Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells


Systems Medicine, School of Medicine, University of Dundee, Dundee, Scotland DD1 9SY, UK
*
Author to whom correspondence should be addressed.
Antioxidants 2019, 8(8), 315; https://doi.org/10.3390/antiox8080315
Submission received: 29 June 2019
/
Revised: 10 August 2019
/
Accepted: 12 August 2019
/
Published: 16 August 2019
(This article belongs to the Special Issue Post-Translational Protein Modifications in Oxidative Stress)
Abstract
:Oxidative post-translational modifications (oxPTM) of receptors, enzymes, ion channels and transcription factors play an important role in cell signaling. oxPTMs are a key way in which oxidative stress can influence cell behavior during diverse pathological settings such as cardiovascular diseases (CVD), cancer, neurodegeneration and inflammatory response. In addition, changes in oxPTM are likely to be ways in which low level reactive oxygen and nitrogen species (RONS) may contribute to redox signaling, exerting changes in physiological responses including angiogenesis, cardiac remodeling and embryogenesis. Among oxPTM, S-glutathionylation of reactive cysteines emerges as an important regulator of vascular homeostasis by modulating endothelial cell (EC) responses to their local redox environment. This review summarizes the latest findings of S-glutathionylated proteins in major EC pathways, and the functional consequences on vascular pathophysiology. This review highlights the diversity of molecules affected by S-glutathionylation, and the complex consequences on EC function, thereby demonstrating an intricate dual role of RONS-induced S-glutathionylation in maintaining vascular homeostasis and participating in various pathological processes.
1. Introduction
Covering vascular lumen walls, endothelial cells (ECs) are a central component building the interface between blood and underlying tissue. ECs act on multiple processes to ensure the maintenance of systemic vascular homeostasis, including the regulation of blood pressure, haemostasis, tissue vascularization and inflammation. Endothelial dysfunction is thus involved in a wide range of pathologies, especially cardiovascular diseases (CVDs) such as diabetes, hypertension and peripheral arterial disease. Importantly, the direct contact of ECs with blood circulation makes them highly sensitive to local oxygen levels. The role of oxidative stress and high levels of reactive oxygen and nitrogen species (RONS), resulting from ischemia/reperfusion, is now widely recognized in the onset of CVDs [1,2]. RONS alter EC functions via inducing post-translational modifications (PTM) on major signalling proteins involved in the main physiological processes. Those modifications significantly modulate protein structure and activity, which highlights the potential importance of local redox environment for driving EC functions in both physiological and pathological processes. Among those modifications, S-glutathionylation emerges as an important modulator of EC behavior under oxidative stress.
The harmful effects of oxPTM have been widely related to pathological processes. However, attempts to develop antioxidant therapies to reduce oxidative damages and resolve CVDs have not met the success expected [3]. Moreover, some antioxidant molecules reversing oxPTM, and more precisely S-glutathionylation, recently appeared to impair key EC functions under oxidative stress such as post-ischemia revascularization [4]. More generally, antioxidants appear to contribute to pathological processes such as lung diseases [5,6]. Further evidence for antioxidant treatment leading to adverse effects comes from a randomized clinical trial for antioxidant use in pregnancy disorder, preeclampsia. This trial reported a surprising secondary outcome, in which antioxidant treatment (Vitamin C and E) caused lower birth weight with no protection against preeclampsia [7]. Taken together these findings reveal an essential underpinning role of basal RONS levels in the maintenance of vascular homeostasis. Our understanding of how S-glutathionylation affects EC function is developing. However, the precise implications of this modification in physiological processes are not fully understood, and often lead to contradictory findings. This complexity underlies a double-edge sword role of S-glutathionylation in response to oxidative stress and suggests an example of how an ox-PTM can contribute to both physiological and pathological processes by altering interlinked signaling pathways. It is then of critical importance to fully understand the mechanisms by which oxPTM like S-glutathionylation can influence EC functions to better appreciate the complex relation between oxidative stress and CVDs, and potentially rethink our therapeutic approaches.
In this context, this review will summarize the latest insights on the role of glutathionylated proteins in the regulation of major EC pathways, and their implications in the main physiological and pathological processes linked with vascular homeostasis.
2. Protein S-Glutathionylation
After translation, proteins undergo various modifications including palmitoylation, phosphorylation or hydroxylation, which alter protein function through changes in cellular location, activity or stability. Cysteinyl residues are especially sensitive to levels of oxygen and nitrogen species (RONS), resulting in a range of oxidative post-translational modifications (PTM) including S-nitrosylation, S-sulfenylation and S-glutathionylation.
Under physiological RONS levels, the antioxidant molecule glutathione (GSH) can bind to specific reactive cysteinyl residues via the creation of reversible disulfide bonds depending on the cysteine position and redox potential. This process of S-glutathionylation can be reversed by thiol modifying enzymes, predominantly glutaredoxin (Grx), and appears as a protective mechanism against permanent protein damage following irreversible cysteine oxidation, summarized in Figure 1.
The different molecular mechanisms involved in S-glutathionylation PTM have been extensively reviewed [8,9]. Beyond its protective functions, S-glutathionylation can directly modulate the structure and activity of redox sensitive proteins by modulating their global molecular mass and charge, or preventing other molecules to bind to their active sites [10]. Targeting a wide-range of proteins involved in key cellular processes acting from transcriptional, structural to post-translational levels, S-glutathionylation is now emerging as a key regulator of major signaling pathways in combination with other posttranslational regulatory mechanisms [11,12]. In this review, we will focus on the literature demonstrating S-glutathionylation involvement in endothelial signaling pathways and thus its importance in cardiovascular diseases. The wide range of proteins with redox sensitive thiols involved in EC function, and the impact of S-glutathionylation on the corresponding protein activity are summarized in Table 1. This gives an overview of the breadth and impact that S-glutathionylation can have on EC function.
3. Role of Endothelium in Vascular Physiology
Endothelial cells (ECs) are a specialized tissue lining the lumen of all blood vessels. At the interface between circulating blood and underlying tissues, ECs have multiple functions (Figure 2) including: (a) maintenance of a selective permeability barrier between both compartments to coordinate the passage of macromolecules, ions or signaling molecules through endothelial junctions or intracellular clefts; (b) maintenance of hemostasis through tight spatial and temporal interplays between pro-thrombotic, anti-coagulant, anti-platelet and fibrinolytic activities and the regulation of blood cell-vessel wall interactions; (c) involvement in innate and adaptive immune responses and inflammation; (d) modulation of vascular smooth muscle tone and thus blood flow distribution and control of blood pressure; (e) formation of new blood vessels, or angiogenesis, through the regulation of vessel wall cell proliferation and apoptosis; and (f) a contribution to the maintenance of a quiescent, differentiated vascular smooth muscle phenotype. This broad array of EC functions is ensured by multiple actors involved in orchestrated signalling pathways to maintain vascular homeostasis (Figure 2). Figure 2 depicts some of the key proteins involved in EC function, which are modification by S-glutathionylation that will be reviewed herein. Since ECs are found in all organs, a proper interplay between these pathways is essential to a wide range of physiological functions. In contrast, EC activation or dysfunction can lead to various pathologies. Endothelial activation is a term used to describe reversible phenotypical changes of the endothelium, notably an increase in adhesion molecule expression, and includes a wide spectrum of events such as variations in signaling cascades, transcription factors, gene expression and the cytoskeleton. Endothelial dysfunction entails pathophysiological dysregulation, which is at the extreme range of endothelial activation where it becomes permanent or maladaptive, contributing to the development of pathological conditions. Endothelial dysfunction is often present many years before the manifestation of clinical disease symptoms, for example in the case of type-2 diabetes and Alzheimer’s disease.
4. Transcription Regulation by S-Glutathionylation: Epigenetics Regulators and Transcription Factors
4.1. Epigenetic Regulators
The importance of epigenetics in EC function and related complex diseases is meeting a growing interest, highlighted by the recent emergence of whole genome studies. The modification of histone methylation state plays a key role in EC function by acting upstream of gene transcription, and its implications in cardiovascular pathologies [76]. Importantly, epigenetics appears as an essential player in EC progenitor function, therefore regulating vascular repair processes [77,78]. Histone modifications are particularly important for regulating the expression of key EC genes as the endothelial nitric oxide synthase (eNOS) and the vascular endothelial growth factor 1 (VEGFR1), therefore playing a pivotal role in vascular homeostasis and angiogenesis (Figure 3a) [79,80,81]. Oxidative stress is known to alter chromatin structure, and S-glutathionylation occurs in histones and histone-modifying enzymes [82]. However, how S-glutathionylation directly relates to epigenetic-induced onset of cardiovascular diseases (CVD) warrants further assessment since the following factors undergo oxPTM.
4.1.1. Histone Proteins
In eukaryotes, the chromatin is packaged in individual units called nucleosomes. Histones, the building bricks of nucleosomes, are subject to various PTM which directly impact the chromatin structure and, as a result, gene expression [83]. Although cysteine residues have been identified in histone H3 [84], studies on redox regulation processes such as S-glutathionylation on histones have been halted by challenging technical requirements. Only recently has S-glutathionylation been identified on histone H3 in cancerous cells [13]. S-glutathionylation of histone H3 has been confirmed and further characterized by Olaso et al., who identified a target cysteine residue on the protein [14]. Despite altering the nucleosome structure and, therefore, chromatin compaction, the exact functional implications of S-glutathionylation on chromatin structure are not widely appreciated. While Garcia et al. suggested that histone H3 S-glutathionylation opens chromatin structure and promotes gene expression [14], another study claimed opposite effects [85]. These discrepancies may be due to the difficulty in studying highly reversible oxidative modifications. Further studies are required to elucidate the functional consequences of histone H3 S-glutathionylation on gene expression, and the potential existence of other S-glutathionylation modifications on other reactive Cys in histone H3 previously identified [14,84].
4.1.2. Histone-Modifying Enzymes
The effect of S-glutathionylation on the modulation of chromatin structure can be appreciated indirectly, via the modification of histone-modifying enzymes. Particularly, RONS are known to decrease histone deacetylase (HDAC) activity, notably via nitrosylation, favoring gene transcription [86,87]. However, the role of S-glutathionylation in the regulation of histone acetylation state and implications in EC physiology is only starting to be elucidated with sirtuins. This family of proteins plays an important role in the regulation of endothelial cell homeostasis via deacetylation of multiple targets [88]. In addition to its role in angiogenesis and diastolic functions [89,90], sirtuin-1 (SIRT1), also called “longevity protein”, appears to play a protective role in EC response to oxidative stress by reducing RONS levels. While p65 gene deacetylation by SIRT1 inhibits nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling [91,92], the same process enhances eNOS activity, therefore promoting nitric oxide (NO) production and vasodilation while reducing RONS production [93,94]. Three cysteines have been identified as susceptible to thiol modifications on SIRT1 [15], and the presence of S-glutathionylation has been confirmed [16]. SIRT1 S-glutathionylation appears to inhibit enzymatic activity by altering protein structure and binding to nicotinamide adenine dinucleotide (NAD+), leading to cell senescence and apoptosis (Figure 3a) [17].
4.2. Transcription Factors
S-glutathionylation regulates various transcription factors in ECs, most of the time by inhibiting their activity and, therefore, silencing the expression of downstream targets (Figure 3a). Interestingly, some exceptions were reported where this modification had opposite effects, enhancing gene expression. The redox regulation of transcription factors illustrates the complexity of S-glutathionylation acting to switch effects in multiple signaling pathways and its potential clinical implications.
4.2.1. S-Glutathionylation-Mediated Inhibition of Transcription Factors
NFκB transcription factor complex is mostly known for its pro-inflammatory properties, characterized in ECs by a promotion of vascular permeability and leukocyte recruitment [95]. Besides promoting inflammation, NFκB also plays a key role in the regulation of angiogenesis and cell survival, notably via Wnt5a signaling pathway [4,96].
S-glutathionylation regulates various components of the NFκB pathway. NFκB major subunits p50 and p65 activity are inhibited by S-glutathionylation, preventing DNA-binding or nuclear translocation and downstream gene transcription [18,19]. S-glutathionylation was also reported in the protein kinase IKKb, which impaired its kinase activity and, therefore, further promotes the p65/p50 sequestration in the cytosol [31]. The consequences of S-glutathionylation-induced inhibition of NFκB have been demonstrated in various inflammatory diseases and have been extended to ECs. Recent studies highlighted an essential role of this process for neovascularization response. This is in accordance with the recognized antiangiogenic properties of NFκB signaling via the production of the antiangiogenic factor soluble Vascular growth factor receptor 1 (sVEGFR1) (Figure 3a). [42,43]. Previous studies, however, suggested that NFκB inhibition by S-glutathionylation in mice fibroblasts promoted cell hypoxic apoptosis [44].
As a component of the AP-1 transcription factor complex, c-Jun protein promotes cell proliferation and angiogenesis in ECs. However, its precise roles in cell survival are still controversial. The signaling cascade of c-Jun is known to be susceptible to redox regulation [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100]. Displaying a particularly high redox potential, S-glutathionylation of its Cys269 residue has been extensively reported (Figure 3a) [20,21]. This oxPTM inhibits the DNA binding of c-jun, with the functional consequence in ECs and may contribute to invasive breast cancer and/or pulmonary hypertension [43,100].
p53 is gatekeeper of a wide range of cellular pathways, mostly known for its antitumorigenic properties preventing abnormal cells proliferating. However, the role of p53 in non-cancerous cells such as ECs has recently attracted interest due to its crucial implications in angiogenesis, apoptosis and vascular dysfunction [101]. p53 S-glutathionylation inhibition of DNA binding and protein dimerization has been reported in cancerous cells [22], having potential implications in EC function and vascular pathologies associated with defects in angiogenesis regulation.
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a basic redox-regulated leucine zipper protein, is another major transcriptional factor that plays fundamental roles in EC functions including essential anti-angiogenic activity. In response to oxidative stress, Nrf2 mediates antioxidant and anti-inflammatory signals providing crucial cytoprotective effects in many cells types including the endothelium(Figure 3a) [102]. The range of cytoprotective proteins induced by Nrf2 activation include NADPH quinone oxidoreductase 1, sulfiredoxin 1, Heme oxygenase-1 and glutathione S-transferases. In addition, Nrf2 regulates angiogenesis post ischemia [103]. A novel role of S-glutathionylation in modulating Nrf2 activity was recently reported in various cell types, where S-glutathionylation of its inhibitor Keap1 results in Nrf2 nuclear translocation and expression of its downstream targets [29,30]. Those findings highlight the importance of S-glutathionylation in cell response to oxidative stress.
4.2.2. S-Glutathionylation-Mediated Activation of Transcription Factors
Hypoxia-inducible factor 1-alpha (HIF-1α) is the major regulator of oxygen homeostasis in ECs, and promotes angiogenic signaling in response to hypoxia [104]. Alterations in the protein activity, therefore, play a crucial role in severe pathological states associated with ischemia [105]. The role of HIF-1a S-glutathionylation was extensively studied in cancerous cells. In the hypoxic tumor environment, this modification stabilizes the protein and, therefore, promotes tumor growth and angiogenesis (Figure 3a) [23]. Accordingly, the promotion of S-glutathionylation of HIF-1α in endothelial cells was found to accelerate ischemic revascularization via VEGF-A production in animal models [24], showing important implications in the mechanism of recovery from ischemic injury.
4.2.3. Opposite Effects of S-Glutathionylation on Various Signal Transducer and Activator of Transcription (STAT) Proteins
The JAK/STAT signaling pathway is activated in response to cytokines and growth factors and stimulates a wide range of physiological processes including inflammation, apoptosis, differentiation, cell migration and proliferation. In this pathway, Signal Transducer and Activator of Transcription (STAT) family proteins are intracellular transcription factors phosphorylated by JACK, and translocated to the nucleus to promote gene expression (Figure 3a) [106]. These include STAT1 and STAT3, activated not only by cytokine receptors but also by Vascular Endothelial Growth Factor (VEGF) signaling in endothelial cells. STAT pathway, therefore, plays a role in angiogenic response and cell growth [107,108]. Beyond their role in angiogenesis, STATs are highly important in promoting EC inflammatory response, and are known to play a role in atherosclerosis progression [109,110].
S-glutathionylation of STAT3 prevents phosphorylation inhibiting DNA-binding capacities [25], and a more recent study identified two reactive cysteine residues subject to S-glutathionylation in STAT3 structure [26]. Carbon monoxide (CO)-mediated S-glutathionylation of the reactive cysteines under mild oxidative conditions exerts a cytoprotective mechanism, modulating pro-inflammatory signals in ECs [27]. S-glutathionylation of STAT1 was observed in microglial cells. Interestingly, the effects of this modification appear contrary to the ones observed with STAT3, as STAT1 phosphorylation was not impaired by S-gluathionylation and activity was enhanced [26]. STAT3 and STAT1 have been shown to have antagonistic effects on angiogenesis and cell proliferation in endothelial cells [111,112]. Although inducing opposite effects on the two STAT proteins, S-glutathionylation seems to exhibit a general inhibitory role on JACK-STAT-mediated angiogenesis.
5. Redox Control of Phosphorylation by S-Glutathionylation: Phosphatases, GTPases and Kinases
Protein phosphorylation is a pivotal post-translational modification regulating cell signaling pathways. Phosphatase and kinase enzymes regulate the phosphorylation state of major proteins in EC signaling as receptors and downstream targets (Figure 3a). While cysteine oxidation is generally considered as an inhibitor of phosphatase activity, the effects on kinases requires further investigation [113,114]. The example of S-glutathionylation seems to follow this pattern and could bring novel insights into the redox regulation of phosphorylation in key EC signaling pathways and effects of oxidative stress in pathophysiological conditions.
5.1. Phosphatases
Protein tyrosine phosphatases (PTP) down-regulate key receptors by dephosphorylating kinase domains.
Low-molecular-weight protein tyrosine phosphatase (LMW-PTP) family proteins inhibit growth factors regulating cell growth and angiogenesis, but also inhibit focal adhesion kinase regulating cell migration. LMW-PTP, characterized by the presence of two reactive cysteines located in their catalytic domain, are widely recognized as a redox-dependent “molecular switche” in many cell types [115,116,117]. More specifically, S-glutathionylation was shown to inhibit LMW-PTP phosphatase activity in ECs and, therefore, appears essential for promoting angiogenesis and EC migration [32]. This same study demonstrated that this modification was mediated by VEGF-dependent ONOO-, highlighting its role in a negative feedback loop central to VEGF signaling.
PTP1B is a major regulator of EC proliferation through its binding and inactivation of the VEGFR2 receptor. Its phosphatase activity on tyrosine domains also applies to the VE-cadherin receptor, stabilizing cell–cell adhesion and, therefore, limiting vascular permeability [118]. S-glutathionylation inhibits PTP1B activity [33,34]. Although the functional consequences of this modification were not studied in directly in ECs, mice lacking PTP1B display enhanced angiogenic and revascularization capacities as well as an increased cardiac perfusion following myocardial infarction [119]. Further studies are needed to investigate the potential implications of PTP1B inhibition by S-glutathionylation in cardiovascular diseases related to VEGF and VE-cadherin signaling. S-glutathionylation could also appear as a key redox switch for the regulation of other PTPs such as HCPTPA, also involved in VEGF receptor inhibition [120].
5.2. GTPases
GTPases, hydrolyze GTP to provide the energy required for central physiological processes, play an important role in the regulation of vascular permeability and RONS production. Defects in vascular barrier function are a hallmark of vascular dysfunction, promoting leukocyte transmigration and chronic inflammation, and have been associated with various severe pathologies such as diabetes [121,122] or pulmonary disorders [123]. Enhanced RONS and S-glutathionylation levels appear to promote vascular permeability, suggesting redox control [35,123]. However, which components are S-glutathionylated and how this process affects vascular permeability is yet to be described in detail.
Small Rho GTPases act in a complex interplay to modulate the dynamics of the actin cytoskeleton, a central feature for EC barrier function and migration [124,125]. GTPases are recognized as redox-sensitive and generally inhibited by oxidative stress [38]. Among them, Ras-related C3 botulinum toxin substrate 1 (Rac1) S-glutathionylation was recently shown to alter the enzyme function, resulting in loss of cortical actin structure, increased stress fibers and cell–cell adherens junction disassembly [35]. On the contrary, another study evidenced the opposite effect on Rac2, on which S-glutathionylation appeared to enhance GTP-binding activity [37]. The Ras subfamily of GTPases S-glutathionylation was observed in ECs and smooth muscle cells (SMC). However, the biological effects on nucleotide exchange and protein activity remain controversial [87,89,90].
Beyond the regulation of vascular barrier integrity, further understanding of Rho GTPase redox regulation could provide important insights on EC response to oxidative stress through NADPH oxidase and eNOS activation [36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126] and, therefore, angiogenesis and cell migration [127].
5.3. Kinases
Kinase proteins are now widely recognized as redox-regulated, and more specifically subject to inhibition by S-glutathionylation due to the presence of a reactive cysteine residue in their catalytic domain [40]. It is notably the case for protein kinase B (PKB), which activity was maintained by Grx in various studies [42,43]. PKA, PKC and mitogen-activated protein kinase (MEK) are other examples of S-glutathionylation-induced inhibition of kinase activity [45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98].
Although precise biological implications of protein kinase redox regulation have not been extensively explored in ECs, the results showing that their enzymatic activities are importantly altered by S-glutathionylation could have crucial impacts for EC physiology. Indeed, the role of various kinases in key EC functions, especially the regulation of vascular barrier function and blood pressure, is well established [128,129].
6. S-Glutathionylation Effects on RONS Production
6.1. NADPH Oxidase Complex
In ECs, the major source of RONS is a NADPH oxidase (NOX) homologue. Upon activation by VEGF, ET-1, Angiotensin II (AngII) or Transforming Growth Factor β (TGFβ), the production of superoxide (O2−) and hydrogen peroxide from NOX regulates multiple redox-dependent pathways, especially those involved in the regulation of vascular tone and angiogenesis through NO inhibition [130,131,132] (Figure 3a). As a result, NOX-dependent RONS expression induces endothelial dysfunction and various cardiovascular pathologies such as hypertension, diabetes and cardiac failure [4,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135]. The activation of the catalytic transmembrane unit relies on interactions between several cytosolic proteins, including cytosolic phox subunits and Rac1 [136,137]. In addition to Rac1 S-glutathionylation mentioned previously, p47phox S-glutathionylation was reported in neutrophils on three cysteines, in which the modification appears essential to drive sustained O2− generation [46]. Interestingly, S-glutathionylation did not alter the protein phosphorylation state of p47phox, essential for its activation. As p47phox was identified as a key factor for RONS production in ECs stimulated by TNF-α [138], p47phox S-glutathionylation could then appear as an upstream redox switch promoting endothelial dysfunction through multiple target oxidation. However, further studies are needed to confirm that a similar effect is observed in EC despite some variations in vascular NOX properties compared to phagocytic cells [139]. A similar effect was observed on mitochondrial Complex I, another major source of O2− in EC (Figure 3b) [47,140].
6.2. Endothelial Nitric Oxide Synthase System
eNOS is the major source of NO in ECs. This soluble gasotransmitter plays a central role in vascular homeostasis by regulating vascular tone, angiogenesis and maintenance of barrier integrity. The effects of RONS on eNOS activity are now generally recognized. Under oxidative stress, eNOS adopts a NADPH oxidase function and switches from NO to O2− generation, which is a hallmark of multiple cardiovascular pathologies including atherosclerosis [141]. The role of eNOS S-glutathionylation in this process has recently met a growing interest. eNOS S-glutathionylation at the reductase site alters electron transfer and, therefore, uncouples eNOS, promoting O2− over NO generation [48]. Since then, the role of S-glutathionylation has been further assessed [142] and shown in different pathologies such as hypoxia/reoxygenation injury [49] or necrotizing enterocolitis [50]. Importantly, eNOS S-glutathionylation is NADPH oxidase dependent [51,52], potentially contributing to NO inhibition resulting from O2− production by NOS (Figure 3a).
7. S-Glutathionylation Effects on Ca2+ Homeostasis
Major functions of EC including angiogenesis, cell migration and growth, all depend on cytosolic calcium ion (Ca2+) concentrations ([Ca]i), tightly regulated by calcium-dependent channels located at the surface of the endoplasmic reticulum (ER), mitochondria and plasma membrane (Figure 3c) [143]. The effects of oxidative stress, and more precisely oxidized glutathione, on [Ca2+]i modulation have been reported [53,144], suggesting S-glutathionylation as a potential regulator of cellular Ca2+ handling.
7.1. Calcium-Dependent IP3R & PMCA Channels
Lock et al. were the first to show that direct modification of inositol trisphosphate receptor (IP3R) and plasma membrane Ca2+ ATPase (PMCA) by S-glutathionylation have an effect on Ca2+ oscillatory patterns and [Ca2+]i [56]. Interestingly, opposite effects were observed on each channel: while S-glutathionylation appeared to inhibit PMCA activity, it was shown to enhance IP3R activity, both increasing Ca2+ entry into aortic EC cytosol [56]. This latter effect was further explored in a later study which suggested that S-glutathionylation enhanced the receptor sensitivity to cytosolic Ca2+, leading to increased Ca2+ leaking into the cytosol [54]. Despite the identification of several cysteines potentially subject to S-glutathionylation in it the receptor structure, [55], whether this enhanced activity is due to direct protein modification or S-glutathionylation of accessory components is not known.
7.2. SERCA2b Calcium Pump
The sarco/endoplasmic reticulum calcium ATPase2 (SERCA2) pump promotes Ca2+ uptake in ER stores in ECs and SMC, directly regulating downstream processes involved in the maintenance of vascular homeostasis [145]. The importance of SERCA2 S-glutathionylation was first identified in SMC, in which NO-induced S-glutathionylation on one reactive cysteine enhanced protein uptake activity and muscle relaxation. S-glutathionylation of SERCA2 was well characterized, yet it is not clear how it leads to increased uptake by the pump [146].
This finding brought novel insights on the redox regulation of EC functions in response to hypoxia. Recent studies pointed out the essential role of SERCA S-glutathionylation in VEGF-induced EC migration via NOX signaling [57,147]. Thompson et al. further confirmed the physiological implications by showing that reversible SERCA2 S-glutathionylation was required for hypoxia-induced angiogenic responses in mouse models [58]. Another study proposed a multiplayer model where SERCA2 S-glutathionylation in EC and macrophages is essential for their interplay leading to angiogenic response [59]. Altogether, those results provide novel insights in the importance of redox-regulated Ca2+ store maintenance in VEGF-induced angiogenic response in EC.
The multiple effects of SERCA2 S-glutathionylation in SMC, EC and macrophages emphasize its importance in CVDs. This reversible modification appears as a protective mechanism against permanent SERCA oxidation, linked to severe conditions including atherosclerosis, cardiac dysfunctions, diabetes and impaired ischemic revascularization [90,132,134,135,136].
7.3. STIM1 Molecule and ORAI1 Channel
Stromal interaction molecule 1 (STIM1) senses Ca2+ contents of ER stores and replenishes them by activating calcium release-activated calcium channel protein 1 (Orai1) promoting Ca2+ entry into the cell [148]. S-glutathionylation was shown on one highly conserved reactive cysteine of STIM1, leading to protein oligomerization in fibroblasts. This process triggered a constitutive activation of Orai1, therefore, increasing global Ca2+ entry into intracellular stores. This sustained mitochondrial Ca2+ overload alters its function, resulting in cell death [63]. Highly expressed in EC, STIM1 is key to the maintenance of cell apoptosis and barrier integrity via both Ca2+ dependent and independent pathways and is, therefore, an important player in inflammation [130,132,133].
8. S-Glutathionylation Effects on Cell Death and Autophagy
8.1. Apoptotic Signalling
EC apoptosis is a central process of vascular homeostasis, as it maintains vasculature turnover and tightly regulates network formation during angiogenesis [149]. In addition to causing defects in vessel network formation, the dysregulation of apoptosis contributes to pathological conditions such as atherosclerosis via factors released by extracellular vesicles throughout the process [150]. Oxidative stress is regarded as a general mediator of EC apoptosis [151,152].
One aspect of the apoptotic process in EC involves the activation of the death receptor Fas [153] also involved in the maintenance of vascular integrity [154,155]. S-glutathionylation is suggested to upregulate the receptor activity and subsequent apoptotic signaling (Figure 3a) [64].
The apoptotic response is mediated by a cascade activation of the proteolytic enzymes cysteine-aspartic proteases (Caspases) downstream death receptors [153]. Caspase active sites, composed of cysteines, by definition are interesting targets for deciphering the role of redox-dependent thiol modifications such as S-glutathionylation in EC death receptor signaling pathway. The importance of thiol modifications in Caspases were first highlighted when a disulfide bond formation on Caspase-1 thiol inhibited its activity [156]. Pan et al. demonstrated that S-glutathionylation of Caspase-3 stabilizes the enzyme, inhibiting its cleavage required for activation, therefore, inhibiting apoptosis in EC [65]. Caspase-3 S-glutathionylation was further characterized in various cell types, in a study revealing two cysteines undergoing S-glutathionylation. While one is located in the enzymatic site, it remains to be discovered how the second one affects enzymatic activity [66]. Similarly, S-glutathionylation of caspase-8 in mice models seems to inhibit apoptosis, suggesting a protective mechanism against pathologies related to cell toxicity [67].
8.2. Autophagy
Growing evidence points out a protective role of EC autophagy in various physiological processes following oxidative stress and RONS production [157,158], as in ischemia/reperfusion injury [159], notably by promoting NO production [160]. Apoptosis and autophagy pathways are interlinked, and S-glutathionylation is likely to promote EC autophagy by upregulating the activity of autophagy-related protein Beclin-1 [68]. The altered function of Beclin-1, a major mediator of this process, was found to participate in various pathological processes such as atherosclerosis or hypertension promoting EC dysfunctions [161,162], pointing out the potential implications of S-glutathionylation in CVDs through autophagy regulation.
9. Redox Regulation of Cell Structure and Dynamics by S-Glutathionylation
9.1. Metalloproteases
Metalloprotease (MMP) enzymes are implicitly involved in angiogenesis by degrading endothelial cell matrix (ECM) components, permitting the endothelial cells to migrate within tissues. MMPs also contribute to angiogenesis releasing proangiogenic stimuli to further promote EC migration (Figure 3a) [163]. In addition to cancer, the dysregulation of MMP activity contributes to inflammation, and multiple cardiovascular conditions especially cardiac hypertrophy and atherosclerosis [164,165]. While previous studies pointed out the mechanisms of MMP redox regulation through the modulation of upstream mitogen-activated protein kinase (MAPK) pathway [166,167], others identified direct redox-dependent modifications of the MMP system. MMP activity is tightly regulated and activated in situ by cleavage of a pro-MMP form by tissue inhibitors of metalloproteinases (TIMPs). Promising results showed that S-glutathionylation of pro-MMP inhibitory domain could trigger the passage from latency to activated state, therefore, enhancing angiogenesis and vascular permeability (Figure 3a) in animal models [69,168].
9.2. Adhesion Proteins
Integrins are the major points of contact between ECs and their extracellular microenvironment, driving cell migration, proliferation and regulating vessel permeability. In addition to promoting cell adhesion and migration within the ECM, integrins act as receptors to trigger specific proangiogenic signaling pathways (Figure 3a) [169]. In leukocytes, thiol-based redox regulation of integrins modulate their structure and activity [170]. More precisely, S-glutathionylation of integrin α4 enhances the binding of neutrophils to ECs [171,172]. Despite Integrins wide range of biological functions, the physiological effects of EC integrins S-glutathionylation remain elusive. However, this is likely to contribute to endothelial activation, therefore, modulating the inflammatory response.
Vascular cell adhesion protein 1 and Intercellular Adhesion Molecule 1 (VCAM-1 and ICAM-1) are mainly expressed in ECs in response to pro-inflammatory cytokines and RONS. Adhesion proteins play a pivotal role in EC activation and binding to leukocytes, mediating leukocyte transmigration during inflammation and are, therefore, involved in inflammatory diseases such as rheumatoid arthritis [173,174]. Growing evidence also suggests a role of VCAM-1 in angiogenesis via VEGF activation, with subsequent implications in cancer progression [175]. A recent study suggested that Tumor necrosis factor α (TNF-α) induced ICAM-1 S-glutathionylation, altering protein folding and promoting its degradation [70]. The multiple cystine residues present on ICAM-1 make it particularly subject to oxidation processes, and a better understanding of redox effects on protein function could provide novel insights on the mechanisms involved in inflammatory diseases and cancer.
9.3. Cytoskeletal Proteins
Cytoskeletal dynamics, a key feature driving vascular angiogenesis and barrier function, are governed by polymerization and depolymerization of the two main component units, actin and tubulin. Both proteins are highly redox-sensitive and S-glutathionylation is now recognized to have a major impact on the regulation of polymerization-depolymerization cycles in various cell types [71,136,176]. Polymerization-depolymerization cycles have been better characterized in actin, on which S-glutathionylation modification on one reactive cysteine residue inhibits polymerization and, therefore, plays a key role in the regulation of cytoskeletal dynamics driving cell motility (Figure 3a) [71,72,73]. Similarly, glutathionylated microtubules appear depolymerized, promoting cell growth arrest [74,75]. Phenotypical effects were not investigated in ECs specifically. However, S-glutathionylation-induced changes in cytoskeletal reorganization could emerge as an important feature altering cell motility under oxidative stress.
10. Conclusions
S-glutathionylation has emerged as a novel redox switch altering the functions of EC through the modulation of key signaling pathways. Its importance and complexity in EC homeostasis and cardiovascular physiopathology is highlighted by recent insights on how S-glutathionylated protein functions are altered in interlinked molecular pathways at multiple levels (Figure 3).
It is now apparent that S-glutathionylation can coordinate gene transcription by modulating epigenetics and transcription factors. Studies investigating the impact of S-glutathionylation in epigenetics are still at their early stage, and further work is required to fully understand how it alters gene expression. However, current insights demonstrate that both histone proteins and histone-modifying enzymes are prone to S-glutathionylation, altering chromatin structure and gene expression. Given the number of various genes regulated by those processes, novel insights on the links between oxidative stress and epigenetics would allow a better understanding of EC signaling in response to RONS, and highlight novel factors involved in complex diseases for which genetic factors alone are not sufficient to explain. The effect of S-glutathionylation on transcription factors suggest differential effects on their activity depending on the protein targeted. Accordingly, it is difficult to define a general effect of this modification on EC physiology. However, the redox regulation of VEGF signaling appears central to the activation of downstream transcription pathways and the alteration of resulting EC functions.
S-glutathionylation exhibits a general inhibitory effect on enzymes by altering the structure of their catalytic site and impairing their activity. This has important repercussions when applied to phosphatases, GTPases and kinases, which are key signal transducers in ECs. The multiplicity of pathways regulated by enzymatic activity underlines the crucial importance of understanding the redox mechanisms dictating their behavior to better understand EC responses to oxidative stress.
As upstream mediators of S-glutathionylation, RONS themselves are subject to redox regulation. The two main RONS sources in EC, NOX and eNOS, are under tight redox control. The different effects of S-glutathionylation on RONS-producing systems have a general tendency to promote RONS production. This suggests a retroactive system, where RONS sustain their own production, which could importantly participate in oxidative stress-mediated EC dysfunction.
In respect to calcium signaling, S-glutathionylation tends to favor ion entry into the cell cytosol through reversible alteration of various player functions, except for the SERCA channel. Those results confirm that this modification plays a central role in Ca2+ homeostasis in EC, and that a tight regulation of RONS levels is required to ensure physiological Ca2+ levels in cellular stores allowing proper EC functions.
The opposing effects of S-glutathionylation on different apoptotic factors may explain the complex and diverse effect of oxidative stress on EC apoptosis.
The role of S-glutathionylation in EC autophagy is less studied, and we expect future work will target this pathway.
Beyond signaling molecules, the influence of S-glutathionylation on cell structure components is another example of the duality of S-glutathionylation effects. While this modification tends to lower cell growth and motility by inhibiting cytoskeleton polymerization as well as surface adhesion molecules, the positive effects observed on metalloprotease proteins favor cell migration within extracellular tissues. S-glutathionylation induced degradation of adhesion molecules might also play an important role in the regulation of leukocyte transmigration and inflammation.
All modifications depicted in this review are integrated into a diagram to reflect the complexity of S-glutathionylation redox switch in the modulation of EC signaling pathways (Figure 3). Altogether, these findings highlight the wide range of proteins targeted for S-glutathionylation and the diversity of EC functions involved. This process is now recognized as a novel signaling “switch” among other more characterized PTM such as phosphorylation.
However, the difficulty lies in finding a consistent effect of S-glutathionylation and appreciating the complex mechanism of RONS/antioxidant balance during physiology and pathophysiology. The importance of basal redox signaling in normal physiology could provide a better understanding of why antioxidant therapies failed to meet expectations. Greater appreciation of redox signaling in physiology is of upmost importance since antioxidant treatments in clinical trials have caused unexpected adverse effects. Studies on S-glutathionylation are still at their relatively early stage compared to other PTMs. Another aspect which requires further investigation to appreciate the implications in cardiovascular pathophysiology is how S-glutathionylation interrelates with other oxPTM. A better understanding of the S-glutathionylation process in EC could illuminate the complicated relations linking oxidative stress/redox signaling with CVDs and may also be relevant for other pathologies such as cancer and inflammatory diseases.
Author Contributions
C.E.M. and A.L. wrote the original document and reviewed and edited the document. C.E.M. was responsible for funding acquisition.
Funding
A.L. and C.E.M. have received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 765274, project iPLACENTA. (This publication reflects only the authors’ view and that the European Commission and the Research Executive Agency is not responsible for any use that may be made of the information it contains); and C.E.M.’s work is supported by Diabetes UK, grant number 16/0005453, Tenovus Scotland, T18-23 and EU Marie Skłodowska-Curie Framework 7, grant agreement No 626633.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| [Ca2+]i | cytosolic calcium ion concentrations |
| AngII | angiotensin II |
| Ca2+ | calcium ion |
| CO | carbon oxide |
| CVD | cardiovascular diseases |
| Cys | cysteine |
| EC | endothelial cell |
| ECM | extracellular matrix |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| Grx | glutaredoxin |
| GSH | glutathione |
| HDAC | histone deacetylase |
| HIF-1α | hypoxia-inducible factor 1α |
| ICAM-1 | intercellular adhesion molecule 1 |
| LMW-PTP | Low-molecular-weight protein tyrosin phosphatase |
| MAPK | mitogen-activated protein kinase |
| MEK | mitogen-activated protein kinase kinase |
| MMP | matrix metalloproteinases |
| NAD+ | nicotinamide adenine dinucleotide |
| NFκB | nuclear factor-kappa B |
| NO | nitric oxide |
| NOX | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| O2 | superoxide |
| oxPTM | oxidative post-translational modifications |
| PKB | protein kinase B |
| PTM | post-translational modifications |
| PTP | protein tyrosine phosphatase |
| Rac1 | ras-related C3 botulinum toxin substrate 1 |
| RONS | reactive oxygen and nitrogen species |
| Sirt-1 | sirtuin 1 |
| STAT | signal transducer and activator of transcription |
| TGF-β | transforming growth factor beta |
| TIMPs | tissue inhibitors of metalloproteinases |
| TNF-α | tumor necrosis factor alpha |
| VCAM-1 | vascular cell adhesion protein 1 |
| VEGF | vascular endothelial growth factor |
| VEGFR1 | vascular endothelial growth factor receptor 1 |
References
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; De Luca, N.; Trimarco, B.; Iaccarino, G. The Antioxidant Therapy: New Insights in the Treatment of Hypertension. Front. Physiol. 2018, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.E.; Shuler, M.; Haeussler, D.J.F.; Kikuchi, R.; Bearelly, P.; Han, J.; Watanabe, Y.; Fuster, J.J.; Walsh, K.; Ho, Y.-S.; et al. Glutaredoxin-1 Up-regulation Induces Soluble Vascular Endothelial Growth Factor Receptor 1, Attenuating Post-ischemia Limb Revascularization. J. Boil. Chem. 2014, 289, 8633–8644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolin, J.D. Redox Control of Allergic Airway Disease: Impact of Glutaredoxin-1 on Epithelial Driven Inflammation and Allergen-Induced Airway Remodeling. Bachelor’s Thesis, The University of Vermon, Burlington, VT, USA, 2015. [Google Scholar]
- Kuipers, I.; Louis, R.; Manise, M.; Dentener, M.A.; Irvin, C.G.; Janssen-Heininger, Y.M.; Brightling, C.E.; Wouters, E.F.; Reynaert, N.L. Increased glutaredoxin-1 and decreased protein S-glutathionylation in sputum of asthmatics. Eur. Respir. J. 2013, 41, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Briley, A.; Seed, P.; Kelly, F.; Shennan, A.; Seed, P. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): Randomised placebo-controlled trial. Lancet 2006, 367, 1145–1154. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From Molecular Mechanisms to Health Outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011, 7, 891–910. [Google Scholar] [CrossRef]
- Dominko, K.; Đikić, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arch. Ind. Hyg. Toxicol. 2018, 69, 1–24. [Google Scholar] [CrossRef]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and Consequences of Cysteine S-Glutathionylation. J. Boil. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Moroni, N.; Serafino, A.; Primavera, A.; Pastore, A.; Pedersen, J.Z.; Petruzzelli, R.; Farrace, M.G.; Pierimarchi, P.; Pierimarchi, G.; et al. Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem. J. 2011, 440, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Giménez, J.L.; Olaso, G.; Hake, S.B.; Bönisch, C.; Wiedemann, S.M.; Markovic, J.; Dasi, F.; Gimeno, A.; Perez-Quilis, C.; Palacios, Ò.; et al. Histone H3 Glutathionylation in Proliferating Mammalian Cells Destabilizes Nucleosomal Structure. Antioxid. Redox Signal. 2013, 19, 1305–1320. [Google Scholar] [CrossRef] [Green Version]
- Autiero, I.; Costantini, S.; Colonna, G. Human Sirt-1: Molecular Modeling and Structure-Function Relationships of an Unordered Protein. PLoS ONE 2009, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox Regulation of Sirtuin-1 by S-Glutathiolation. Antioxid. Redox Signal. 2010, 13, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A Redox-resistant Sirtuin-1 Mutant Protects against Hepatic Metabolic and Oxidant Stress. J. Boil. Chem. 2014, 289, 7293–7306. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.-C.; Hsieh, C.-W.; Lin, Y.-C.; Wung, B.-S. The Glutaredoxin/Glutathione System Modulates NF-κB Activity by Glutathionylation of p65 in Cinnamaldehyde-Treated Endothelial Cells. Toxicol. Sci. 2010, 116, 151–163. [Google Scholar] [CrossRef]
- Pineda-Molina, E.; Klatt, P.; Vázquez, J.; Marina, A.; De Lacoba, M.G.; Pérez-Sala, D.; Lamas, S. Glutathionylation of the p50 Subunit of NF-κB: A Mechanism for Redox-Induced Inhibition of DNA Binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef]
- Klatt, P.; Molina, E.P.; De Lacoba, M.G.; Padilla, C.A.; Martínez-Galisteo, E.; Bárcena, J.A.; Lamas, S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999, 13, 1481–1490. [Google Scholar] [CrossRef]
- Klatt, P.; Molina, E.P.; Lamas, S. Nitric Oxide Inhibits c-Jun DNA Binding by Specifically TargetedS-Glutathionylation. J. Boil. Chem. 1999, 274, 15857–15864. [Google Scholar] [CrossRef] [Green Version]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is Inhibited by Glutathionylation of Cysteines Present in the Proximal DNA-Binding Domain During Oxidative Stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Park, H.J.; Kim, H.S. Protein S-glutathionylation induced by hypoxia increases hypoxia-inducible factor-1α in human colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 495, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Murdoch, C.E.; Sano, S.; Ido, Y.; Bachschmid, M.M.; Cohen, R.A.; Matsui, R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc. Natl. Acad. Sci. USA 2016, 113, 6011–6016. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S-Glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 2009, 150, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Pucci, P.; Dell’Orco, D.; Monti, M.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 Impairs STAT3 Phosphorylation. ACS Chem. Boil. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-C.; Huang, Y.-T.; Hsieh, C.-W.; Yang, P.-M.; Wung, B.-S. Carbon Monoxide Induces Heme Oxygenase-1 to Modulate STAT3 Activation in Endothelial Cells via S-Glutathionylation. PLoS ONE 2014, 9, e100677. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Cozzolino, F.; Boriero, D.; Carcereri de Prati, A.; Monti, M.; Rossin, M.; Canetti, D.; Cellini, B.; Pucci, P.; Mariotto, S. S-glutathionylation exerts opposing roles in the regulation of STAT1 and STAT3 signaling in reactive microglia. Free Radic Biol. Med. 2018, 590, 191–201. [Google Scholar]
- Carvalho, A.N.; Marques, C.; Guedes, R.C.; Castro-Caldas, M.; Rodrigues, E.; Van Horssen, J.; Gama, M.J.; Castro-Caldas, M.; Horssen, J. S-Glutathionylation of Keap1: A new role for glutathioneS-transferase pi in neuronal protection. FEBS Lett. 2016, 590, 1455–1466. [Google Scholar] [CrossRef]
- Wang, L.; Qu, G.; Gao, Y.; Su, L.; Ye, Q.; Jiang, F.; Zhao, B.; Miao, J. A small molecule targeting glutathione activates Nrf2 and inhibits cancer cell growth through promoting Keap-1 S-glutathionylation and inducing apoptosis. RSC Adv. 2018, 8, 792–804. [Google Scholar] [CrossRef]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.-S.; Matthews, D.E.; et al. Dynamic redox control of NF- kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory B kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef]
- Abdelsaid, M.A.; El-Remessy, A.B. S-glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. J. Cell Sci. 2012, 125, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Barrett, W.C.; DeGnore, J.P.; Keng, Y.-F.; Zhang, Z.-Y.; Yim, M.B.; Chock, P.B. Roles of Superoxide Radical Anion in Signal Transduction Mediated by Reversible Regulation of Protein-tyrosine Phosphatase 1B. J. Boil. Chem. 1999, 274, 34543–34546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, W.C.; DeGnore, J.P.; König, S.; Fales, H.M.; Keng, Y.-F.; Zhang, Z.-Y.; Yim, M.B.; Chock, P.B. Regulation of PTP1B via Glutathionylation of the Active Site Cysteine 215. Biochemistry 1999, 38, 6699–6705. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Weisbrod, R.M.; Shao, D.; Watanabe, Y.; Yin, X.; Bachschmid, M.M.; Seta, F.; Janssen-Heininger, Y.M.; Matsui, R.; Zang, M.; et al. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: Glutathionylation of Rac1 in endothelial cells. Redox Boil. 2016, 9, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, G.A.; Zhou, B.; Cox, A.D.; Campbell, S.L. Rho GTPases, oxidation, and cell redox control. Small GTPases 2014, 5, e28579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kil, I.S.; Shin, S.W.; Park, J.-W. S-glutathionylation regulates GTP-binding of Rac2. Biochem. Biophys. Res. Commun. 2012, 425, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.; Hobbs, G.A.; Aghajanian, A.; Campbell, S.L. Redox Regulation of Ras and Rho GTPases: Mechanism and Function. Antioxid. Redox Signal. 2013, 18, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavreul, N.; Pimental, D.R.; Adachi, T.; Ido, Y.; Schöneich, C.; Cohen, R.A. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006, 20, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.N.; Cobb, M.H. Protein kinase function and glutathionylation. Biochem. J. 2004, 381, e1. [Google Scholar] [CrossRef] [PubMed]
- Humphries, K.M.; Juliano, C.; Taylor, S.S. Regulation of cAMP-dependent Protein Kinase Activity by Glutathionylation. J. Boil. Chem. 2002, 277, 43505–43511. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pan, S.; Berk, B.C. Glutaredoxin Mediates Akt and eNOS Activation by Flow in a Glutathione Reductase-Dependent Manner. Arter. Thromb. Vasc. Boil. 2007, 27, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jann, J.; Xavier, C.; Wu, H. Glutaredoxin 1 (Grx1) Protects Human Retinal Pigment Epithelial Cells from Oxidative Damage by Preventing AKT Glutathionylation. Investig. Opthalmol. Vis. Sci. 2015, 56, 2821. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.V.; Templeton, D.J. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 2004, 381, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, N.E.; Stewart, J.R.; Ioannides, C.G.; O’Brian, C.A. Oxidant-InducedS-Glutathiolation Inactivates Protein Kinase C-α (PKC-α): A Potential Mechanism of PKC Isozyme Regulation. Biochemistry 2000, 39, 10319–10329. [Google Scholar] [CrossRef] [PubMed]
- Nagarkoti, S.; Dubey, M.; Awasthi, D.; Kumar, V.; Chandra, T.; Kumar, S.; Dikshit, M. S-Glutathionylation of p47phox sustains superoxide generation in activated neutrophils. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.R.; Hurrell, F.; Murphy, M.P.; Shannon, R.J.; Lin, T.-K.; Hirst, J. Reversible Glutathionylation of Complex I Increases Mitochondrial Superoxide Formation. J. Boil. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Hassan Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.-A.; Zweier, J.L. Hypoxia and Reoxygenation Induce Endothelial Nitric Oxide Synthase Uncoupling in Endothelial Cells through Tetrahydrobiopterin Depletion and S-Glutathionylation. Biochemistry 2014, 53, 3679–3688. [Google Scholar] [CrossRef]
- Shang, Q.; Bao, L.; Guo, H.; Hao, F.; Luo, Q.; Chen, J.; Guo, C. Contribution of glutaredoxin-1 to S-glutathionylation of endothelial nitric oxide synthase for mesenteric nitric oxide generation in experimental necrotizing enterocolitis. Transl. Res. 2017, 188, 92–105. [Google Scholar] [CrossRef]
- Galougahi, K.K.; Liu, C.; Gentile, C.; Kok, C.; Nunez, A.; Garcia, A.; Fry, N.A.S.; Davies, M.J.; Hawkins, C.L.; Rasmussen, H.H.; et al. Glutathionylation Mediates Angiotensin II–Induced eNOS Uncoupling, Amplifying NADPH Oxidase—Dependent Endothelial Dysfunction. J. Am. Heart Assoc. 2014, 3, 1–11. [Google Scholar] [CrossRef]
- Wu, F.; Szczepaniak, W.S.; Shiva, S.; Liu, H.; Wang, Y.; Wang, L.; Wang, Y.; Kelley, E.E.; Chen, A.F.; Gladwin, M.T.; et al. Nox2-dependent glutathionylation of endothelial NOS leads to uncoupled superoxide production and endothelial barrier dysfunction in acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L987–L997. [Google Scholar] [CrossRef] [PubMed]
- Henschke, P.N.; Elliott, S.J. Oxidized glutathione decreases luminal Ca2+ content of the endothelial cell ins(1,4,5) P3 -sensitive Ca2+ store. Biochem. J. 2015, 312, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J. Physiol. 2012, 590, 3431–3447. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kang, J.; Kwon, H.; Frueh, D.; Yoo, S.H.; Wagner, G.; Park, S. Effects of Redox Potential and Ca2+ on the Inositol 1,4,5-Trisphosphate Receptor L3-1 Loop Region. J. Biol. Chem. 2008, 283, 25567–25575. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. AJP Heart Circ. Physiol. 2011, 300, H493–H506. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Bolotina, V.M.; Tong, X.; Cohen, R.A. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radic. Boil. Med. 2012, 53, 2327–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.D.; Mei, Y.; Weisbrod, R.M.; Silver, M.; Shukla, P.C.; Bolotina, V.M.; Cohen, R.A.; Tong, X. Glutathione adducts on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J. Biol. Chem. 2014, 289, 19907–19916. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Shiraishi, Y.; Cohen, R.A.; Tong, X. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase C674 promotes ischemia- and hypoxia-induced angiogenesis via coordinated endothelial cell and macrophage function. J. Mol. Cell. Cardiol. 2014, 76, 275–282. [Google Scholar] [CrossRef]
- Sharov, V.S.; Dremina, E.S.; Galeva, N.A.; Williams, T.D.; Schöneich, C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC–electrospray-tandem MS: Selective protein oxidation during biological aging. Biochem. J. 2006, 394, 605–615. [Google Scholar] [CrossRef]
- Tong, X.Y.; Hou, X.; Jourd’Heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGFβ1 Oxidizes SERCA and Inhibits NO in Arterial Smooth Muscle of the Prediabetic Zucker Rat. Circ. Res. 2010, 107, 975–983. [Google Scholar] [CrossRef]
- Qin, F.; Siwik, D.A.; Pimentel, D.R.; Morgan, R.J.; Biolo, A.; Tu, V.H.; Kang, Y.J.; Cohen, R.A.; Colucci, W.S. Cytosolic H2O2 mediates hypertrophy, apoptosis, and decreased SERCA activity in mice with chronic hemodynamic overload. Am. J. Physiol. Circ. Physiol. 2014, 306, H1453–H1463. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.-C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Boil. 2010, 190, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anathy, V.; Aesif, S.W.; Guala, A.S.; Havermans, M.; Reynaert, N.L.; Ho, Y.-S.; Budd, R.C.; Janssen-Heininger, Y.M. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J. Cell Boil. 2009, 184, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, S.; Berk, B.C. Glutathiolation Regulates Tumor Necrosis Factor-α–Induced Caspase-3 Cleavage and Apoptosis. Circ. Res. 2006, 100, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P. Inhibition of Caspase-3 Activity and Activation by Protein Glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- McGarry, D.J.; Chakravarty, P.; Wolf, C.R.; Henderson, C.J. Altered Protein S-Glutathionylation Identifies a Potential Mechanism of Resistance to Acetaminophen-Induced Hepatotoxicity. J. Pharmacol. Exp. Ther. 2015, 355, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; Del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative Stress, Redox Signaling, and Autophagy: Cell Death Versus Survival. Antioxid. Redox Signal. 2014, 21, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akaike, T.; Sawa, T.; Miyamoto, Y.; Van Der Vliet, A.; Maeda, H. Activation of Matrix Metalloproteinases by Peroxynitrite-induced ProteinS-Glutathiolation via DisulfideS-Oxide Formation. J. Boil. Chem. 2001, 276, 29596–29602. [Google Scholar] [CrossRef]
- Mukherjee, T.K.; Mishra, A.K.; Mukhopadhyay, S.; Hoidal, J.R. High Concentration of Antioxidants N-Acetylcysteine and Mitoquinone-Q Induces Intercellular Adhesion Molecule 1 and Oxidative Stress by Increasing Intracellular Glutathione. J. Immunol. 2014, 178, 1835–1844. [Google Scholar] [CrossRef]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.-U.; Yousefi, S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Boil. 2017, 216, 4073–4090. [Google Scholar] [CrossRef]
- Fiaschi, T.; Cozzi, G.; Raugei, G.; Formigli, L.; Ramponi, G.; Chiarugi, P. Redox regulation of β-actin during integrin-mediated cell adhesion. J. Biol. Chem. 2006, 281, 22983–22991. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; English, S.; Mieyal, J.J.; Chock, P.B.; Fales, H.M. Reversible Glutathionylation Regulates Actin Polymerization in A431 Cells. J. Boil. Chem. 2001, 276, 47763–47766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Seefeldt, T.; Young, A.; Zhang, X.; Zhao, Y.; Ruffolo, J.; Kaushik, R.S.; Guan, X. Microtubule S-glutathionylation as a potential approach for antimitotic agents. BMC Cancer 2012, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Landino, L.M.; Moynihan, K.L.; Todd, J.V.; Kennett, K.L. Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun. 2004, 314, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.S.; Marsden, P.A. Epigenetics in the Vascular Endothelium: Looking from a different perspective in the epigenomics era. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E. Epigenetic Mechanisms of Endothelial Progenitor Cell Dysfunction. J. Clin. Epigenet. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Fraineau, S.; Brand, M.; Palii, C.G.; Allan, D.S. Epigenetic regulation of endothelial-cell-mediated vascular repair. FEBS J. 2015, 282, 1605–1629. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Ray, S.; Home, P.; Saha, B.; Wang, S.; Sheibani, N.; Tawfik, O.; Cheng, N.; Paul, S. Regulation of Angiogenesis by Histone Chaperone HIRA-mediated Incorporation of Lysine 56-acetylated Histone H3.3 at Chromatin Domains of Endothelial Genes. J. Boil. Chem. 2010, 285, 41567–41577. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.E.; Matouk, C.C.; Rachlis, A.; Lin, S.; Tai, S.C.; D’Abreo, C.; Marsden, P.A. The Expression of Endothelial Nitric-oxide Synthase Is Controlled by a Cell-specific Histone Code. J. Boil. Chem. 2005, 280, 24824–24838. [Google Scholar] [CrossRef] [Green Version]
- Urbich, C.; Dimmeler, S.; Zeiher, A.M.; Rossig, L.; Diehl, F. The histone methyltransferase MLL is an upstream regulator of endothelial-cell sprout formation. Blood 2006, 109, 1472–1478. [Google Scholar]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs, J.A.; Nussenzweig, M.C.; Nussenzweig, A. Chromatin dynamics and the preservation of genetic information. Nature 2007, 447, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Panyim, S.; Sommer, K.R.; Chalkley, R. Oxidation of the cysteine-containing histone F3. Detection of an evolutionary mutation in a conservative histone. Biochemistry 1971, 10, 3911–3917. [Google Scholar] [CrossRef] [PubMed]
- Blackshire, B. The effects of histone glutathionylation on chromatin structure. FASEB J. 2014, 28, 942–945. [Google Scholar]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.D.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wu, Y.; Yang, P. High glucose-induced oxidative stress represses sirtuin deacetylase expression and increases histone acetylation leading to neural tube defects. J. Neurochem. 2016, 137, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Escande, C.; Denicola, A. Potential modulation of sirtuins by oxidative stress. Oxid. Med. Cell. Longev. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto-Komatsu, A.; Hirase, T.; Asaka, M.; Node, K. Angiotensin II induces microtubule reorganization mediated by a deacetylase SIRT2 in endothelial cells. Hypertens. Res. 2011, 34, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maizel, J.; Xavier, S.; Chen, J.; Lin, C.H.S.; Vasko, R.; Goligorsky, M.S. Sirtuin 1 ablation in endothelial cells is associated with impaired angiogenesis and diastolic dysfunction. Am. J. Physiol. Circ. Physiol. 2014, 307, H1691–H1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghisays, F.; Brace, C.S.; Yackly, S.M.; Kwon, H.J.; Mills, K.F.; Kashentseva, E.; Dimitriev, I.P.; Curiel, D.T.; Imai, S.-I.; Ellenberger, T. The N-terminal Domain of SIRT1 Is a Positive Regulator of Endogenous SIRT1-dependent Deacetylation and Transcriptional Outputs. Cell Rep. 2015, 10, 1665–1673. [Google Scholar] [CrossRef]
- Yeung, F.; E Hoberg, J.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; A Frye, R.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Strand, S.; Schlufter, F.; Siuda, D.; Reifenberg, G.; Kleinert, H.; Förstermann, U.; Li, H. Role of SIRT1 and FOXO factors in eNOS transcriptional activation by resveratrol. Nitric Oxide 2013, 32, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Kim, C.-S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.-B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-κB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-N.; Zhu, N.; Liu, C.; Wu, H.-T.; Gui, Y.; Liao, D.-F.; Qin, L. Wnt5a and its signaling pathway in angiogenesis. Clin. Chim. Acta 2017, 471, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Qanungo, S.; Starke, D.W.; Pai, H.V.; Mieyal, J.J.; Nieminen, A.-L. Glutathione Supplementation Potentiates Hypoxic Apoptosis by S-Glutathionylation of p65-NFκB. J. Boil. Chem. 2007, 282, 18427–18436. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis and c-Jun. J. Natl. Cancer Inst. 2004, 96, 644. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Han, W.; Shen, T.; Ma, C.; Liu, Y.; Nie, X.; Liu, M.; Ran, Y.; Zhu, D. Activation of JNK/c-Jun is required for the proliferation, survival, and angiogenesis induced by EET in pulmonary artery endothelial cells. J. Lipid Res. 2012, 53, 1093–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; Van Der Wall, E.; Van Diest, P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 2006, 37, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Panta, S.; Hashiguchi, T. p53 and Vascular Dysfunction: MicroRNA in Endothelial Cells, Vasculitis in Practice—An Update on Special Situations—Clinical and Therapeutic Considerations. IntechOpen 2018. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florczyk, U.; Jazwa, A.; Maleszewska, M.; Mendel, M.; Szade, K.; Kozakowska, M.; Grochot-Przeczek, A.; Viscardi, M.; Czauderna, S.; Bukowska-Strakova, K.; et al. Nrf2 Regulates Angiogenesis: Effect on Endothelial Cells, Bone Marrow-Derived Proangiogenic Cells and Hind Limb Ischemia. Antioxid. Redox Signal. 2013, 20, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Shui, Q.Y.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genome Res. 1998, 12, 149–162. [Google Scholar]
- Rawlings, J.S. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, M.; Gu, X.; Tsai, N.T.; Venema, R.C.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. Vascular Endothelial Growth Factor Activates STAT Proteins in Aortic Endothelial Cells. J. Boil. Chem. 2000, 275, 33189–33192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-H.; Murphy, D.A.; Lassoued, W.; Thurston, G.; Feldman, M.D.; Lee, W.M. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Boil. Ther. 2008, 7, 1994–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Laso, V.; Sastre, C.; Méndez-Barbero, N.; Egido, J.; Martín-Ventura, J.L.; Gómez-Guerrero, C.; Blanco-Colio, L.M. TWEAK blockade decreases atherosclerotic lesion size and progression through suppression of STAT1 signaling in diabetic mice. Sci. Rep. 2017, 7, 46679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhang, Y.; Xu, L.; Lin, Y.; Yang, X.; Bai, L.; Chen, Y.; Zhao, S.; Fan, J.; Cheng, X.; et al. Protein Inhibitor of Activated STAT3 Suppresses Oxidized LDL-induced Cell Responses during Atherosclerosis in Apolipoprotein E-deficient Mice. Sci. Rep. 2016, 6, 36790. [Google Scholar] [CrossRef]
- Xue, C.; Xie, J.; Zhao, D.; Lin, S.; Zhou, T.; Shi, S.; Shao, X.; Lin, Y.; Zhu, B.; Cai, X. The JAK/STAT3 signalling pathway regulated angiogenesis in an endothelial cell/adipose-derived stromal cell co-culture, 3D gel model. Cell Prolif. 2017, 50, 1–10. [Google Scholar] [CrossRef]
- Battle, T.E.; Lynch, R.A.; Frank, D.A. Signal Transducer and Activator of Transcription 1 Activation in Endothelial Cells Is a Negative Regulator of Angiogenesis. Cancer Res. 2006, 66, 3649–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Bae, Y.S.; Lee, S.-R.; Kwon, J. Hydrogen Peroxide: A Key Messenger That Modulates Protein Phosphorylation Through Cysteine Oxidation. Sci. Signal. 2000, 2000, pe1. [Google Scholar] [CrossRef] [PubMed]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-Based Redox Switches in Enzymes. Antioxid. Redox Signal. 2010, 14, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P. The Redox Regulation of LMW-PTP During Cell Proliferation or Growth Inhibition. IUBMB Life 2001, 52, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Raugei, G.; Chiarugi, P.; Ramponi, G. A novel redox-based switch: LMW-PTP oxidation enhances Grb2 binding and leads to ERK activation. Biochem. Biophys. Res. Commun. 2006, 348, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Xing, K.; Raza, A.; Löfgren, S.; Fernando, M.R.; Ho, Y.S.; Lou, M.F. LMW-PTP and Its Possible Physiological Functions of redox signaling in the eye lens. Biochim. Biophys. Acta. 2007, 1774, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Patrushev, N.; Inomata, H.; Mehta, D.; Urao, N.; Kim, H.W.; Razvi, M.; Kini, V.; Mahadev, K.; Goldstein, B.J.; et al. Role of Protein Tyrosine Phosphatase 1B in VEGF Signaling and Cell-Cell Adhesions in Endothelial Cells. Circ. Res. 2008, 102, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Besnier, M.; Galaup, A.; Nicol, L.; Henry, J.-P.; Coquerel, D.; Gueret, A.; Mulder, P.; Brakenhielm, E.; Thuillez, C.; Germain, S.; et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 2014, 28, 3351–3361. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Sankar, S.; Lin, C.; Kontos, C.D.; Schroff, A.D.; Cha, E.H.; Feng, S.-M.; Li, S.-F.; Yu, Z.; Van Etten, R.L.; et al. HCPTPA, a Protein Tyrosine Phosphatase That Regulates Vascular Endothelial Growth Factor Receptor-mediated Signal Transduction and Biological Activity. J. Boil. Chem. 1999, 274, 38183–38188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilzer, P.; Chang, K.; Marvel, J.; Rowold, E.; Jaudes, P.; Ullensvang, S.; Kilo, C.; Williamson, J.R. Albumin Permeation of New Vessels Is Increased in Diabetic Rats. Diabetes 1985, 34, 333–336. [Google Scholar] [CrossRef]
- Williamson, J.R.; Chang, K.; Tilton, R.G.; Prater, C.; Jeffrey, J.R.; Weigel, C.; Sherman, W.R.; Eades, D.M.; Kilo, C. Increased Vascular Permeability in Spontaneously Diabetic BB/W Rats and in Rats with Mild Versus Severe Streptozocin-Induced Diabetes - Prevention by Aldose Reductase Inhibitors and Castration. Diabetes 1987, 36, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; Vepa, S.; Watkins, T.; He, D.; Parinandi, N.L.; Natarajan, V. Redox Regulation of Reactive Oxygen Species-Induced p38 MAP Kinase Activation and Barrier Dysfunction in Lung Microvascular Endothelial Cells. Antioxid. Redox Signal. 2003, 5, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of Endothelial Junctional Permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Boil. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvakumar, B.; Hess, D.T.; Goldschmidt-clermont, P.J.; Stamler, J.S. Co-regulation of constitutive nitric oxide synthase and NADPH oxidase by the small GTPase Rac. Measurement 2009, 582, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, D.J.; Pimentel, D.R.; Hou, X.; Burgoyne, J.R.; Cohen, R.A.; Bachschmid, M.M. Endomembrane H-Ras Controls Vascular Endothelial Growth Cell Migration. J. Biol. Chem. 2013, 288, 15380–15389. [Google Scholar] [CrossRef] [PubMed]
- Küppers, V.; Vockel, M.; Nottebaum, A.F.; Vestweber, D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res. 2014, 355, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Bogatcheva, N.V.; Dudek, S.M.; Garcia, J.G.N.; Verin, A.D. Mitogen-Activated Protein Kinases in Endothelial Pathophysiology. J. Investig. Med. 2003, 51, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Shah, A.M. NADPH oxidase and endothelial cell function. Clin. Sci. 2005, 109, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, P.; Dussault, S.; Groleau, J.; Turgeon, J.; Michaud, S.-E.; Ménard, C.; Perez, G.; Maingrette, F.; Rivard, A.; Ménard, C. Nox2-Containing NADPH Oxidase Deficiency Confers Protection from Hindlimb Ischemia in Conditions of Increased Oxidative Stress. Arter. Thromb. Vasc. Boil. 2009, 29, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Cave, A.C.; Zhang, M.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirker, A.; Murdoch, C.E.; Protti, A.; Sawyer, G.J.; Santos, C.X.; Martin, D.; Zhang, X.; Brewer, A.C.; Zhang, M.; Shah, A.M. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell. Cardiol. 2016, 98, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH Oxidase-2 Promotes Interstitial Cardiac Fibrosis and Diastolic Dysfunction Through Proinflammatory Effects and Endothelial-Mesenchymal Transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybyłowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary Artery Superoxide Production and Nox Isoform Expression in Human Coronary Artery Disease. Arter. Thromb. Vasc. Boil. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mythreye, K.; Goldschmidt-Clermont, P.J.; Satterwhite, L.L.; Moldovan, L. Reactive oxygen species in vascular endothelial cell motility. Roles of NAD(P)H oxidase and Rac1. Cardiovasc. Res. 2006, 71, 236–246. [Google Scholar]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-M.; Mullen, A.M.; Yun, S.; Wientjes, F.; Brouns, G.Y.; Thrasher, A.J.; Shah, A.M. Essential Role of the NADPH Oxidase Subunit p47 phox in Endothelial Cell Superoxide Production in Response to Phorbol Ester and Tumor Necrosis Factor-α. Circ. Res. 2002, 90, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Bernard, L.; Clempus, R. Vascular NAD (P) H oxidases: Specific features, expression, and regulation. Am. J. Physiol. 2003, 285, R277–R297. [Google Scholar]
- Tang, X.; Luo, Y.-X.; Chen, H.-Z.; Liu, D.-P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 1–17. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris Nikolaos Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2011, 10, 4–18. [Google Scholar] [CrossRef]
- Crabtree, M.J.; Brixey, R.; Batchelor, H.; Hale, A.B.; Channon, K.M. Integrated Redox Sensor and Effector Functions for Endothelial Nitric-oxide Synthase (eNOS) Uncoupling. J. Biol. Chem. 2013, 288, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.-K.; Ohashi, K.; Watanabe, H. Calcium signalling in endothelial cells. Cardiovasc. Res. 2000, 48, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Raturi, A.; Ortiz-Sandoval, C.; Simmen, T. Redox dependence of endoplasmic reticulum (ER) Ca2+ signaling. Histol. Histopathol. 2014, 29, 543–552. [Google Scholar] [PubMed]
- Tong, X.Y.; Evangelista, A.; Cohen, R.A. Targeting the Redox Regulation of SERCA in Vascular Physiology and Disease. Curr. Opin. Pharmacol. 2010, 10, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.Y.; Bolotina, V.M.; Cohen, R.A. Redox Regulation of SERCA2 Is Required for Vascular Endothelial Growth Factor-Induced Signaling and Endothelial Cell Migration. Antioxid. Redox Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.R.; Sheikh, R.; Feske, S.; Komarova, K.; Mehta, D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca2+ Entry. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Watson, E.C.; Grant, Z.L.; Coultas, L. Endothelial cell apoptosis in angiogenesis and vessel regression. Cell. Mol. Life Sci. 2017, 74, 4387–4403. [Google Scholar] [CrossRef]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2018, 76, 1093–1106. [Google Scholar] [CrossRef]
- Warren, M.C.; A Bump, E.; Medeiros, D.; Braunhut, S.J. Oxidative stress–induced apoptosis of endothelial cells. Free Radic. Boil. Med. 2000, 29, 537–547. [Google Scholar] [CrossRef]
- Kobayashi, N.; Delano, F.A.; Schmid-Schoönbein, G.W. Oxidative Stress Promotes Endothelial Cell Apoptosis and Loss of Microvessels in the Spontaneously Hypertensive Rats. Arter. Thromb. Vasc. Boil. 2005, 25, 2114–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A. Death receptor-induced cell killing. Cell. Signal. 2004, 16, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Sata, M.; Suhara, T.; Walsh, K. Vascular endothelial cells and smooth muscle cells differ in expression of Fas and Fas ligand and in sensitivity to Fas ligand-induced cell death: Implications for vascular disease and therapy. Arter. Thromb. Vasc. Boil. 2000, 20, 309–316. [Google Scholar] [CrossRef]
- Kamei, R.; Tanaka, H.Y.; Kawano, T.; Morii, C.; Tanaka, S.; Nishihara, H.; Iwata, C.; Kano, M.R. Regulation of endothelial Fas expression as a mechanism of promotion of vascular integrity by mural cells in tumors. Cancer Sci. 2017, 108, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Scheer, J.M.; Romanowski, M.J.; Wells, J.A. A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. USA 2006, 103, 7595–7600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Houbaert, D.; Meçe, O.; Agostinis, P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 2019, 26, 665–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boteon, Y.L.; Laing, R.; Mergental, H.; Reynolds, G.M.; Mirza, D.F.; Afford, S.C.; Bhogal, R.H. Mechanisms of autophagy activation in endothelial cell and their targeting during normothermic machine liver perfusion. World J. Gastroenterol. 2017, 23, 8443–8451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F. Autophagy in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Hao, Y.; Chong, X.; Zhong, W. Overexpression of microRNA-506-3p aggravates the injury of vascular endothelial cells in patients with hypertension by downregulating Beclin1 expression. Exp. Ther. Med. 2018, 15, 2844–2850. [Google Scholar] [CrossRef]
- Geng, Z.; Xu, F.; Zhang, Y. MiR-129-5p-mediated Beclin-1 suppression inhibits endothelial cell autophagy in atherosclerosis. Am. J. Transl. Res. 2016, 8, 1886–1894. [Google Scholar] [PubMed]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis Angiogenesis Review Series. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Allen, T.C.; Kurdowska, A.K. Metalloproteinase-9 contributes to endothelial dysfunction in atherosclerosis via protease activated receptor-1. PLoS ONE 2017, 12, e0171427. [Google Scholar] [CrossRef] [PubMed]
- Papazafiropoulou, A.; Tentolouris, N. Matrix metalloproteinases and cardiovascular diseases. Hippokratia 2009, 13, 76–82. [Google Scholar] [PubMed]
- Nelson, K.K.; Subbaram, S.; Connor, K.M.; Dasgupta, J.; Ha, X.-F.; Meng, T.-C.; Tonks, N.K.; Melendez, J.A. Redox-dependent Matrix Metalloproteinase-1 Expression Is Regulated by JNK through Ets and AP-1 Promoter Motifs. J. Boil. Chem. 2006, 281, 14100–14110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, S.; Subbaram, S.; Carrico, P.M.; Melendez, J.A. Redox-control of Matrix Metalloproteinase-1: A critical link between free radicals, matrix remodeling and degenerative disease. Respir. Physiol. Neurobiol. 2010, 174, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Stupack, D.G.; Cheresh, D.A. Integrins and Angiogenesis. Curr. Top. Dev. Biol. 2004, 64, 207–238. [Google Scholar] [PubMed]
- Eble, J.A.; de Rezende, F.F. Redox-Relevant Aspects of the Extracellular Matrix and Its Cellular Contacts via Integrins. Antioxid. Redox Signal. 2013, 20, 1977–1993. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Tsai, M.-Y.; Chuang, K.-P.; Huang, Y.-F.; Shieh, C.-C. Ligand binding of leukocyte integrin very late antigen-4 involves exposure of sulfhydryl groups and is subject to redox modulation. Eur. J. Immunol. 2008, 38, 410–423. [Google Scholar] [CrossRef]
- You, Y.; Chen, J.; Zhu, F.; Xu, Q.; Han, L.; Gao, X.; Zhang, X.; Luo, H.R.; Miao, J.; Sun, X.; et al. Glutaredoxin 1 up-regulates deglutathionylation of α4 integrin and thereby restricts neutrophil mobilization from bone marrow. J. Biol. Chem. 2019, 294, 2616–2627. [Google Scholar] [CrossRef] [PubMed]
- Deem, T.L.; Cook-Mills, J.M. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: Role of reactive oxygen species. Blood 2004, 104, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, G.W.; Odell, A.F.; Latham, A.M.; Mughal, N.A.; Bruns, A.F.; Burgoyne, N.J.; Homer-Vanniasinkam, S.; Zachary, I.C.; Hollstein, M.C.; Wheatcroft, S.B.; et al. VEGF-A isoforms differentially regulate ATF-2–dependent VCAM-1 gene expression and endothelial–leukocyte interactions. Mol. Boil. Cell 2014, 25, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Gonzalez-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell. Neurosci. 2015, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
General mechanisms of reversible protein S-glutathionylation. Free thiols on reactive cysteinyl residues can be modified after the formation of an intermediate thiol derivative, or more rarely undergo direct thiol-disulfide exchange. S-glutathionylation can be reversed by the action of thiol-modifying enzymes, e.g., glutaredoxin (Grx). Cys = cysteine, GSH = glutathione.
Figure 1.
General mechanisms of reversible protein S-glutathionylation. Free thiols on reactive cysteinyl residues can be modified after the formation of an intermediate thiol derivative, or more rarely undergo direct thiol-disulfide exchange. S-glutathionylation can be reversed by the action of thiol-modifying enzymes, e.g., glutaredoxin (Grx). Cys = cysteine, GSH = glutathione.

Figure 2.
Schematic representation of the main endothelial cell functions and associated molecular pathways which are suspectable to S-glutathionylation.
Figure 2.
Schematic representation of the main endothelial cell functions and associated molecular pathways which are suspectable to S-glutathionylation.

Figure 3.
Overview of the interlinked effects of S-glutathionylation in major EC functions via the alteration of (a) major signaling pathways in cytosol and nucleus (b) RONS formation in mitochondria (c) calcium-dependent signaling in the endoplasmic reticulum.
Figure 3.
Overview of the interlinked effects of S-glutathionylation in major EC functions via the alteration of (a) major signaling pathways in cytosol and nucleus (b) RONS formation in mitochondria (c) calcium-dependent signaling in the endoplasmic reticulum.


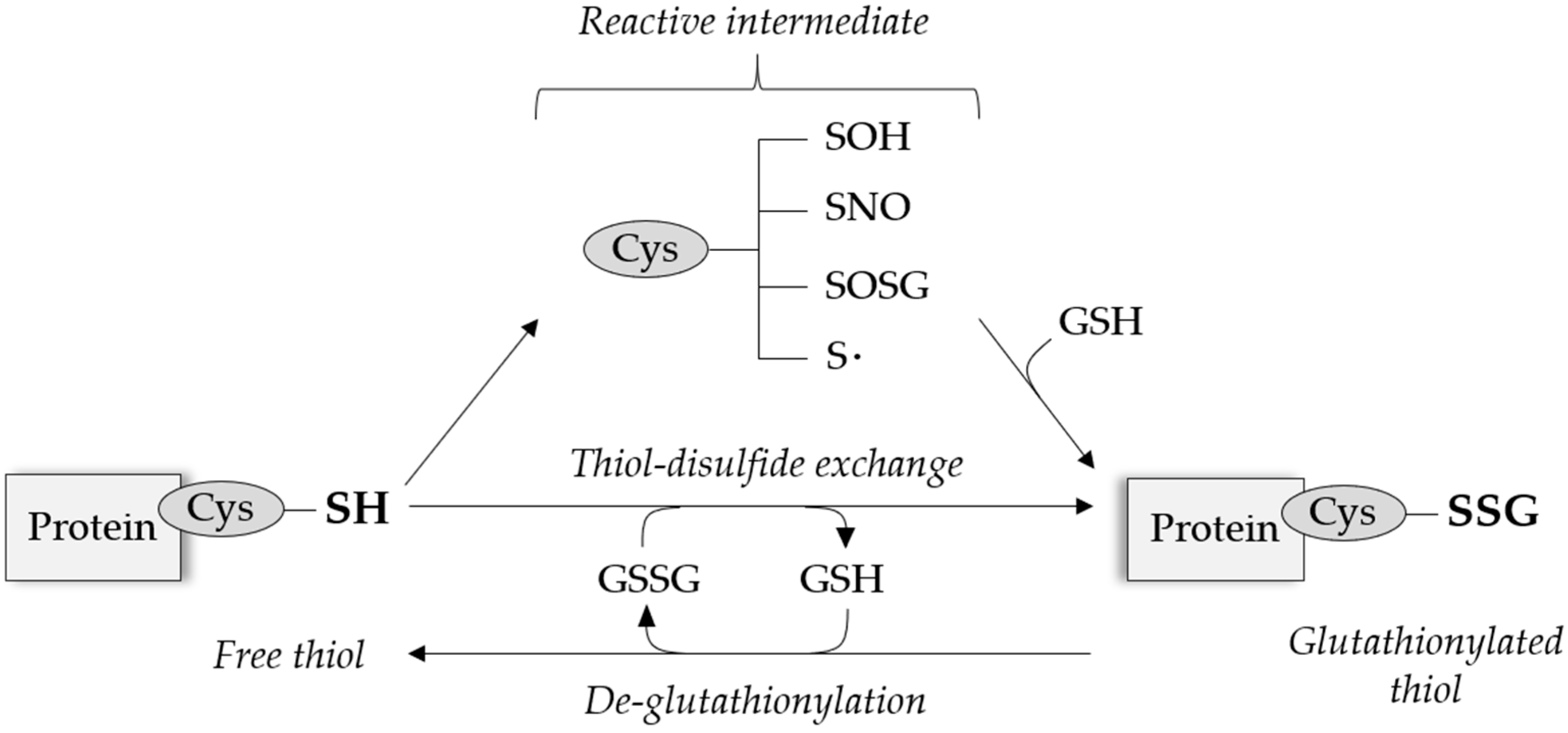{kind=link}
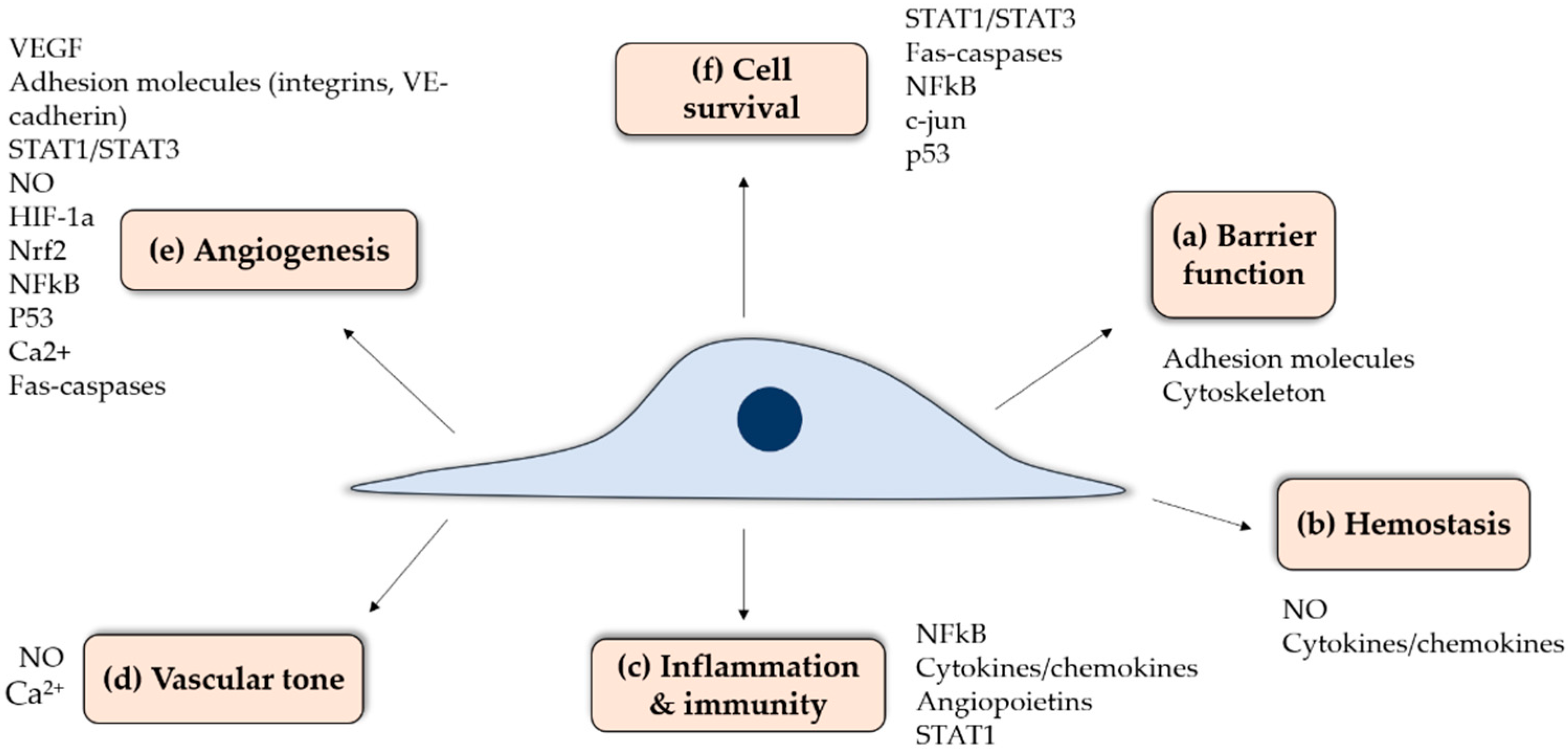{kind=link}
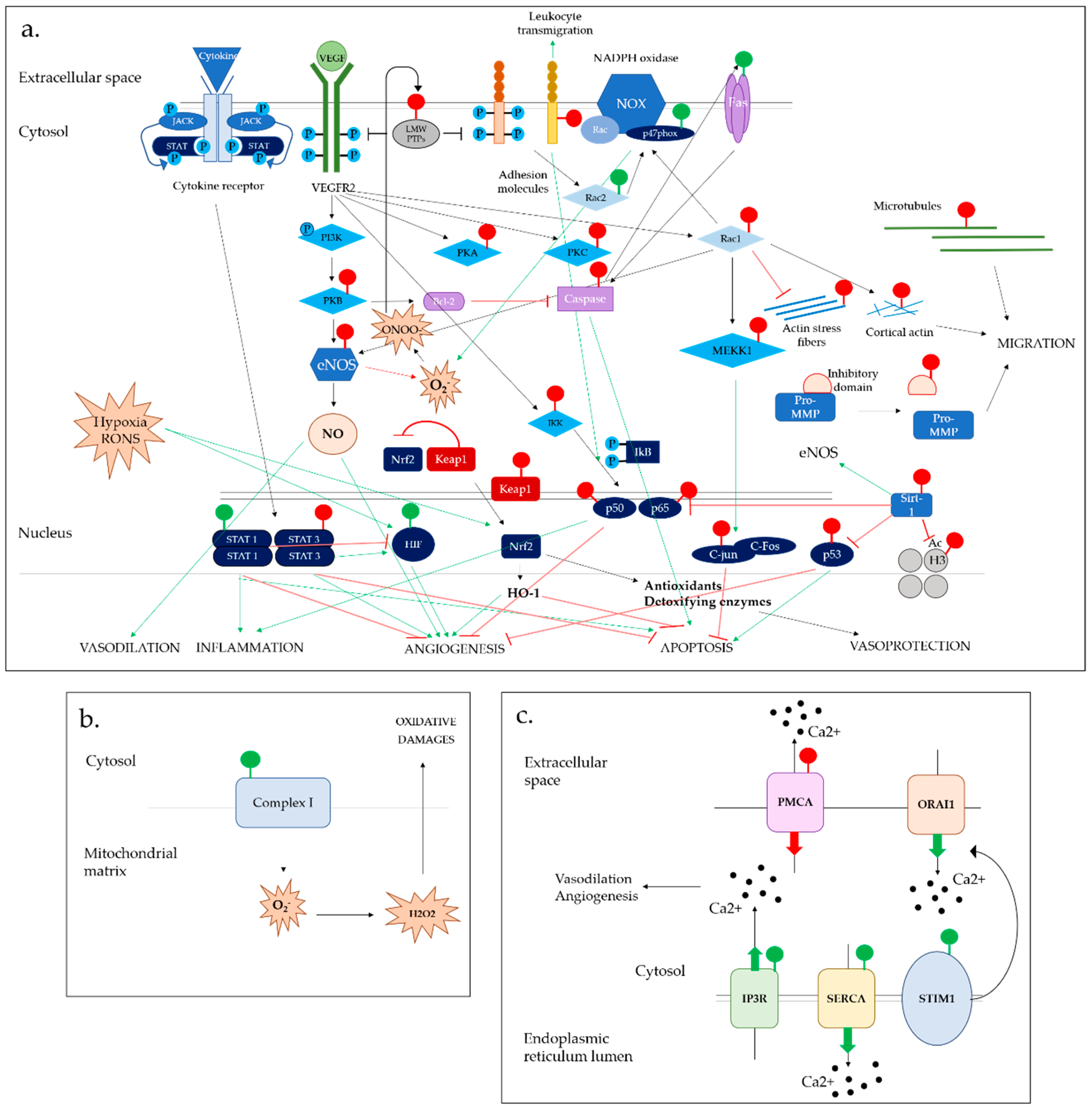{kind=link}
Table 1.
Summary of S-glutathionylation activation (green) or inhibition (red) effects on major molecular players in endothelial cell (EC) function and their physiological consequences.
Table 1.
Summary of S-glutathionylation activation (green) or inhibition (red) effects on major molecular players in endothelial cell (EC) function and their physiological consequences.
| Protein Name | Protein Type | Glutathionylated Cysteine(s) | Direct Effect(s) on Protein | Physiological Effect(s) in ECs | Reference(s) |
|---|---|---|---|---|---|
| 4.1. Epigenetics regulators | |||||
| Histone H3 | Nucleosomal | C110 | Not confirmed | Regulation of gene expression via modulating chromatin structure | [13,14] |
| Sirtuin1 | Histone deacetylase | C67 C268 C623 | Inhibition of enzymatic activity | Apoptosis - Senescence | [15,16,17] |
| 4.2. Transcription factors | |||||
| p65 | Transcription factor | Unknown | Inhibition of nuclear translocation | Angiogenesis & cell survival | [18] |
| p50 | C62 | Inhibition of DNA-binding activity | [19] | ||
| c-jun | C269 | Unknown | [20,21] | ||
| p53 | C124 C141 C182 | Inhibition of DNA-binding and protein dimerization | Angiogenesis & cell survival (supposed) | [22] | |
| HIF-1a | C520 | Protein stabilization | Angiogenesis & ischemic revascularisation | [23,24] | |
| STAT3 | C328 C542 | Inhibition of phosphorylation and activity | Anti-angiogenesis and reduced inflammation | [25,26,27,28] | |
| STAT1 | C324 C492 | Protein activation | Unknown | [28] | |
| Keap1 | Nrf2 inhibitor | C434 | Inhibition of Nrf2 binding | Antioxidant and anti-inflammatory response via Nrf2 signalling | [29,30] |
| IKKb | Kinase | C179 | Inhibition of kinase activity | Angiogenesis & neovascularisation | [31] |
| 5. Kinases & phosphatases | |||||
| LMW PTP | Phosphatase | Unknown | Inhibition of activity | Cell migration and angiogenesis | [32] |
| PTP1B | C215 | Not confirmed | [33,34] | ||
| Rac1 | Small Rho GTPase | C81 C157 | Altered actin structure and barrier function | [35,36] | |
| Rac2 | C157 | Increased GTP-binding activity | Unknown | [36,37] | |
| Ras | GTPase | C118 | Not confirmed | Unknown | [36,38,39] |
| PKA | Kinase | C199 | Inhibition of activity | Alteration of barrier function and blood pressure regulation (supposed) | [40,41] |
| PKB | Unknown | [40,42,43] | |||
| MEKK1 | C1238 | [40,44] | |||
| PKC | Unknown | [40,45] | |||
| 6. RONS production | |||||
| p47 phox | NADPH oxidase | C98 C111 C196 | Enhanced protein function | Sustained superoxide production Endothelial dysfunction (supposed) | [46] |
| Complex I | NADH-ubiquinone oxidoreductase | Unknown | [47] | ||
| eNOS | Oxide synthase | C689 C908 | Protein uncoupling | Sustained superoxide production Impaired vasodilation and endothelial dysfunction | [48,49,50,51,52] |
| 7. Calcium-dependent channels | |||||
| IP3R | Ca2+ channel | Unknown (C34 C42 C65?) | Protein activation | Increased [Ca2+]i Regulation of Ca2+ homeostasis | [53,54,55] |
| PMCA | Ca2+ ATPase pump | Unknown | Protein inhibition | [56] | |
| SERCA2b | C674 | Protein activation | Increased Ca2+ uptake in ER stores Cell migration and angiogenesis | [57,58,59,60,61,62] | |
| STIM1 | Ca2+ sensor | C56 | Protein oligomerization | Increased [Ca]i via Orai1 activation Mitochondrial dysfunction | [63] |
| 8. Apoptosis and autophagy | |||||
| Fas | Death receptor | C294 | Enhanced activity | Cell death | [64] |
| Caspase-3 | Protease | C45 (p12) C135 (p17) | Inhibition of proteolytic activity | Cell survival | [65,66] |
| Caspase-8 | Unknown | [67] | |||
| Beclin-1 | Autophagy-related protein | Unknown | Upregulation of protein activity | Not confirmed | [68] |
| 9. Cell structure and dynamics | |||||
| ProMMPs | Metalloprotease | PRCGVPD motif on inhibitory domain | Activation | Angiogenesis and vascular permeability | [69] |
| ICAM-1 | Adhesion receptor | Unknown | Protein degradation | Cell junction disassembly | [70] |
| Actin | Cytoskeletal | C374 | Inhibition of polymerization | Inhibition of cell motility | [71,72,73] |
| Microtubules | Unknown | Cell growth arrest and apoptosis | [74,75] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lermant, A.; Murdoch, C.E. Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants 2019, 8, 315. https://doi.org/10.3390/antiox8080315
AMA Style
Lermant A, Murdoch CE. Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants. 2019; 8(8):315. https://doi.org/10.3390/antiox8080315
Chicago/Turabian StyleLermant, Agathe, and Colin E. Murdoch. 2019. "Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells" Antioxidants 8, no. 8: 315. https://doi.org/10.3390/antiox8080315
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.