
Microcarrier-Based Culture of Human Pluripotent Stem-Cell-Derived Retinal Pigmented Epithelium

 , , ,
, , ,
Abstract
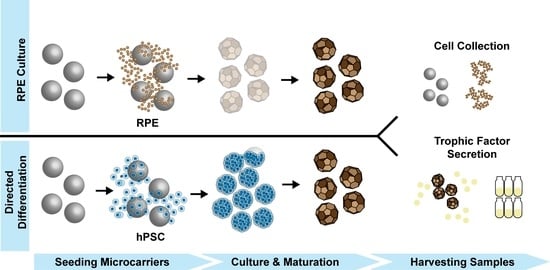
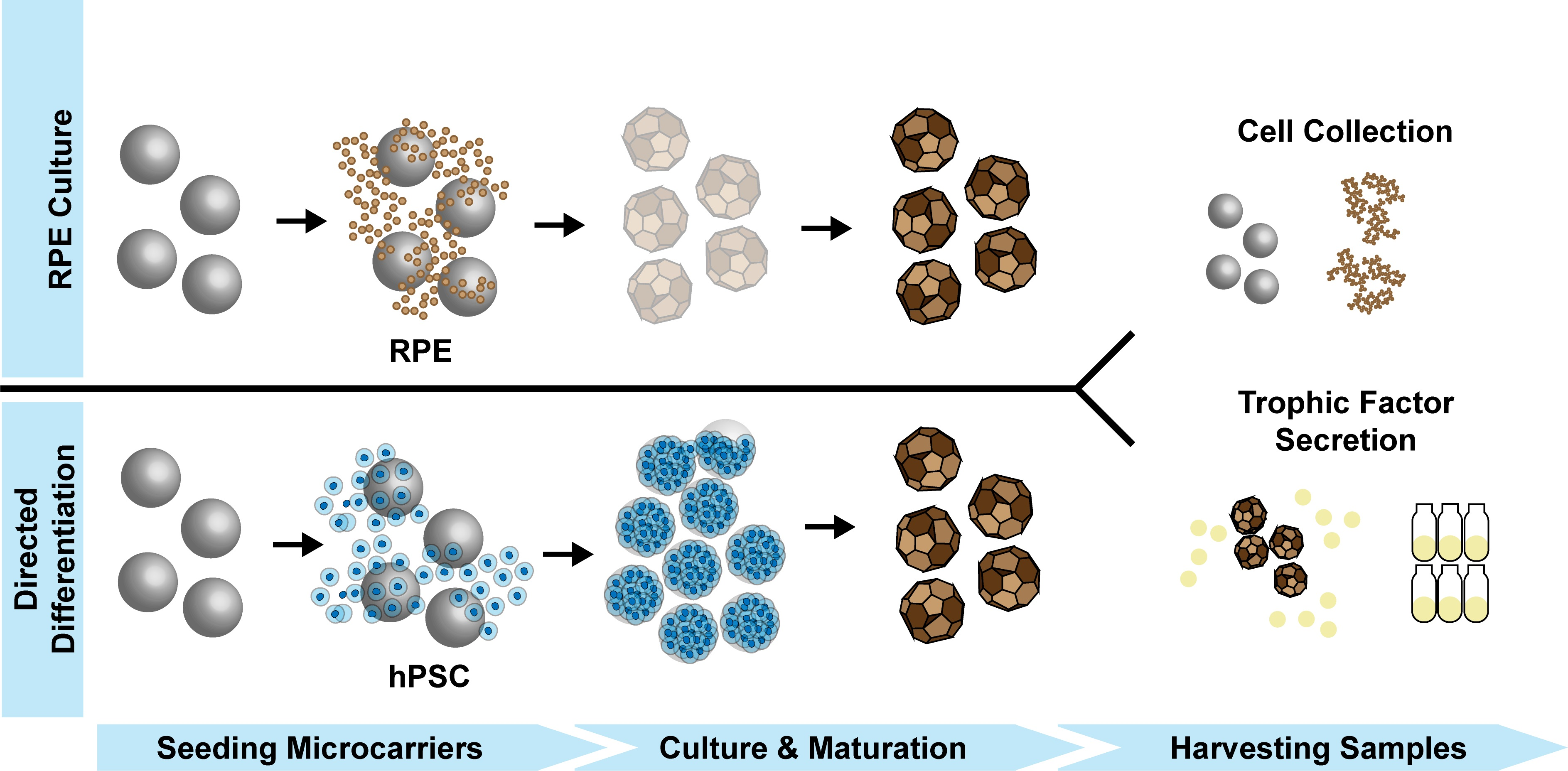
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
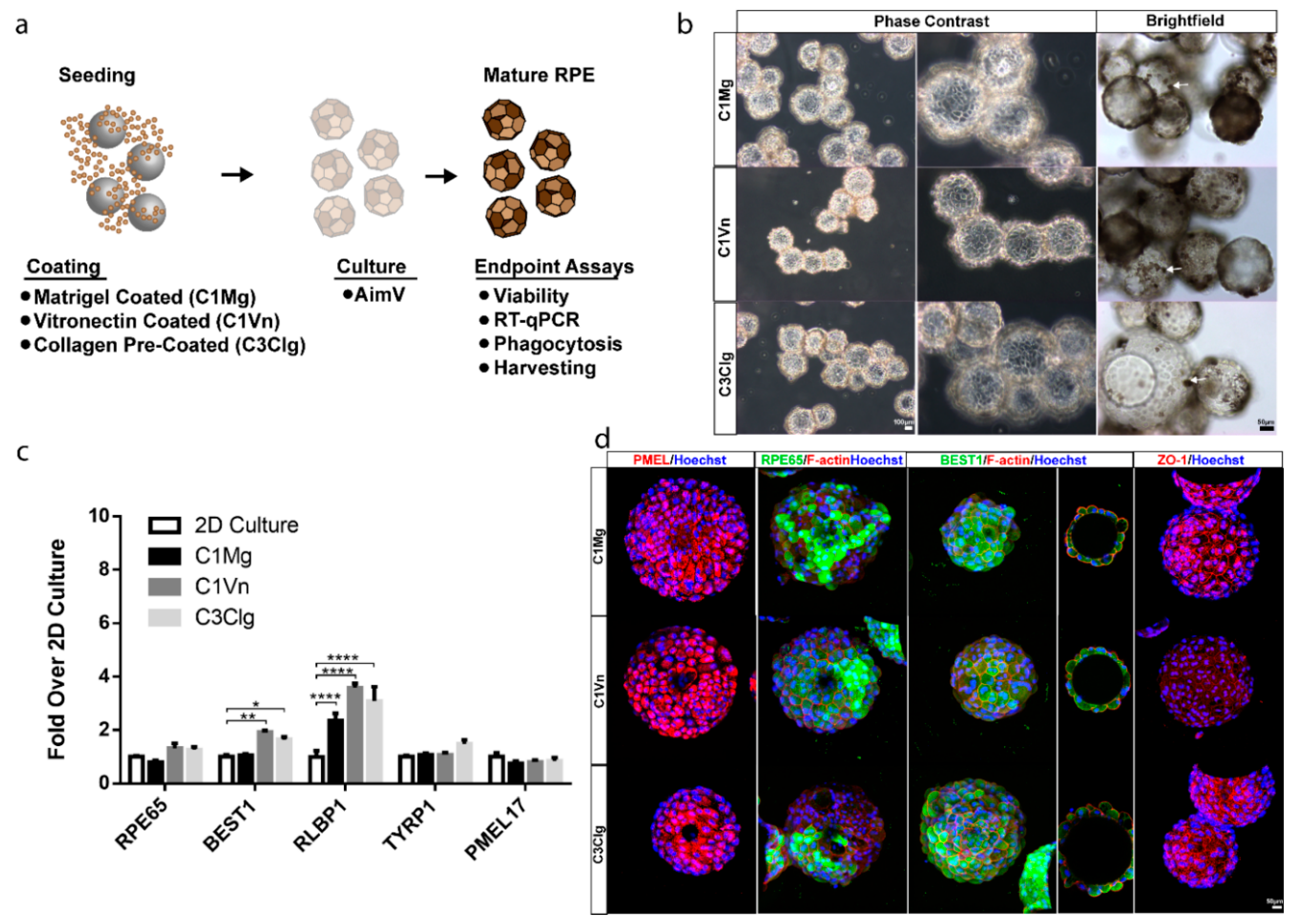
2.1. Microcarrier Preparation
2.2. Microcarrier Coating and Seeding
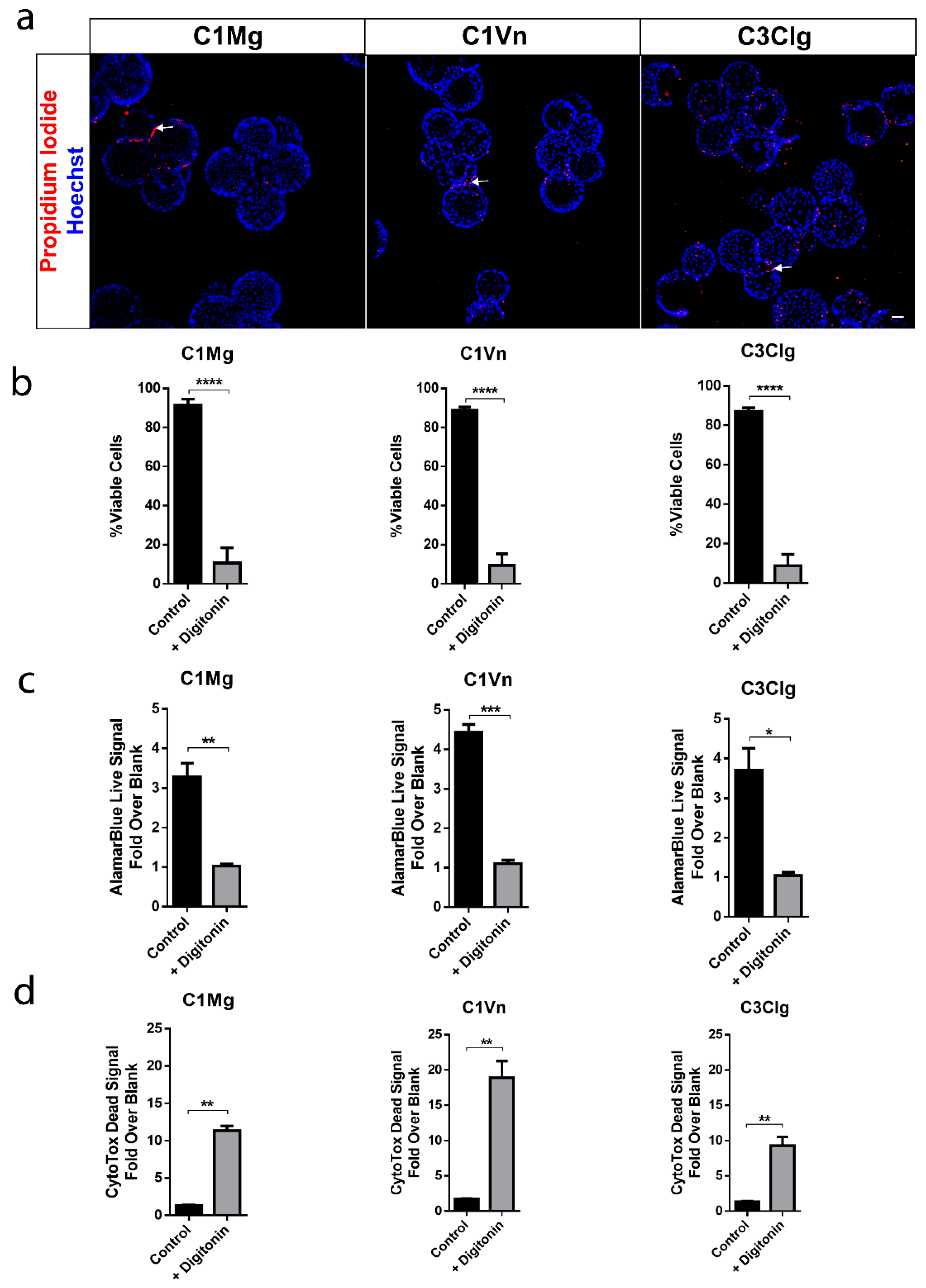
2.3. Cell Viability Assay
2.4. AlamarBlue Metabolic Assay and CytoTox-Fluor Cytotoxicity Assay
2.5. PEDF and VEGF Supernatant ELISA
2.6. mcRPE Photoreceptor Outersegment Phagocytosis
2.7. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.8. Immunocytochemistry
2.9. hPSC Microcarrier Differentiation
2.10. Statistical Analysis and Data Display
3. Results
3.1. hESC-RPE Attach and Mature on Microcarriers Coated with Extracellular Matrix Proteins
3.2. mcRPE areHighly Viable and Metabolically Active
3.3. mcRPE Can Be Harvested from Microcarriers Using Xeno-Free Methods
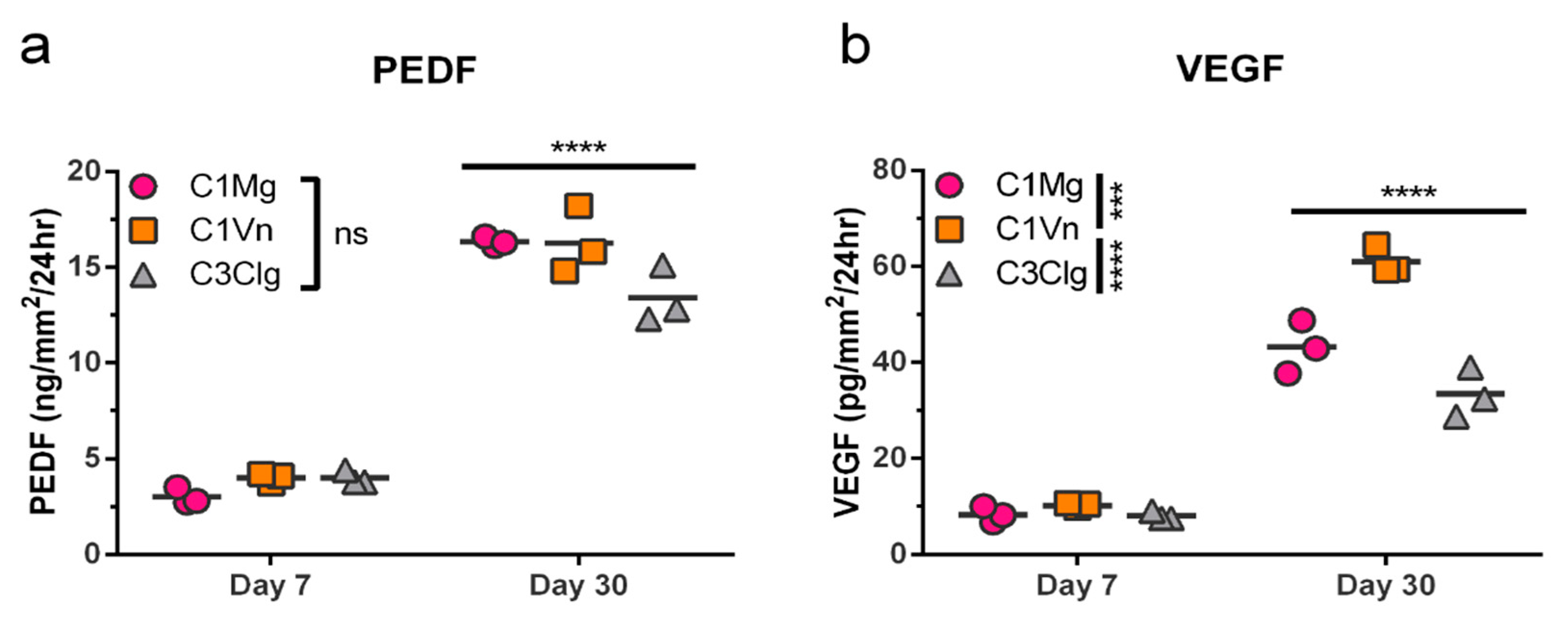
3.4. mcRPE Secretes Increasing Amounts of PEDF and VEGF
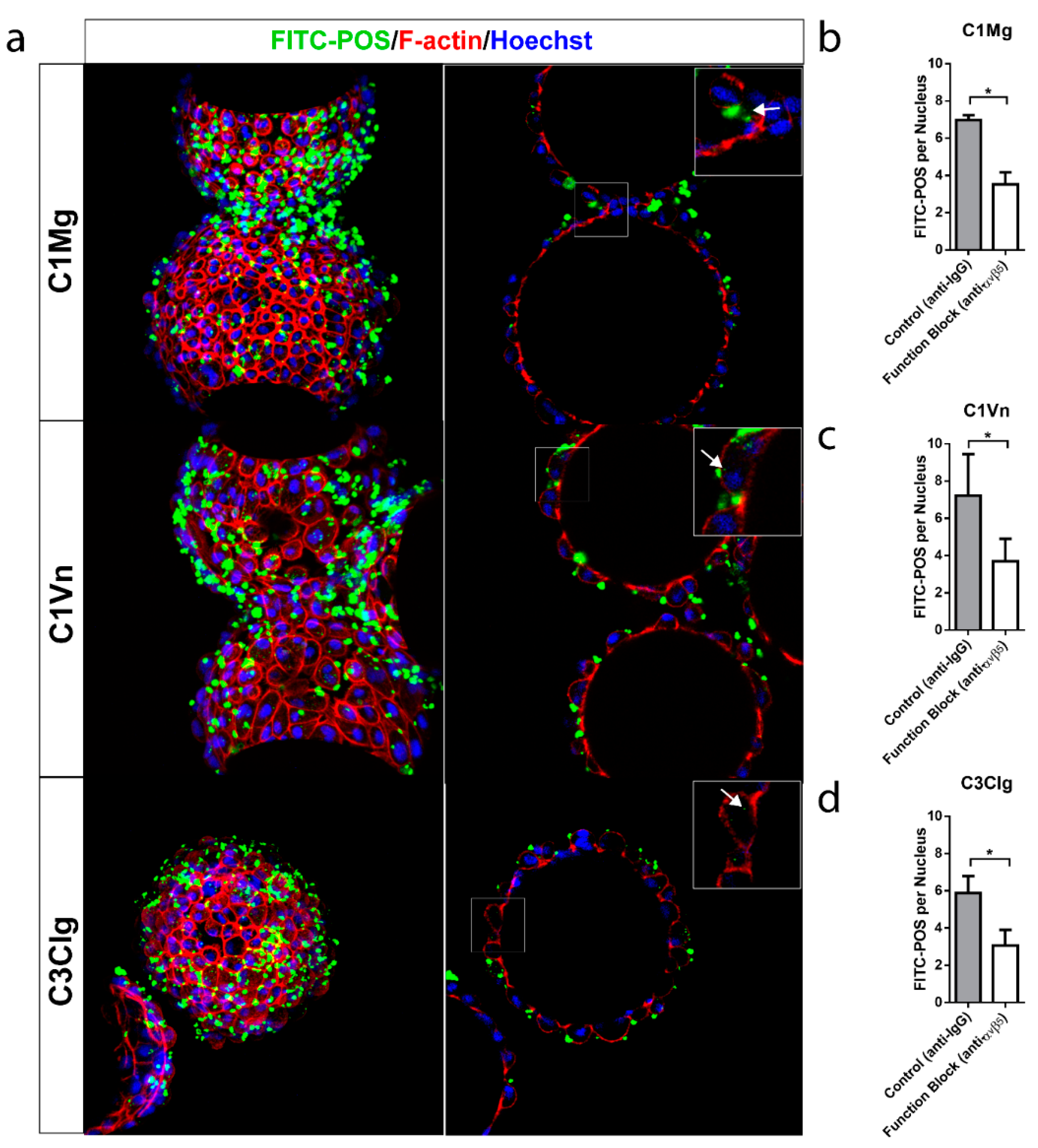
3.5. mcRPE Phagocytose Photoreceptor Outer Segments
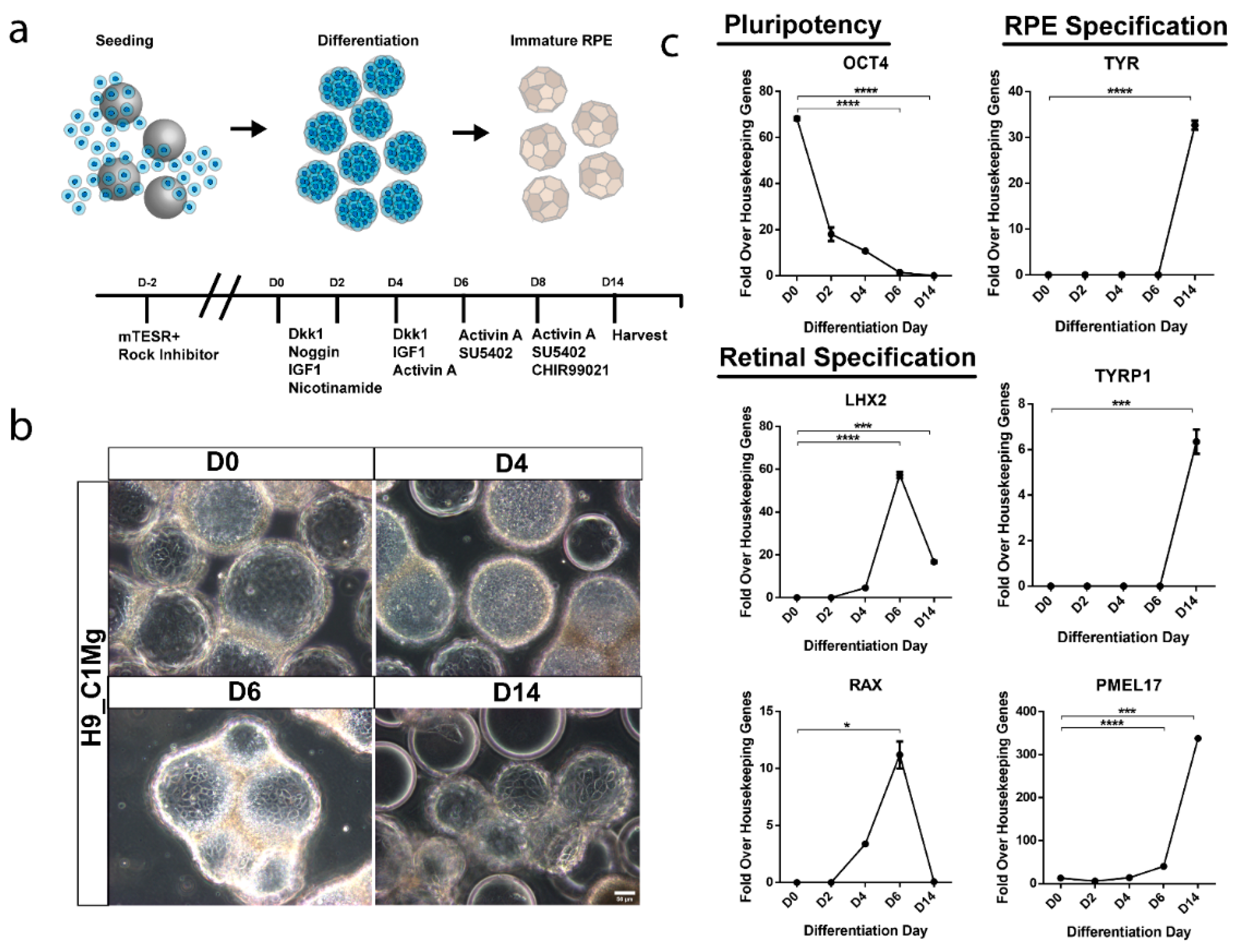
3.6. hESCs Can Be Differentiated into RPE-Progenitors on Microcarriers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global Prevalence of Age-Related Macular Degeneration and Disease Burden Projection for 2020 and 2040: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Shu, Y.K.; Noureddine, M.; Gilbert, J.R.; et al. Complement Factor H Variant Increases the Risk of Age-Related Macular Degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrrow, J.R.; Hicks, D.; Hamel, C.P. The Retinal Pigment Epithelium in Health and Disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal Pigment Epithelium and Age-Related Macular Degeneration: A Review of Major Disease Mechanisms. Clin. Exp. Ophthalmol. 2020, 48, 1043–1056. [Google Scholar] [CrossRef]
- Kim, H.J.; Koh, H.C. Chaperon-Mediated Autophagy Can Regulate Diquat-Induced Apoptosis by Inhibiting α-Synuclein Accumulation Cooperatively with Macroautophagy. Food Chem. Toxicol. 2021, 158, 112706. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.J.F.; Smart, M.J.K.; Ramsden, C.M.; Powner, M.B.; da Cruz, L.; Coffey, P.J. Development of Human Embryonic Stem Cell Therapies for Age-Related Macular Degeneration. Trends Neurosci. 2013, 36, 385–395. [Google Scholar] [CrossRef]
- Sharma, R.; Bose, D.; Maminishkis, A.; Bharti, K. Retinal Pigment Epithelium Replacement Therapy for Age-Related Macular Degeneration: Are We There Yet? Annu. Rev. Pharmacol. Toxicol. 2020, 60, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, M.; Sugino, I.; Townes-Anderson, E. Concise Review: Update on Retinal Pigment Epithelium Transplantation for Age-Related Macular Degeneration. Stem Cells Transl. Med. 2019, 8, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashani, A.H.; Lebkowski, J.S.; Rahhal, F.M.; Avery, R.L.; Salehi-Had, H.; Dang, W.; Lin, C.M.; Mitra, D.; Zhu, D.; Thomas, B.B.; et al. A Bioengineered Retinal Pigment Epithelial Monolayer for Advanced, Dry Age-Related Macular Degeneration. Sci. Transl. Med. 2018, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human Embryonic Stem Cell-Derived Retinal Pigment Epithelium in Patients with Age-Related Macular Degeneration and Stargardt’s Macular Dystrophy: Follow-up of Two Open-Label Phase 1/2 Studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 Clinical Study of an Embryonic Stem Cell–Derived Retinal Pigment Epithelium Patch in Age-Related Macular Degeneration. Nat. Publ. Gr. 2018, 36, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, T.; Gonzalez, R.T.; Sherman, S.J. The Yin and Yang of VEGF and PEDF: Multifaceted Neurotrophic Factors and Their Potential in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2010, 11, 2875–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.X.; Wang, J.J.; Gao, G.; Shao, C.; Mott, R.; Ma, J. Pigment Epithelium-derived Factor (PEDF) Is an Endogenous Antiinflammatory Factor. FASEB J. 2006, 20, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.L.; Mao, X.O.; Greenberg, D.A. Vascular Endothelial Growth Factor: Direct Neuroprotective Effect in in Vitro Ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar] [CrossRef] [Green Version]
- Yasuhara, T.; Shingo, T.; Muraoka, K.; Kameda, M.; Agari, T.; Yuan, W.J.; Hayase, H.; Hamada, H.; Borlongan, C.V.; Date, I. Neurorescue Effects of VEGF on a Rat Model of Parkinson’s Disease. Brain Res. 2005, 1053, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Regent, F.; Morizur, L.; Lesueur, L.; Habeler, W.; Plancheron, A.; M’Barek, K.B.; Monville, C. Automation of Human Pluripotent Stem Cell Differentiation toward Retinal Pigment Epithelial Cells for Large-Scale Productions. Sci. Rep. 2019, 9, 10646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.A.V.; Silva, T.P.; Nogueira, D.E.S.; Fernandes, T.G.; Hashimura, Y.; Wesselschmidt, R.; Diogo, M.M.; Lee, B.; Cabral, J.M.S. Scalable Culture of Human Induced Pluripotent Cells on Microcarriers under Xeno-Free Conditions Using Single-Use Vertical-WheelTM Bioreactors. J. Chem. Technol. Biotechnol. 2018, 93, 3597–3606. [Google Scholar] [CrossRef]
- Tavassoli, H.; Alhosseini, S.N.; Tay, A.; Chan, P.P.Y.; Weng Oh, S.K.; Warkiani, M.E. Large-Scale Production of Stem Cells Utilizing Microcarriers: A Biomaterials Engineering Perspective from Academic Research to Commercialized Products. Biomaterials 2018, 181, 333–346. [Google Scholar] [CrossRef]
- Neto, M.D.; Oliveira, M.B.; Mano, J.F. Microparticles in Contact with Cells: From Carriers to Multifunctional Tissue Modulators. Trends Biotechnol. 2019, 37, 1011–1028. [Google Scholar] [CrossRef]
- Simaria, A.S.; Hassan, S.; Varadaraju, H.; Rowley, J.; Warren, K.; Vanek, P.; Farid, S.S. Allogeneic Cell Therapy Bioprocess Economics and Optimization: Single-Use Cell Expansion Technologies. Biotechnol. Bioeng. 2014, 111, 69–83. [Google Scholar] [CrossRef] [Green Version]
- GE Health Care Bio-Sciences Cytodex 1, Cytodex 3; GE Healthcare Bio-Sciences AB: Uppsala, Sweden, 2006; pp. 3–6.
- Blüml, G. Microcarrier cell culture technology. In Animal Cell Biotechnology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 149–178. [Google Scholar]
- Kuriyama, S.; Nakano, T.; Yoshimura, N.; Ohuchi, T.; Moritera, T.; Honda, Y. Mass Cultivation of Human Retinal Pigment Epithelial Cells with Microcarrier. Ophthalmologica 1992, 205, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Stover, N.P.; Watts, R.L. Spheramine for Treatment of Parkinson’s Disease. Neurotherapeutics 2008, 5, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, H.A.; Treharne, A.J.; Backholer, L.S.; Cuda, F.; Grossel, M.C.; Lotery, A.J. Biodegradable Poly(α-Hydroxy Ester) Blended Microspheres as Suitable Carriers for Retinal Pigment Epithelium Cell Transplantation. J. Biomed. Mater. Res.-Part A 2010, 95, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Charniga, C.; Temple, S.; Finnemann, S.C.; Mu, C. Quantified F-Actin Morphology Is Predictive of Phagocytic Capacity of StemCell-Derived Retinal Pigment Epithelium. Stem Cell Rep. 2018, 10, 1075–1087. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, D.E.; Pennington, B.O.; Croze, R.H.; Hinman, C.R.; Coffey, P.J.; Clegg, D.O. Rapid and Efficient Directed Differentiation of Human Pluripotent Stem Cells Into Retinal Pigmented Epithelium. Stem Cells Transl. Med. 2013, 2, 384–393. [Google Scholar] [CrossRef]
- Leach, L.L.; Buchholz, D.E.; Nadar, V.P.; Lowenstein, S.E.; Clegg, D.O. Canonical / b-Catenin Wnt Pathway Activation Improves Embryonic Stem Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1002–1013. [Google Scholar] [CrossRef]
- Falk, T.; Zhang, S.; Sherman, S.J. Pigment Epithelium Derived Factor (PEDF) Is Neuroprotective in Two in Vitro Models of Parkinson’s Disease. Neurosci. Lett. 2009, 458, 49–52. [Google Scholar] [CrossRef]
- Finnemann, S.C.; Bonilha, V.L.; Marmorstein, A.D.; Rodriguez-Boulan, E. Phagocytosis of Rod Outer Segments by Retinal Pigment Epithelial Cells Requires Avβ5 Integrin for Binding but Not for Internalization. Proc. Natl. Acad. Sci. USA 1997, 94, 12932–12937. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, F.; Safa, H.; Finnemann, C.S. Understanding Photoreceptor Outer Segment Phagocytosis: Use and Utility of RPE Cells in Culture. Exp. Eye Res. 2014, 126, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Nandrot, E.F.; Anand, M.; Almeida, D.; Atabai, K.; Sheppard, D.; Finnemann, S.C. Essential Role for MFG-E8 as Ligand for Avβ5 Integrin in Diurnal Retinal Phagocytosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12005–12010. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Brickman, J.M.; Serup, P. Properties of Embryoid Bodies. Dev. Biol. 2016, 6, e259. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, A.R.; Kapetanakis, V.V.; Jarrar, Z.; Wathern, A.K.; Wormald, R.; Fletcher, A.E.; Cook, D.G.; Owen, C.G. Incidence of Late-Stage Age-Related Macular Degeneration in American Whites: Systematic Review and Meta-Analysis. Am. J. Ophthalmol. 2015, 160, 85–93.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badenes, S.M.; Fernandes, T.G.; Miranda, C.C.; Pusch-Klein, A.; Haupt, S.; Rodrigues, C.A.V.; Diogo, M.M.; Brüstle, O.; Cabral, J.M.S. Long-Term Expansion of Human Induced Pluripotent Stem Cells in a Microcarrier-Based Dynamic System. J. Chem. Technol. Biotechnol. 2017, 92, 492–503. [Google Scholar] [CrossRef]
- Srinivasan, G.; Morgan, D.; Varun, D.; Brookhouser, N.; Brafman, D.A. An Integrated Biomanufacturing Platform for the Large-Scale Expansion and Neuronal Differentiation of Human Pluripotent Stem Cell-Derived Neural Progenitor Cells. Acta Biomater. 2018, 74, 168–179. [Google Scholar] [CrossRef]
- Sousa, M.F.Q.; Silva, M.M.; Giroux, D.; Hashimura, Y.; Wesselschmidt, R.; Lee, B.; Roldão, A.; Carrondo, M.J.T.; Alves, P.M.; Serra, M. Production of Oncolytic Adenovirus and Human Mesenchymal Stem Cells in a Single-Use, Vertical-Wheel Bioreactor System: Impact of Bioreactor Design on Performance of Microcarrier-Based Cell Culture Processes. Biotechnol. Prog. 2015, 31, 1600–1612. [Google Scholar] [CrossRef]
- Olaf, S. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Pennington, B.O.; Bailey, J.K.; Faynus, M.A.; Hinman, C.; Hee, M.N.; Ritts, R.; Nadar, V.; Zhu, D.; Mitra, D.; Martinez-Camarillo, J.C.; et al. Xeno-Free Cryopreservation of Adherent Retinal Pigmented Epithelium Yields Viable and Functional Cells in Vitro and in Vivo. Sci. Rep. 2021, 11, 6286. [Google Scholar] [CrossRef]
- Bilak, M.M.; Corse, A.M.; Bilak, S.R. Pigment Epithelium-Derived Factor (PEDF) Protects Motor Neurons from Chronic Glutamate-Mediated Neurodegeneration. J. Neuropathol. Exp. Neurol. 1991, 58, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Deng, X.; Spee, C.; Sonoda, S.; Hsieh, C.L.; Barron, E.; Pera, M.; Hinton, D.R. Polarized Secretion of PEDF from Human Embryonic Stem Cell-Derived RPE Promotes Retinal Progenitor Cell Survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1573–1585. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faynus, M.A.; Bailey, J.K.; Pennington, B.O.; Katsura, M.; Proctor, D.A.; Yeh, A.K.; Menon, S.; Choi, D.G.; Lebkowski, J.S.; Johnson, L.V.; et al. Microcarrier-Based Culture of Human Pluripotent Stem-Cell-Derived Retinal Pigmented Epithelium. Bioengineering 2022, 9, 297. https://doi.org/10.3390/bioengineering9070297
Faynus MA, Bailey JK, Pennington BO, Katsura M, Proctor DA, Yeh AK, Menon S, Choi DG, Lebkowski JS, Johnson LV, et al. Microcarrier-Based Culture of Human Pluripotent Stem-Cell-Derived Retinal Pigmented Epithelium. Bioengineering. 2022; 9(7):297. https://doi.org/10.3390/bioengineering9070297
Chicago/Turabian StyleFaynus, Mohamed A., Jeffrey K. Bailey, Britney O. Pennington, Mika Katsura, Duncan A. Proctor, Ashley K. Yeh, Sneha Menon, Dylan G. Choi, Jane S. Lebkowski, Lincoln V. Johnson, and et al. 2022. "Microcarrier-Based Culture of Human Pluripotent Stem-Cell-Derived Retinal Pigmented Epithelium" Bioengineering 9, no. 7: 297. https://doi.org/10.3390/bioengineering9070297