Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration

 ,
, 
 ,
,
Abstract
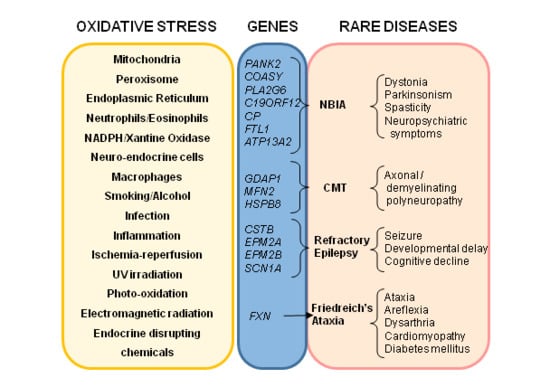
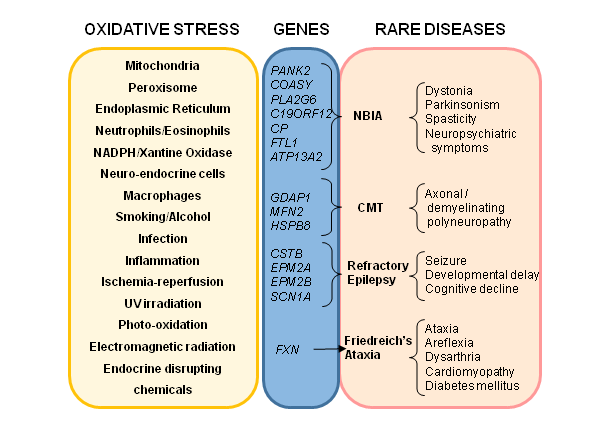
:
1. Introduction
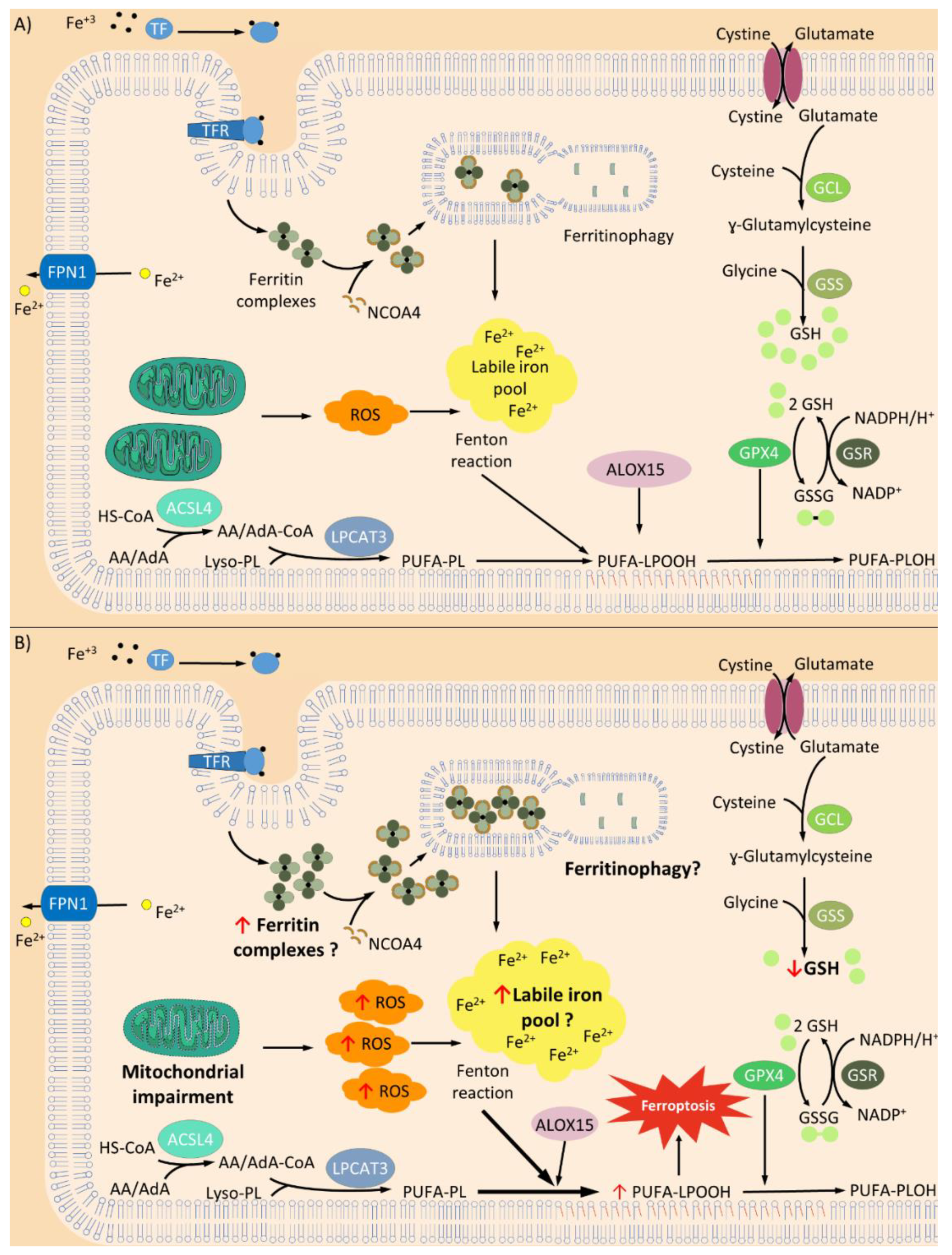
2. Friedreich’s Ataxia
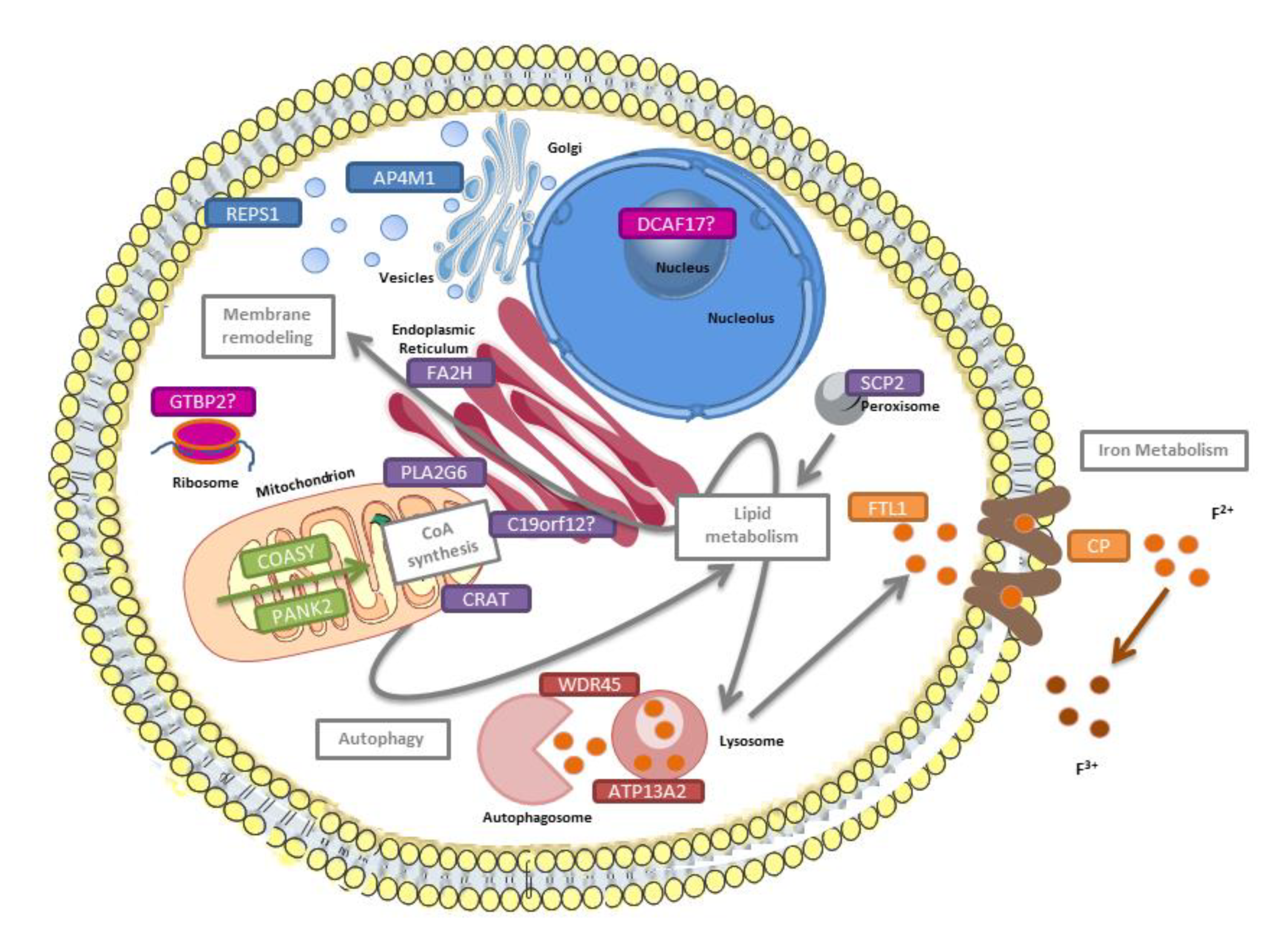
3. Neurodegenerative Disorders with Brain Iron Accumulation
4. Neuromuscular Diseases
5. Inherited Retinal Dystrophies
6. Rare Epilepsies
6.1. Progressive Myoclonus Epilepsies (PMEs)
6.1.1. Unverricht–Lundborg Disease (ULD)
6.1.2. Lafora Disease (LD)
6.2. Dravet Syndrome
6.3. Oxidative Stress and Neuroinflammation as Therapeutic Targets in Rare Epilepsies
7. Conclusions
Funding
Conflicts of Interest
References
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019, 26, 1544–1556. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Bidichandani, S.I.; Delatycki, M.B. Friedreich Ataxia. In GeneReviews®[Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1998; [updated 2017 June 1]. [Google Scholar]
- Musselman, K.E.; Stoyanov, C.T.; Marasigan, R.; Jenkins, M.E.; Konczak, J.; Morton, S.M.; Bastian, A.J. Prevalence of ataxia in children: A systematic review. Neurology 2014, 82, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Delatycki, M.B.; Corben, L.A. Clinical features of Friedreich ataxia. J. Child. Neurol 2012, 27, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Cady, R.B.; Bobechko, W.P. Incidence, natural history, and treatment of scoliosis in Friedreich’s ataxia. J. Pediatr. Orthop. 1984, 4, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E.; Hewer, R.L. The heart disease of Friedreich’s ataxia: A clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q. J. Med. 1983, 52, 489–502. [Google Scholar] [PubMed]
- Korner, A.; Barta, L. Association of diabetes mellitus with Friedreich’s ataxia. Orv. Hetil. 1983, 124, 1391–1392. [Google Scholar]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol 2009, 256, 3–8. [Google Scholar] [CrossRef]
- Sharma, R.; De Biase, I.; Gomez, M.; Delatycki, M.B.; Ashizawa, T.; Bidichandani, S.I. Friedreich ataxia in carriers of unstable borderline GAA triplet-repeat alleles. Ann. Neurol. 2004, 56, 898–901. [Google Scholar] [CrossRef]
- Cossee, M.; Puccio, H.; Gansmuller, A.; Koutnikova, H.; Dierich, A.; LeMeur, M.; Fischbeck, K.; Dolle, P.; Koenig, M. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 2000, 9, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Jasoliya, M.J.; McMackin, M.Z.; Henderson, C.K.; Perlman, S.L.; Cortopassi, G.A. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum. Mol. Genet. 2017, 26, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell Neurosci 2013, 55, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef]
- Martelli, A.; Puccio, H. Dysregulation of cellular iron metabolism in Friedreich ataxia: From primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front. Pharmacol. 2014, 5, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gimenez, J.L.; Gimeno, A.; Gonzalez-Cabo, P.; Dasi, F.; Bolinches-Amoros, A.; Molla, B.; Palau, F.; Pallardo, F.V. Differential expression of PGC-1alpha and metabolic sensors suggest age-dependent induction of mitochondrial biogenesis in Friedreich ataxia fibroblasts. PLoS ONE 2011, 6, e20666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantrel-Groussard, K.; Geromel, V.; Puccio, H.; Koenig, M.; Munnich, A.; Rotig, A.; Rustin, P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 2001, 10, 2061–2067. [Google Scholar] [CrossRef]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [Green Version]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Geng, N.; Shi, B.J.; Li, S.L.; Zhong, Z.Y.; Li, Y.C.; Xua, W.L.; Zhou, H.; Cai, J.H. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals (Basel) 2018, 11, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Codazzi, F.; Hu, A.; Rai, M.; Donatello, S.; Salerno Scarzella, F.; Mangiameli, E.; Pelizzoni, I.; Grohovaz, F.; Pandolfo, M. Friedreich ataxia-induced pluripotent stem cell-derived neurons show a cellular phenotype that is corrected by a benzamide HDAC inhibitor. Hum. Mol. Genet. 2016, 25, 4847–4855. [Google Scholar] [CrossRef] [Green Version]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett 2018, 592, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Planamente, S.; Jornea, L.; Dur, A.; Lesuisse, E.; Camadro, J.M.; Auchere, F. Changes in mitochondrial glutathione levels and protein thiol oxidation in yfh1 yeast cells and the lymphoblasts of patients with Friedreich’s ataxia. Biochim. Biophys. Acta 2012, 1822, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Llorens, J.V.; Soriano, S.; Calap-Quintana, P.; Gonzalez-Cabo, P.; Molto, M.D. The Role of Iron in Friedreich’s Ataxia: Insights from Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 2019, 13, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Bolinches-Amoros, A.; Molla, B.; Pla-Martin, D.; Palau, F.; Gonzalez-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell Neurosci. 2014, 8, 124. [Google Scholar]
- Edenharter, O.; Schneuwly, S.; Navarro, J.A. Mitofusin-Dependent ER Stress Triggers Glial Dysfunction and Nervous System Degeneration in a Drosophila Model of Friedreich’s Ataxia. Front. Mol. Neurosci. 2018, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef]
- Bai, T.; Liang, R.; Zhu, R.; Wang, W.; Zhou, L.; Sun, Y. MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J. Cell Physiol. 2020. [Google Scholar] [CrossRef]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wu, L.; Zhang, P.; Luo, M.; Du, J.; Gao, T.; O’Connell, D.; Wang, G.; Wang, H.; Yang, Y. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol. Carcinog. 2018, 57, 1566–1576. [Google Scholar] [CrossRef]
- Seco-Cervera, M.; Gonzalez-Rodriguez, D.; Ibanez-Cabellos, J.S.; Peiro-Chova, L.; Gonzalez-Cabo, P.; Garcia-Lopez, E.; Vilchez, J.J.; Sanz-Gallego, I.; Pallardo, F.V.; Garcia-Gimenez, J.L. Circulating miR-323-3p is a biomarker for cardiomyopathy and an indicator of phenotypic variability in Friedreich’s ataxia patients. Sci. Rep. 2017, 7, 5237. [Google Scholar] [CrossRef] [PubMed]
- Seco-Cervera, M.; Gonzalez-Rodriguez, D.; Ibanez-Cabellos, J.S.; Peiro-Chova, L.; Pallardo, F.V.; Garcia-Gimenez, J.L. Small RNA-seq analysis of circulating miRNAs to identify phenotypic variability in Friedreich’s ataxia patients. Sci. Data 2018, 5, 180021. [Google Scholar] [CrossRef] [Green Version]
- Dantham, S.; Srivastava, A.K.; Gulati, S.; Rajeswari, M.R. Differentially Regulated Cell-Free MicroRNAs in the Plasma of Friedreich’s Ataxia Patients and Their Association with Disease Pathology. Neuropediatrics 2018, 49, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tello, C.; Darling, A.; Lupo, V.; Pérez-Dueñas, B.; Espinós, C. On the complexity of clinical and molecular bases of neurodegeneration with brain iron accumulation. Clin. Genet. 2018, 93, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals (Basel) 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Bosveld, F.; Rana, A.; van der Wouden, P.E.; Lemstra, W.; Ritsema, M.; Kampinga, H.H.; Sibon, O.C. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum. Mol. Genet. 2008, 17, 2058–2069. [Google Scholar] [CrossRef] [Green Version]
- Perry, T.L.; Norman, M.G.; Yong, V.W.; Whiting, S.; Crichton, J.U.; Hansen, S.; Kish, S.J. Hallervorden-Spatz disease: Cysteine accumulation and cysteine dioxygenase deficiency in the globus pallidus. Ann. Neurol. 1985, 18, 482–489. [Google Scholar] [CrossRef]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; van der Want, J.J.; Hayflick, S.; Reijngoud, D.J.; Kayser, O.; Sibon, O.C. Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; d’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate kinase-associated neurodegeneration: Altered mitochondria membrane potential and defective respiration in Pank2 knock-out mouse model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial iron and energetic dysfunction distinguish fibroblasts and induced neurons from pantothenate kinase-associated neurodegeneration patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanella, A.; Privitera, D.; Guaraldo, M.; Rovelli, E.; Barzaghi, C.; Garavaglia, B.; Santambrogio, P.; Cozzi, A.; Levi, S. Skin fibroblasts from pantothenate kinase-associated neurodegeneration patients show altered cellular oxidative status and have defective iron-handling properties. Hum. Mol. Genet. 2012, 21, 4049–4059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. iPSC-derived neuronal models of PANK2-associated neurodegeneration reveal mitochondrial dysfunction contributing to early disease. PLoS ONE 2017, 12, e0184104. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, X.; Zhao, C.; Choi, J.; Shi, J.; Song, K.; Turk, J.; Ma, Z.A. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology 2010, 151, 3038–3048. [Google Scholar] [CrossRef] [Green Version]
- Venco, P.; Bonora, M.; Giorgi, C.; Papaleo, E.; Iuso, A.; Prokisch, H.; Pinton, P.; Tiranti, V. Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca(2)(+). Front. Genet. 2015, 6, 185. [Google Scholar] [CrossRef] [Green Version]
- Gutteridge, J.M. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loreal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta. 2012, 1820, 403–410. [Google Scholar] [CrossRef]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzi, A.; Rovelli, E.; Frizzale, G.; Campanella, A.; Amendola, M.; Arosio, P.; Levi, S. Oxidative stress and cell death in cells expressing L-ferritin variants causing neuroferritinopathy. Neurobiol. Dis. 2010, 37, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, A.; Santambrogio, P.; Corsi, B.; Campanella, A.; Arosio, P.; Levi, S. Characterization of the l-ferritin variant 460InsA responsible of a hereditary ferritinopathy disorder. Neurobiol. Dis. 2006, 23, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Vidal, R.; Englander, E.W. Accumulation of oxidative DNA damage in brain mitochondria in mouse model of hereditary ferritinopathy. Neurosci. Lett. 2010, 479, 44–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capoccia, S.; Maccarinelli, F.; Buffoli, B.; Rodella, L.F.; Cremona, O.; Arosio, P.; Cirulli, F. Behavioral characterization of mouse models of neuroferritinopathy. PLoS ONE 2015, 10, e0118990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccarinelli, F.; Pagani, A.; Cozzi, A.; Codazzi, F.; Di Giacomo, G.; Capoccia, S.; Rapino, S.; Finazzi, D.; Politi, L.S.; Cirulli, F.; et al. A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiol. Dis. 2015, 81, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Rane, A.; Chinta, S.J.; Andersen, J.K. Regulation of ATP13A2 via PHD2-HIF1alpha Signaling Is Critical for Cellular Iron Homeostasis: Implications for Parkinson’s Disease. J. Neurosci. 2016, 36, 1086–1095. [Google Scholar] [CrossRef]
- Rinaldi, D.E.; Corradi, G.R.; Cuesta, L.M.; Adamo, H.P.; de Tezanos Pinto, F. The Parkinson-associated human P5B-ATPase ATP13A2 protects against the iron-induced cytotoxicity. Biochim. Biophys. Acta 2015, 1848, 1646–1655. [Google Scholar] [CrossRef] [Green Version]
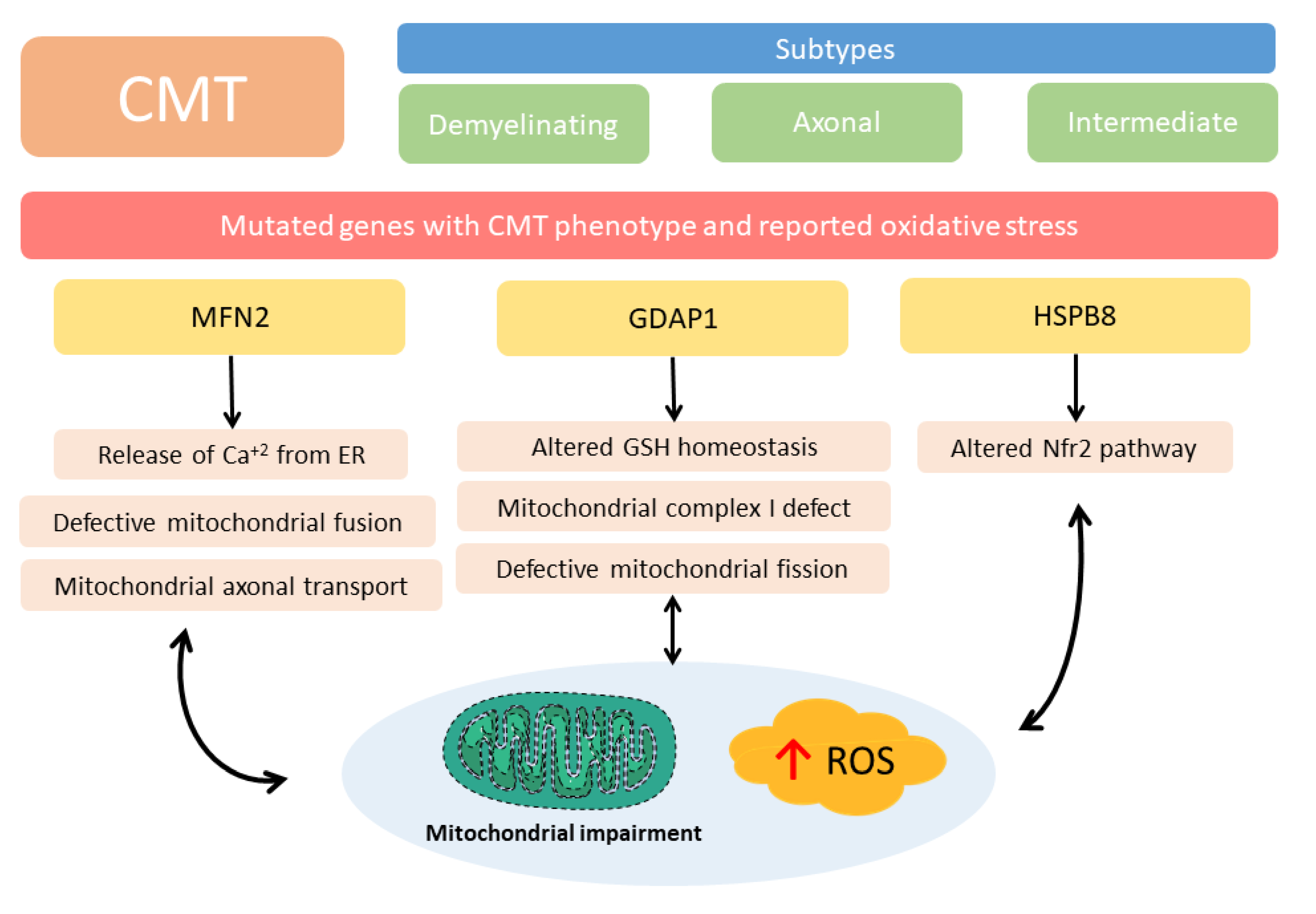
- Cartoni, R.; Martinou, J.C. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp. Neurol. 2009, 218, 268–273. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Langer, T. Mitofusin 2 builds a bridge between ER and mitochondria. Cell 2008, 135, 1165–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacin, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef]
- Pareyson, D.; Saveri, P.; Sagnelli, A.; Piscosquito, G. Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci. Lett. 2015, 596, 66–77. [Google Scholar] [CrossRef]
- Han, S.; Nandy, P.; Austria, Q.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Wang, W.; Zhu, X. Mfn2 Ablation in the Adult Mouse Hippocampus and Cortex Causes Neuronal Death. Cells 2020, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef]
- Marco, A.; Cuesta, A.; Pedrola, L.; Palau, F.; Marin, I. Evolutionary and structural analyses of GDAP1, involved in Charcot-Marie-Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol. Biol. Evol. 2004, 21, 176–187. [Google Scholar] [CrossRef]
- Noack, R.; Frede, S.; Albrecht, P.; Henke, N.; Pfeiffer, A.; Knoll, K.; Dehmel, T.; Meyer Zu Horste, G.; Stettner, M.; Kieseier, B.C.; et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum. Mol. Genet. 2012, 21, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassereau, J.; Chevrollier, A.; Codron, P.; Goizet, C.; Gueguen, N.; Verny, C.; Reynier, P.; Bonneau, D.; Lenaers, G.; Procaccio, V. Oxidative stress contributes differentially to the pathophysiology of Charcot-Marie-Tooth disease type 2K. Exp. Neurol. 2020, 323, 113069. [Google Scholar] [CrossRef] [PubMed]
- Barneo-Munoz, M.; Juarez, P.; Civera-Tregon, A.; Yndriago, L.; Pla-Martin, D.; Zenker, J.; Cuevas-Martin, C.; Estela, A.; Sanchez-Arago, M.; Forteza-Vila, J.; et al. Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of charcot-marie-tooth neuropathy. PLoS Genet. 2015, 11, e1005115. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Lizarbe, S.; Civera-Tregon, A.; Cantarero, L.; Herrer, I.; Juarez, P.; Hoenicka, J.; Palau, F. Neuroinflammation in the pathogenesis of axonal Charcot-Marie-Tooth disease caused by lack of GDAP1. Exp. Neurol. 2019, 320, 113004. [Google Scholar] [CrossRef] [PubMed]
- Lopez Del Amo, V.; Seco-Cervera, M.; Garcia-Gimenez, J.L.; Whitworth, A.J.; Pallardo, F.V.; Galindo, M.I. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: Implications for the Charcot-Marie-Tooth neuropathy. Hum. Mol. Genet. 2015, 24, 21–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pla-Martin, D.; Calpena, E.; Lupo, V.; Marquez, C.; Rivas, E.; Sivera, R.; Sevilla, T.; Palau, F.; Espinos, C. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 2015, 24, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.D.; Cen, Z.D.; Cheng, H.P.; Shi, K.; Bai, J.; Xie, F.; Wu, H.W.; Li, B.B.; Luo, W. L-3-n-Butylphthalide Protects HSPB8 K141N Mutation-Induced Oxidative Stress by Modulating the Mitochondrial Apoptotic and Nrf2 Pathways. Front. Neurosci. 2017, 11, 402. [Google Scholar] [CrossRef]
- Cohen, J. Diseases of the retina: 1975-76 review. Am. J. Optom. Physiol. Opt. 1977, 54, 485–494. [Google Scholar] [CrossRef]
- Bunker, C.H.; Berson, E.L.; Bromley, W.C.; Hayes, R.P.; Roderick, T.H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 1984, 97, 357–365. [Google Scholar] [CrossRef]
- Fishman, G.A. Retinitis pigmentosa. Genetic percentages. Arch. Ophthalmol. 1978, 96, 822–826. [Google Scholar] [CrossRef]
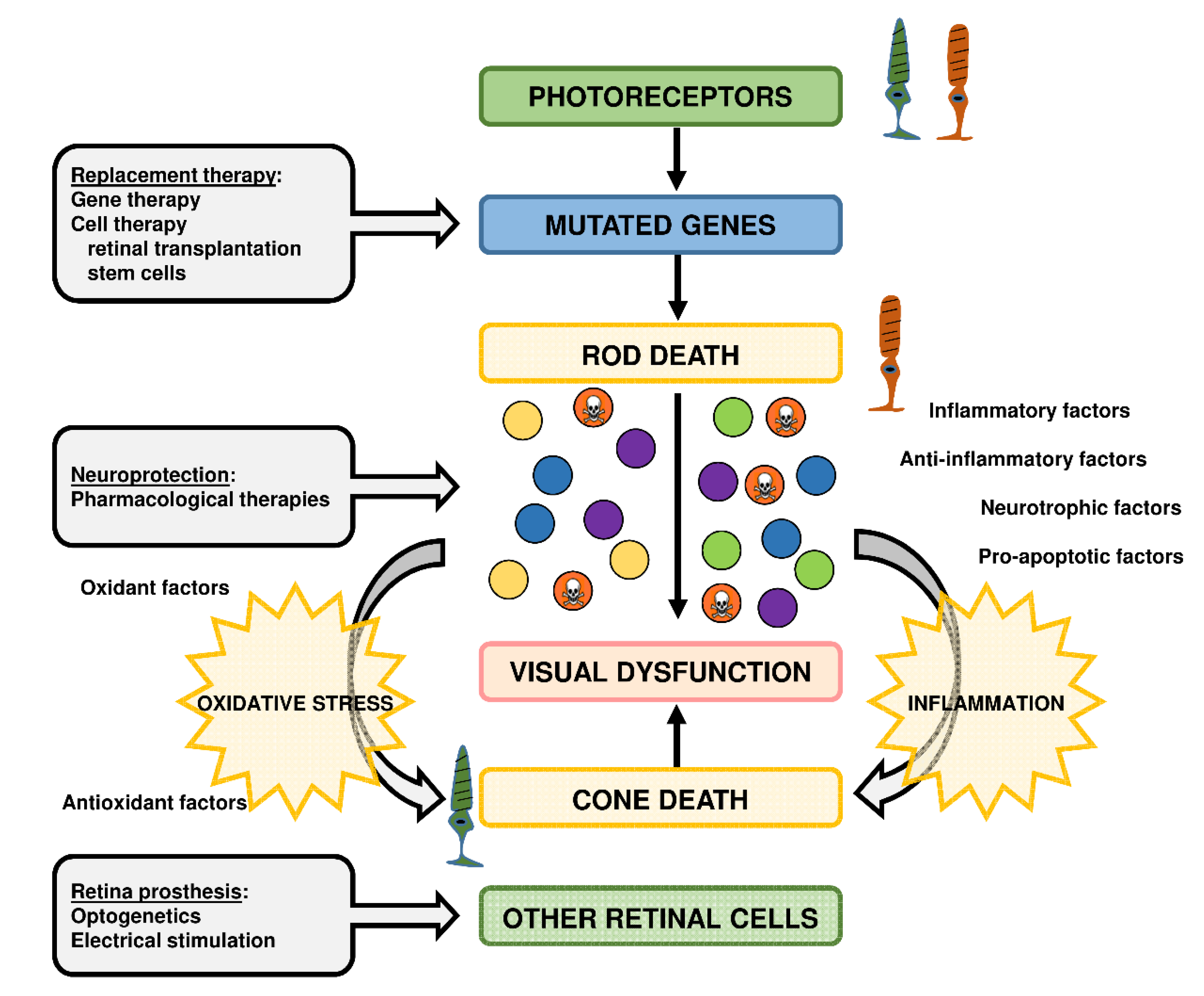
- den Hollander, A.I.; Black, A.; Bennett, J.; Cremers, F.P. Lighting a candle in the dark: Advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Investig. 2010, 120, 3042–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasiou, D.; Aguila, M.; Bevilacqua, D.; Novoselov, S.S.; Parfitt, D.A.; Cheetham, M.E. The cell stress machinery and retinal degeneration. FEBS Lett. 2013, 587, 2008–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Yu, D.Y.; Cringle, S.; Valter, K.; Walsh, N.; Lee, D.; Stone, J. Photoreceptor death, trophic factor expression, retinal oxygen status, and photoreceptor function in the P23H rat. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2013–2019. [Google Scholar] [CrossRef]
- Stone, J.; Maslim, J.; Valter-Kocsi, K.; Mervin, K.; Bowers, F.; Chu, Y.; Barnett, N.; Provis, J.; Lewis, G.; Fisher, S.K.; et al. Mechanisms of photoreceptor death and survival in mammalian retina. Prog. Retin. Eye Res. 1999, 18, 689–735. [Google Scholar] [CrossRef]
- Shen, J.; Yang, X.; Dong, A.; Petters, R.M.; Peng, Y.W.; Wong, F.; Campochiaro, P.A. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J. Cell Physiol. 2005, 203, 457–464. [Google Scholar] [CrossRef]
- Stringham, J.M.; Stringham, N.T. Nitric Oxide and Lutein: Function, Performance, and Protection of Neural Tissue. Foods 2015, 4, 678–689. [Google Scholar] [CrossRef]
- Yu, D.Y.; Cringle, S.J.; Su, E.N.; Yu, P.K. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3999–4006. [Google Scholar]
- Yu, D.Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef]
- Olivares-Gonzalez, L.; Martinez-Fernandez de la Camara, C.; Hervas, D.; Millan, J.M.; Rodrigo, R. HIF-1alpha stabilization reduces retinal degeneration in a mouse model of retinitis pigmentosa. FASEB J. 2018, 32, 2438–2451. [Google Scholar] [CrossRef] [Green Version]
- Eysteinsson, T.; Hardarson, S.H.; Bragason, D.; Stefansson, E. Retinal vessel oxygen saturation and vessel diameter in retinitis pigmentosa. Acta Ophthalmol. 2014, 92, 449–453. [Google Scholar] [CrossRef]
- Zong, Y.; Lin, L.; Yi, C.; Huang, X.; Fu, Y.; Dong, Y.; Qian, X.; Li, Y.; Gao, Q. Retinal vessel oxygen saturation and vessel diameter in retinitis pigmentosa at various ages. Graefes. Arch. Clin. Exp. Ophthalmol. 2016, 254, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez de la Camara, C.; Hernandez-Pinto, A.M.; Olivares-Gonzalez, L.; Cuevas-Martin, C.; Sanchez-Arago, M.; Hervas, D.; Salom, D.; Cuezva, J.M.; de la Rosa, E.J.; Millan, J.M.; et al. Adalimumab Reduces Photoreceptor Cell Death in A Mouse Model of Retinal Degeneration. Sci. Rep. 2015, 5, 11764. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez de la Camara, C.; Salom, D.; Sequedo, M.D.; Hervas, D.; Marin-Lambies, C.; Aller, E.; Jaijo, T.; Diaz-Llopis, M.; Millan, J.M.; Rodrigo, R. Altered antioxidant-oxidant status in the aqueous humor and peripheral blood of patients with retinitis pigmentosa. PLoS ONE 2013, 8, e74223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komeima, K.; Rogers, B.S.; Campochiaro, P.A. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J. Cell Physiol. 2007, 213, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is There Excess Oxidative Stress and Damage in Eyes of Patients with Retinitis Pigmentosa? Antioxid. Redox Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, Y.; Ikeda, Y.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Oka, S.; De Luca, G.; Yonemitsu, Y.; Bignami, M.; Nakabeppu, Y.; et al. MutT homolog-1 attenuates oxidative DNA damage and delays photoreceptor cell death in inherited retinal degeneration. Am. J. Pathol. 2012, 181, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Narayan, D.S.; Wood, J.P.; Chidlow, G.; Casson, R.J. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016, 94, 748–754. [Google Scholar] [CrossRef]
- Bramall, A.N.; Wright, A.F.; Jacobson, S.G.; McInnes, R.R. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu. Rev. Neurosci. 2010, 33, 441–472. [Google Scholar] [CrossRef] [Green Version]
- Komeima, K.; Usui, S.; Shen, J.; Rogers, B.S.; Campochiaro, P.A. Blockade of neuronal nitric oxide synthase reduces cone cell death in a model of retinitis pigmentosa. Free Radic. Biol. Med. 2008, 45, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Perdices, L.; Fuentes-Broto, L.; Segura, F.; Ben Gdara, N.; Sanchez-Cano, A.I.; Insa, G.; Orduna, E.; Pinilla, I. Hepatic oxidative stress in pigmented P23H rhodopsin transgenic rats with progressive retinal degeneration. Free Radic. Biol. Med. 2018, 124, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Mir, T.A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 62, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Ames, A., III; Li, Y.Y.; Heher, E.C.; Kimble, C.R. Energy metabolism of rabbit retina as related to function: High cost of Na+ transport. J. Neurosci. 1992, 12, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Vlachantoni, D.; Bramall, A.N.; Murphy, M.P.; Taylor, R.W.; Shu, X.; Tulloch, B.; Van Veen, T.; Turnbull, D.M.; McInnes, R.R.; Wright, A.F. Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration. Hum. Mol. Genet. 2011, 20, 322–335. [Google Scholar] [CrossRef] [Green Version]
- Maleki, S.; Gopalakrishnan, S.; Ghanian, Z.; Sepehr, R.; Schmitt, H.; Eells, J.; Ranji, M. Optical imaging of mitochondrial redox state in rodent model of retinitis pigmentosa. J. Biomed. Opt. 2013, 18, 16004. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Roux, M.J.; Merl, J.; Giangrande, A.; Hauck, S.M.; Aron, L.; Ueffing, M. Proteomic survey reveals altered energetic patterns and metabolic failure prior to retinal degeneration. J. Neurosci. 2014, 34, 2797–2812. [Google Scholar] [CrossRef] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- McKie, A.B.; McHale, J.C.; Keen, T.J.; Tarttelin, E.E.; Goliath, R.; van Lith-Verhoeven, J.J.; Greenberg, J.; Ramesar, R.S.; Hoyng, C.B.; Cremers, F.P.; et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum. Mol. Genet. 2001, 10, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Vithana, E.N.; Abu-Safieh, L.; Allen, M.J.; Carey, A.; Papaioannou, M.; Chakarova, C.; Al-Maghtheh, M.; Ebenezer, N.D.; Willis, C.; Moore, A.T.; et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol. Cell 2001, 8, 375–381. [Google Scholar] [CrossRef]
- Xu, G.; Li, T.; Chen, J.; Li, C.; Zhao, H.; Yao, C.; Dong, H.; Wen, K.; Wang, K.; Zhao, J.; et al. Autosomal dominant retinitis pigmentosa-associated gene PRPF8 is essential for hypoxia-induced mitophagy through regulating ULK1 mRNA splicing. Autophagy 2018, 14, 1818–1830. [Google Scholar] [CrossRef] [Green Version]
- Peter, V.G.; Nikopoulos, K.; Quinodoz, M.; Granse, L.; Farinelli, P.; Superti-Furga, A.; Andreasson, S.; Rivolta, C. A novel missense variant in IDH3A causes autosomal recessive retinitis pigmentosa. Ophthalmic Genet. 2019, 40, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metodiev, M.D.; Gerber, S.; Hubert, L.; Delahodde, A.; Chretien, D.; Gerard, X.; Amati-Bonneau, P.; Giacomotto, M.C.; Boddaert, N.; Kaminska, A.; et al. Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J. Med. Genet. 2014, 51, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Nagar, S.; Noveral, S.M.; Trudler, D.; Lopez, K.M.; McKercher, S.R.; Han, X.; Yates, J.R., III; Pina-Crespo, J.C.; Nakanishi, N.; Satoh, T.; et al. MEF2D haploinsufficiency downregulates the NRF2 pathway and renders photoreceptors susceptible to light-induced oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, E4048–E4056. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.M.; Ruiz-Lopez, A.M.; Roche, S.L.; Moloney, J.N.; Wyse-Jackson, A.C.; Cotter, T.G. The synthetic progestin norgestrel modulates Nrf2 signaling and acts as an antioxidant in a model of retinal degeneration. Redox Biol. 2016, 10, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, Y.; Hatano, E.; Inoue, T.; Yoshida, K.; Kondo, M.; Terasaki, H. Cytoprotective Effects of a Novel Nrf2 Activator, RS9, in Rhodopsin Pro347Leu Rabbits. Curr. Eye Res. 2016, 41, 1123–1126. [Google Scholar] [CrossRef]
- Campello, L.; Kutsyr, O.; Noailles, A.; Michalska, P.; Fernandez-Sanchez, L.; Martinez-Gil, N.; Ortuno-Lizaran, I.; Sanchez-Saez, X.; de Juan, E.; Lax, P.; et al. New Nrf2-Inducer Compound ITH12674 Slows the Progression of Retinitis Pigmentosa in the Mouse Model rd10. Cell Physiol. Biochem. 2020, 54, 142–159. [Google Scholar]
- Wubben, T.J.; Zacks, D.N.; Besirli, C.G. Retinal neuroprotection: Current strategies and future directions. Curr. Opin. Ophthalmol. 2019, 30, 199–205. [Google Scholar] [CrossRef]
- Benati, D.; Patrizi, C.; Recchia, A. Gene editing prospects for treating inherited retinal diseases. J. Med. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ben M’Barek, K.; Habeler, W.; Regent, F.; Monville, C. Developing Cell-Based Therapies for RPE-Associated Degenerative Eye Diseases. Adv. Exp. Med. Biol. 2019, 1186, 55–97. [Google Scholar] [PubMed]
- Sahaboglu, A.; Miranda, M.; Canjuga, D.; Avci-Adali, M.; Savytska, N.; Secer, E.; Feria-Pliego, J.A.; Kayik, G.; Durdagi, S. Drug repurposing studies of PARP inhibitors as a new therapy for inherited retinal degeneration. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Usui, S.; Zafar, A.B.; Oveson, B.C.; Jo, Y.J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J. Cell Physiol. 2011, 226, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Brockhurst, R.J.; Hayes, K.C.; Johnson, E.J.; Anderson, E.J.; Johnson, C.A.; Gaudio, A.R.; et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch. Ophthalmol. 2010, 128, 403–411. [Google Scholar]
- Berson, E.L. Long-term visual prognoses in patients with retinitis pigmentosa: The Ludwig von Sallmann lecture. Exp. Eye Res. 2007, 85, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Sanz, M.M.; Johnson, L.E.; Ahuja, S.; Ekstrom, P.A.; Romero, J.; van Veen, T. Significant photoreceptor rescue by treatment with a combination of antioxidants in an animal model for retinal degeneration. Neuroscience 2007, 145, 1120–1129. [Google Scholar] [CrossRef]
- Shintani, K.; Shechtman, D.L.; Gurwood, A.S. Review and update: Current treatment trends for patients with retinitis pigmentosa. Optometry 2009, 80, 384–401. [Google Scholar] [CrossRef]
- Emoto, Y.; Yoshizawa, K.; Uehara, N.; Kinoshita, Y.; Yuri, T.; Shikata, N.; Tsubura, A. Curcumin suppresses N-methyl-N-nitrosourea-induced photoreceptor apoptosis in Sprague-Dawley rats. In Vivo 2013, 27, 583–590. [Google Scholar]
- Piano, I.; D’Antongiovanni, V.; Testai, L.; Calderone, V.; Gargini, C. A Nutraceutical Strategy to Slowing Down the Progression of Cone Death in an Animal Model of Retinitis Pigmentosa. Front. Neurosci. 2019, 13, 461. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers. 2018, 4, 18024. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.P.; Patel, M. Seizure-induced changes in mitochondrial redox status. Free Radic. Biol. Med. 2006, 40, 316–322. [Google Scholar] [CrossRef]
- Ravizza, T.; Vezzani, A. Pharmacological targeting of brain inflammation in epilepsy: Therapeutic perspectives from experimental and clinical studies. Epilepsia Open 2018, 3, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalviainen, R. Progressive Myoclonus Epilepsies. Semin Neurol. 2015, 35, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knupp, K.; Wirrell, E. Progressive myoclonic epilepsies: It takes a village to make a diagnosis. Neurology 2014, 82, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Orsini, A.; Valetto, A.; Bertini, V.; Esposito, M.; Carli, N.; Minassian, B.A.; Bonuccelli, A.; Peroni, D.; Michelucci, R.; Striano, P. The best evidence for progressive myoclonic epilepsy: A pathway to precision therapy. Seizure 2019, 71, 247–257. [Google Scholar] [CrossRef]
- Shahwan, A.; Farrell, M.; Delanty, N. Progressive myoclonic epilepsies: A review of genetic and therapeutic aspects. Lancet Neurol. 2005, 4, 239–248. [Google Scholar] [CrossRef]
- Zupanc, M.L.; Legros, B. Progressive myoclonic epilepsy. Cerebellum 2004, 3, 156–171. [Google Scholar] [CrossRef]
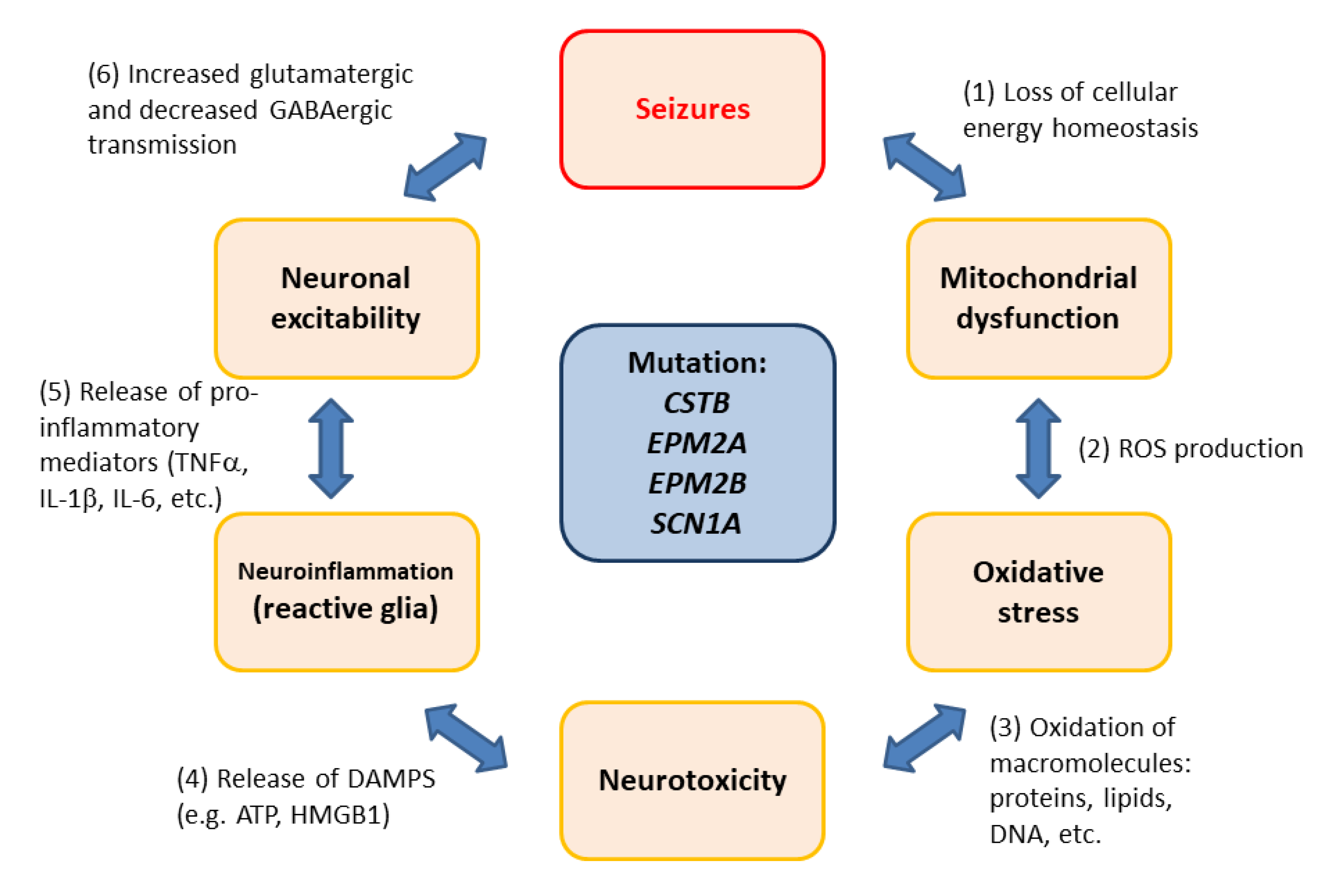
- Joensuu, T.; Tegelberg, S.; Reinmaa, E.; Segerstrale, M.; Hakala, P.; Pehkonen, H.; Korpi, E.R.; Tyynela, J.; Taira, T.; Hovatta, I.; et al. Gene expression alterations in the cerebellum and granule neurons of Cstb−/− mouse are associated with early synaptic changes and inflammation. PLoS ONE 2014, 9, e89321. [Google Scholar] [CrossRef] [Green Version]
- Lahuerta, M.; Gonzalez, D.; Aguado, C.; Fathinajafabadi, A.; Garcia-Gimenez, J.L.; Moreno-Estelles, M.; Roma-Mateo, C.; Knecht, E.; Pallardo, F.V.; Sanz, P. Reactive Glia-Derived Neuroinflammation: A Novel Hallmark in Lafora Progressive Myoclonus Epilepsy That Progresses with Age. Mol. Neurobiol. 2020, 57, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Parviainen, L.; Dihanich, S.; Anderson, G.W.; Wong, A.M.; Brooks, H.R.; Abeti, R.; Rezaie, P.; Lalli, G.; Pope, S.; Heales, S.J.; et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol. Commun. 2017, 5, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespel, A.; Ferlazzo, E.; Franceschetti, S.; Genton, P.; Gouider, R.; Kalviainen, R.; Korja, M.; Lehtinen, M.K.; Mervaala, E.; Simonato, M.; et al. Unverricht-Lundborg disease. Epileptic Disord. 2016, 18, 28–37. [Google Scholar] [CrossRef]
- Lasek-Bal, A.; Lukasik, M.; Zak, A.; Sulek, A.; Bosak, M. Unverricht-Lundborg disease: Clinical course and seizure management based on the experience of polish centers. Seizure 2019, 69, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N. Innate Immune Response in Brain, NF-Kappa B Signaling and Cystatins. Front. Mol. Neurosci. 2015, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gimeno, M.A.; Knecht, E.; Sanz, P. Lafora Disease: A Ubiquitination-Related Pathology. Cells 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.Y.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef]
- Rao, S.N.; Maity, R.; Sharma, J.; Dey, P.; Shankar, S.K.; Satishchandra, P.; Jana, N.R. Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum. Mol. Genet. 2010, 19, 4726–4734. [Google Scholar] [CrossRef] [Green Version]
- Sinadinos, C.; Valles-Ortega, J.; Boulan, L.; Solsona, E.; Tevy, M.F.; Marquez, M.; Duran, J.; Lopez-Iglesias, C.; Calbo, J.; Blasco, E.; et al. Neuronal glycogen synthesis contributes to physiological aging. Aging Cell 2014, 13, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gimenez, J.L.; Seco-Cervera, M.; Aguado, C.; Roma-Mateo, C.; Dasi, F.; Priego, S.; Markovic, J.; Knecht, E.; Sanz, P.; Pallardo, F.V. Lafora disease fibroblasts exemplify the molecular interdependence between thioredoxin 1 and the proteasome in mammalian cells. Free Radic. Biol. Med. 2013, 65, 347–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernia, S.; Rubio, T.; Heredia, M.; Rodriguez de Cordoba, S.; Sanz, P. Increased endoplasmic reticulum stress and decreased proteasomal function in lafora disease models lacking the phosphatase laforin. PLoS ONE 2009, 4, e5907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, R.; Suzuki, T.; Yamakawa, K.; Ganesh, S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 2012, 21, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, P.; Lahuerta, M.; Viana, R.; Knecht, E.; Sanz, P. Regulation of the autophagic PI3KC3 complex by laforin/malin E3-ubiquitin ligase, two proteins involved in Lafora disease. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1867, 118613. [Google Scholar] [CrossRef]
- Garyali, P.; Siwach, P.; Singh, P.K.; Puri, R.; Mittal, S.; Sengupta, S.; Parihar, R.; Ganesh, S. The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum. Mol. Genet. 2009, 18, 688–700. [Google Scholar] [CrossRef]
- Lahuerta, M.; Aguado, C.; Sanchez-Martin, P.; Sanz, P.; Knecht, E. Degradation of altered mitochondria by autophagy is impaired in Lafora disease. FEBS J. 2018, 285, 2071–2090. [Google Scholar] [CrossRef] [Green Version]
- Roma-Mateo, C.; Aguado, C.; Garcia-Gimenez, J.L.; Ibanez-Cabellos, J.S.; Seco-Cervera, M.; Pallardo, F.V.; Knecht, E.; Sanz, P. Increased oxidative stress and impaired antioxidant response in Lafora disease. Mol. Neurobiol. 2015, 51, 932–946. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gonzalez, I.; Viana, R.; Sanz, P.; Ferrer, I. Inflammation in Lafora Disease: Evolution with Disease Progression in Laforin and Malin Knock-out Mouse Models. Mol. Neurobiol. 2017, 54, 3119–3130. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Sharma, S.; Tripathi, M. Diagnosis and management of epileptic encephalopathies in children. Epilepsy Res. Treat. 2013, 2013, 501981. [Google Scholar] [CrossRef]
- Dravet, C. The core Dravet syndrome phenotype. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Gataullina, S.; Dulac, O. From genotype to phenotype in Dravet disease. Seizure 2017, 44, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, R. Dravet syndrome: The main issues. Eur. J. Paediatr. Neurol. 2012, 16, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Claes, L.; Del-Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogiwara, I.; Miyamoto, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K.; et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Avbersek, A.; Bast, T.; Chiron, C.; Guerrini, R.; Kaminski, R.M.; Lagae, L.; Muglia, P.; Cross, J.H. Drug Development for Rare Paediatric Epilepsies: Current State and Future Directions. Drugs 2019, 79, 1917–1935. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Okumura, A.; Uematsu, M.; Imataka, G.; Tanaka, M.; Okanishi, T.; Kubota, T.; Sudo, A.; Tohyama, J.; Tsuji, M.; Ohmori, I.; et al. Acute encephalopathy in children with Dravet syndrome. Epilepsia 2012, 53, 79–86. [Google Scholar] [CrossRef]
- Tang, S.; Lin, J.P.; Hughes, E.; Siddiqui, A.; Lim, M.; Lascelles, K. Encephalopathy and SCN1A mutations. Epilepsia 2011, 52, e26–e30. [Google Scholar] [CrossRef]
- Boguszewicz, L.; Jamroz, E.; Ciszek, M.; Emich-Widera, E.; Kijonka, M.; Banasik, T.; Skorupa, A.; Sokol, M. NMR-based metabolomics in pediatric drug resistant epilepsy—Preliminary results. Sci. Rep. 2019, 9, 15035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, J.; Laan, L.; Klar, J.; Jin, Z.; Huss, M.; Korol, S.; Noraddin, F.H.; Sobol, M.; Birnir, B.; Dahl, N. Transcriptomes of Dravet syndrome iPSC derived GABAergic cells reveal dysregulated pathways for chromatin remodeling and neurodevelopment. Neurobiol. Dis. 2019, 132, 104583. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Mammana, S.; Cavalli, E.; Bramanti, P.; Mazzon, E. Use of Cannabidiol in the Treatment of Epilepsy: Efficacy and Security in Clinical Trials. Molecules 2019, 24, 1459. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; De Filippis, D.; Maiuri, M.C.; De Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea Meira, I.; Romao, T.T.; Pires do Prado, H.J.; Kruger, L.T.; Pires, M.E.P.; da Conceiçao, P.O. Ketogenic Diet and Epilepsy: What We Know So Far. Front. Neurosci. 2019, 13, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, N.; Curatolo, N.; Benoist, J.F.; Auvin, S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 2015, 56, e95–e98. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreasson, K.I.; Bachstetter, A.D.; Colonna, M.; Ginhoux, F.; Holmes, C.; Lamb, B.; Landreth, G.; Lee, D.C.; Low, D.; Lynch, M.A.; et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J. Neurochem. 2016, 138, 653–693. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NBIA form | MIM # | Frequency Inheritance | Gene MIM * | Protein Location | Pathway |
|---|---|---|---|---|---|
| Pantothenate kinase-associated neurodegeneration (PKAN) | 234200 | 35%–50% AR | PANK2 606157 | Mitochondria | CoA synthesis (fatty acid metabolism) |
| Phospholipase 2, group VI-associated neurodegeneration (PLAN) | 610217 | 20% AR | PLA2G6 603604 | Mitochondria, ER, cytosol | Membrane phospholipids turnover |
| Mitochondrial membrane protein-associated neurodegeneration (MPAN) | 614298 | 6%–10% AR | C19ORF12 614297 | Mitochondria, ER, MAM | Lipid metabolism ? Membrane remodeling ? |
| β-propeller-associated neurodegeneration (BPAN) | 300894 | 1%–2% XD | WDR45 300526 | ER | Autophagy |
| Fatty acid hydroxylase-associated neurodegeneration (FA2H) | 612319 | Rare AR | FA2H 611026 | ER | Lipid metabolism Membrane remodeling |
| Neuroferritinopathy (NF) | 606159 | Rare AD | FTL1 134790 | Cytosol | Iron homeostasis |
| Aceruloplasminemia | 604290 | Rare AR | CP 117700 | Plasma membrane | Iron homeostasis |
| Woodhouse-Sakati syndrome | 241080 | Rare AR | DCAF17 612515 | Nucleolus | Unknown |
| Kufor-Rakeb syndrome | 606693 | 2 probands AR | ATP13A2 610513 | Lysosome, mitochondria | Autophagy |
| COASY protein-associated neurodegeneration (CoPAN) | 615643 | 4 probands AR | COASY 609855 | Mitochondria, cytosol | CoA synthesis (fatty acid metabolism) |
| Jaberi-Elahi syndrome (JABELS) + NBIA | 617988 | 1 family AR | GTPBP2 607434 | Cytoplasm | Unknown |
| Leukoencephalopathy with dystonia and motor neuropathy + NBIA | 613724 | 1 proband AR | SCP2 184755 | Peroxisomes | Lipid metabolism Membrane remodeling |
| NBIA7 | 617916 | 1 family AR | REPS1 614825 | Cytoplasm, endosome | Endocytosis Vesicle transport |
| Hereditary spastic paraplegia + NBIA | 612936 | 1 family AR | AP4M1 602296 | Endosome | Vesicle formation |
| NBIA8 | 617917 | 1 proband AR | CRAT 600184 | Mitochondria | Lipid metabolism |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinós, C.; Galindo, M.I.; García-Gimeno, M.A.; Ibáñez-Cabellos, J.S.; Martínez-Rubio, D.; Millán, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration. Antioxidants 2020, 9, 313. https://doi.org/10.3390/antiox9040313
Espinós C, Galindo MI, García-Gimeno MA, Ibáñez-Cabellos JS, Martínez-Rubio D, Millán JM, Rodrigo R, Sanz P, Seco-Cervera M, Sevilla T, et al. Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration. Antioxidants. 2020; 9(4):313. https://doi.org/10.3390/antiox9040313
Chicago/Turabian StyleEspinós, Carmen, Máximo Ibo Galindo, María Adelaida García-Gimeno, José Santiago Ibáñez-Cabellos, Dolores Martínez-Rubio, José María Millán, Regina Rodrigo, Pascual Sanz, Marta Seco-Cervera, Teresa Sevilla, and et al. 2020. "Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration" Antioxidants 9, no. 4: 313. https://doi.org/10.3390/antiox9040313