Epigallocatechin-3-Gallate and PEDF 335 Peptide, 67LR Activators, Attenuate Vasogenic Edema, and Astroglial Degeneration Following Status Epilepticus

Abstract
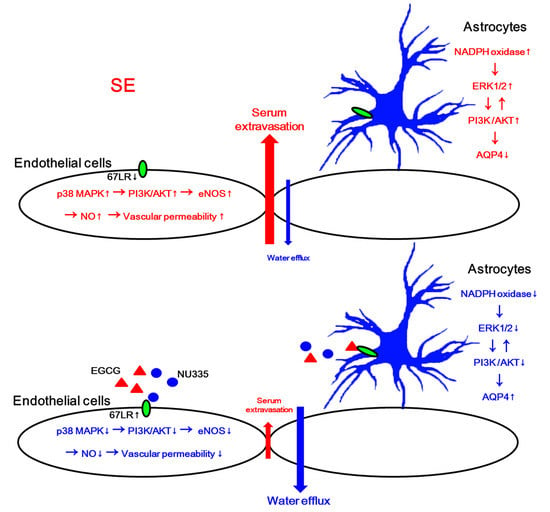
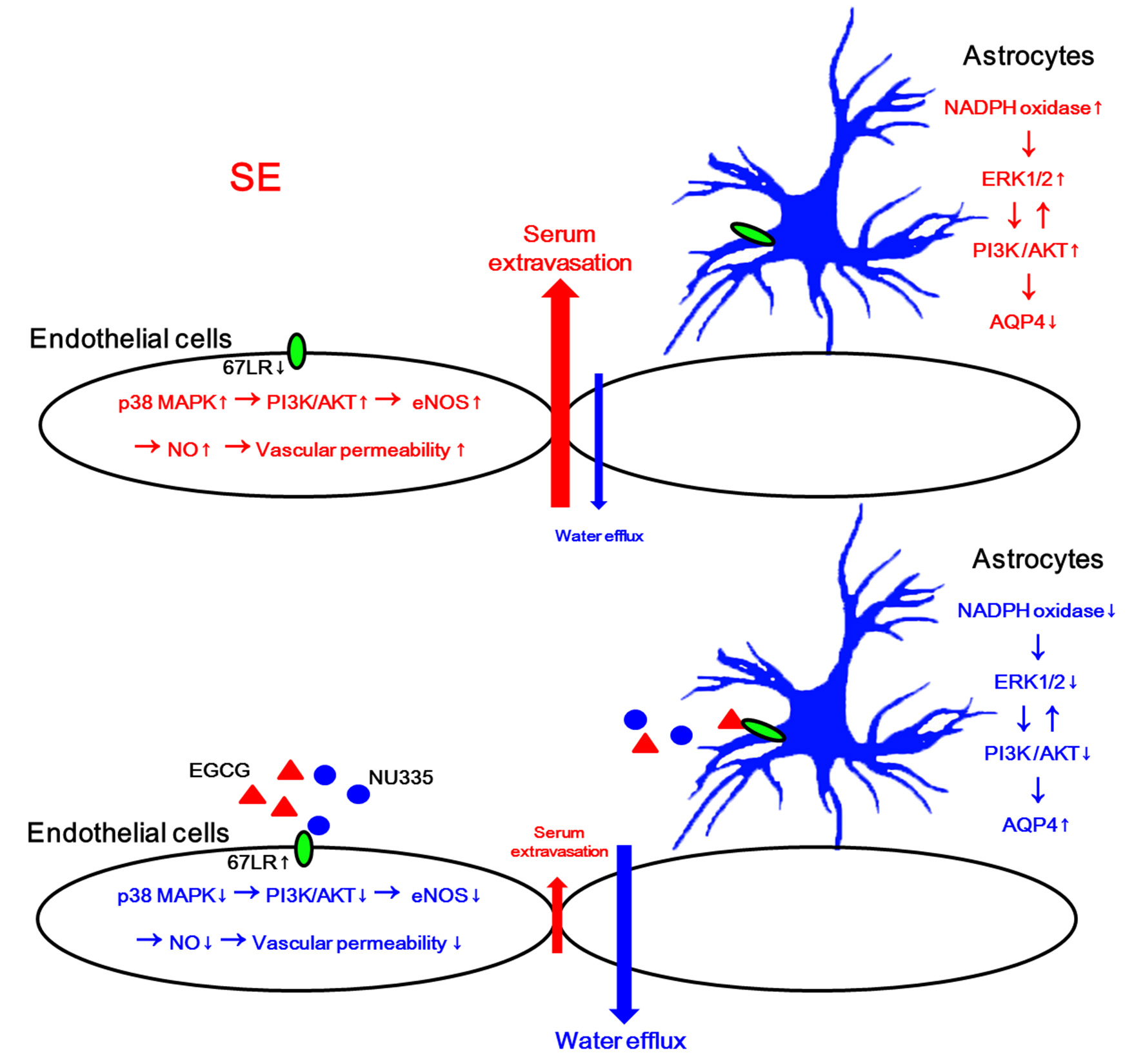
:
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. Surgery
2.3. SE Induction and EEG Recording
2.4. Tissue Processing
2.5. Western Blot
2.6. Immunohistochemistry
2.7. Measurements of Serum Extravasation, GFAP-Deleted Area, and Fluorescent Intensity
2.8. Statistical Analysis
3. Results
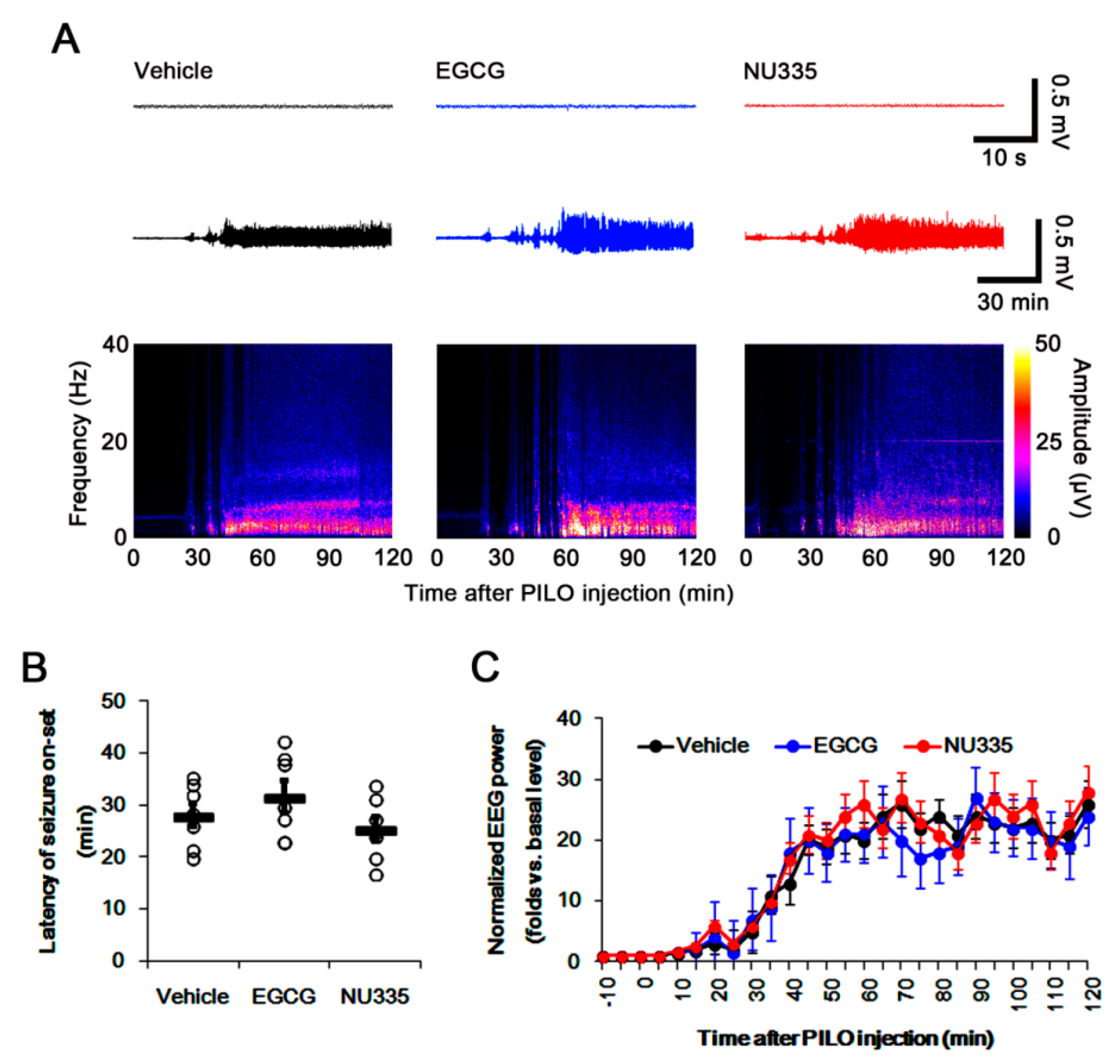
3.1. Effects of EGCG and NU335 Peptide on Seizure Susceptibility in Response to Pilocarpine
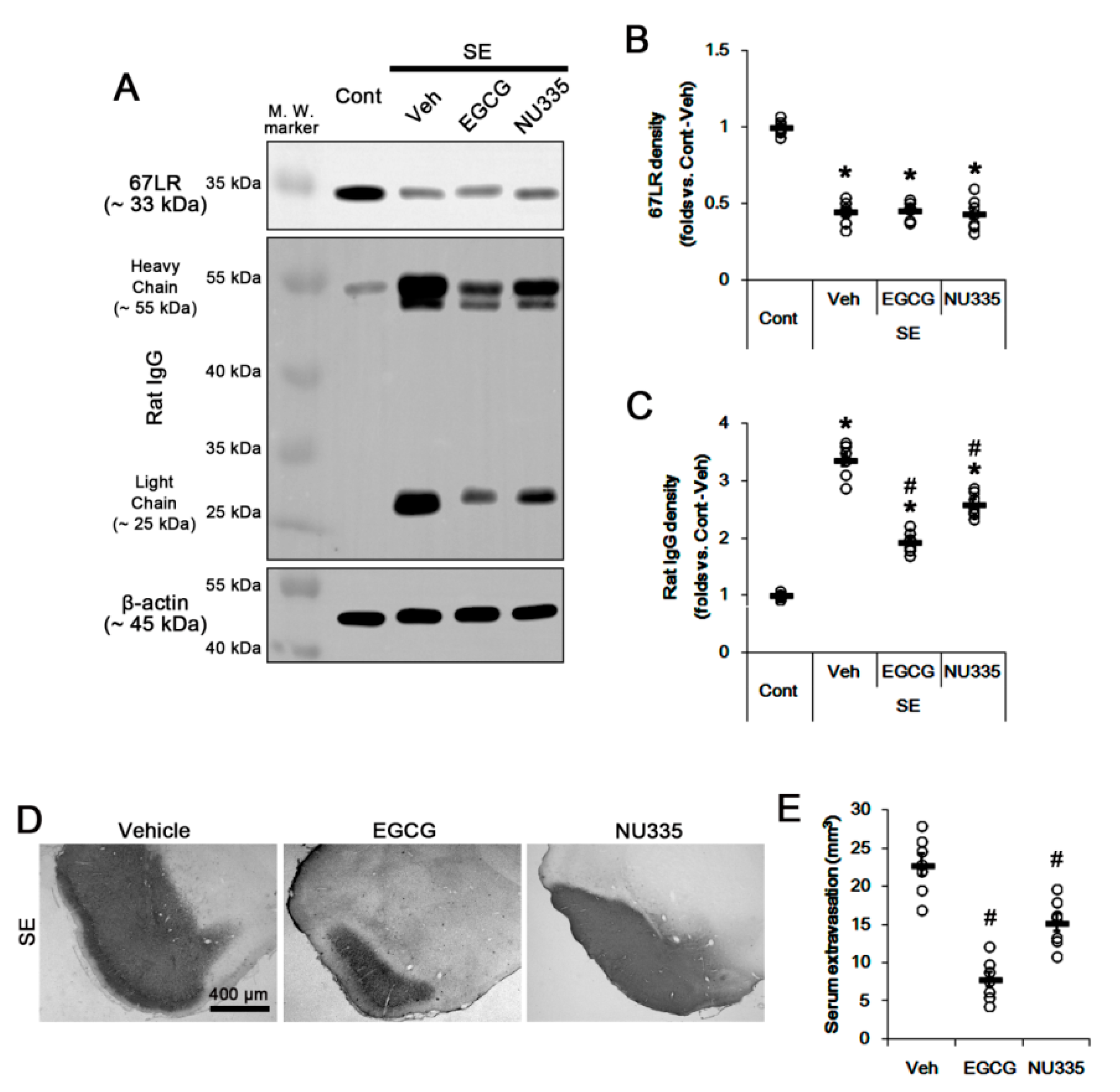
3.2. Effects of EGCG and NU335 Peptide on Serum Extravasation Following SE
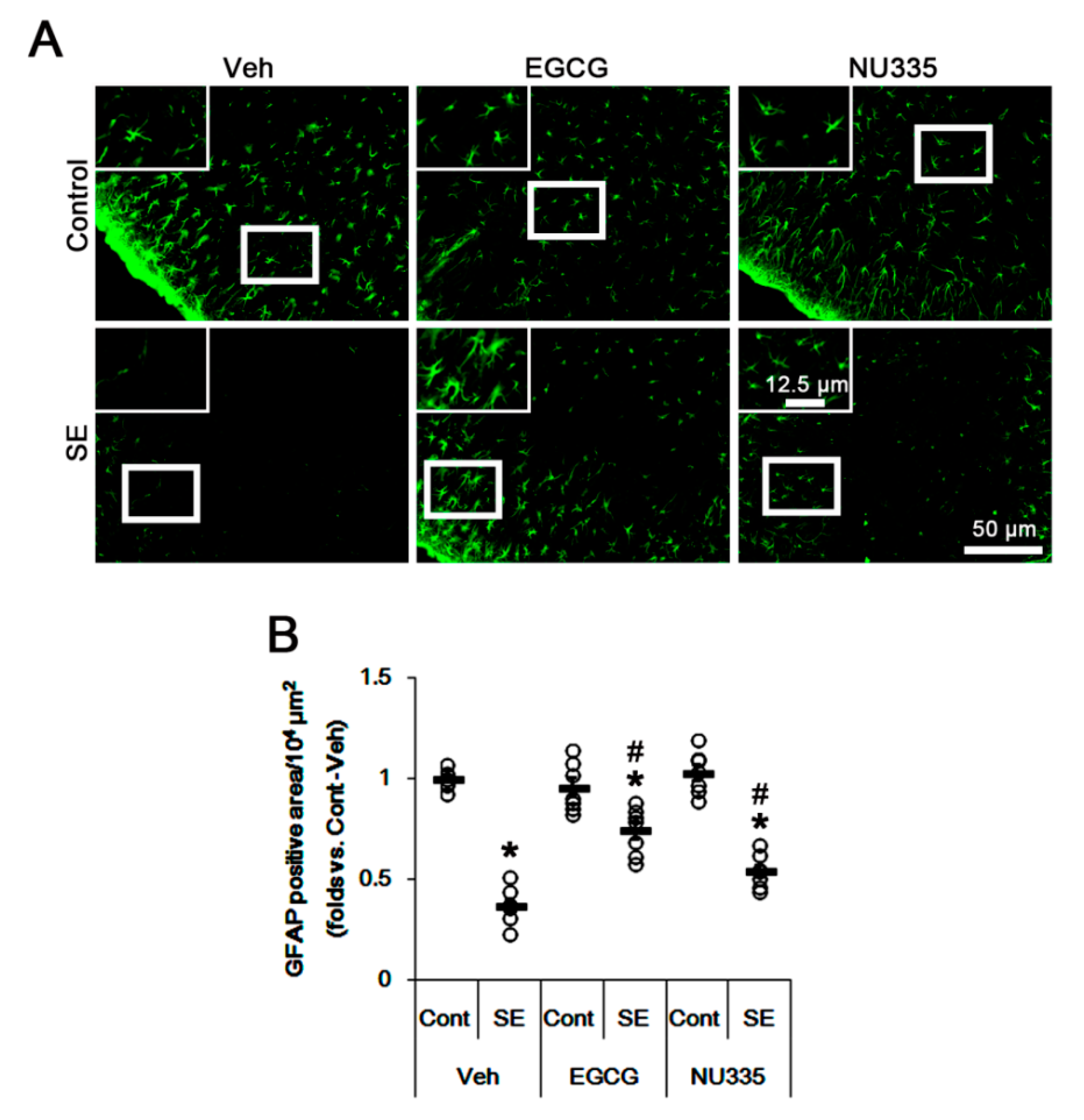
3.3. Effects of EGCG and NU335 Peptide on Astroglial Damage under Physiological and Post-SE Conditions
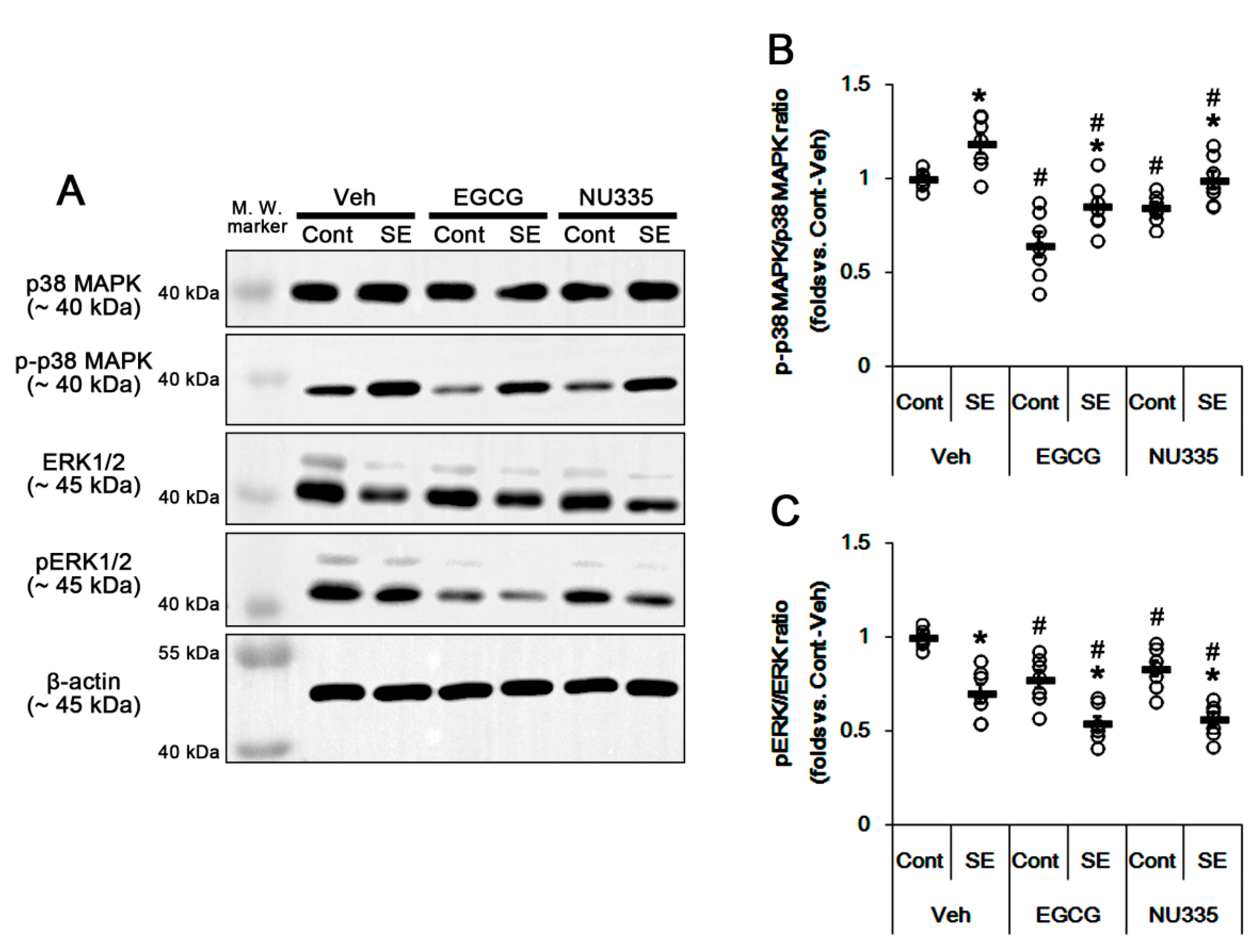
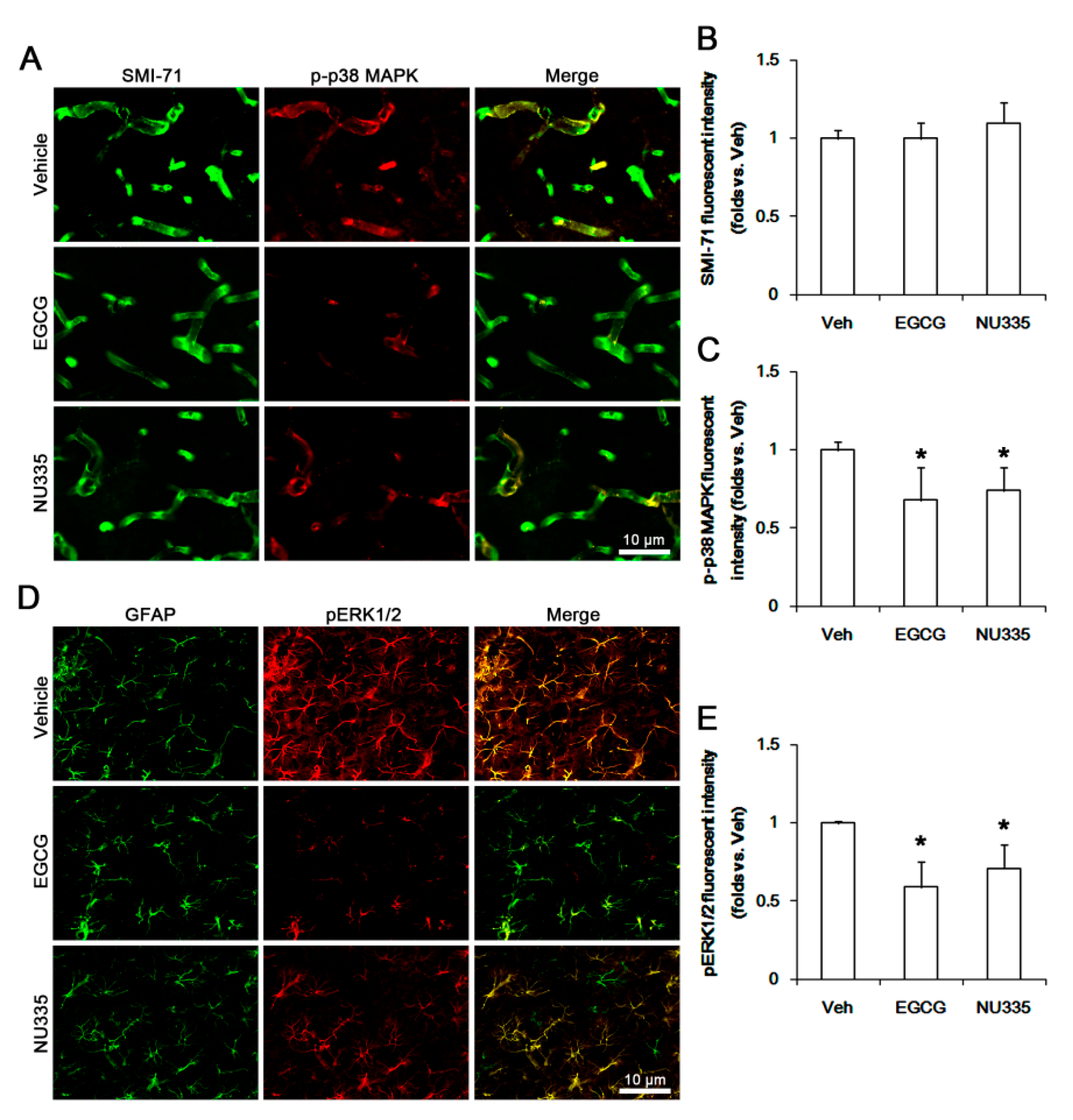
3.4. The Effects of EGCG and NU335 on Phosphorylations of p38 MAPK and ERK1/2
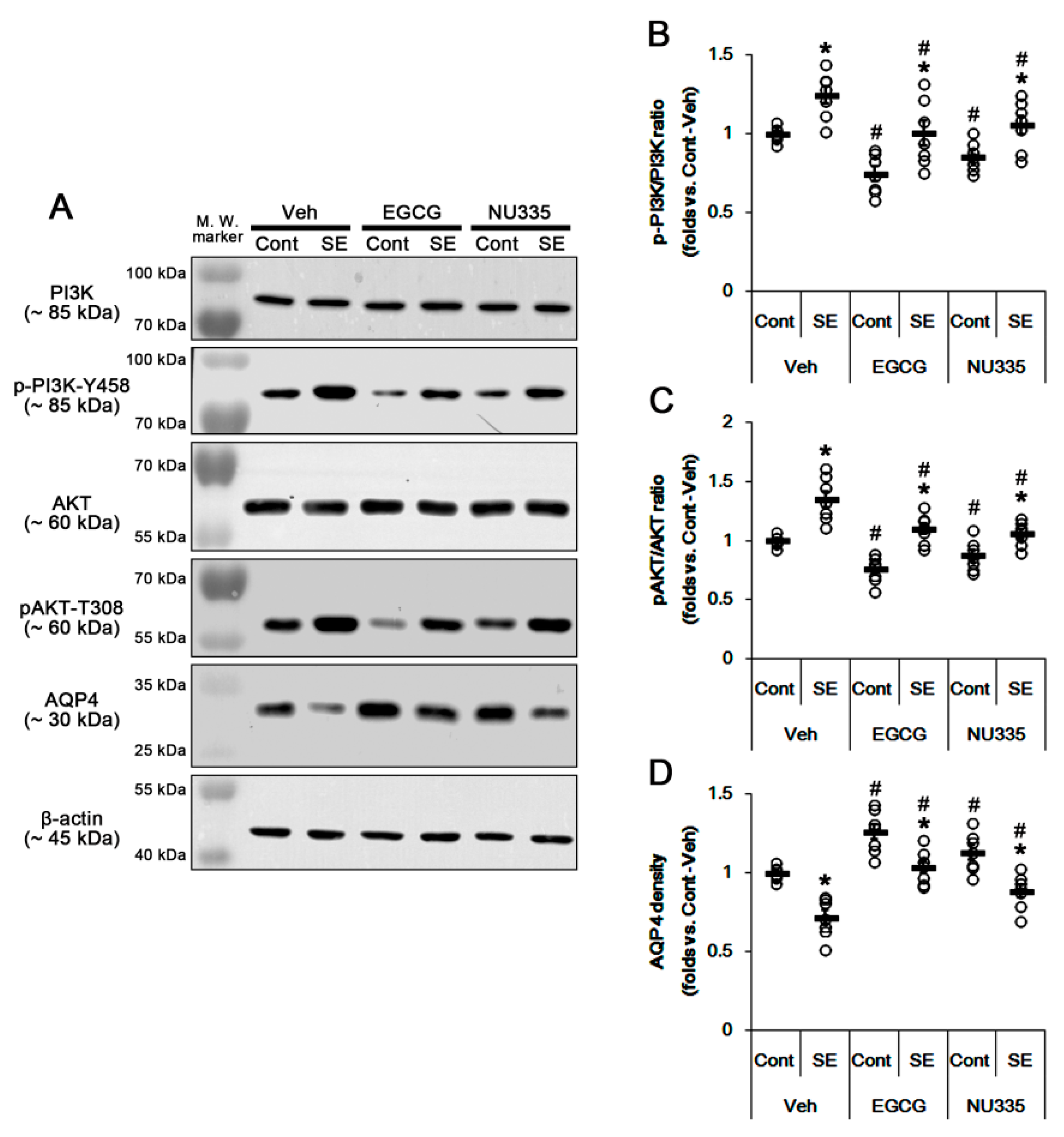
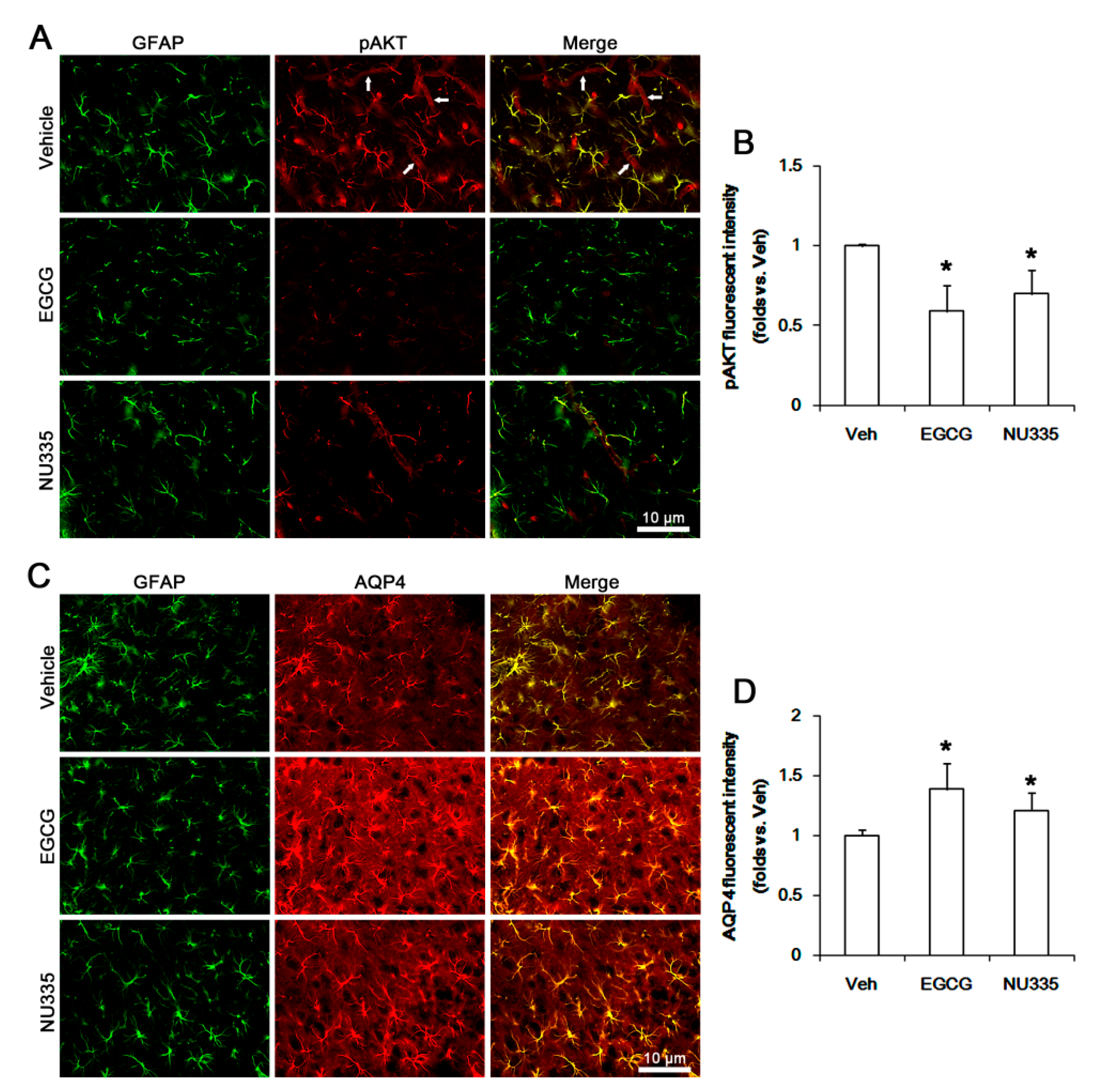
3.5. The Effects of EGCG and NU335 on PI3K/AKT Phosphorylation and AQP4 Expression
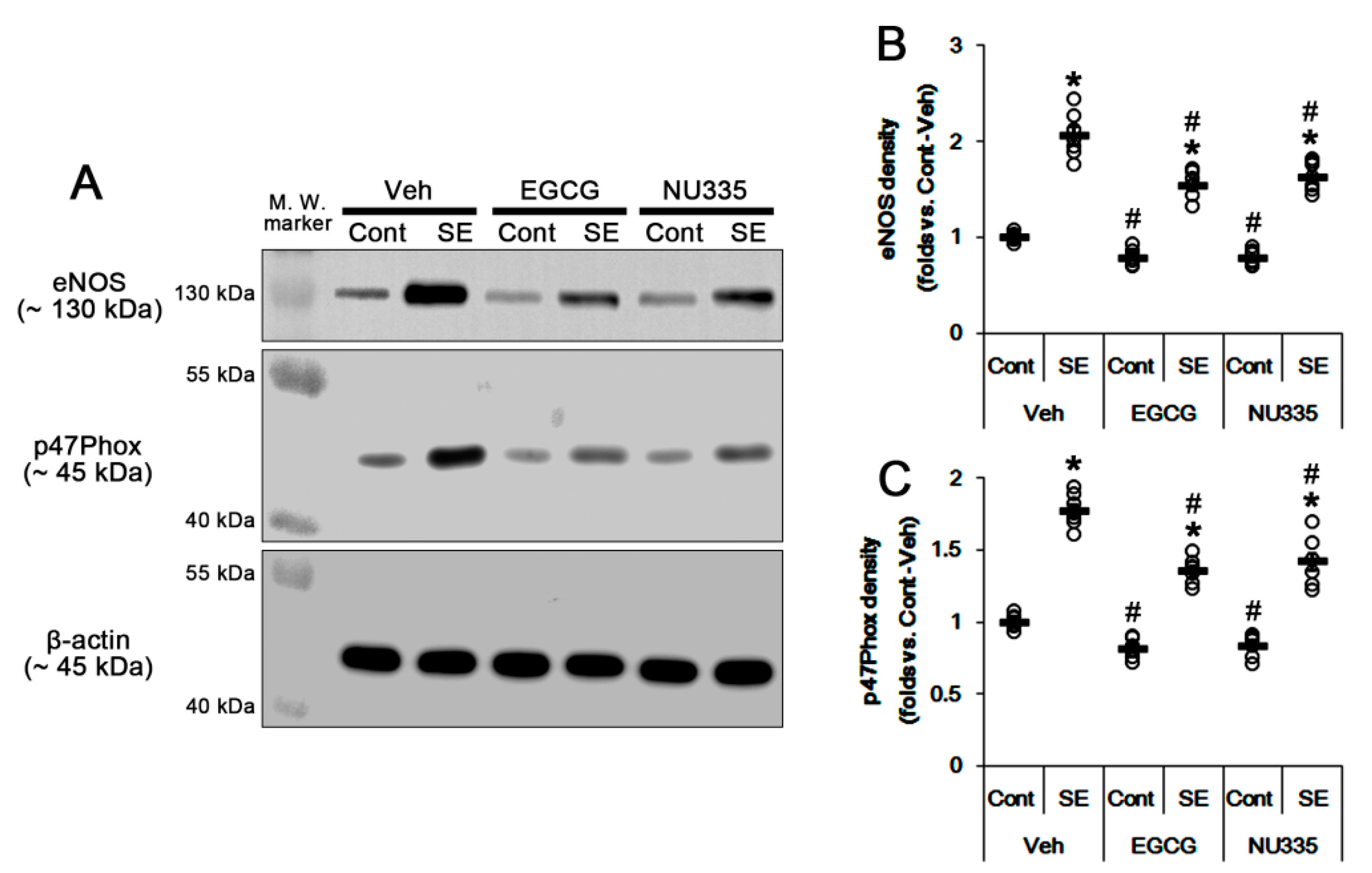
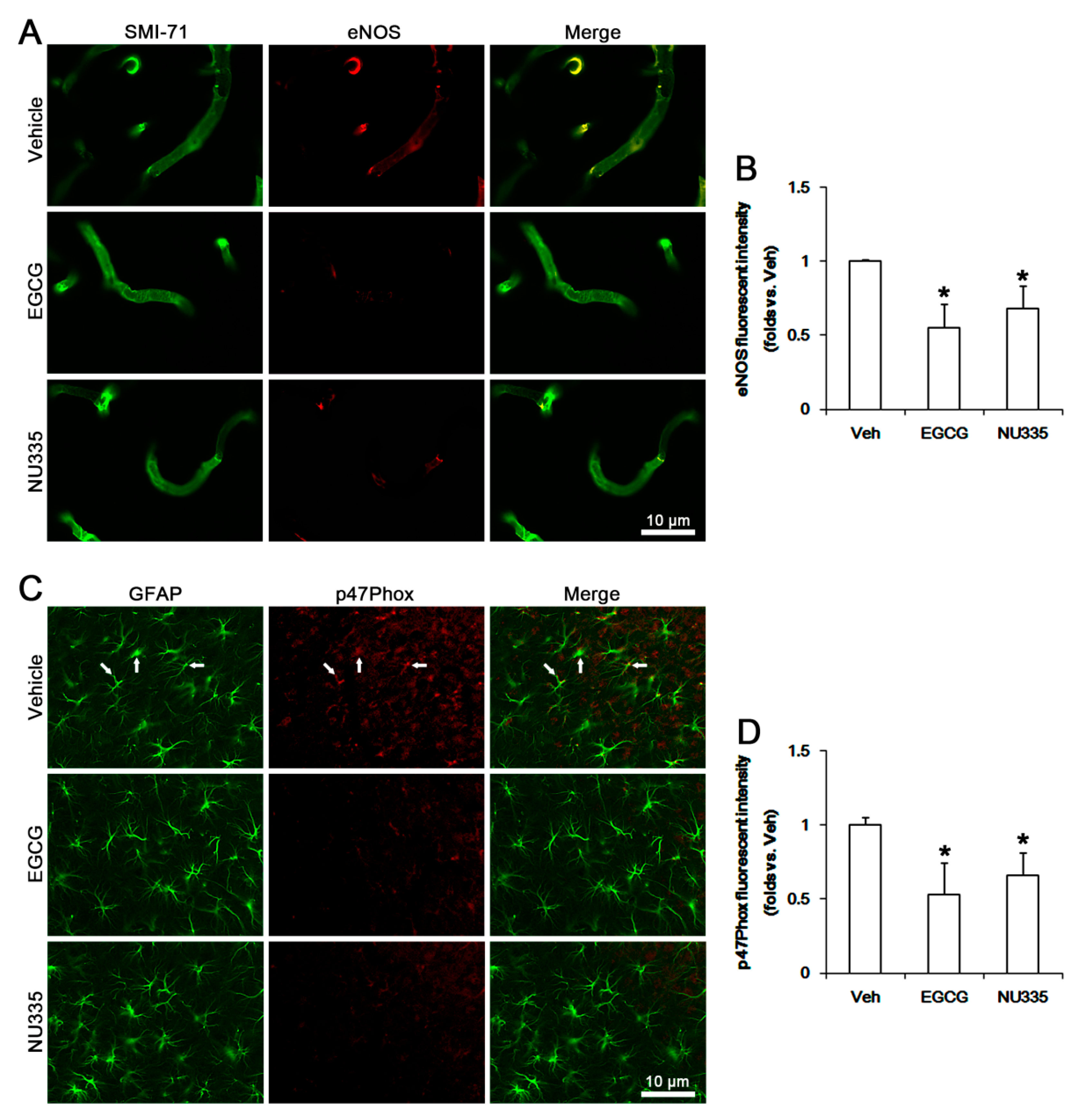
3.6. The Effects of EGCG and NU335 on eNOS and NADPH Oxidase Expression
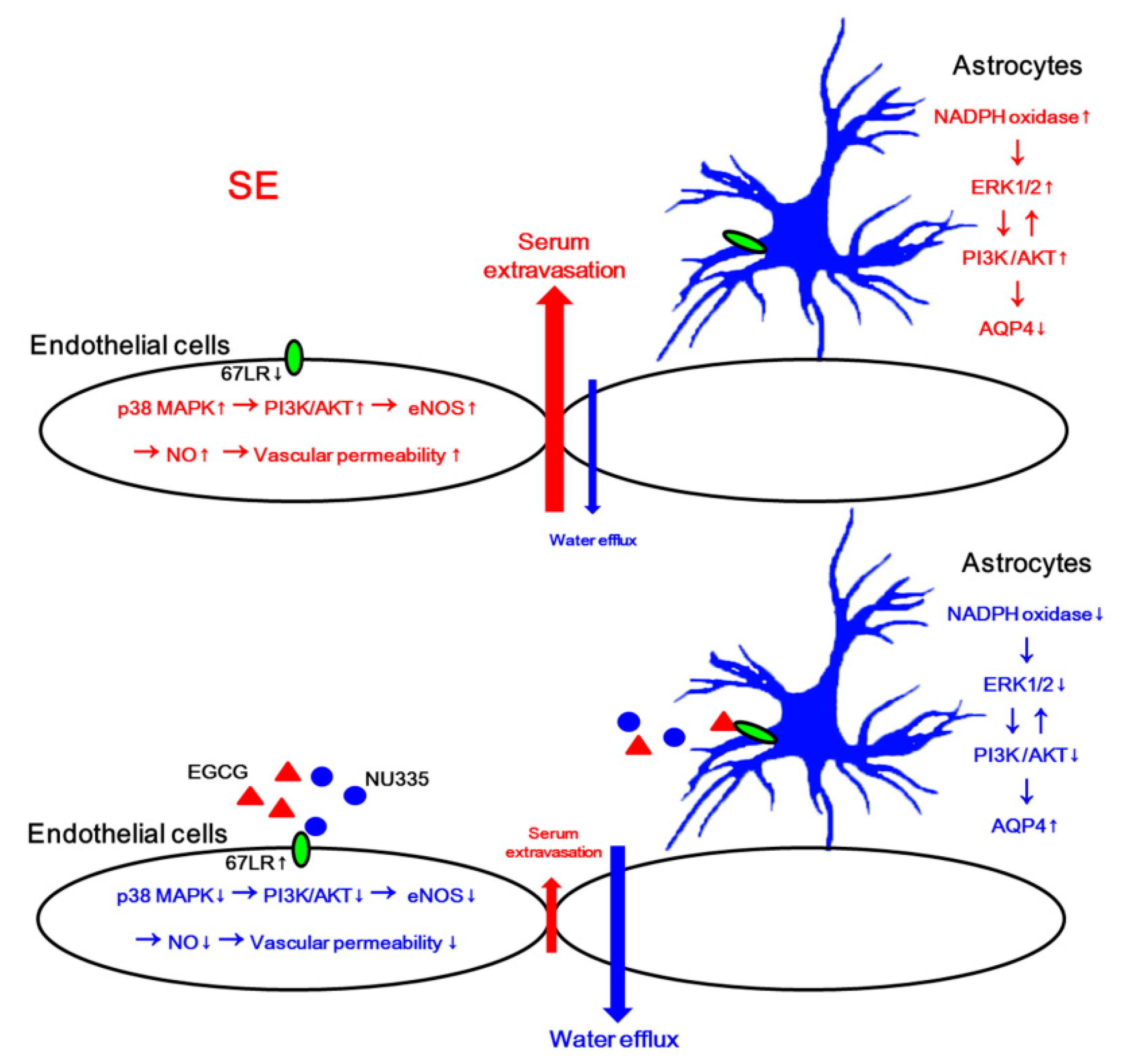
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hesdorffer, D.C.; Logroscino, G.; Cascino, G.; Annegers, J.F.; Hauser, W.A. Risk of unprovoked seizure after acute symptomatic seizure: Effect of status epilepticus. Ann. Neurol. 1998, 44, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.; Kaufer, D.; Heinemann, U. Blood-brain barrier breakdown-inducing astrocytic transformation: Novel targets for the prevention of epilepsy. Epilepsy Res. 2009, 85, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Vliet, E.A.V.; Araújo, S.D.C.; Redeker, S.; Schaik, R.V.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [CrossRef]
- Chiang, C.W.; Wang, Y.; Sun, P.; Lin, T.H.; Trinkaus, K.; Cross, A.H.; Song, S.K. Quantifying white matter tract diffusion parameters in the presence of increased extra-fiber cellularity and vasogenic edema. Neuroimage 2014, 101, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, K.; Kamintsky, L.; Kim, S.Y.; Lublinsky, S.; Prager, O.; Nichtweiss, J.F.; Salar, S.; Kaufer, D.; Heinemann, U.; Friedman, A. Epileptiform activity and spreading depolarization in the blood-brain barrier-disrupted peri-infarct hippocampus are associated with impaired GABAergic inhibition and synaptic plasticity. J. Cereb. Blood Flow Metab. 2017, 37, 1803–1819. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, Q.; Wang, Z.; Qi, C.; Han, X.; Lan, X.; Wan, J.; Wang, W.; Zhao, X.; Hou, Z.; et al. Multimodality MRI assessment of grey and white matter injury and blood-brain barrier disruption after intracerebral haemorrhage in mice. Sci. Rep. 2017, 7, 40358. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Ryu, H.J.; Kang, T.C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/endothelin-1-mediated two different pathways. PLoS ONE 2013, 8, e74458. [Google Scholar] [CrossRef] [Green Version]
- Thyboll, J.; Kortesmaa, J.; Cao, R.; Soininen, R.; Wang, L.; Iivanainen, A.; Sorokin, L.; Risling, M.; Cao, Y.; Tryggvason, K. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol. Cell Biol. 2002, 22, 1194–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, J.H. Laminins and their roles in mammals. Microsc. Res. Tech. 2008, 71, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Lesot, H.; Kuhl, U.; Mark, K. Isolation of a laminin-binding protein from muscle cell membranes. EMBO J. 1983, 2, 861–865. [Google Scholar] [CrossRef] [PubMed]
- McCaffery, P.; Neve, R.L.; Drager, U.C. A dorso-ventral asymmetry in the embryonic retina defined by protein conformation. Proc. Natl. Acad. Sci. USA. 1990, 87, 8570–8574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohgo, A.; Takasawa, S.; Munakata, H.; Yonekura, H.; Hayashi, N.; Okamoto, H. Structural determination and characterization of a 40 kDa protein isolated from rat 40 S ribosomal subunit. FEBS Lett. 1994, 340, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Menard, S.; Castronovo, V.; Tagliabue, E.; Sobel, M.E. New insights into the metastasis-associated 67 kD laminin receptor. J. Cell Biochem. 1997, 67, 155–165. [Google Scholar] [CrossRef]
- Pellegrini, R.; Martignone, S.; Menard, S.; Colnaghi, M.I. Laminin receptor expression and function in small-cell lung carcinoma. Int. J. Cancer. 1994, 8, 116–120. [Google Scholar] [CrossRef]
- Ardini, E.; Sporchia, B.; Pollegioni, L.; Modugno, M.; Ghirelli, C.; Castiglioni, F.; Tagliabue, E.; Ménard, S. Identification of a novel function for 67-kDa laminin receptor: Increase in laminin degradation rate and release of motility fragments. Cancer Res. 2002, 62, 1321–1325. [Google Scholar]
- Nelson, J.; McFerran, N.V.; Pivato, G.; Chambers, E.; Doherty, C.; Steele, D.; Timson, D.J. The 67 kDa laminin receptor: Structure, function and role in disease. Biosci. Rep. 2008, 28, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Givant-Horwitz, V.; Davidson, B.; Reich, R. Laminin-induced signaling in tumor cells: The role of the M(r) 67,000 laminin receptor. Cancer Res. 2004, 64, 3572–3579. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. Dysfunction of 67-kDa laminin receptor disrupts BBB integrity via impaired dystrophin/AQP4 complex and p38 MAPK/VEGF activation following status epilepticus. Front. Cell. Neurosci. 2019, 13, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Kang, T.C. The regional specific alterations in BBB permeability are relevant to the differential responses of 67-kDa LR expression in endothelial cells and astrocytes following status epilepticus. Int. J. Mol. Sci. 2019, 20, 6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Park, H.; Lee, J.E.; Kang, T.C. Blockade of 67-kDa laminin receptor facilitates AQP4 down-regulation and BBB disruption via ERK1/2- and p38 MAPK-mediated PI3K/AKT activations. Cells 2020, 9, 1670. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Kumazoe, M.; Sugihara, K.; Tsukamoto, S.; Huang, Y.; Tsurudome, Y.; Suzuki, T.; Suemasu, Y.; Ueda, N.; Yamashita, S.; Kim, Y.; et al. 67-kDa laminin receptor increases cGMP to induce cancer-selective apoptosis. J. Clin. Investig. 2013, 123, 787–799. [Google Scholar] [CrossRef]
- Li, H.; Li, Z.; Xu, Y.M.; Wu, Y.; Yu, K.K.; Zhang, C.; Ji, Y.H.; Ding, G.; Chen, F.X. Epigallocatechin-3-gallate induces apoptosis, inhibits proliferation and decreases invasion of glioma cell. Neurosci. Bull. 2014, 30, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Bernard, A.; Gao-Li, J.; Franco, C.A.; Bouceba, T.; Huet, A.; Li, Z. Laminin receptor involvement in the anti-angiogenic activity of pigment epithelium-derived factor. J. Biol. Chem. 2009, 284, 10480–10490. [Google Scholar] [CrossRef] [Green Version]
- Sheibani, N.; Wang, S.; Darjatmoko, S.R.; Fisk, D.L.; Shahi, P.K.; Pattnaik, B.R.; Sorenson, C.M.; Bhowmick, R.; Volpert, O.V.; Albert, D.M.; et al. Novel anti-angiogenic PEDF-derived small peptides mitigate choroidal neovascularization. Exp. Eye Res. 2019, 188, 107798. [Google Scholar] [CrossRef]
- Zhou, X.; Liang, L.; Zhao, Y.; Zhang, H. Epigallocatechin-3-Gallate ameliorates angiotensin II-induced oxidative stress and apoptosis in human umbilical vein endothelial cells through the activation of Nrf2/Caspase-3 signaling. J. Vasc. Res. 2017, 54, 299–308. [Google Scholar] [CrossRef]
- Liu, S.; Sun, Z.; Chu, P.; Li, H.; Ahsan, A.; Zhou, Z.; Zhang, Z.; Sun, B.; Wu, J.; Xi, Y.; et al. EGCG protects against homocysteine-induced human umbilical vein endothelial cells apoptosis by modulating mitochondrial-dependent apoptotic signaling and PI3K/Akt/eNOS signaling pathways. Apoptosis 2017, 22, 672–680. [Google Scholar] [CrossRef]
- Li, J.; Ye, L.; Wang, X.; Liu, J.; Wang, Y.; Zhou, Y.; Ho, W. Epigallocatechin gallate inhibits endotoxin-induced expression of inflammatory cytokines in human cerebral microvascular endothelial cells. J. Neuroinflamm. 2012, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S.; Jun, J.H.; Jung, E.H.; Koo, B.A.; Kim, Y.S. Epigalloccatechin-3-gallate inhibits ocular neovascularization and vascular permeability in human retinal pigment epithelial and human retinal microvascular endothelial cells via suppression of MMP-9 and VEGF activation. Molecules 2014, 19, 12150–12172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, D.R.; Shanbhag, N.C.; Dittmar, M.S.; Bogdahn, U.; Schlachetzki, F. Neurovascular protection by targeting early blood-brain barrier disruption with neurotrophic factors after ischemia-reperfusion in rats. J. Cereb. Blood Flow Metab. 2013, 33, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalski, D.; Pitsch, R.; Pillai, D.R.; Mages, B.; Aleithe, S.; Grosche, J.; Martens, H.; Schlachetzki, F.; Härtig, W. Delayed histochemical alterations within the neurovascular unit due to transient focal cerebral ischemia and experimental treatment with neurotrophic factors. PLoS ONE 2017, 12, e0174996. [Google Scholar] [CrossRef] [Green Version]
- Riabinska, A.; Zille, M.; Terzi, M.Y.; Cordell, R.; Nieminen-Kelhä, M.; Klohs, J.; Piña, A.L. Pigment Epithelium-Derived Factor Improves Paracellular Blood-Brain Barrier Integrity in the Normal and Ischemic Mouse Brain. Cell. Mol. Neurobiol. 2020, 40, 751–764. [Google Scholar] [CrossRef]
- Kim, J.E.; Yeo, S.I.; Ryu, H.J.; Kim, M.J.; Kim, D.S.; Jo, S.M.; Kang, T.C. Astroglial loss and edema formation in the rat piriform cortex and hippocampus following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2010, 518, 4612–4628. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. Roscovitine attenuates microglia activation and monocyte infiltration via p38 MAPK inhibition in the rat frontoparietal cortex following status epilepticus. Cells 2019, 8, 746. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Park, H.; Choi, S.H.; Kong, M.J.; Kim, J.E.; Kang, T.C. CDDO-Me Attenuates Vasogenic Edema and Astroglial Death by Regulating NF-κB p65 Phosphorylations and Nrf2 Expression Following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 4862. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Min, S.J.; Kim, M.J.; Kim, J.E.; Kang, T.C. Leptomycin B ameliorates vasogenic edema formation induced by status epilepticus via inhibiting p38 MAPK/VEGF pathway. Brain Res. 2016, 1651, 27–35. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. TRPC3- and ET(B) receptor-mediated PI3K/AKT activation induces vasogenic edema formation following status epilepticus. Brain Res. 2017, 1672, 58–64. [Google Scholar] [CrossRef]
- Yang, L.; Guo, W.; Zhang, Q.; Li, H.; Liu, X.; Yang, Y.; Zuo, J.; Liu, W. Crosstalk between Raf/MEK/ERK and PI3K/AKT in suppression of Bax conformational change by Grp75 under glucose deprivation conditions. J. Mol. Biol. 2011, 414, 654–666. [Google Scholar] [CrossRef]
- Yu, L.N.; Zhou, X.L.; Yu, J.; Huang, H.; Jiang, L.S.; Zhang, F.J.; Cao, J.L.; Yan, M. PI3K contributed to modulation of spinal nociceptive information related to ephrinBs/EphBs. PLoS ONE 2012, 7, e40930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.H.; Fu, Q.C.; Shi, D.; Bu, H.L.; Song, Z.P.; Xiong, B.R.; Shu, B.; Xiang, H.B.; Xu, B.; Manyande, A.; et al. Activation of spinal chemokine receptor CXCR3 mediates bone cancer pain through an Akt-ERK crosstalk pathway in rats. Exp. Neurol. 2015, 263, 39–49. [Google Scholar] [CrossRef]
- Kapoor, S.; Kim, S.M.; Farook, J.M.; Mir, S.; Saha, R.; Sen, N. Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. J. Neurosci. 2013, 33, 17398–17403. [Google Scholar] [CrossRef]
- Sharma, H.S.; Drieu, K.; Alm, P.; Westman, J. Role of nitric oxide in blood-brain barrier permeability, brain edema and cell damage following hyperthermic brain injury. An experimental study using EGB-761 and Gingkolide B pretreatment in the rat. Acta Neurochir. Suppl. 2000, 76, 81–86. [Google Scholar]
- Wu, M.; Tsirka, S.E. Endothelial NOS-deficient mice reveal dual roles for nitric oxide during experimental autoimmune encephalomyelitis. Glia 2009, 57, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Elbim, C. Phagocyte NADPH oxidase: A multicomponent enzyme essential for host defenses. Arch. Immunol. Ther. Exp. 2005, 53, 199–206. [Google Scholar]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Ranaivo, H.R.; Wainwright, M.S. Albumin activates astrocytes and microglia through mitogen-activated protein kinase pathways. Brain Res. 2010, 1313, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, J.L.; Ranaivo, H.R.; Patel, F.; Chrzaszcz, M.; Venkatesan, C.; Wainwright, M.S. Albumin causes increased myosin light chain kinase expression in astrocytes via p38 mitogen-activated protein kinase. J. Neurosci. Res. 2011, 89, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranaivo, H.R.; Hodge, J.N.; Choi, N.; Wainwright, M.S. Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J. Neuroinflamm. 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Ehrich, M. Transient alterations of the blood-brain barrier tight junction and receptor potential channel gene expression by chlorpyrifos. J. Appl. Toxicol. 2013, 33, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- Gundimeda, U.; McNeill, T.H.; Elhiani, A.A.; Schiffman, J.E.; Hinton, D.R.; Gopalakrishna, R. Green tea polyphenols precondition against cell death induced by oxygen-glucose deprivation via stimulation of laminin receptor, generation of reactive oxygen species, and activation of protein kinase Cε. J. Biol. Chem. 2012, 287, 34694–34708. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Tsukamoto, S.; Huang, Y.; Makio, A.; Kumazoe, M.; Yamashita, S.; Tachibana, H. Epigallocatechin-3-O-gallate up-regulates microRNA-let-7b expression by activating 67-kDa laminin receptor signaling in melanoma cells. Sci. Rep. 2016, 6, 19225. [Google Scholar] [CrossRef]
- Barnstable, C.J.; Tombran-Tink, J. Neuroprotective and antiangiogenic actions of PEDF in the eye: Molecular targets and therapeutic potential. Prog. Retin. Eye Res. 2004, 23, 561–577. [Google Scholar] [CrossRef]
- Bouck, N. PEDF: Anti-angiogenic guardian of ocular function. Trends Mol. Med. 2002, 8, 330–334. [Google Scholar] [CrossRef]
- Tombran-Tink, J.; Barnstable, C.J. PEDF: A multifaceted neurotrophic factor. Nat. Rev. Neurosci. 2003, 4, 628–636. [Google Scholar] [CrossRef]
- Becerra, S.P. Focus on Molecules: Pigment epithelium-derived factor (PEDF). Exp. Eye Res. 2006, 82, 739–740. [Google Scholar] [CrossRef]
- Ek, E.T.; Dass, C.R.; Choong, P.F. PEDF: A potential molecular therapeutic target with multiple anti-cancer activities. Trends Mol. Med. 2006, 12, 497–502. [Google Scholar] [CrossRef]
- Zhang, S.X.; Wang, J.J.; Gao, G.; Shao, C.; Mott, R.; Ma, J.X. Pigment epithelium-derived factor (PEDF) is an endogenous antiinflammatory factor. FASEB J. 2006, 20, 323–325. [Google Scholar] [CrossRef]
- Chavan, S.S.; Hudson, L.K.; Li, J.H.; Ochani, M.; Harris, Y.; Patel, N.B.; Katz, D.; Scheinerman, J.A.; Pavlov, V.A.; Tracey, K.J. Identification of pigment epithelium-derived factor as an adipocyte-derived inflammatory factor. Mol. Med. 2012, 18, 1161–1168. [Google Scholar] [CrossRef]
- Li, F.; Armstrong, G.B.; Tombran-Tink, J.; Niyibizi, C. Pigment epithelium derived factor upregulates expression of vascular endothelial growth factor by human mesenchymal stem cells: Possible role in PEDF regulated matrix mineralization. Biochem. Biophys. Res. Commun. 2016, 478, 1106–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.; Zhao, L.; Zhang, D.; Zhang, Q.; Jia, J.; Hu, J.; Huang, Y. Pigment Epithelium-Derived Factor Induces Endothelial Barrier Dysfunction via p38/MAPK Phosphorylation. Biomed. Res. Int. 2015, 2015, 791825. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Zhou, T.; Fang, S.; Jia, M.; Xu, Z.; Dai, Z.; Li, C.; Li, S.; Li, L.; Zhang, T.; et al. Pigment epithelium-derived factor (PEDF) inhibits breast cancer metastasis by down-regulating fibronectin. Breast Cancer Res. Treat. 2014, 148, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, C.; Zhu, Y.; Wang, Y.; Sui, A.; Zhong, Y.; Xie, B.; Shen, X. PEDF mediates pathological neovascularization by regulating macrophage recruitment and polarization in the mouse model of oxygen-induced retinopathy. Sci. Rep. 2017, 7, 42846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalczyk, E.R.; Chen, L.; Fine, D.; Zhao, Y.; Mascarinas, E.; Grippo, P.J.; DiPietro, L.A. Pigment Epithelium-Derived Factor (PEDF) as a Regulator of Wound Angiogenesis. Sci. Rep. 2018, 8, 11142. [Google Scholar] [CrossRef]
- Cai, J.; Jiang, W.G.; Grant, M.B.; Boulton, M. Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J. Biol. Chem. 2006, 281, 3604–3613. [Google Scholar] [CrossRef] [Green Version]
- Notari, L.; Baladron, V.; Aroca-Aguilar, J.D.; Balko, N.; Heredia, R.; Meyer, C.; Notario, P.M.; Saravanamuthu, S.; Nueda, M.L.; Sanchez-Sanchez, F.; et al. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J. Biol. Chem. 2006, 281, 38022–38037. [Google Scholar] [CrossRef] [Green Version]
- Shih, S.C.; Ho, T.C.; Chen, S.L.; Tsao, Y.P. Pigment epithelium derived factor peptide protects murine hepatocytes from carbon tetrachloride-induced injury. PLoS ONE 2016, 11, e0157647. [Google Scholar] [CrossRef] [PubMed]
- Manalo, K.B.; Choong, P.F.; Becerra, S.P.; Dass, C.R. Pigment epithelium-derived factor as an anticancer drug and new treatment methods following the discovery of its receptors: A patent perspective. Expert Opin. Ther. Pat. 2011, 21, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, H.C.; Chang, H.H.; Liu, H.C.; Hsiao, C.H.; Lee, M.J.; Hu, Y.J.; Hung, P.F.; Liu, C.W.; Kao, Y.H. Green tea (-)-epigallocatechin gallate inhibits insulin stimulation of 3T3-L1 preadipocyte mitogenesis via the 67-kDa laminin receptor pathway. Am. J. Physiol. Cell. Physiol. 2009, 297, C121–C132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, H.C.; Liu, H.S.; Hung, P.F.; Chen, C.L.; Liu, H.C.; Chang, H.H.; Tsuei, Y.W.; Shih, L.J.; Lin, C.L.; Lin, C.M.; et al. Green tea (-)-epigallocatechin gallate inhibits IGF-I and IGF-II stimulation of 3T3-L1 preadipocyte mitogenesis via the 67-kDa laminin receptor, but not AMP-activated protein kinase pathway. Mol. Nutr. Food Res. 2012, 56, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Byun, E.B.; Choi, H.G.; Sung, N.Y.; Byun, E.H. Green tea polyphenol epigallocatechin-3-gallate inhibits TLR4 signaling through the 67-kDa laminin receptor on lipopolysaccharide-stimulated dendritic cells. Biochem. Biophys. Res. Commun. 2012, 426, 480–485. [Google Scholar] [CrossRef]
- Wang, Q.M.; Wang, H.; Li, Y.F.; Xie, Z.Y.; Ma, Y.; Yan, J.J.; Gao, Y.F.; Wang, Z.M.; Wang, L.S. Inhibition of EMMPRIN and MMP-9 Expression by Epigallocatechin-3-Gallate through 67-kDa Laminin Receptor in PMA-Induced Macrophages. Cell Physiol. Biochem. 2016, 39, 2308–2319. [Google Scholar] [CrossRef]
- Shi, Z.F.; Zhao, W.J.; Xu, L.X.; Dong, L.P.; Yang, S.H.; Yuan, F. Downregulation of aquaporin 4 expression through extracellular signal-regulated kinases1/2 activation in cultured astrocytes following scratch-injury. Biomed. Environ. Sci. 2015, 28, 199–205. [Google Scholar]
- Qi, L.L.; Fang, S.H.; Shi, W.Z.; Huang, X.Q.; Zhang, X.Y.; Lu, Y.B.; Zhang, W.P.; Wei, E.Q. CysLT2 receptor-mediated AQP4 up-regulation is involved in ischemic-like injury through activation of ERK and p38 MAPK in rat astrocytes. Life Sci. 2011, 88, 50–56. [Google Scholar] [CrossRef]
- Salman, M.M.; Sheilabi, M.A.; Bhattacharyya, D.; Kitchen, P.; Conner, A.C.; Bill, R.M.; Woodroofe, M.N.; Conner, M.T.; Princivalle, A.P. Transcriptome analysis suggests a role for the differential expression of cerebral aquaporins and the MAPK signalling pathway in human temporal lobe epilepsy. Eur. J. Neurosci. 2017, 46, 2121–2132. [Google Scholar] [CrossRef]
- Bloch, O.; Papadopoulos, M.C.; Manley, G.T.; Verkman, A.S. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J. Neurochem. 2005, 95, 254–262. [Google Scholar] [CrossRef]
- Askarova, S.; Yang, X.; Sheng, W.; Sun, G.Y.; Lee, J.C. Role of Aβ-receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase A2 activation in astrocytes and cerebral endothelial cells. Neuroscience 2011, 199, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, H.L.; Wang, H.H.; Wu, C.Y.; Yang, C.M. Reactive Oxygen Species-Dependent c-Fos/Activator Protein 1 Induction Upregulates Heme Oxygenase-1 Expression by Bradykinin in Brain Astrocytes. Antioxid. Redox Signal. 2010, 13, 1829–1844. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Yao, H.; Peng, F.; Yang, Y.; Bethel-Brown, C.; Buch, S. Cooperative induction of CXCL10 involves NADPH oxidase: Implications for HIV dementia. Glia 2010, 58, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, J.; Chakraborti, T.; Chowdhury, A.; Bhuyan, R.; Chakraborti, S. Protective role of epigallocatechin-3-gallate in NADPH oxidase-MMP2-Spm-Cer-S1P signalling axis mediated ET-1 induced pulmonary artery smooth muscle cell proliferation. J. Cell Commun. Signal. 2019, 13, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Wang, H.; Fan, Y.; Shi, H.J.; Wang, Q.M.; Chen, B.R.; Khurwolah, M.R.; Long, Q.Q.; Wang, S.B.; Wang, Z.M.; et al. Epigallocatechin-3-Gallate Inhibits Matrix Metalloproteinase-9 and Monocyte Chemotactic Protein-1 Expression Through the 67-κDa Laminin Receptor and the TLR4/MAPK/NF-κB Signalling Pathway in Lipopolysaccharide-Induced Macrophages. Cell. Physiol. Biochem. 2017, 43, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Yafai, Y.; Lange, J.; Wiedemann, P.; Reichenbach, A.; Eichler, W. Pigment epithelium-derived factor acts as an opponent of growth-stimulatory factors in retinal glial-endothelial cell interactions. Glia 2007, 55, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liu, Z.; Huang, B.; Yuan, Y.; Liu, X.; Zhang, H.; Qiu, F.; Zhang, Y.; Li, Y.; Miao, H.; et al. PEDF improves cardiac function in rats subjected to myocardial ischemia/reperfusion injury by inhibiting ROS generation via PEDF-R. Int. J. Mol. Med. 2018, 41, 3243–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Yamagishi, S.; Matsui, T.; Yoshida, T.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Ueda, S.; Adachi, H.; Imaizumi, T. Pigment epithelium-derived factor inhibits neointimal hyperplasia after vascular injury by blocking NADPH oxidase-mediated reactive oxygen species generation. Am. J. Pathol. 2007, 170, 2159–2170. [Google Scholar] [CrossRef] [Green Version]
- Covolan, L.; Mello, L.E. Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res. 2000, 39, 133–152. [Google Scholar] [CrossRef]
- Sztriha, L.; Joó, F.; Szerdahelyi, P.; Eck, E.; Koltai, M. Effects of dexamethasone on brain edema induced by kainic acid seizures. Neuroscience 1986, 17, 107–114. [Google Scholar] [CrossRef]
- Ding, M.; Haglid, K.G.; Hamberger, A. Quantitative immunochemistry on neuronal loss, reactive gliosis and BBB damage in cortex/striatum and hippocampus/amygdala after systemic kainic acid administration. Neurochem. Int. 2000, 36, 313–318. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, G.A.; Mason, M.; Christie, L.A.; Hansen, C.; Hernandez, L.M.; Burke, J.; Luhrs, K.A.; Hohman, T.C. Functional characterization of Abicipar-Pegol, an anti-VEGF DARPin therapeutic that potently inhibits angiogenesis and vascular permeability. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5836–5846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Noguchi, T.; Inoue, Y.; Sakurai, S.; Nishinaka, A.; Hida, Y.; Masuda, T.; Nakagami, Y.; Horai, N.; Tsusaki, H.; et al. Nrf2 activator RS9 suppresses pathological ocular angiogenesis and hyperpermeability. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Bosma, E.K.; Noorden, C.J.F.V.; Schlingemann, R.O.; Klaassen, I. The role of plasmalemma vesicle-associated protein in pathological breakdown of blood-brain and blood-retinal barriers: Potential novel therapeutic target for cerebral edema and diabetic macular edema. Fluids Barriers CNS 2018, 15, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood brain barrier permeability of (-)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem. Biophys. Rep. 2017, 9, 180–186. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| 67LR | Rabbit | Abcam (#ab133645) | 1:1000 (WB) |
| AKT | Rabbit | Cell signaling (#9272) | 1:1000 (WB) |
| AQP4 | Rabbit | Alomone labs (#AQP-004) | 1:5000 (WB) 1:200 (IH) |
| eNOS | Rabbit | Abcam (ab66127) | 1:1000 (WB) 1:500 (IH) |
| ERK1/2 | Rabbit | Biorbyt (Orb160960) | 1:2000 (WB) |
| GFAP | Mouse | Millipore (#MAB3402) | 1:5000 (IH) |
| p-p38 MAPK | Rabbit | Abbiotec (#251256) | 1:200 (WB) 1:50 (IH) |
| p38 MAPK | Rabbit | Cell signaling (#9212) | 1:1000 (WB) |
| p47Phox | Rabbit | Abbiotec (252159) | 1:1000 (WB) 1:200 (IH) |
| pAKT-T308 | Rabbit | Cell signalling (#9275) | 1:1000 (WB) 1:50 (IH) |
| pERK1/2 | Rabbit | Bioss (bs-3330R) | 1:1000 (WB) 1:100 (IH) |
| PI3K | Rabbit | Cell signaling (#4292) | 1:1000 (WB) |
| pPI3K-Y458 | Rabbit | Cell signaling (#4228) | 1:1000 (WB) |
| Rat IgG | Goat | Vector (#BA-9400) | 1:2000 (WB) 1:200 (IH) |
| SMI-71 | Mouse | Covance (#SMI-71R) | 1:1000 (IH) |
| β-actin | Mouse | Sigma (#A5316) | 1:5000 (WB) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Park, H.; Jeong, M.-J.; Kang, T.-C. Epigallocatechin-3-Gallate and PEDF 335 Peptide, 67LR Activators, Attenuate Vasogenic Edema, and Astroglial Degeneration Following Status Epilepticus. Antioxidants 2020, 9, 854. https://doi.org/10.3390/antiox9090854
Kim J-E, Park H, Jeong M-J, Kang T-C. Epigallocatechin-3-Gallate and PEDF 335 Peptide, 67LR Activators, Attenuate Vasogenic Edema, and Astroglial Degeneration Following Status Epilepticus. Antioxidants. 2020; 9(9):854. https://doi.org/10.3390/antiox9090854
Chicago/Turabian StyleKim, Ji-Eun, Hana Park, Min-Jeong Jeong, and Tae-Cheon Kang. 2020. "Epigallocatechin-3-Gallate and PEDF 335 Peptide, 67LR Activators, Attenuate Vasogenic Edema, and Astroglial Degeneration Following Status Epilepticus" Antioxidants 9, no. 9: 854. https://doi.org/10.3390/antiox9090854