Peroxiredoxins in Cancer and Response to Radiation Therapies

 , , and
, , and
Abstract
:1. Introduction
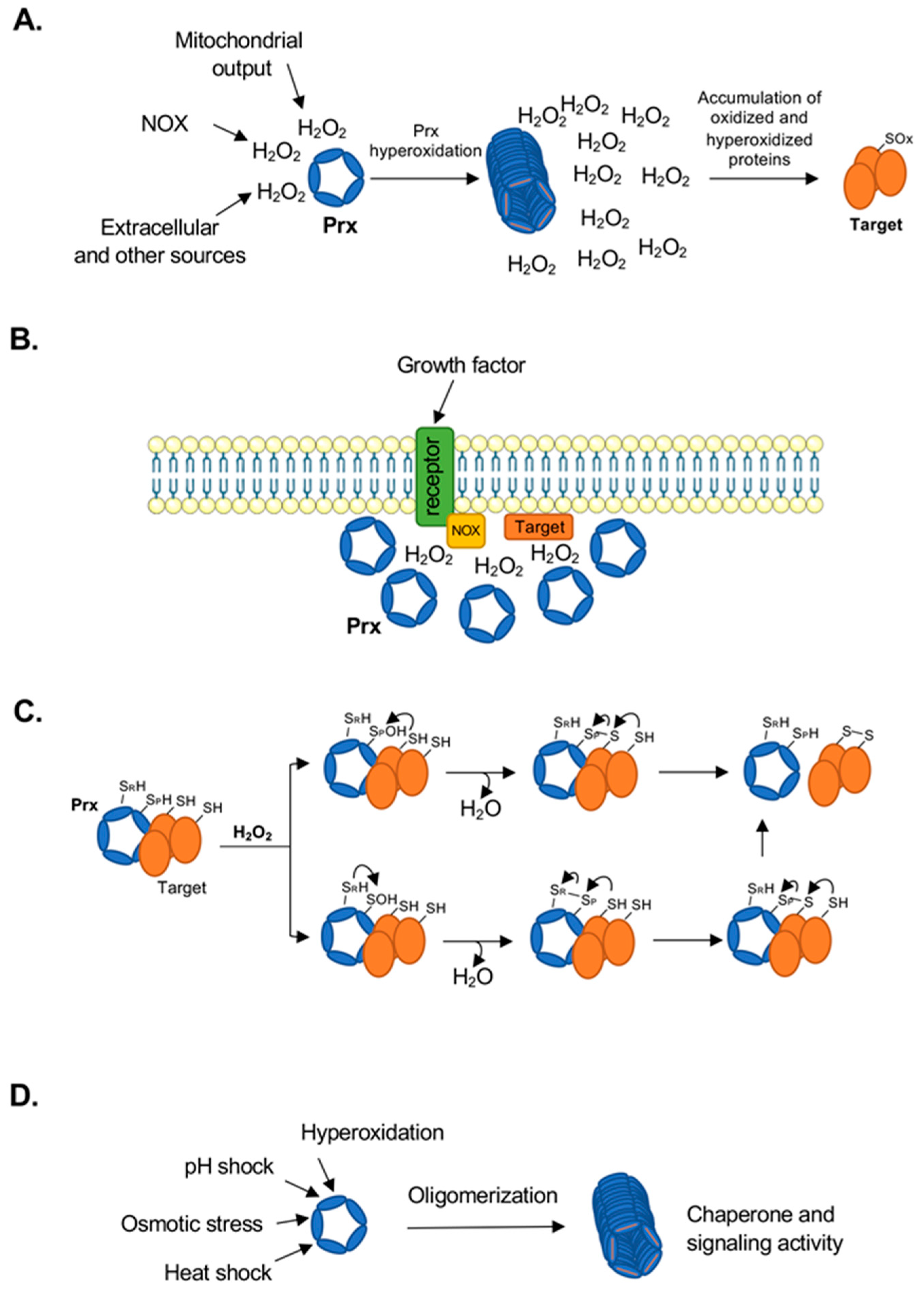
2. Peroxiredoxins as Cellular Antioxidant and Signaling Proteins
2.1. Subcellular Distribution of Prx Isoforms and Catalytic Cycle
2.2. Prx Oligomeric State
3. Peroxiredoxins in H2O2 Sensing, Signaling and Protein Folding
3.1. Sensors of H2O2 Levels
3.2. Regulators of Signaling and Metabolism
3.3. Propagators of Redox Signaling
3.4. Protein Folding/Chaperone Activity
4. Peroxiredoxins in Cancer
4.1. Prx Expression in Cancer and Potential for Targeted Therapeutics
4.2. Peroxiredoxins as Protectors Against DNA Damage
4.3. Peroxiredoxins in Oncogenic Signaling
4.4. Peroxiredoxins and Hypoxia in Cancer
5. Peroxiredoxins in Radiation Treatment of Cancer
5.1. Ionizing Radiation
5.2. Mechanisms of Resistance to IR
5.3. Peroxiredoxins and Response to Cancer Radiation Therapy
5.4. Prx Expression and Radiation Resistance in Cancer Databases
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Idelchik, M.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Pelletier, M.; Lepow, T.S.; Billingham, L.K.; Murphy, M.P.; Siegel, R.M. New tricks from an old dog: Mitochondrial redox signaling in cellular inflammation. Semin. Immunol. 2012, 24, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Roos, G.; Messens, J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic. Biol. Med. 2011, 51, 314–326. [Google Scholar] [CrossRef]
- Devarie-Baez, N.O.; Silva Lopez, E.I.; Furdui, C.M. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 2016, 50, 172–194. [Google Scholar] [CrossRef]
- Chae, H.Z.; Kim, H.J.; Kang, S.W.; Rhee, S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999, 45, 101–112. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Lehtonen, S.T.; Svensk, A.M.; Soini, Y.; Paakko, P.; Hirvikoski, P.; Kang, S.W.; Saily, M.; Kinnula, V.L. Peroxiredoxins, a novel protein family in lung cancer. Int. J. Cancer 2004, 111, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith-Pearson, P.S.; Kooshki, M.; Spitz, D.R.; Poole, L.B.; Zhao, W.; Robbins, M.E. Decreasing peroxiredoxin II expression decreases glutathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H2O2. Free Radic. Biol. Med. 2008, 45, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Su, Y. Peroxiredoxins, a novel target in cancer radiotherapy. Cancer Lett. 2009, 286, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Mims, J.; Kuremsky, J.G.; Olex, A.L.; Zhao, W.; Yin, L.; Wani, R.; Qian, J.; Center, B.; Marrs, G.S.; et al. Broad phenotypic changes associated with gain of radiation resistance in head and neck squamous cell cancer. Antioxid. Redox Signal. 2014, 21, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Iwao-Koizumi, K.; Matoba, R.; Ueno, N.; Kim, S.J.; Ando, A.; Miyoshi, Y.; Maeda, E.; Noguchi, S.; Kato, K. Prediction of docetaxel response in human breast cancer by gene expression profiling. J. Clin. Oncol. 2005, 23, 422–431. [Google Scholar] [CrossRef]
- Harper, A.F.; Leuthaeuser, J.B.; Babbitt, P.C.; Morris, J.H.; Ferrin, T.E.; Poole, L.B.; Fetrow, J.S. An atlas of peroxiredoxins created using an active site profile-based approach to functionally relevant clustering of proteins. PLoS Comput. Biol. 2017, 13, e1005284. [Google Scholar] [CrossRef]
- Nelson, K.J.; Knutson, S.T.; Soito, L.; Klomsiri, C.; Poole, L.B.; Fetrow, J.S. Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins 2011, 79, 947–964. [Google Scholar] [CrossRef]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef]
- Schroder, E.; Littlechild, J.A.; Lebedev, A.A.; Errington, N.; Vagin, A.A.; Isupov, M.N. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 A resolution. Structure 2000, 8, 605–615. [Google Scholar] [CrossRef]
- Jonsson, T.J.; Johnson, L.C.; Lowther, W.T. Structure of the sulphiredoxin-peroxiredoxin complex reveals an essential repair embrace. Nature 2008, 451, 98–101. [Google Scholar] [CrossRef]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Pearson, A.G.; Pullar, J.M.; Jonsson, T.J.; Lowther, W.T.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J. 2009, 421, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Haynes, A.C.; Qian, J.; Reisz, J.A.; Furdui, C.M.; Lowther, W.T. Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J. Biol. Chem. 2013, 288, 29714–29723. [Google Scholar] [CrossRef] [PubMed]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef]
- Cao, Z.; Roszak, A.W.; Gourlay, L.J.; Lindsay, J.G.; Isaacs, N.W. Bovine mitochondrial peroxiredoxin III forms a two-ring catenane. Structure 2005, 13, 1661–1664. [Google Scholar] [CrossRef]
- Morais, M.A.; Giuseppe, P.O.; Souza, T.A.; Alegria, T.G.; Oliveira, M.A.; Netto, L.E.; Murakami, M.T. How pH modulates the dimer-decamer interconversion of 2-Cys peroxiredoxins from the Prx1 subfamily. J. Biol. Chem. 2015, 290, 8582–8590. [Google Scholar] [CrossRef]
- Phillips, A.J.; Littlejohn, J.; Yewdall, N.A.; Zhu, T.; Valery, C.; Pearce, F.G.; Mitra, A.K.; Radjainia, M.; Gerrard, J.A. Peroxiredoxin is a versatile self-assembling tecton for protein nanotechnology. Biomacromolecules 2014, 15, 1871–1881. [Google Scholar] [CrossRef]
- Rivera-Santiago, R.F.; Harper, S.L.; Zhou, S.; Sriswasdi, S.; Feinstein, S.I.; Fisher, A.B.; Speicher, D.W. Solution structure of the reduced form of human peroxiredoxin-6 elucidated using zero-length chemical cross-linking and homology modelling. Biochem. J. 2015, 468, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, W.; Kim, E.E. Crystal structures of human peroxiredoxin 6 in different oxidation states. Biochem. Biophys. Res. Commun. 2016, 477, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sorokina, E.M.; Harper, S.; Li, H.; Ralat, L.; Dodia, C.; Speicher, D.W.; Feinstein, S.I.; Fisher, A.B. Peroxiredoxin 6 homodimerization and heterodimerization with glutathione S-transferase pi are required for its peroxidase but not phospholipase A2 activity. Free Radic. Biol. Med. 2016, 94, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.C.; Hah, Y.S.; Kim, W.Y.; Jung, B.G.; Jang, H.H.; Lee, J.R.; Kim, S.Y.; Lee, Y.M.; Jeon, M.G.; Kim, C.W.; et al. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J. Biol. Chem. 2005, 280, 28775–28784. [Google Scholar] [CrossRef]
- Noichri, Y.; Palais, G.; Ruby, V.; D’Autreaux, B.; Delaunay-Moisan, A.; Nystrom, T.; Molin, M.; Toledano, M.B. In vivo parameters influencing 2-Cys Prx oligomerization: The role of enzyme sulfinylation. Redox Biol. 2015, 6, 326–333. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Nelson, K.J.; Haynes, A.C.; Lee, J.; Reisz, J.A.; Graff, A.H.; Clodfelter, J.E.; Parsonage, D.; Poole, L.B.; Furdui, C.M.; et al. Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J. Biol. Chem. 2018, 293, 11901–11912. [Google Scholar] [CrossRef] [Green Version]
- Portillo-Ledesma, S.; Randall, L.M.; Parsonage, D.; Dalla Rizza, J.; Karplus, P.A.; Poole, L.B.; Denicola, A.; Ferrer-Sueta, G. Differential kinetics of two-cysteine peroxiredoxin disulfide formation reveal a novel model for peroxide sensing. Biochemistry 2018, 57, 3416–3424. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Jeong, W.; Choi, S.Y.; Yu, S.; Kang, S.W.; Rhee, S.G. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J. Biol. Chem. 2002, 277, 25370–25376. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J. Cell Biol. 2015, 210, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Kil, I.S.; Lee, S.K.; Ryu, K.W.; Woo, H.A.; Hu, M.C.; Bae, S.H.; Rhee, S.G. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 2012, 46, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Karplus, P.A.; Poole, L.B. Typical 2-Cys peroxiredoxins—Structures, mechanisms and functions. FEBS J. 2009, 276, 2469–2477. [Google Scholar] [CrossRef] [PubMed]
- Travasso, R.D.M.; Sampaio Dos Aidos, F.; Bayani, A.; Abranches, P.; Salvador, A. Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 2017, 12, 233–245. [Google Scholar] [CrossRef]
- Stocker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef]
- Calvo, I.A.; Boronat, S.; Domenech, A.; Garcia-Santamarina, S.; Ayte, J.; Hidalgo, E. Dissection of a redox relay: H2O2-dependent activation of the transcription factor Pap1 through the peroxidatic Tpx1-thioredoxin cycle. Cell Rep. 2013, 5, 1413–1424. [Google Scholar] [CrossRef]
- Fernandez-Caggiano, M.; Schroder, E.; Cho, H.J.; Burgoyne, J.; Barallobre-Barreiro, J.; Mayr, M.; Eaton, P. Oxidant-induced interprotein disulfide formation in cardiac protein DJ-1 occurs via an interaction with peroxiredoxin 2. J. Biol. Chem. 2016, 291, 10399–10410. [Google Scholar] [CrossRef] [PubMed]
- Furdui, C.M.; Poole, L.B. Chemical approaches to detect and analyze protein sulfenic acids. Mass Spectrom. Rev. 2014, 33, 126–146. [Google Scholar] [CrossRef] [PubMed]
- Holmila, R.J.; Vance, S.A.; Chen, X.; Wu, H.; Shukla, K.; Bharadwaj, M.S.; Mims, J.; Wary, Z.; Marrs, G.; Singh, R.; et al. Mitochondria-targeted probes for imaging protein sulfenylation. Sci. Rep. 2018, 8, 6635. [Google Scholar] [CrossRef]
- Lim, J.C.; Choi, H.I.; Park, Y.S.; Nam, H.W.; Woo, H.A.; Kwon, K.S.; Kim, Y.S.; Rhee, S.G.; Kim, K.; Chae, H.Z. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J. Biol. Chem. 2008, 283, 28873–28880. [Google Scholar] [CrossRef] [PubMed]
- Barranco-Medina, S.; Lazaro, J.J.; Dietz, K.J. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 2009, 583, 1809–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.H.; Kim, S.Y.; Park, S.K.; Jeon, H.S.; Lee, Y.M.; Jung, J.H.; Lee, S.Y.; Chae, H.B.; Jung, Y.J.; Lee, K.O.; et al. Phosphorylation and concomitant structural changes in human 2-Cys peroxiredoxin isotype I differentially regulate its peroxidase and molecular chaperone functions. FEBS Lett. 2006, 580, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Piszczek, G.; Rhee, S.G.; Chock, P.B. Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry 2011, 50, 3204–3210. [Google Scholar] [CrossRef]
- Yewdall, N.A.; Venugopal, H.; Desfosses, A.; Abrishami, V.; Yosaatmadja, Y.; Hampton, M.B.; Gerrard, J.A.; Goldstone, D.C.; Mitra, A.K.; Radjainia, M. Structures of human peroxiredoxin 3 suggest self-chaperoning assembly that maintains catalytic state. Structure 2016, 24, 1120–1129. [Google Scholar] [CrossRef]
- Day, A.M.; Brown, J.D.; Taylor, S.R.; Rand, J.D.; Morgan, B.A.; Veal, E.A. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell 2012, 45, 398–408. [Google Scholar] [CrossRef]
- Bayer, S.B.; Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Interactions between peroxiredoxin 2, hemichrome and the erythrocyte membrane. Free Radic. Res. 2016, 50, 1329–1339. [Google Scholar] [CrossRef]
- Engelman, R.; Weisman-Shomer, P.; Ziv, T.; Xu, J.; Arner, E.S.; Benhar, M. Multilevel regulation of 2-Cys peroxiredoxin reaction cycle by S-nitrosylation. J. Biol. Chem. 2013, 288, 11312–11324. [Google Scholar] [CrossRef] [PubMed]
- Randall, L.; Manta, B.; Nelson, K.J.; Santos, J.; Poole, L.B.; Denicola, A. Structural changes upon peroxynitrite-mediated nitration of peroxiredoxin 2; nitrated Prx2 resembles its disulfide-oxidized form. Arch. Biochem. Biophys. 2016, 590, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Barrett, J.C. Reactive oxygen species as double-edged swords in cellular processes: Low-dose cell signaling versus high-dose toxicity. Hum. Exp. Toxicol. 2002, 21, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lutz, W.K. Endogenous genotoxic agents and processes as a basis of spontaneous carcinogenesis. Mutat. Res. 1990, 238, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Freese, E.B.; Gerson, J.; Taber, H.; Rhaese, H.J.; Freese, E. Inactivating DNA alterations induced by peroxides and peroxide-producing agents. Mutat. Res. 1967, 4, 517–531. [Google Scholar] [CrossRef]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Egler, R.A.; Fernandes, E.; Rothermund, K.; Sereika, S.; de Souza-Pinto, N.; Jaruga, P.; Dizdaroglu, M.; Prochownik, E.V. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene 2005, 24, 8038–8050. [Google Scholar] [CrossRef] [Green Version]
- Argyropoulou, V.; Goemaere, J.; Clippe, A.; Lefort, C.; Tissir, F.; Schakman, O.; Gailly, P.; Ahn, M.-T.; Guiot, Y.; Galant, C.; et al. Peroxiredoxin-5 as a Novel Actor in Inflammation and Tumor Suppression. Free Radic Biol. Med. 2016, 100, S92. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, re308. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox paradox: A novel approach to therapeutics-resistant cancer. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Vick, K.A.; Skoko, J.J.; Neumann, C.A. Peroxiredoxin involvement in the initiation and progression of human cancer. Antioxid. Redox Signal. 2018, 28, 591–608. [Google Scholar] [CrossRef]
- Chang, X.Z.; Li, D.Q.; Hou, Y.F.; Wu, J.; Lu, J.S.; Di, G.H.; Jin, W.; Ou, Z.L.; Shen, Z.Z.; Shao, Z.M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, R76. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, J.; Xie, J.; Niu, Y.; Ye, S.; Wan, Q.; Ye, Q. Detection and identification of peroxiredoxin 3 as a biomarker in hepatocellular carcinoma by a proteomic approach. Int. J. Mol. Med. 2012, 29, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Baek, J.Y.; Goo, J.; Shin, Y.; Park, J.K.; Jang, J.Y.; Wang, S.B.; Jeong, W.; Lee, H.J.; Um, H.D.; et al. Effective killing of cancer cells through ROS-mediated mechanisms by AMRI-59 targeting peroxiredoxin I. Antioxid. Redox Signal. 2016, 24, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, B.; Newick, K.; Nelson, K.J.; Wozniak, A.N.; Beuschel, S.; Leavitt, B.; Bhave, A.; Butnor, K.; Koenig, A.; Chouchani, E.T.; et al. Disabling mitochondrial peroxide metabolism via combinatorial targeting of peroxiredoxin 3 as an effective therapeutic approach for malignant mesothelioma. PLoS ONE 2015, 10, e0127310. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; Cunniff, B.; Preston, K.; Held, P.; Arbiser, J.; Pass, H.; Mossman, B.; Shukla, A.; Heintz, N. Peroxiredoxin 3 is a redox-dependent target of thiostrepton in malignant mesothelioma cells. PLoS ONE 2012, 7, e39404. [Google Scholar] [CrossRef]
- Li, L.; Shoji, W.; Takano, H.; Nishimura, N.; Aoki, Y.; Takahashi, R.; Goto, S.; Kaifu, T.; Takai, T.; Obinata, M. Increased susceptibility of MER5 (peroxiredoxin III) knockout mice to LPS-induced oxidative stress. Biochem. Biophys. Res. Commun. 2007, 355, 715–721. [Google Scholar] [CrossRef]
- Bruner, S.D.; Norman, D.P.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313 Pt 1, 17–29. [Google Scholar] [CrossRef]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 protects telomeres from oxidative damage and preserves telomeric DNA for extension by telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Lingner, J. PRDX1 and MTH1 cooperate to prevent ROS-mediated inhibition of telomerase. Genes Dev. 2018, 32, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Vallabhaneni, H.; Yin, J.; Liu, Y. Deletion of the major peroxiredoxin Tsa1 alters telomere length homeostasis. Aging Cell 2013, 12, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somyajit, K.; Gupta, R.; Sedlackova, H.; Neelsen, K.J.; Ochs, F.; Rask, M.B.; Choudhary, C.; Lukas, J. Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science 2017, 358, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Mauney, C.H.; Rogers, L.C.; Harris, R.S.; Daniel, L.W.; Devarie-Baez, N.O.; Wu, H.; Furdui, C.M.; Poole, L.B.; Perrino, F.W.; Hollis, T. The SAMHD1 dNTP triphosphohydrolase is controlled by a redox switch. Antioxid. Redox Signal. 2017, 27, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- West, J.D.; Roston, T.J.; David, J.B.; Allan, K.M.; Loberg, M.A. Piecing together how peroxiredoxins maintain genomic stability. Antioxidants 2018, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Banmeyer, I.; Marchand, C.; Clippe, A.; Knoops, B. Human mitochondrial peroxiredoxin 5 protects from mitochondrial DNA damages induced by hydrogen peroxide. FEBS Lett. 2005, 579, 2327–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banmeyer, I.; Marchand, C.; Verhaeghe, C.; Vucic, B.; Rees, J.F.; Knoops, B. Overexpression of human peroxiredoxin 5 in subcellular compartments of Chinese hamster ovary cells: Effects on cytotoxicity and DNA damage caused by peroxides. Free Radic. Biol. Med. 2004, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Kim, S.U.; Lee, B.K.; Kim, H.S.; Song, I.S.; Shin, H.J.; Han, Y.H.; Chang, K.T.; Kim, J.M.; Lee, D.S.; et al. Prx I suppresses K-ras-driven lung tumorigenesis by opposing redox-sensitive ERK/cyclin D1 pathway. Antioxid. Redox Signal. 2013, 19, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolfs, F.; Huber, M.; Gruber, F.; Bohm, F.; Pfister, H.J.; Bochkov, V.N.; Tschachler, E.; Dummer, R.; Hohl, D.; Schafer, M.; et al. Dual role of the antioxidant enzyme peroxiredoxin 6 in skin carcinogenesis. Cancer Res. 2013, 73, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Frijhoff, J.; Sandin, A.; Bohmer, F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011, 150, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.T.; Van Etten, R.A. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997, 11, 2456–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morell, M.; Espargaro, A.; Aviles, F.X.; Ventura, S. Detection of transient protein-protein interactions by bimolecular fluorescence complementation: The Abl-SH3 case. Proteomics 2007, 7, 1023–1036. [Google Scholar] [CrossRef]
- Wong, C.M.; Zhou, Y.; Ng, R.W.; Kung Hf, H.F.; Jin, D.Y. Cooperation of yeast peroxiredoxins Tsa1p and Tsa2p in the cellular defense against oxidative and nitrosative stress. J. Biol. Chem. 2002, 277, 5385–5394. [Google Scholar] [CrossRef]
- Takeuchi, K.; Morizane, Y.; Kamami-Levy, C.; Suzuki, J.; Kayama, M.; Cai, W.; Miller, J.W.; Vavvas, D.G. AMP-dependent kinase inhibits oxidative stress-induced caveolin-1 phosphorylation and endocytosis by suppressing the dissociation between c-Abl and Prdx1 proteins in endothelial cells. J. Biol. Chem. 2013, 288, 20581–20591. [Google Scholar] [CrossRef]
- Turner-Ivey, B.; Manevich, Y.; Schulte, J.; Kistner-Griffin, E.; Jezierska-Drutel, A.; Liu, Y.; Neumann, C.A. Role for Prdx1 as a specific sensor in redox-regulated senescence in breast cancer. Oncogene 2013, 32, 5302–5314. [Google Scholar] [CrossRef] [Green Version]
- Schindler, E.M.; Hindes, A.; Gribben, E.L.; Burns, C.J.; Yin, Y.; Lin, M.H.; Owen, R.J.; Longmore, G.D.; Kissling, G.E.; Arthur, J.S.; et al. p38delta Mitogen-activated protein kinase is essential for skin tumor development in mice. Cancer Res. 2009, 69, 4648–4655. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.M.; Yin, X.Y.; Prochownik, E.V. Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J. Biol. Chem. 2002, 277, 43175–43184. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Wang, N.; Chen, Q.; Xu, J.; Cheng, W.; Di, M.; Xia, W.; Gao, W.Q. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget 2015, 6, 26494–26507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Yin, Q.; Fujio, Y.; Walsh, K.; Griendling, K.K. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 22699–22704. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Tian, D.; He, J.; Wang, Y.; Zhang, L.; Cui, L.; Jia, L.; Zhang, L.; Li, L.; Shu, Y.; et al. Repeated PM2.5 exposure inhibits BEAS-2B cell P53 expression through ROS-Akt-DNMT3B pathway-mediated promoter hypermethylation. Oncotarget 2016, 7, 20691–20703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, R.; Qian, J.; Yin, L.; Bechtold, E.; King, S.B.; Poole, L.B.; Paek, E.; Tsang, A.W.; Furdui, C.M. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc. Natl. Acad. Sci. USA 2011, 108, 10550–10555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, A.L.; Zeng, W.; Cai, W.L.; Liu, J.L.; Zheng, X.W.; Liu, Y.; Yang, X.C.; Long, Y.; Li, J. Peroxiredoxin-1 promotes cell proliferation and metastasis through enhancing Akt/mTOR in human osteosarcoma cells. Oncotarget 2018, 9, 8290–8302. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, S.; Wang, R.; Wu, X.; Zeng, L.; Fu, Z. Knockdown of PRDX2 sensitizes colon cancer cells to 5-FU by suppressing the PI3K/AKT signaling pathway. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Ho, J.N.; Yoon, S.H.; Kang, G.Y.; Hwang, S.G.; Um, H.D. Peroxiredoxin 6 promotes lung cancer cell invasion by inducing urokinase-type plasminogen activator via p38 kinase, phosphoinositide 3-kinase, and Akt. Mol. Cells 2009, 28, 583–588. [Google Scholar] [CrossRef]
- He, Y.; Xu, W.; Xiao, Y.; Pan, L.; Chen, G.; Tang, Y.; Zhou, J.; Wu, J.; Zhu, W.; Zhang, S.; et al. Overexpression of peroxiredoxin 6 (PRDX6) promotes the aggressive phenotypes of esophageal squamous cell carcinoma. J. Cancer 2018, 9, 3939–3949. [Google Scholar] [CrossRef]
- Riddell, J.R.; Bshara, W.; Moser, M.T.; Spernyak, J.A.; Foster, B.A.; Gollnick, S.O. Peroxiredoxin 1 controls prostate cancer growth through Toll-like receptor 4-dependent regulation of tumor vasculature. Cancer Res. 2011, 71, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Kashiwagi, E.; Takeuchi, A.; Fujimoto, N.; Uchiumi, T.; Naito, S. Peroxiredoxin 2 in the nucleus and cytoplasm distinctly regulates androgen receptor activity in prostate cancer cells. Free Radic. Biol. Med. 2011, 51, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, H.C.; Patel, D.; Howat, W.J.; Warren, A.Y.; Kay, J.D.; Sangan, T.; Marioni, J.C.; Mitchell, J.; Aldridge, S.; Luxton, H.J.; et al. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br. J. Cancer 2013, 109, 983–993. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Banerjee, H.; Rojas, H.; Martinez, S.R.; Roy, S.; Jia, Z.; Lilly, M.B.; De Leon, M.; Casiano, C.A. Differential expression of peroxiredoxins in prostate cancer: Consistent upregulation of PRDX3 and PRDX4. Prostate 2011, 71, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Raatikainen, S.; Aaaltomaa, S.; Karja, V.; Soini, Y. Increased peroxiredoxin 6 expression predicts biochemical recurrence in prostate cancer patients after radical prostatectomy. Anticancer Res. 2015, 35, 6465–6470. [Google Scholar]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 2016, 57 (Suppl. 1), i90–i98. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 2005, 1, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Waypa, G.B.; Marks, J.D.; Schumacker, P.T. Peroxiredoxin-5 targeted to the mitochondrial intermembrane space attenuates hypoxia-induced reactive oxygen species signalling. Biochem. J. 2013, 456, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Ahn, J.Y.; Liang, P.; Ip, C.; Zhang, Y.; Park, Y.M. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: Implication to tumor biology. Cancer Res. 2007, 67, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Chae, H.Z.; Kim, Y.J.; Kim, Y.H.; Hwangs, T.S.; Park, E.M.; Park, Y.M. Preferential elevation of Prx I and Trx expression in lung cancer cells following hypoxia and in human lung cancer tissues. Cell Biol. Toxicol. 2003, 19, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hou, M.; Ge, L.; Miao, C.; Zhang, J.; Jing, X.; Shi, N.; Chen, T.; Tang, X. Induction of peroxiredoxin 1 by hypoxia regulates heme oxygenase-1 via NF-kappaB in oral cancer. PLoS ONE 2014, 9, e105994. [Google Scholar] [CrossRef]
- Huh, J.Y.; Kim, Y.; Jeong, J.; Park, J.; Kim, I.; Huh, K.H.; Kim, Y.S.; Woo, H.A.; Rhee, S.G.; Lee, K.J.; et al. Peroxiredoxin 3 is a key molecule regulating adipocyte oxidative stress, mitochondrial biogenesis, and adipokine expression. Antioxid. Redox Signal. 2012, 16, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.K.; Zhu, J.Y.; Wang, W.; Lv, Y.; Zhou, H.C.; Yu, J.H.; Xu, G.L.; Ma, J.L.; Zhong, W.; Jia, W.D. Diagnostic and prognostic significance of peroxiredoxin 1 expression in human hepatocellular carcinoma. Med. Oncol. 2014, 31, 786. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Y.; Zhai, L.L.; Wu, Y.; Tang, Z.G. Expression and clinical value of peroxiredoxin-1 in patients with pancreatic cancer. Eur. J. Surg. Oncol. 2015, 41, 228–235. [Google Scholar] [CrossRef]
- Nonn, L.; Berggren, M.; Powis, G. Increased expression of mitochondrial peroxiredoxin-3 (thioredoxin peroxidase-2) protects cancer cells against hypoxia and drug-induced hydrogen peroxide-dependent apoptosis. Mol. Cancer Res. 2003, 1, 682–689. [Google Scholar]
- Xi, H.; Gao, Y.H.; Han, D.Y.; Li, Q.Y.; Feng, L.J.; Zhang, W.; Ji, G.; Xiao, J.C.; Zhang, H.Z.; Wei, Q. Hypoxia inducible factor-1alpha suppresses Peroxiredoxin 3 expression to promote proliferation of CCRCC cells. FEBS Lett. 2014, 588, 3390–3394. [Google Scholar] [CrossRef]
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5’ enhancer. Circ. Res. 1995, 77, 638–643. [Google Scholar] [CrossRef]
- Kang, D.H.; Lee, D.J.; Lee, K.W.; Park, Y.S.; Lee, J.Y.; Lee, S.H.; Koh, Y.J.; Koh, G.Y.; Choi, C.; Yu, D.Y.; et al. Peroxiredoxin II is an essential antioxidant enzyme that prevents the oxidative inactivation of VEGF receptor-2 in vascular endothelial cells. Mol. Cell 2011, 44, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.W.; Lin, S.C.; Chen, K.F.; Lai, Y.Y.; Tsai, S.J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef]
- Xia, Y.; Warshaw, J.B.; Haddad, G.G. Effect of chronic hypoxia on glucose transporters in heart and skeletal muscle of immature and adult rats. Am. J. Physiol. 1997, 273, R1734–R1741. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, J.L.; Garcia-Cazarin, M.L.; Andrade, F.H. Chronic hypoxia increases insulin-stimulated glucose uptake in mouse soleus muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R85–R91. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Chen, I.; Chen, Y.; Alkam, D.; Wang, Y.; Semenza, G.L. PRDX2 and PRDX4 are negative regulators of hypoxia-inducible factors under conditions of prolonged hypoxia. Oncotarget 2016, 7, 6379–6397. [Google Scholar] [CrossRef] [Green Version]
- Nulton-Persson, A.C.; Szweda, L.I. Modulation of mitochondrial function by hydrogen peroxide. J. Biol. Chem. 2001, 276, 23357–23361. [Google Scholar] [CrossRef]
- Chen, L.; Na, R.; Gu, M.; Salmon, A.B.; Liu, Y.; Liang, H.; Qi, W.; Van Remmen, H.; Richardson, A.; Ran, Q. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell 2008, 7, 866–878. [Google Scholar] [CrossRef]
- Overgaard, J. Hypoxic radiosensitization: Adored and ignored. J. Clin. Oncol. 2007, 25, 4066–4074. [Google Scholar] [CrossRef]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Thoday, J.M.; Read, J. Effect of oxygen on the frequency of chromosome aberrations produced by X-rays. Nature 1947, 160, 608. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.M.; Koch, C.J. Prognostic significance of tumor oxygenation in humans. Cancer Lett. 2003, 195, 1–16. [Google Scholar] [CrossRef]
- Howard-Flanders, P.; Alper, T. The sensitivity of microorganisms to irradiation under controlled gas conditions. Radiat. Res. 1957, 7, 518–540. [Google Scholar] [CrossRef]
- Ewing, D. The oxygen fixation hypothesis: A reevaluation. Am. J. Clin. Oncol. 1998, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Pena-Rico, M.A.; Calvo-Vidal, M.N.; Villalonga-Planells, R.; Martinez-Soler, F.; Gimenez-Bonafe, P.; Navarro-Sabate, A.; Tortosa, A.; Bartrons, R.; Manzano, A. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother Oncol. 2011, 101, 132–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Kim, T.N.; Kim, S.; Baek, S.H.; Kim, J.H.; Lee, S.R.; Kim, J.R. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res. 2002, 22, 3331–3335. [Google Scholar]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Qi, F.; Kobayashi, J. Potential relationship between the biological effects of low-dose irradiation and mitochondrial ROS production. J. Radiat. Res. 2018, 59, ii91–ii97. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; Wang, H.J.; Qian, H.L. Biological effects of radiation on cancer cells. Mil. Med. Res. 2018, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef]
- Prise, K.M.; O’Sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, W.K.; Morton, R.A. X-ray sensitivity during the cell generation cycle of cultured Chinese hamster cells. Radiat. Res. 1966, 29, 450–474. [Google Scholar] [CrossRef]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Tang, F.R.; Loke, W.K. Molecular mechanisms of low dose ionizing radiation-induced hormesis, adaptive responses, radioresistance, bystander effects, and genomic instability. Int. J. Radiat. Biol. 2015, 91, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.; Grantham, V. Radiation hormesis: Historical and current perspectives. J. Nucl. Med. Technol. 2015, 43, 242–246. [Google Scholar] [CrossRef] [PubMed]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Chung, Y.M.; Lee, Y.S.; Kim, H.J.; Kim, J.S.; Chae, H.Z.; Yoo, Y.D. Antisense of human peroxiredoxin II enhances radiation-induced cell death. Clin. Cancer Res. 2000, 6, 4915–4920. [Google Scholar] [PubMed]
- Lee, Y.S.; Chang, H.W.; Jeong, J.E.; Lee, S.W.; Kim, S.Y. Proteomic analysis of two head and neck cancer cell lines presenting different radiation sensitivity. Acta. Otolaryngol. 2008, 128, 86–92. [Google Scholar] [CrossRef]
- Zhang, B.; Su, Y.P.; Ai, G.P.; Liu, X.H.; Wang, F.C.; Cheng, T.M. Differentially expressed proteins of gamma-ray irradiated mouse intestinal epithelial cells by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. World J. Gastroenterol. 2003, 9, 2726–2731. [Google Scholar] [CrossRef]
- Chen, W.C.; McBride, W.H.; Iwamoto, K.S.; Barber, C.L.; Wang, C.C.; Oh, Y.T.; Liao, Y.P.; Hong, J.H.; de Vellis, J.; Shau, H. Induction of radioprotective peroxiredoxin-I by ionizing irradiation. J. Neurosci. Res. 2002, 70, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Park, J.S.; Kim, Y.J.; Soo Lee, Y.S.; Sook Hwang, T.S.; Kim, D.J.; Park, E.M.; Park, Y.M. Differential expression of Prx I and II in mouse testis and their up-regulation by radiation. Biochem. Biophys. Res. Commun. 2002, 296, 337–342. [Google Scholar] [CrossRef]
- Di Pietro, R.; Fang, H.; Fields, K.; Miller, S.; Flora, M.; Petricoin, E.C.; Dveksler, G.; Rana, R.A.; Grimley, P.M. Peroxiredoxin genes are not induced in myeloid leukemia cells exposed to ionizing radiation. Int. J. Immunopathol. Pharmacol. 2006, 19, 517–524. [Google Scholar] [CrossRef]
- Park, J.J.; Chang, H.W.; Jeong, E.J.; Roh, J.L.; Choi, S.H.; Jeon, S.Y.; Ko, G.H.; Kim, S.Y. Peroxiredoxin IV protects cells from radiation-induced apoptosis in head-and-neck squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tamae, D.; LeBon, T.; Shively, J.E.; Yen, Y.; Li, J.J. The role of peroxiredoxin II in radiation-resistant MCF-7 breast cancer cells. Cancer Res. 2005, 65, 10338–10346. [Google Scholar] [CrossRef]
- Zhang, B.; Su, Y.; Ai, G.; Wang, Y.; Wang, T.; Wang, F. Involvement of peroxiredoxin I in protecting cells from radiation-induced death. J. Radiat. Res. 2005, 46, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Chen, W.C.; Wu, C.T.; Lin, P.Y.; Shau, H.; Liao, S.K.; Yang, C.T.; Lee, K.D. p53 status is a major determinant of effects of decreasing peroxiredoxin I expression on tumor growth and response of lung cancer cells to treatment. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Lee, W.S.; Ip, C.; Chae, H.Z.; Park, E.M.; Park, Y.M. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006, 66, 7136–7142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, W.; Gu, Q.; Xue, J.; Cao, H.; Tang, Y.; Xu, X.; Cao, J.; Zhou, J.; Wu, J.; et al. Protein and miRNA profiling of radiation-induced skin injury in rats: The protective role of peroxiredoxin-6 against ionizing radiation. Free Radic. Biol. Med. 2014, 69, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Jackson, L.K.; Johnson, W.E.; Li, D.Y.; Bild, A.H.; Piccolo, S.R. Alternative preprocessing of RNA-Sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics 2015, 31, 3666–3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholami, A.M.; Hahne, H.; Wu, Z.; Auer, F.J.; Meng, C.; Wilhelm, M.; Kuster, B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013, 4, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Schneider, T. Analysis of incomplete climate data: Estimation of mean values and covariance matrices and imputation of missing values. J. Clim. 2001, 14, 853–871. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, S.C.; Kim, S.J.; Park, C.H.; Jeung, H.C.; Kim, Y.B.; Ahn, J.B.; Chung, H.C.; Rha, S.Y. Identification of a radiosensitivity signature using integrative metaanalysis of published microarray data for NCI-60 cancer cells. BMC Genom. 2012, 13, 348. [Google Scholar] [CrossRef]
- Balliet, R.M.; Capparelli, C.; Guido, C.; Pestell, T.G.; Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Pestell, R.G.; Howell, A.; et al. Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: Understanding the aging and cancer connection. Cell Cycle 2011, 10, 4065–4073. [Google Scholar] [CrossRef] [Green Version]
- Seifried, H.E.; McDonald, S.S.; Anderson, D.E.; Greenwald, P.; Milner, J.A. The antioxidant conundrum in cancer. Cancer Res. 2003, 63, 4295–4298. [Google Scholar] [PubMed]
- Ladas, E.J.; Jacobson, J.S.; Kennedy, D.D.; Teel, K.; Fleischauer, A.; Kelly, K.M. Antioxidants and cancer therapy: A systematic review. J. Clin. Oncol. 2004, 22, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Dietary antioxidants during cancer chemotherapy: Impact on chemotherapeutic effectiveness and development of side effects. Nutr. Cancer 2000, 37, 1–18. [Google Scholar] [CrossRef]
- D’Andrea, G.M. Use of antioxidants during chemotherapy and radiotherapy should be avoided. CA Cancer J. Clin. 2005, 55, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, H.; Hershman, D.L.; Jacobson, J.S. Use of antioxidant supplements during breast cancer treatment: A comprehensive review. Breast Cancer Res. Treat. 2009, 115, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prx Protein | Prx1 | Prx2 | Prx3 | Prx4 | Prx5 | Prx6 |
|---|---|---|---|---|---|---|
| Class | Prx1 | Prx5 | Prx6 | |||
| Mechanism | Typical 2-Cys | Atypical 2-Cys | 1-Cys | |||
| Localization | Cytosol Nucleus | Cytosol Lipid membranes Nucleus | Mitochondria | ER/Golgi Extracellular | Cytosol Mitochondria Peroxisome | Cytosol |
| Prx Class | Prx Protein | Oxidation State | Favored Oligomeric State in Solution | Size on Non-Reducing SDS-PAGE | References |
|---|---|---|---|---|---|
| Prx1 | Prx1, 2, & 4 | SH (reduced) | Decamer | Monomer | [18,29] |
| Prx3 | SH (reduced) | Dodecamer | Monomer | [30] | |
| Prx1, 2, 3, & 4 | SS (oxidized) | Dimer | Dimer | [18,29] | |
| Prx1, 2, 3, & 4 | SOH (oxidized) | n.d. | Monomer 1 | ||
| Prx1, 2, 3, & 4 | SO2H (hyperoxidized) | Decamer & high order oligomers 2 | Can run as either dimer (1 SO2H, 1SS in dimer) or monomer (2 SO2H per dimer) | [19] | |
| Prx5 | Prx5 | SH (reduced) | Dimer | Monomer | [18] |
| Prx5 | SS (oxidized) | Dimer | Monomer | [18] | |
| Prx6 | Prx6 | SH (reduced) | Dimer | Monomer | [31,32] |
| Prx6 | SOH, SO2H (oxidized) | Dimer and monomer | Monomer | [32,33,34] |
| Protein | Modification | Effect on Oligomeric State | References |
|---|---|---|---|
| Prx1 | Glutathionylation-C83 (Non-catalytic Cys at dimer/dimer interface) | Destabilizes decamer | [57] |
| p-Thr90 | Favors decamer and higher order oligomers | [56] | |
| p-Tyr194 | No change in SS oligomers | [43] | |
| CP-S-nitrosylation | Destabilizes decamer | [61] | |
| Prx2 | Tyr nitration by ONOO− | Destabilizes decamer | [62] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forshaw, T.E.; Holmila, R.; Nelson, K.J.; Lewis, J.E.; Kemp, M.L.; Tsang, A.W.; Poole, L.B.; Lowther, W.T.; Furdui, C.M. Peroxiredoxins in Cancer and Response to Radiation Therapies. Antioxidants 2019, 8, 11. https://doi.org/10.3390/antiox8010011
Forshaw TE, Holmila R, Nelson KJ, Lewis JE, Kemp ML, Tsang AW, Poole LB, Lowther WT, Furdui CM. Peroxiredoxins in Cancer and Response to Radiation Therapies. Antioxidants. 2019; 8(1):11. https://doi.org/10.3390/antiox8010011
Chicago/Turabian StyleForshaw, Tom E., Reetta Holmila, Kimberly J. Nelson, Joshua E. Lewis, Melissa L. Kemp, Allen W. Tsang, Leslie B. Poole, W. Todd Lowther, and Cristina M. Furdui. 2019. "Peroxiredoxins in Cancer and Response to Radiation Therapies" Antioxidants 8, no. 1: 11. https://doi.org/10.3390/antiox8010011