nrf2 and SOD2 by H2O2 was significantly recovered by CI treatment (Figure 6B,C). Moreover, the expression levels of HO-1 and NQO1, major Nrf2-dependent enzymes, were increased in the H2O2-treated cultures and were further increased when treated with CI (Figure 6D,E). Hence, these findings imply that activation of Nrf2 and its downstream antioxidant enzymes is a possible mechanism through which cofilin inhibition blocks oxidative damage in neuronal cells.
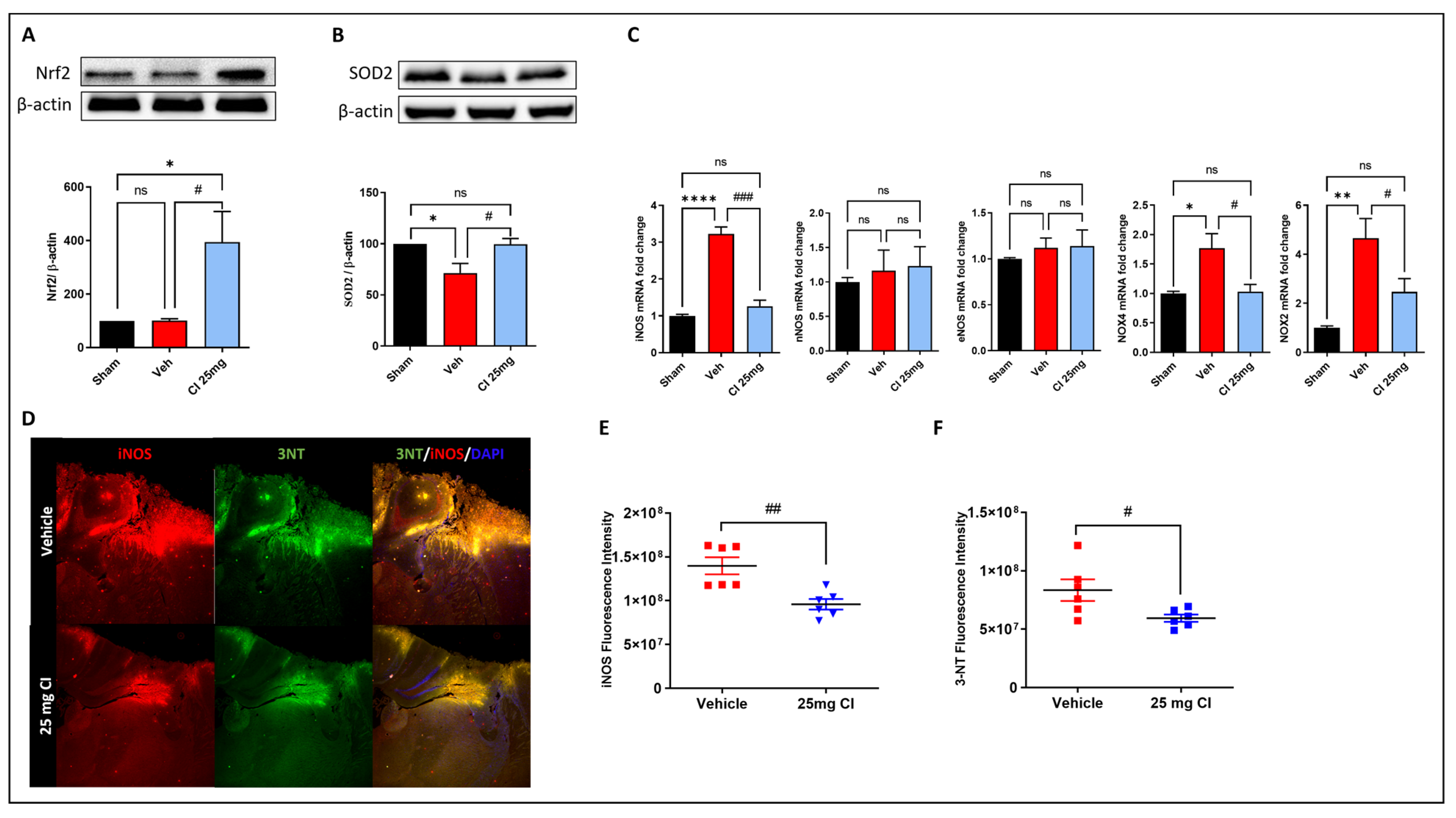
3.7. CI treatment Attenuates TBI-Induced Oxidative and Nitrosative Stress in Mice
Oxidative stress is one of the major mechanisms that contributes to post-traumatic brain injury [8]. Thus, to further validate the efficacy of CI in rescuing oxidative stress in vivo, we tested the gene and protein expression of many oxidative stress regulators in mouse brain lysate 3 days following controlled cortical impact brain injury with or without 25 mg/kg of CI treatment. The results showed that the expression of Nrf2 was dramatically increased in the CI-treated mice compared to the vehicle and sham mice (Figure 7A). The TBI-induced reduction in SOD2 expression was rescued in the CI-treated mice (Figure 7B). Furthermore, we analyzed the gene expression of NADPH oxidases (NOX), nitric oxide synthases (NOS), and 3-nitrotyrosine (3-NT). qPCR analysis shows a significant elevation in the mRNA levels of inducible NOS (iNOS), NOX2, and NOX4, while no change was found in neuronal NOS (nNOS) and endothelial NOS (eNOS) (Figure 7C).
CI treatment significantly reduced the mRNA levels of iNOS, NOX2, NOX4 compared to the vehicle-treated mice. Additionally, immunofluorescence staining of iNOS, and 3-NT confirmed those results on the protein level and showed a significant reduction in the mean fluorescence intensity of iNOS and 3-NT in the traumatized cortex area of the CI-treated group in comparison with the vehicle group (Figure 7D–F). These observations confirm a relevant involvement of cofilin in brain injury-induced oxidative damage in mice.
4. Discussion
In the present study, we evaluated the role of cofilin in mediating oxidative stress-induced microglial activation and neurotoxicity and the potential therapeutic benefits of a novel small molecule cofilin inhibitor in vitro and in vivo. We demonstrated that microglial stimulation with H2O2 activates the cofilin pathway and increases the expression of inflammatory mediators such as TNFα and HMGB1, an effect reversed by treatment with CI. Moreover, we showed that CI increases the viability of SH-SY5Y cells and attenuates oxidative damage by altering the expression of apoptotic modulators and by activating the Nrf2 pathway, respectively. As a major TBI-induced secondary brain injury mechanism, oxidative damage was reduced in CI-treated mice on the gene and protein levels. Results from this study highlight the role of cofilin in mediating oxidative damage and propose the cofilin inhibitor as a promising therapeutic alternative for neurodegenerative diseases such as TBI.
The pathophysiology of traumatic brain injury is intrinsically heterogeneous and is mediated by multiple mechanisms, including ROS formation and oxidative damage, glutamate excitotoxicity, neuroinflammation, blood–brain barrier disruption, and cellular apoptosis [31]. Among these, neuroinflammation is considered one of the most critical mediators that orchestrates the pathology and chronic neurodegeneration processes during TBI [32]. Following TBI, microglia respond within minutes by becoming activated, migrating to the site of injury and mediating pathological responses, mainly the overexpression of inflammatory and oxidative/nitrosative stress factors, which aggravate the initial injury [32,33]. Thus, interventions targeting microglial activation are conceived to be potential neuroprotective therapeutic strategies for the treatment of TBI [34].
Cofilin is expressed in all cell types, and disturbance of cofilin dynamics has been involved in the pathology of many neurodegenerative disorders [35]. Cofilin is regulated by phosphatases such as SSH-1 that are activated in response to stress signals, such as ROS accumulation and ATP reduction, leading to cofilin dephosphorylation and activation [36,37]. In accordance with this, the present study shows that microglial stimulation with H2O2 activates cofilin through SSH-1 overexpression. Our previous studies on cofilin dynamics in microglia identified cofilin as a stress-associated protein that plays a pivotal role in microglial activation during pathological states. In this paradigm, cofilin knockdown by siRNA reduces LPS-induced microglial activation, proliferation, phagocytosis, and inflammatory cytokines production [16]. Moreover, treatment with a first-in-class cofilin inhibitor attenuates microglial activation and nitric oxide and TNFα expression in an in vitro model of hemorrhagic stroke [26]. In microglia, H2O2 insult promotes inflammatory phenotype represented by increased inflammatory protein levels such as TNFα, HMGB1, and IL-17 [17]. HMGB1 binds to microglial cellular receptors such as toll-like receptors (TLRs) that trigger the activation and translocation of NF-κB to the nucleus and the subsequent release of cytokines such as TNFα [38]. Similarly, the increase in HMGB1 and TNFα expression by H2O2 in this study confirms microglial activation, which was attenuated by CI treatment. However, there was no change in the level of total NF-κB. These findings are in agreement with other reports that suggest during a neuroinflammatory response, cofilin might intervene and promote the cytoskeleton dynamics of microglia and change its morphology into the activated ameboid phenotype [35,39].
We previously reported that inhibiting cofilin restores the viability of neuronal cells subjected to oxygen-glucose deprivation (OGD) or thrombin-induced cell death [25,26]. Furthermore, a study showed recently that cofilin knockout in primary cortical neurons enhances cell viability in a glutamate-induced excitotoxicity model [40]. In this study, although cofilin expression was not increased in SH-SY5Y cells stimulated with H2O2, the cofilin/p-cofilin ratio increased, indicating cofilin activation. To further explore whether inhibiting cofilin protects against oxidative-induced neuronal cell death, we investigated the effect of CI in H2O2-stimulated SH-SY5Y cells. We confirmed the neuroprotective effect of CI against H2O2-induced SH-SY5Y cell death by CCK-8 assay.
It is evident that treating cells with H2O2 increases ROS production, which is associated with oxidative stress-induced DNA and mitochondrial damage, leading to cell apoptosis [41]. Here, we found that CI reduces ROS production in SH-SY5Y cells. These findings are consistent with a recent report proposing that inhibiting ROS accumulation by inhibiting cofilin is an effective intervention to salvage neurons under pathophysiological conditions such as glutamate or erastin stimulation [40]. Cofilin was identified as a key modulator in the apoptotic cell death cascade and was involved in transducing apoptotic signals to mitochondria during oxidative damage [42]. In this context, cofilin knockdown using siRNA inhibits H2O2-induced apoptosis in fibroblast cells [42]. Another study suggested that Aβ oligomers induce apoptosis by activating and translating cofilin to mitochondria regulated by SSH-1 [43]. A large body of evidence demonstrated that Akt promotes survival signals by regulating different protein kinases, transcription factors, and the expression of pro- and anti-apoptotic proteins, such as the Bcl-2 family [44]. Additionally, Akt activation has been suggested to be linked to apoptosis inhibition in SH-SY5Y cells under H2O2-induced oxidative damage [45]. As a member of the Bcl-2 family, BAX acts as a pro-apoptotic protein that induces the release of cytochrome c by impairing the mitochondrial outer membrane barrier function. On the opposite side, Bcl-2 is an anti-apoptotic protein that promotes mitochondrial membrane stabilization and inhibits the release of apoptogenic factors [46]. In the present study, H2O2 treatment upregulated BAX and cleaved caspase-3 expression, downregulated Bcl-2 expression, and reduced Akt phosphorylation in SH-SY5Y cells, of which the findings are consistent with previous studies [47,48]. However, these changes were significantly blunted in the presence of CI, proposing that CI may protect neuronal cell apoptosis by inhibiting mitochondrial damage-associated apoptotic regulators activated by oxidative stress.
As an adaptive cellular response to ROS accumulation and oxidative stress, Nrf2 is activated and induces the transcription of several antioxidant enzymes [13]. Several studies have shown that the activation of the Nrf2 signaling pathway is mainly responsible for neuroprotective effects from cytotoxic damage [47]. Additionally, we have previously shown that cofilin knockdown increases the expression of Nrf2 and HO-1 in hemin-treated microglial cells [24]. In line with these results, our data showed that CI induces the expression of Nrf2 and downstream antioxidant enzymes, including HO-1, SOD2, and NQO1 in H2O2-stimulated SH-SY5Y cells, which correlate with the ROS reduction shown earlier.
To further explore the potential therapeutic effect of CI against oxidative damage, we tested its efficacy in a TBI mouse model. The activation of Nrf2 is suggested to be crucial to offset oxidative damage after TBI through modulating mitochondrial and NAPDH oxidase functions [49]. Given this, an exacerbated neurological impairment accompanied by increased levels of oxidative stress and neuroinflammatory markers was reported in Nrf2 knockout mice following CCI injury [50]. On the other hand, TBI studies on NOX2 knockout mice showed reduced lesion volume and neuronal cell death, which were associated with downregulated gene expression of oxidative and pro-inflammatory markers such as iNOS, 3-NT, CD16, and CD86 [51,52]. Consistent with these lines of evidence and the in vitro data presented in this study, CI treatment enhanced the expression of Nrf2 and SOD2 and suppressed oxidative stress damage exhibited by the reduction of iNOS, NOX2, and NOX4 at the gene level, and iNOS and 3-NT at the protein level. These results align with an earlier study from our lab where localized knockdown of cofilin improved outcomes following intracerebral hemorrhage in mice partly through reducing oxidative and nitrosative stress [53]. These findings reveal that one of the potential mechanisms underlying the antioxidative effect of CI is through activating the Nrf2 signaling pathway.
5. Conclusions
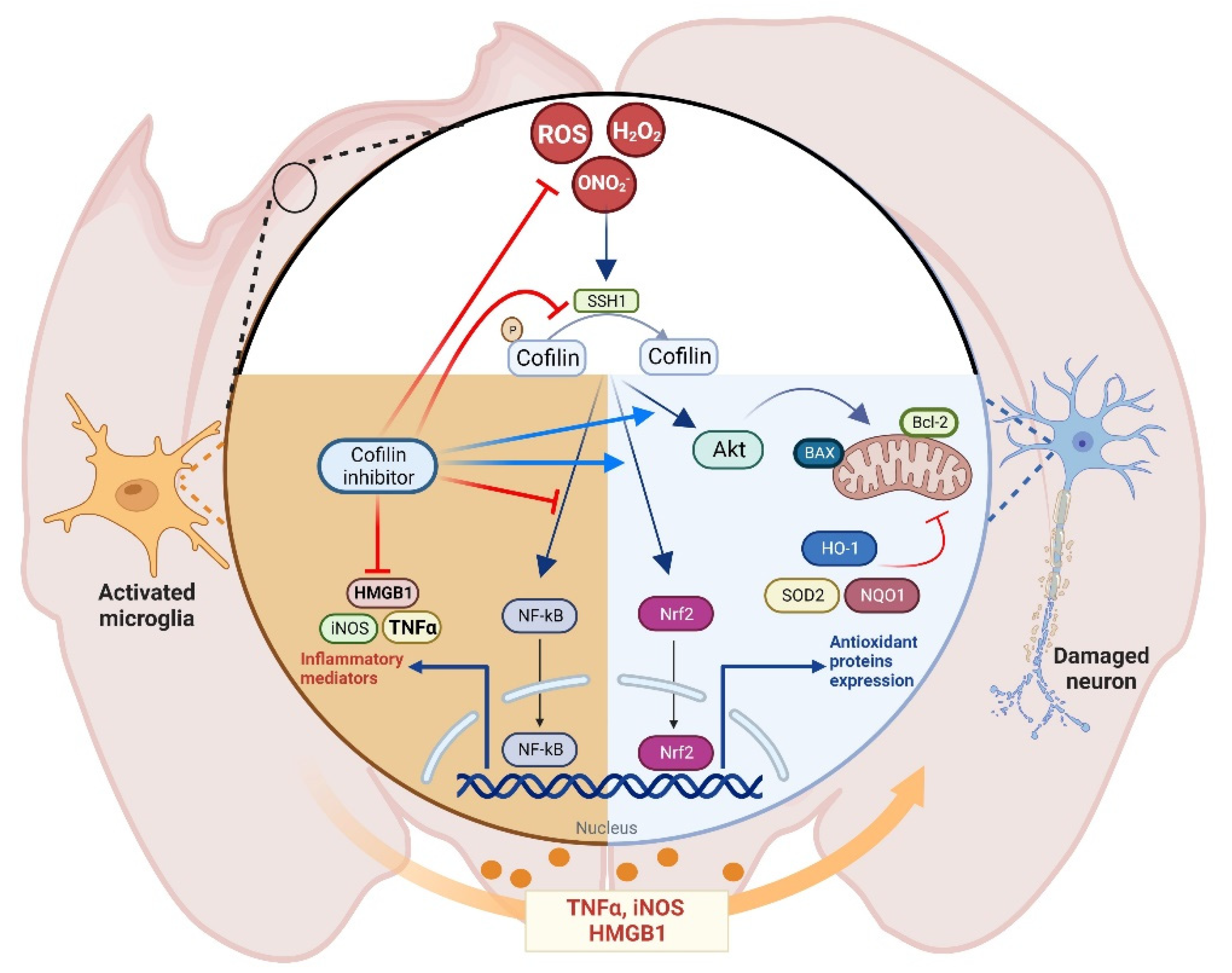
In conclusion, in the present study, the protective effect of CI against H2O2-induced microglial activation and neuronal cell death was elucidated, and some potential mechanisms were explored in vitro and in vivo (Figure 8). CI suppressed the oxidative-mediated microglial activation, represented by increased neuroinflammatory markers expression, via downregulating cofilin and SSH1. Additionally, CI retrieved neuronal viability and reversed ROS generation and mitochondrial dysfunction. In this process, CI modulated mitochondrial-related survival and apoptotic markers and activated Nrf2 and its related antioxidant enzymes, likely contributing to mitigating oxidative stress caused by H2O2 or TBI. Accordingly, the current findings confirm the role of cofilin and its targeting as a potential therapeutic strategy for the treatment and prevention of oxidative stress associated with different neurodegenerative diseases, particularly TBI. Future animal studies are required to investigate the role of cofilin in secondary TBI-induced brain damage and functional recovery and to evaluate the long-term effects of CI as a potential treatment of TBI.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biology12040630/s1, Figure S1: Original western blot for Figure 1 showing the bands with molecular weight markers. Figure S2: Original western blot for Figure 2 showing the bands with molecular weight markers. Figure S3: Original western blot for Figure 3 showing the bands with molecular weight markers. Figure S4: Original western blot for Figure 5 showing the bands with molecular weight markers. Figure S5: Original western blot for Figure 6 showing the bands with molecular weight markers. Figure S6: Original western blot for Figure 7 showing the bands with molecular weight markers.
Author Contributions
G.A.B. and Z.A.S. conceived the idea and designed the experimental plan. G.A.B. and A.W.J. performed the in vitro studies. G.A.B. performed the in vivo study and wrote the manuscripts. D.A.A. performed the immunofluorescence staining study. Z.A.S. supervised the project, provided critical feedback on the manuscript and research, and approved the final draft. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by a grant from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health #R01NS112642 to Z.A.S.
Institutional Review Board Statement
The animal study protocol was reviewed and approved by the University of Toledo’s Institutional Animal Care and Use Committee (IACUC) (protocol number: 400095, date of approval: 2 December 2020). The animal use procedures comply with University guidelines, State and Federal regulations, and the standards of the “Guide for the Care and Use of Laboratory Animals”.
Informed Consent Statement
Not applicable.
Data Availability Statement
No data were used for the research described in the article.
Acknowledgments
Figure 8 was created using BioRender.com, accessed on 5 November 2022.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Georges, A.; Das, J.M. Traumatic Brain Injury; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Phillips, S.; Woessner, D. Sports-related traumatic brain injury. Prim. Care Clin. Off. Pract. 2015, 42, 243–248. [Google Scholar] [CrossRef]
- Wojcik, B.E.; Stein, C.R.; Bagg, K.; Humphrey, R.J.; Orosco, J. Traumatic brain injury hospitalizations of US army soldiers deployed to Afghanistan and Iraq. Am. J. Prev. Med. 2010, 38, S108–S116. [Google Scholar] [CrossRef]
- Miller, G.F.; DePadilla, L.; Xu, L. Costs of nonfatal traumatic brain injury in the United States, 2016. Med. Care 2021, 59, 451–455. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S. Traumatic brain injury: Oxidative stress and novel anti-oxidants such as mitoquinone and edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- Eastman, C.L.; D’Ambrosio, R.; Ganesh, T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 2020, 172, 107907. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef]
- A Ma, Z. The role of peroxidation of mitochondrial membrane phospholipids in pancreatic β-cell failure. Curr. Diabetes Rev. 2012, 8, 69–75. [Google Scholar] [CrossRef]
- Cheng, W.-Y.; Tong, H.; Miller, E.W.; Chang, C.J.; Remington, J.; Zucker, R.M.; Bromberg, P.A.; Samet, J.M.; Hofer, T.P. An integrated imaging approach to the study of oxidative stress generation by mitochondrial dysfunction in living cells. Environ. Health Perspect. 2010, 118, 902–908. [Google Scholar] [CrossRef]
- Suematsu, N.; Hosoda, M.; Fujimori, K. Protective effects of quercetin against hydrogen peroxide-induced apoptosis in human neuronal SH-SY5Y cells. Neurosci. Lett. 2011, 504, 223–227. [Google Scholar] [CrossRef]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef]
- Park, J.-H.; Choi, J.W.; Ju, E.J.; Pae, A.N.; Park, K.D. Antioxidant and anti-inflammatory activities of a natural compound, shizukahenriol, through Nrf2 activation. Molecules 2015, 20, 15989–16003. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Shah, Z.A. Cofilin mediates LPS-induced microglial cell activation and associated neurotoxicity through activation of NF-κB and JAK–STAT pathway. Mol. Neurobiol. 2018, 55, 1676–1691. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018, 25, 180–189. [Google Scholar] [CrossRef]
- Simpson, D.S.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Bin Sayeed, M.S.; Shah, Z.A. Cofilin as a promising therapeutic target for ischemic and hemorrhagic stroke. Transl. Stroke Res. 2016, 7, 33–41. [Google Scholar] [CrossRef]
- Posadas, I.; Pérez-Martínez, F.C.; Guerra, J.; Sánchez-Verdú, P.; Ceña, V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J. Neurochem. 2012, 120, 515–527. [Google Scholar] [CrossRef]
- Bamburg, J.R.; Bernstein, B.W. Actin dynamics and cofilin-actin rods in Alzheimer disease. Cytoskeleton 2016, 73, 477–497. [Google Scholar] [CrossRef]
- Won, S.J.; Minnella, A.M.; Wu, L.; Eun, C.H.; Rome, E.; Herson, P.S.; Shaw, A.E.; Bamburg, J.R.; Swanson, R.A. Cofilin-actin rod formation in neuronal processes after brain ischemia. PLoS ONE 2018, 13, e0198709. [Google Scholar] [CrossRef]
- Sayeed, M.S.B.; Alhadidi, Q.; Shah, Z.A. Cofilin signaling in hemin-induced microglial activation and inflammation. J. Neuroimmunol. 2017, 313, 46–55. [Google Scholar] [CrossRef]
- Madineni, A.; Alhadidi, Q.; Shah, Z.A. Cofilin inhibition restores neuronal cell death in oxygen–glucose deprivation model of ischemia. Mol. Neurobiol. 2016, 53, 867–878. [Google Scholar] [CrossRef]
- Alaqel, S.I.; Dlamini, S.; Almarghalani, D.A.; Shettigar, A.; Alhadidi, Q.; Kodithuwakku, S.H.; Stary, C.; Tillekeratne, L.V.; Shah, Z.A. Synthesis and Development of a Novel First-in-Class Cofilin Inhibitor for Neuroinflammation in Hemorrhagic Brain Injury. ACS Chem. Neurosci. 2022, 13, 1014–1029. [Google Scholar] [CrossRef]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef]
- Lee, H.-S.; Kim, E.-N.; Jeong, G.-S. Lupenone protects neuroblastoma SH-SY5y cells against methamphetamine-induced apoptotic cell death via PI3K/Akt/mTOR signaling pathway. Int. J. Mol. Sci. 2020, 21, 1617. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Cruz-Haces, M.; Tang, J.; Acosta, G.; Fernandez, J.; Shi, R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 20. [Google Scholar] [CrossRef]
- Shao, F.; Wang, X.; Wu, H.; Wu, Q.; Zhang, J. Microglia and Neuroinflammation: Crucial Pathological Mechanisms in Traumatic Brain Injury-Induced Neurodegeneration. Front. Aging Neurosci. 2022, 14, 825086. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of traumatic brain injury: Time for a paradigm shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.; Yenari, M.A. Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci. Ther. 2015, 21, 309–319. [Google Scholar] [CrossRef]
- Lapeña-Luzón, T.; Rodríguez, L.R.; Beltran-Beltran, V.; Benetó, N.; Pallardó, F.V.; Gonzalez-Cabo, P. Cofilin and Neurodegeneration: New Functions for an Old but Gold Protein. Brain Sci. 2021, 11, 954. [Google Scholar] [CrossRef]
- Kim, J.-S.; Huang, T.Y.; Bokoch, G.M. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell 2009, 20, 2650–2660. [Google Scholar] [CrossRef]
- Huang, T.Y.; Minamide, L.S.; Bamburg, J.R.; Bokoch, G.M. Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev. Cell 2008, 15, 691–703. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Reichert, F.; Rotshenker, S. Galectin-3 (MAC-2) controls microglia phenotype whether amoeboid and phagocytic or branched and non-phagocytic by regulating the cytoskeleton. Front. Cell. Neurosci. 2019, 13, 90. [Google Scholar] [CrossRef]
- Hoffmann, L.; Waclawczyk, M.S.; Tang, S.; Hanschmann, E.-M.; Gellert, M.; Rust, M.B.; Culmsee, C. Cofilin1 oxidation links oxidative distress to mitochondrial demise and neuronal cell death. Cell Death Dis. 2021, 12, 953. [Google Scholar] [CrossRef]
- Ding, X.; Wang, D.; Li, L.; Ma, H. Dehydroepiandrosterone ameliorates H2O2-induced Leydig cells oxidation damage and apoptosis through inhibition of ROS production and activation of PI3K/Akt pathways. Int. J. Biochem. Cell Biol. 2016, 70, 126–139. [Google Scholar] [CrossRef]
- Klamt, F.; Zdanov, S.; Levine, R.L.; Pariser, A.; Zhang, Y.; Zhang, B.; Yu, L.-R.; Veenstra, T.D.; Shacter, E. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol. 2009, 11, 1241–1246. [Google Scholar] [CrossRef]
- Woo, J.; Zhao, X.; Khan, H.; Penn, C.; Wang, X.; Joly-Amado, A.; Weeber, E.; Morgan, D.; Kang, D. Slingshot-Cofilin activation mediates mitochondrial and synaptic dysfunction via Aβ ligation to β1-integrin conformers. Cell Death Differ. 2015, 22, 921–934. [Google Scholar] [CrossRef]
- Liu, C.-M.; Ma, J.-Q.; Sun, Y.-Z. Puerarin protects rat kidney from lead-induced apoptosis by modulating the PI3K/Akt/eNOS pathway. Toxicol. Appl. Pharmacol. 2012, 258, 330–342. [Google Scholar] [CrossRef]
- Heo, S.R.; Han, A.M.; Kwon, Y.K.; Joung, I. p62 protects SH-SY5Y neuroblastoma cells against H2O2-induced injury through the PDK1/Akt pathway. Neurosci. Lett. 2009, 450, 45–50. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Huang, B.; Liu, J.; Fu, S.; Zhang, Y.; Li, Y.; He, D.; Ran, X.; Yan, X.; Du, J.; Meng, T. α-Cyperone attenuates H2O2-induced oxidative stress and apoptosis in SH-SY5Y cells via activation of Nrf2. Front. Pharmacol. 2020, 11, 281. [Google Scholar] [CrossRef]
- Liu, J.; Wu, P.; Xu, Z.; Zhang, J.; Liu, J.; Yang, Z. Ginkgolide B inhibits hydrogen peroxide-induced apoptosis and attenuates cytotoxicity via activating the PI3K/Akt/mTOR signaling pathway in H9c2 cells. Mol. Med. Rep. 2020, 22, 310–316. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Chandran, R.; Kim, T.; Mehta, S.L.; Udho, E.; Chanana, V.; Cengiz, P.; Kim, H.; Kim, C.; Vemuganti, R. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1818–1827. [Google Scholar] [CrossRef]
- Wang, J.; Ma, M.W.; Dhandapani, K.M.; Brann, D.W. Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic. Biol. Med. 2017, 113, 119–131. [Google Scholar] [CrossRef]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Nash, K.M.; Alaqel, S.; Sayeed, M.S.B.; Shah, Z.A. Cofilin knockdown attenuates hemorrhagic brain injury-induced oxidative stress and microglial activation in mice. Neuroscience 2018, 383, 33–45. [Google Scholar] [CrossRef]
Figure 1.
The expression levels of cofilin, p-cofilin, and SSH-1 in response to H2O2 treatment in microglial cells. HMC3 microglial cells were treated with 100 and 200 μM H2O2 for 24 h, and then, the protein expression level was evaluated by WB. (A,C) While there was no change in cofilin expression in the 100 μM H2O2 treated group, it was significantly upregulated in 200 μM concentration of H2O2; hence, 200 μM was used for the following experiments. (B) Immunoblots of cofilin, p-cofilin and SSH-1 were analyzed by WB. The expression levels of cofilin (D), cofilin/p-cofilin ratio (E), and SSH1 (G) but not p-cofilin (F) were significantly increased in the H2O2 group and significantly reduced when treated with CI. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 relative to control group. # p < 0.05, ## p < 0.01 relative to H2O2 group. (See Figure S1 for original Western blot images).
Figure 1.
The expression levels of cofilin, p-cofilin, and SSH-1 in response to H2O2 treatment in microglial cells. HMC3 microglial cells were treated with 100 and 200 μM H2O2 for 24 h, and then, the protein expression level was evaluated by WB. (A,C) While there was no change in cofilin expression in the 100 μM H2O2 treated group, it was significantly upregulated in 200 μM concentration of H2O2; hence, 200 μM was used for the following experiments. (B) Immunoblots of cofilin, p-cofilin and SSH-1 were analyzed by WB. The expression levels of cofilin (D), cofilin/p-cofilin ratio (E), and SSH1 (G) but not p-cofilin (F) were significantly increased in the H2O2 group and significantly reduced when treated with CI. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 relative to control group. # p < 0.05, ## p < 0.01 relative to H2O2 group. (See Figure S1 for original Western blot images).

Figure 2.
The effect of CI treatment on the H2O2-induced microglial cell activation. HMC3 microglial cells were treated with 200 μM H2O2 with or without 10 μM CI for 24 h, and then, the protein expression level was evaluated by WB. (A) WB of different proteins in the microglial cell lysate 24 h following H2O2 treatment. Analysis of WB shows that relative to H2O2 stimulated control, treatment with CI reduced the H2O2-induced HMGB1 (B) and TNFα (C) expression significantly, while there was no significant change in the expression of NF-κB (D). Data are expressed as mean ± SEM of three independent experiments, where p < 0.05 was considered significant. * p < 0.05, *** p < 0.001 relative to control group. # p < 0.05, ## p < 0.01 relative to H2O2 group. (See Figure S2 for original Western blot images).
Figure 2.
The effect of CI treatment on the H2O2-induced microglial cell activation. HMC3 microglial cells were treated with 200 μM H2O2 with or without 10 μM CI for 24 h, and then, the protein expression level was evaluated by WB. (A) WB of different proteins in the microglial cell lysate 24 h following H2O2 treatment. Analysis of WB shows that relative to H2O2 stimulated control, treatment with CI reduced the H2O2-induced HMGB1 (B) and TNFα (C) expression significantly, while there was no significant change in the expression of NF-κB (D). Data are expressed as mean ± SEM of three independent experiments, where p < 0.05 was considered significant. * p < 0.05, *** p < 0.001 relative to control group. # p < 0.05, ## p < 0.01 relative to H2O2 group. (See Figure S2 for original Western blot images).

Figure 3.
The effect of H2O2 stimulation and CI treatment on SH-SY5Y cell viability and cofilin signaling. (A) SH-SY5Y cells were treated with increasing concentrations of H2O2 for 24 h followed by the CCK8 viability assay to measure the neurotoxic potential of H2O2. SH-SY5Y cell viability was significantly reduced with concentrations of 100, 150, and 200 μM of H2O2. (B) SH-SY5Y cells were treated with 200 μm H2O2 alone or with an increasing concentration of CI for 24 h, and cell viability was measured using the CCK8 assay. Cell viability was significantly reduced in the H2O2-treated group; however, treatment with 5, 7, and 10 μM CI significantly protected the cells from the H2O2-induced neurotoxic effect. (C,D) The expression level of cofilin and p-cofilin was tested by WB. H2O2 significantly reduced the p-cofilin expression and increased cofilin dephosphorylation, which was rescued by CI treatment. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, **** p < 0.0001 relative to control group. # p < 0.05, ### p < 0.001 relative to H2O2 group. (See Figure S3 for original Western blot images).
Figure 3.
The effect of H2O2 stimulation and CI treatment on SH-SY5Y cell viability and cofilin signaling. (A) SH-SY5Y cells were treated with increasing concentrations of H2O2 for 24 h followed by the CCK8 viability assay to measure the neurotoxic potential of H2O2. SH-SY5Y cell viability was significantly reduced with concentrations of 100, 150, and 200 μM of H2O2. (B) SH-SY5Y cells were treated with 200 μm H2O2 alone or with an increasing concentration of CI for 24 h, and cell viability was measured using the CCK8 assay. Cell viability was significantly reduced in the H2O2-treated group; however, treatment with 5, 7, and 10 μM CI significantly protected the cells from the H2O2-induced neurotoxic effect. (C,D) The expression level of cofilin and p-cofilin was tested by WB. H2O2 significantly reduced the p-cofilin expression and increased cofilin dephosphorylation, which was rescued by CI treatment. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, **** p < 0.0001 relative to control group. # p < 0.05, ### p < 0.001 relative to H2O2 group. (See Figure S3 for original Western blot images).

Figure 4.
The effect of CI treatment on H2O2-induced ROS production in SH-SY5Y cells. SH-SY5Y cells were treated with 200 μm H2O2 with or without 10 μM concentration of CI for 24 h, and ROS level was determined by fluorescence microscopy after DCF-DA staining. (A) Representative fluorescence images of DCF-DA staining SH-SY5Y cells. (B) Quantifying green fluorescence intensity showed a significant reduction of ROS levels in the CI-treated group compared to the H2O2-stimulated group. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. ** p < 0.01 relative to the control group. # p < 0.05 relative to H2O2 group.
Figure 4.
The effect of CI treatment on H2O2-induced ROS production in SH-SY5Y cells. SH-SY5Y cells were treated with 200 μm H2O2 with or without 10 μM concentration of CI for 24 h, and ROS level was determined by fluorescence microscopy after DCF-DA staining. (A) Representative fluorescence images of DCF-DA staining SH-SY5Y cells. (B) Quantifying green fluorescence intensity showed a significant reduction of ROS levels in the CI-treated group compared to the H2O2-stimulated group. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. ** p < 0.01 relative to the control group. # p < 0.05 relative to H2O2 group.

Figure 5.
The effect of CI on BAX, Bcl-2, Caspases, and p-Akt expression in H2O2-stimulated SH-SY5Y cells. SH-SY5Y cells were treated with 200 μM H2O2 with or without 10 μM CI for 5 h; then, the cell culture medium was replaced with a fresh one with or without 10 μM CI for another 24 h. (A) WB immunoblots of BAX, Bcl-2, and p-Akt proteins in the Sh-SY5Y cell lysate. (B,D) Analysis of WB shows that the expression of BAX and cleaved caspase-3 were significantly upregulated in the H2O2 group compared to the control and were downregulated in the CI-treated group. (E) The expression of total caspase-3 showed no significant change in all groups. (C,F) Treatment with CI increases both Bcl-2 and p-Akt protein expression significantly compared to the H2O2-stimulated control. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 relative to control group. ## p < 0.01, ### p < 0.001, #### p < 0.0001 relative to H2O2 group. (See Figure S4 for original Western blot images).
Figure 5.
The effect of CI on BAX, Bcl-2, Caspases, and p-Akt expression in H2O2-stimulated SH-SY5Y cells. SH-SY5Y cells were treated with 200 μM H2O2 with or without 10 μM CI for 5 h; then, the cell culture medium was replaced with a fresh one with or without 10 μM CI for another 24 h. (A) WB immunoblots of BAX, Bcl-2, and p-Akt proteins in the Sh-SY5Y cell lysate. (B,D) Analysis of WB shows that the expression of BAX and cleaved caspase-3 were significantly upregulated in the H2O2 group compared to the control and were downregulated in the CI-treated group. (E) The expression of total caspase-3 showed no significant change in all groups. (C,F) Treatment with CI increases both Bcl-2 and p-Akt protein expression significantly compared to the H2O2-stimulated control. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 relative to control group. ## p < 0.01, ### p < 0.001, #### p < 0.0001 relative to H2O2 group. (See Figure S4 for original Western blot images).

Figure 6.
The effect of CI on the H2O2-induced oxidative stress in SH-SY5Y cells. SH-SY5Y cells were treated with 200 μM H2O2 with or without 10 μM CI for 5 h, then after the cell culture medium was replaced with a fresh one with or without 10 μM CI for another 24 h. (A) WB of different oxidative stress and antioxidant-related proteins in the Sh-SY5Y cell lysate was assessed. H2O2 significantly reduces the expression of Nrf2 (B) and SOD2 (C) and increases the expression of HO-1 (D) and NQO1 (E) compared to the unstimulated control group. However, in CI treatment groups, the levels of Nrf2, SOD2, HO-1, and NQO1 were significantly increased as compared to the H2O2 group. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, *** p < 0.001, **** p < 0.0001 relative to control group. # p < 0.05, ### p < 0.001, #### p < 0.0001 relative to H2O2 group. (See Figure S5 for original Western blot images).
Figure 6.
The effect of CI on the H2O2-induced oxidative stress in SH-SY5Y cells. SH-SY5Y cells were treated with 200 μM H2O2 with or without 10 μM CI for 5 h, then after the cell culture medium was replaced with a fresh one with or without 10 μM CI for another 24 h. (A) WB of different oxidative stress and antioxidant-related proteins in the Sh-SY5Y cell lysate was assessed. H2O2 significantly reduces the expression of Nrf2 (B) and SOD2 (C) and increases the expression of HO-1 (D) and NQO1 (E) compared to the unstimulated control group. However, in CI treatment groups, the levels of Nrf2, SOD2, HO-1, and NQO1 were significantly increased as compared to the H2O2 group. Data are expressed as mean ± SEM of three independent experiments where p < 0.05 was considered significant. * p < 0.05, *** p < 0.001, **** p < 0.0001 relative to control group. # p < 0.05, ### p < 0.001, #### p < 0.0001 relative to H2O2 group. (See Figure S5 for original Western blot images).

Figure 7.
The effect of CI treatment on oxidative stress damage after TBI. CI increased the expression of Nrf2 (A) and SOD2 (B) in the perilesional cortex after TBI. (C) mRNA expression of oxidative stress markers determined by RT-qPCR showed a significant reduction in the gene expression of iNOS, NOX2, and NOX4, while there was no change in the levels of nNOS and eNOS. (D) Representative immunofluorescence images and quantitative analysis showed that CI significantly reduced the fluorescence intensity of iNOS (E) and 3-NT (F) in the perilesional cortex after TBI. (n = 6 per group) Data are expressed as mean ± SEM where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, **** p < 0.0001 relative to control group. # p < 0.05, ## p < 0.01, ### p < 0.001 relative to H2O2 group. (See Figure S6 for original Western blot images).
Figure 7.
The effect of CI treatment on oxidative stress damage after TBI. CI increased the expression of Nrf2 (A) and SOD2 (B) in the perilesional cortex after TBI. (C) mRNA expression of oxidative stress markers determined by RT-qPCR showed a significant reduction in the gene expression of iNOS, NOX2, and NOX4, while there was no change in the levels of nNOS and eNOS. (D) Representative immunofluorescence images and quantitative analysis showed that CI significantly reduced the fluorescence intensity of iNOS (E) and 3-NT (F) in the perilesional cortex after TBI. (n = 6 per group) Data are expressed as mean ± SEM where p < 0.05 was considered significant. * p < 0.05, ** p < 0.01, **** p < 0.0001 relative to control group. # p < 0.05, ## p < 0.01, ### p < 0.001 relative to H2O2 group. (See Figure S6 for original Western blot images).

Figure 8.
The proposed mechanism of cofilin inhibitor in TBI. Cofilin signaling is activated during oxidative stress conditions in microglial and neuronal cells. In microglia, ROS activates the NF-kB pathway, which regulates the expression of different cytotoxic mediators such as TNFα, HMGB1, and iNOS, an effect that is augmented by cofilin inhibitor treatment. In neurons, cofilin inhibitor activates the Nrf2 pathway, which regulates the expression of antioxidant enzymes and increases the Akt phosphorylation, leading to reduced expression of mitochondrial apoptotic markers and reduced neurotoxicity.
Figure 8.
The proposed mechanism of cofilin inhibitor in TBI. Cofilin signaling is activated during oxidative stress conditions in microglial and neuronal cells. In microglia, ROS activates the NF-kB pathway, which regulates the expression of different cytotoxic mediators such as TNFα, HMGB1, and iNOS, an effect that is augmented by cofilin inhibitor treatment. In neurons, cofilin inhibitor activates the Nrf2 pathway, which regulates the expression of antioxidant enzymes and increases the Akt phosphorylation, leading to reduced expression of mitochondrial apoptotic markers and reduced neurotoxicity.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Quantitative RT-PCR primers sequences.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | CTGGTGCGAAGTGTGCAAG | TGAGATTAGCGTGGCCCGAA |
| iNOS | CCAAATCCAACGTTCTCCGT | CCAAATCCAACGTTCTCCGT |
| eNOS | CAGATGCCCAACCCAAACCT | ACAGAGAGGTGTCTGGGACT |
| nNOS | CCTTCACAGGGGATGGAACC | AGATCGACAGCTTTGGTGGG |
| NOX2 | CCCTCCCTGTCTAGGTAATGCATGG | GCATTTGCCTTCGGTGATGTGCT |
| NOX4 | CACCAAATGTTGGGCGATTGT | GATGAGGCTGCAGTTGAGGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bahader, G.A.; James, A.W.; Almarghalani, D.A.; Shah, Z.A. Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation. Biology 2023, 12, 630. https://doi.org/10.3390/biology12040630
AMA Style
Bahader GA, James AW, Almarghalani DA, Shah ZA. Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation. Biology. 2023; 12(4):630. https://doi.org/10.3390/biology12040630
Chicago/Turabian StyleBahader, Ghaith A., Antonisamy William James, Daniyah A. Almarghalani, and Zahoor A. Shah. 2023. "Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation" Biology 12, no. 4: 630. https://doi.org/10.3390/biology12040630
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.