
Gene Flow and Diversification in Himalopsyche martynovi Species Complex (Trichoptera: Rhyacophilidae) in the Hengduan Mountains

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
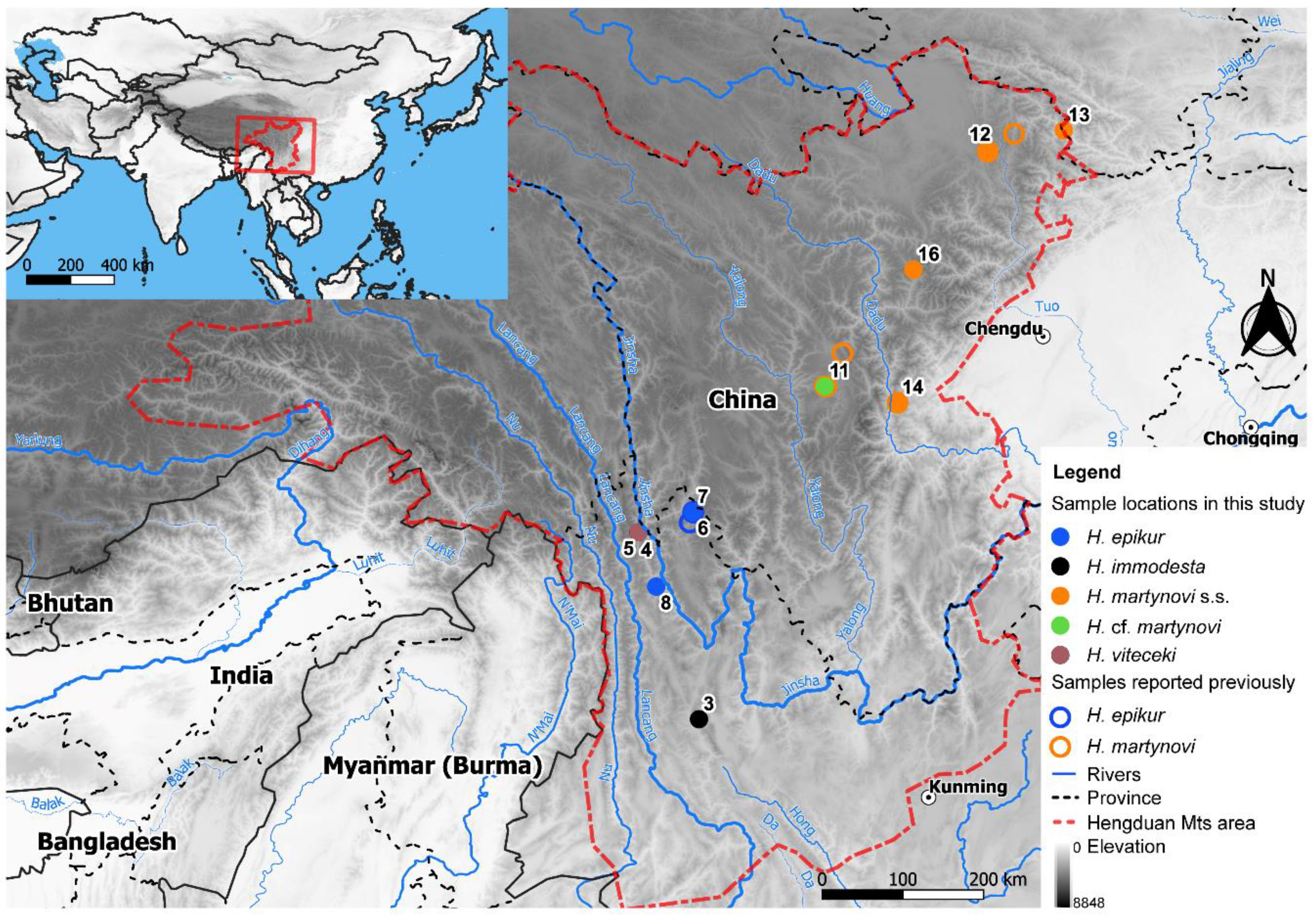
2.1. Taxon Sampling
2.2. DNA Extraction, Library Preparation, Enrichment and Sequencing
2.3. Probe Design
2.4. Raw Data Processing and Assembly
2.5. Alignment and Trimming
2.6. Phylogenetic Analyses
2.7. Gene Flow Analysis Using ExDFOIL
3. Results
3.1. AHE Target Characteristics
3.2. Read and Assembly Characteristics
3.3. Sequence Characteristics after Filtering
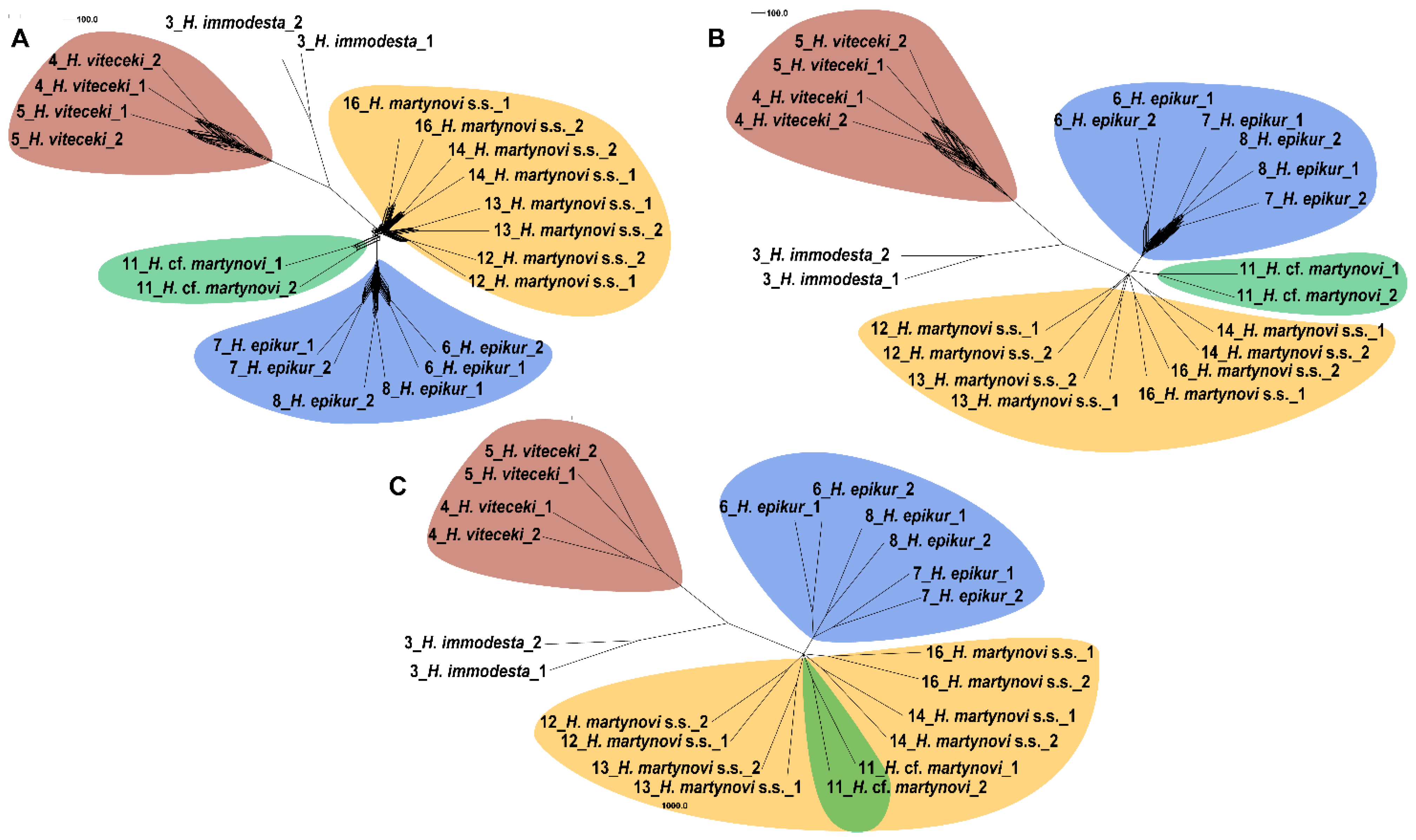
3.4. Phylogeny
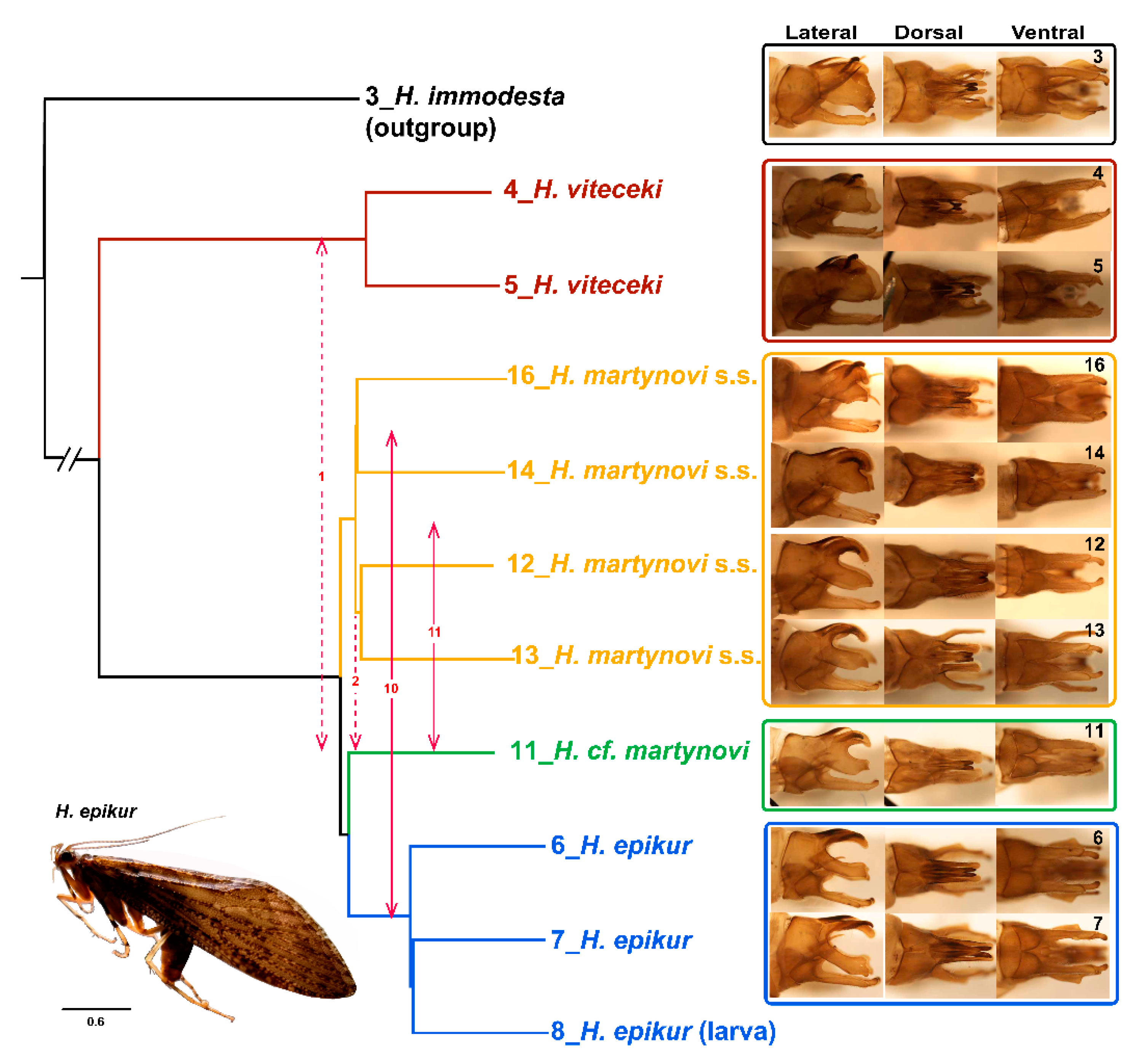
3.5. Introgression
3.6. Morphological Sorting
4. Discussion
4.1. Genomic and Morphological Evidence and Their Taxonomic Implications
4.2. Gene Flow and Speciation of H. martynovi Complex
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meier, R.; Dikow, T. Significance of specimen databases from taxonomic revisions for estimating and mapping the global species diversity of invertebrates and repatriating reliable specimen data. Conserv. Biol. 2004, 18, 478–488. [Google Scholar] [CrossRef]
- Kier, G.; Mutke, J.; Dinerstein, E.; Ricketts, T.H.; Küper, W.; Kreft, H.; Barthlott, W. Global patterns of plant diversity and floristic knowledge. J. Biogeogr. 2005, 32, 1107–1116. [Google Scholar] [CrossRef]
- Bastida, F.; Eldridge, D.J.; Abades, S.; Alfaro, F.D.; Gallardo, A.; García-Velázquez, L.; García, C.; Hart, S.C.; Pérez, C.A.; Santos, F. Climatic vulnerabilities and ecological preferences of soil invertebrates across biomes. Mol. Ecol. 2020, 29, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; Da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Marchese, C. Biodiversity hotspots: A shortcut for a more complicated concept. Glob. Ecol. Conserv. 2015, 3, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Boufford, D.E. Biodiversity hotspot: China’s Hengduan Mountains. Arnoldia 2014, 72, 24–35. [Google Scholar]
- Li, J.; Milne, R.I.; Ru, D.; Miao, J.; Tao, W.; Zhang, L.; Xu, J.; Liu, J.; Mao, K. Allopatric divergence and hybridization within Cupressus chengiana (Cupressaceae), a threatened conifer in the northern Hengduan Mountains of western China. Mol. Ecol. 2020, 29, 1250–1266. [Google Scholar] [CrossRef]
- Liu, C.; Fischer, G.; Garcia, F.H.; Yamane, S.; Liu, Q.; Peng, Y.Q.; Economo, E.P.; Guénard, B.; Pierce, N.E. Ants of the Hengduan Mountains: A new altitudinal survey and updated checklist for Yunnan Province highlight an understudied insect biodiversity hotspot. ZooKeys 2020, 978, 1. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.J.; Excoffier, L. Gene flow and species delimitation. Trends Ecol. Evol. 2009, 24, 386–393. [Google Scholar] [CrossRef]
- Muellner-Riehl, A.N.; Schnitzler, J.; Kissling, W.D.; Mosbrugger, V.; Rijsdijk, K.F.; Seijmonsbergen, A.C.; Versteegh, H.; Favre, A. Origins of global mountain plant biodiversity: Testing the ‘mountain-geobiodiversity hypothesis’. J. Biogeogr. 2019, 46, 2826–2838. [Google Scholar] [CrossRef] [Green Version]
- Mosbrugger, V.; Favre, A.; Muellner-Riehl, A.N.; Päckert, M.; Mulch, A. Cenozoic evolution of geo-biodiversity in the Tibeto-Himalayan region. Mt. Clim. Biodivers. 2018, 429, 448. [Google Scholar]
- Ding, W.-N.; Ree, R.H.; Spicer, R.A.; Xing, Y.-W. Ancient orogenic and monsoon-driven assembly of the world’s richest temperate alpine flora. Science 2020, 369, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.K.; House, M.; Royden, L.; Whipple, K.; Burchfiel, B.; Zhang, X.; Tang, W. Late Cenozoic uplift of southeastern Tibet. Geology 2005, 33, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.-B.; Randle, C.P.; Lu, L.; Wang, H.; Yang, J.-B.; de Pamphilis, C.W.; Corlett, R.T.; Li, D.-Z. The hemiparasitic plant Phtheirospermum (Orobanchaceae) is polyphyletic and contains cryptic species in the Hengduan Mountains of southwest China. Front. Plant. Sci. 2018, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Lu, L.; Cheng, J.; Xia, L.; Chang, Y.; Wen, Z.; Lv, X.; Du, Y.; Liu, Q.; Yang, Q. An endemic rat species complex is evidence of moderate environmental changes in the terrestrial biodiversity centre of China through the late Quaternary. Sci. Rep. 2017, 7, 46127. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhao, Q.; Xu, J.; Qin, J.; Yang, Z.L. Drainage isolation and climate change-driven population expansion shape the genetic structures of Tuber indicum complex in the Hengduan Mountains region. Sci. Rep. 2016, 6, 21811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjalmarsson, A.E.; Graf, W.; Jähnig, S.C.; Vitecek, S.; Pauls, S.U. Molecular association and morphological characterisation of Himalopsyche larval types (Trichoptera, Rhyacophilidae). ZooKeys 2018, 773, 79. [Google Scholar] [CrossRef] [Green Version]
- Hjalmarsson, A.E.; Graf, W.; Vitecek, S.; Jähnig, S.C.; Cai, Q.; Sharma, S.; Tong, X.; Li, F.; Shah, D.N.; Shah, R.D.T. Molecular phylogeny of Himalopsyche (Trichoptera, Rhyacophilidae). Syst. Entomol. 2019, 44, 973–984. [Google Scholar] [CrossRef]
- Shapiro, A.M.; Porter, A.H. The lock-and-key hypothesis: Evolutionary and biosystematic interpretation of insect genitalia. Annu. Rev. Entomol. 1989, 34, 231–245. [Google Scholar] [CrossRef]
- Banks, N. Report on Certain Groups of Neuropteroid Insects from Szechwan, China. Proc. U. S. Natl. Mus. 1940. Available online: https://repository.si.edu/bitstream/handle/10088/16322/1/USNMP-88_3079_1940.pdf (accessed on 17 August 2021). [CrossRef]
- Ross, H.H. Evolution and Classification of the Mountain Caddisflies; The University of Illinois Press: Urbana, IL, USA, 1956; p. 213. [Google Scholar]
- Schmid, F. Le genre Himalopsyche Banks (Trichoptera: Rhyacophilidae). Ann. Ent. Soc. Que. 1966, 11, 123–176. [Google Scholar]
- Malicky, H. Neue Trichopteren aus Europa und Asien. Braueria 2011, 38, 23–43. [Google Scholar]
- Hjalmarsson, A.E. Delimitation and description of three new species of Himalopsyche (Trichoptera: Rhyacophilidae) from the Hengduan Mountains, China. Zootaxa 2019, 4638, 419–441. [Google Scholar] [CrossRef] [PubMed]
- Malicky, H.; Pauls, S.U. Cross-breeding of Chaetopteryx morettii and related species, with molecular and eidonomical results (Trichoptera, Limnephilidae). Ann. Limnol. Int. J. Limnol. 2012, 48, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, A.R.; Emme, S.A.; Lemmon, E.M. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst. Biol. 2012, 61, 727–744. [Google Scholar] [CrossRef] [Green Version]
- Truett, G.; Heeger, P.; Mynatt, R.; Truett, A.; Walker, J.; Warman, M. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 2000, 29, 52–54. [Google Scholar] [CrossRef]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Breinholt, J.W.; Earl, C.; Lemmon, A.R.; Lemmon, E.M.; Xiao, L.; Kawahara, A.Y. Resolving relationships among the megadiverse butterflies and moths with a novel pipeline for anchored phylogenomics. Syst. Biol. 2018, 67, 78–93. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Lemmon, A.R.; Lemmon, E.M.; Bond, J.E. Expanding anchored hybrid enrichment to resolve both deep and shallow relationships within the spider tree of life. BMC Evol. Biol. 2016, 16, 212. [Google Scholar] [CrossRef] [Green Version]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Arnow, K. The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genom. 2012, 13, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyron, R.A.; Hendry, C.R.; Hsieh, F.; Lemmon, A.R.; Lemmon, E.M. Integrating phylogenomic and morphological data to assess candidate species-delimitation models in Brown and Red-bellied snakes (Storeria). Zool. J. Linn. Soc. 2016, 177, 937–949. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Misof, B.; Misof, K. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: A more objective means of data exclusion. Syst. Biol. 2009, 58, 21–34. [Google Scholar] [CrossRef]
- Andermann, T.; Fernandes, A.M.; Olsson, U.; Töpel, M.; Pfeil, B.; Oxelman, B.; Aleixo, A.; Faircloth, B.C.; Antonelli, A. Allele phasing greatly improves the phylogenetic utility of ultraconserved elements. Syst. Biol. 2019, 68, 32–46. [Google Scholar] [CrossRef]
- Stamatakis, A. The RAxML v8.2.X Manual. Heidleberg Institute for Theoretical Studies. 2016. Available online: https://cme.h-its.org/exelixis/resource/download/NewManual.pdf (accessed on 10 August 2021).
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Barton, N.H.; Hewitt, G.M. Analysis of hybrid zones. Annu. Rev. Ecol. Syst. 1985, 16, 113–148. [Google Scholar] [CrossRef]
- Twyford, A.; Ennos, R. Next-generation hybridization and introgression. Heredity 2012, 108, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Slarkin, M. Gene flow in natural populations. Annu. Rev. Ecol. Syst. 1985, 16, 393–430. [Google Scholar] [CrossRef]
- Slatkin, M. Gene flow and the geographic structure of natural populations. Science 1987, 236, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Pease, J.B.; Hahn, M.W. Detection and Polarization of Introgression in a Five-Taxon Phylogeny. Syst. Biol. 2015, 64, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.M.; Streicher, J.W.; Fisher-Reid, M.C.; Mendez de la Cruz, F.R.; Martinez-Mendez, N.; Garcia-Vazquez, U.O.; Nieto Montes de Oca, A.; Wiens, J.J. Inferring introgression using RADseq and DFOIL: Power and pitfalls revealed in a case study of spiny lizards (Sceloporus). Mol. Ecol. Resour. 2019, 19, 818–837. [Google Scholar] [CrossRef]
- Haddad, S.; Shin, S.; Lemmon, A.R.; Lemmon, E.M.; Svacha, P.; Farrell, B.; ŚLIPIŃSKI, A.; Windsor, D.; McKenna, D.D. Anchored hybrid enrichment provides new insights into the phylogeny and evolution of longhorned beetles (Cerambycidae). Syst. Entomol. 2018, 43, 68–89. [Google Scholar] [CrossRef]
- Buenaventura, E.; Szpila, K.; Cassel, B.K.; Wiegmann, B.M.; Pape, T. Anchored hybrid enrichment challenges the traditional classification of flesh flies (Diptera: Sarcophagidae). Syst. Entomol. 2020, 45, 281–301. [Google Scholar] [CrossRef] [Green Version]
- Dowdy, N.J.; Keating, S.; Lemmon, A.R.; Lemmon, E.M.; Conner, W.E.; Scott Chialvo, C.H.; Weller, S.J.; Simmons, R.B.; Sisson, M.S.; Zaspel, J.M. A deeper meaning for shallow-level phylogenomic studies: Nested anchored hybrid enrichment offers great promise for resolving the tiger moth tree of life (Lepidoptera: Erebidae: Arctiinae). Syst. Entomol. 2020, 45, 874–893. [Google Scholar] [CrossRef]
- Nosil, P. Speciation with gene flow could be common. Mol. Ecol. 2008, 17, 2103–2106. [Google Scholar] [CrossRef]
- Fitzpatrick, B.; Fordyce, J.; Gavrilets, S. Pattern, process and geographic modes of speciation. J. Evol. Biol. 2009, 22, 2342–2347. [Google Scholar] [CrossRef]
- Fu, P.-C.; Gao, Q.-B.; Zhang, F.-Q.; Xing, R.; Wang, J.-L.; Liu, H.-R.; Chen, S.-L. Gene flow results in high genetic similarity between Sibiraea (Rosaceae) species in the Qinghai–Tibetan Plateau. Front. Plant. Sci. 2016, 7, 1596. [Google Scholar] [CrossRef] [Green Version]
- Mullen, S.P.; Dopman, E.B.; Harrison, R.G. Hybrid zone origins, species boundaries, and the evolution of wing-pattern diversity in a polytypic species complex of North American admiral butterflies (Nymphalidae: Limenitis). Evol. Int. J. Org. Evol. 2008, 62, 1400–1417. [Google Scholar] [CrossRef]
- Kumar, V.; Lammers, F.; Bidon, T.; Pfenninger, M.; Kolter, L.; Nilsson, M.A.; Janke, A. The evolutionary history of bears is characterized by gene flow across species. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Árnason, Ú.; Lammers, F.; Kumar, V.; Nilsson, M.A.; Janke, A. Whole-genome sequencing of the blue whale and other rorquals finds signatures for introgressive gene flow. Sci. Adv. 2018, 4, eaap9873. [Google Scholar] [CrossRef] [Green Version]
- Malinsky, M.; Svardal, H.; Tyers, A.M.; Miska, E.A.; Genner, M.J.; Turner, G.F.; Durbin, R. Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nat. Ecol. Evol. 2018, 2, 1940–1955. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.A.; Zheng, Y.; Kumar, V.; Phillips, M.J.; Janke, A. Speciation generates mosaic genomes in kangaroos. Genome Biol. Evol. 2018, 10, 33–44. [Google Scholar] [CrossRef]
- Patton, A.H.; Margres, M.J.; Epstein, B.; Eastman, J.; Harmon, L.J.; Storfer, A. Hybridizing salamanders experience accelerated diversification. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Liu, J.W.; Xu, J.; Zhao, K.; Ge, Z.W.; Yang, Z.L. Ecological and physical barriers shape genetic structure of the Alpine Porcini (Boletus reticuloceps). Mycorrhiza 2017, 27, 261–272. [Google Scholar] [CrossRef]
- Li, Y.; Ludwig, A.; Peng, Z. Geographical differentiation of the Euchiloglanis fish complex (Teleostei: Siluriformes) in the Hengduan Mountain Region, China: Phylogeographic evidence of altered drainage patterns. Ecol. Evol. 2017, 7, 928–940. [Google Scholar] [CrossRef]
- Deng, B.; Chew, D.; Mark, C.; Liu, S.; Cogné, N.; Jiang, L.; O’Sullivan, G.; Li, Z.; Li, J. Late Cenozoic drainage reorganization of the paleo-Yangtze river constrained by multi-proxy provenance analysis of the Paleo-lake Xigeda. GSA Bull. 2021, 133, 199–211. [Google Scholar] [CrossRef]
- Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–417. [Google Scholar] [CrossRef]
- Fujita, M.K.; Leaché, A.D.; Burbrink, F.T.; McGuire, J.A.; Moritz, C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 2012, 27, 480–488. [Google Scholar] [CrossRef]
- Oláh, J.; Kiss, O. Splitting by adaptive traits in the Rhyacophila obscura species group (Trichoptera, Rhyacophilidae). Opusc. Zool. 2018, 49, 152–161. [Google Scholar] [CrossRef]
- Oláh, J.; Kovács, T.; Ibrahimi, H. Agaphylax, a new limnephilid genus (Trichoptera) from the Balkan: Lineage ranking by adaptive paramere. Opusc. Zool. 2018, 49, 77–89. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, X.-L.; Favre, A.; Lemmon, E.M.; Lemmon, A.R.; Pauls, S.U. Gene Flow and Diversification in Himalopsyche martynovi Species Complex (Trichoptera: Rhyacophilidae) in the Hengduan Mountains. Biology 2021, 10, 816. https://doi.org/10.3390/biology10080816
Deng X-L, Favre A, Lemmon EM, Lemmon AR, Pauls SU. Gene Flow and Diversification in Himalopsyche martynovi Species Complex (Trichoptera: Rhyacophilidae) in the Hengduan Mountains. Biology. 2021; 10(8):816. https://doi.org/10.3390/biology10080816
Chicago/Turabian StyleDeng, Xi-Ling, Adrien Favre, Emily Moriarty Lemmon, Alan R. Lemmon, and Steffen U. Pauls. 2021. "Gene Flow and Diversification in Himalopsyche martynovi Species Complex (Trichoptera: Rhyacophilidae) in the Hengduan Mountains" Biology 10, no. 8: 816. https://doi.org/10.3390/biology10080816