
Dipeptide Repeat Pathology in C9orf72-ALS Is Associated with Redox, Mitochondrial and NRF2 Pathway Imbalance
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Targeted qRT-PCR Analysis in Human Samples
2.2. Cell Culture and Reagents
2.3. Transfection, Lentiviral Production and Transduction
2.4. Immunofluorescence Analysis
2.5. Superoxide Generation and Mitochondrial Membrane Potential Measurement
2.6. Cell Viability Assessed by MTT Reduction and Annexin-V/Propidium Iodide
2.7. Cell Cycle Analysis
2.8. Immunoblotting
2.9. mRNA Quantification by qRT-PCR
2.10. Luciferase Assays
2.11. RNA Immunoprecipitation
2.12. Statistical Analyses
3. Results
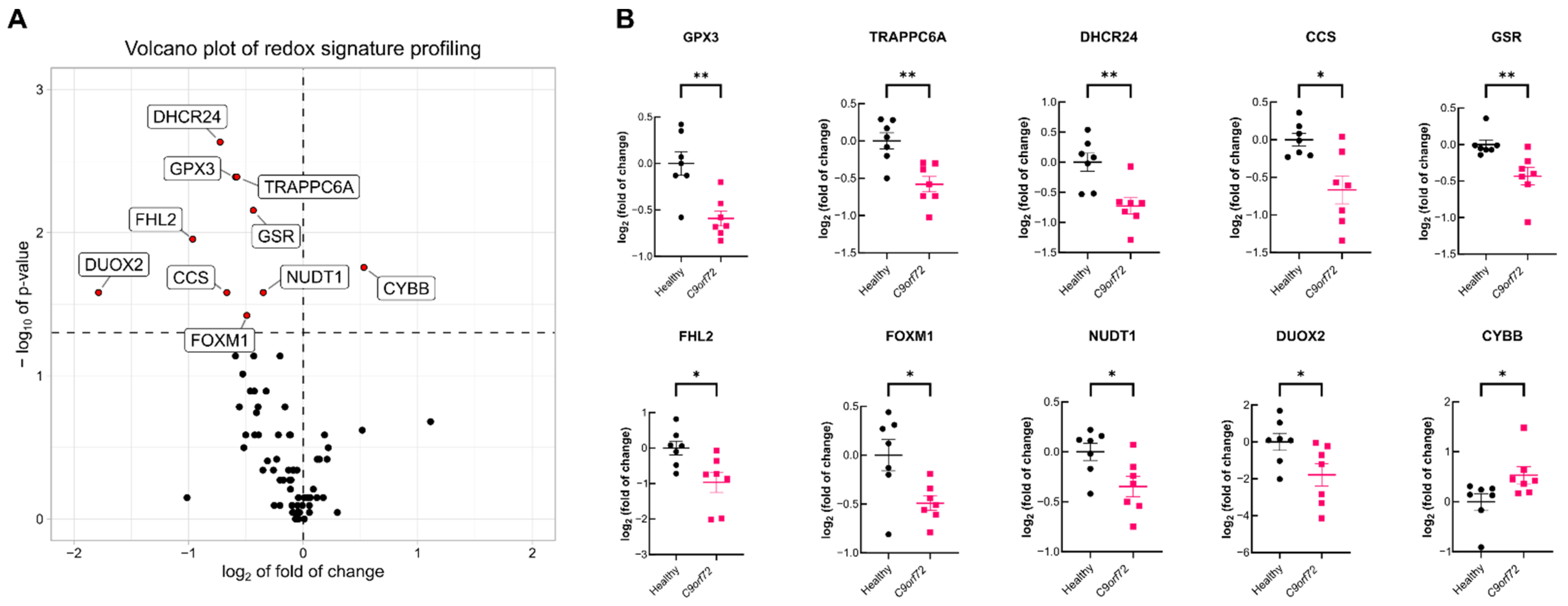
3.1. C9orf72 Patient Leukocytes Exhibit Dysfunctional Expression of Several Redox Genes
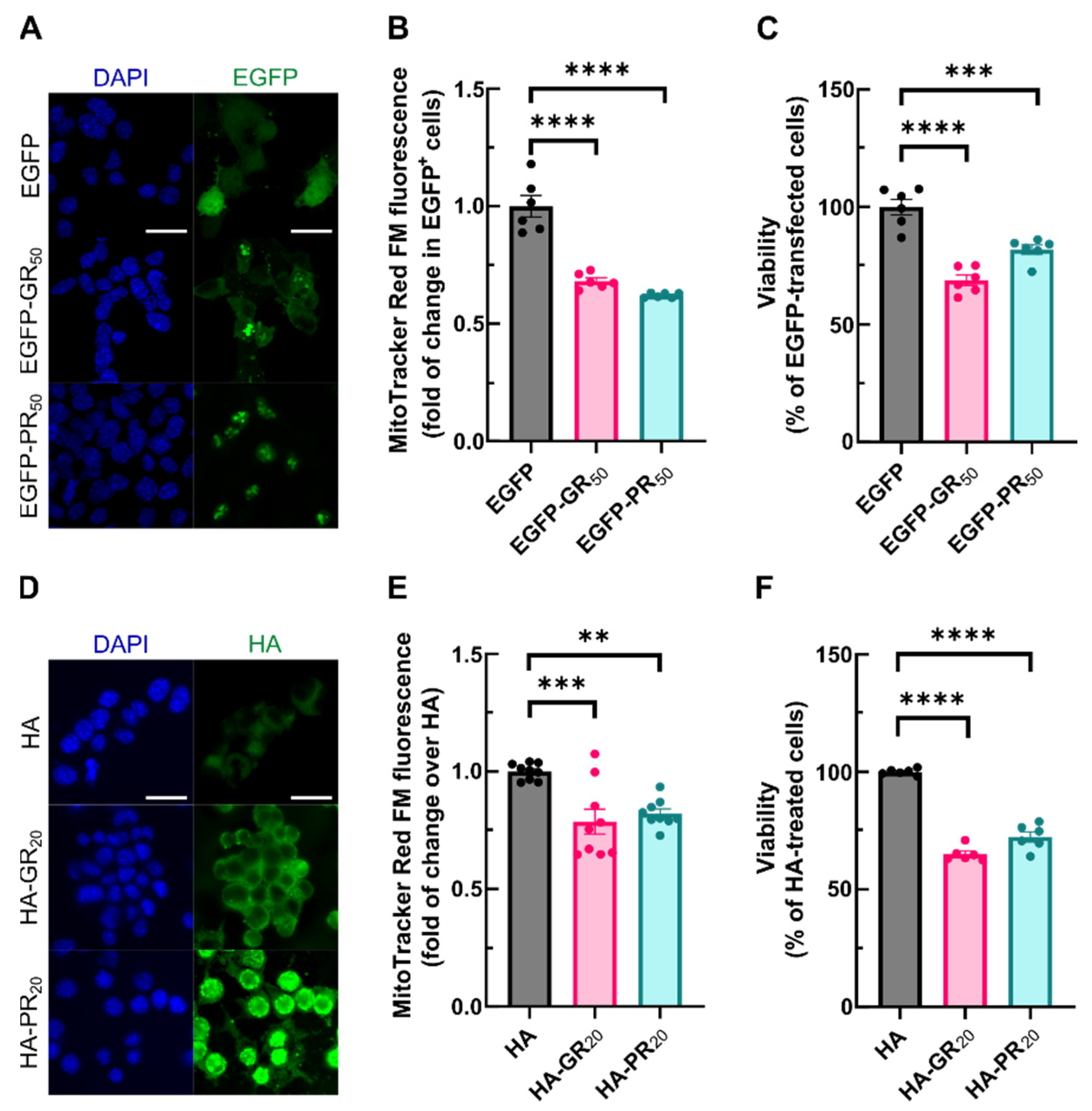
3.2. Arg-DPRs Lead to Decreased Mitochondrial Membrane Potential and Cell Death
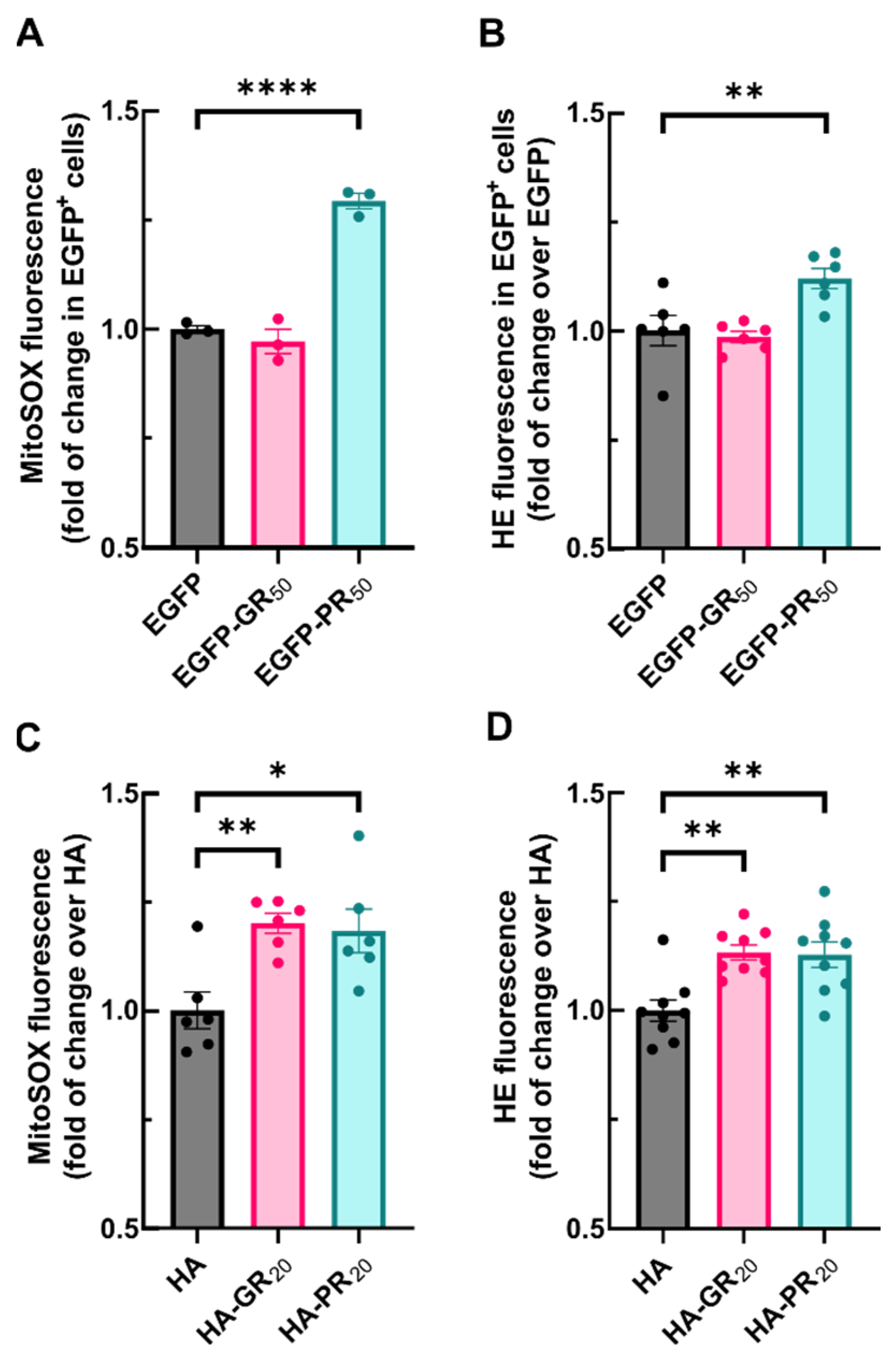
3.3. Arg-Containing Dipeptide Repeats from the C9orf72 Expansion Disrupt Redox Homeostasis
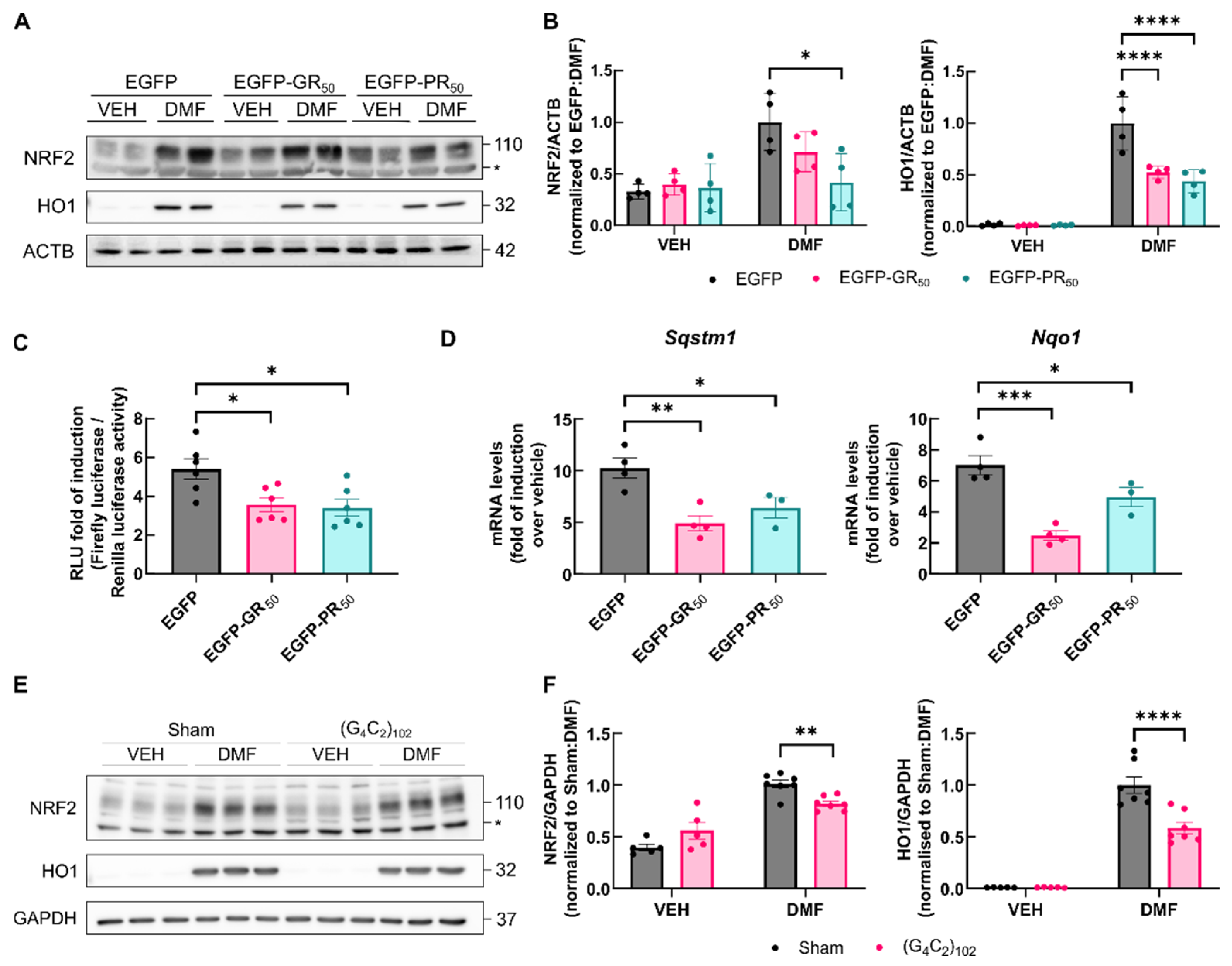
3.4. Activation of NRF2 Is Impeded by DPRs
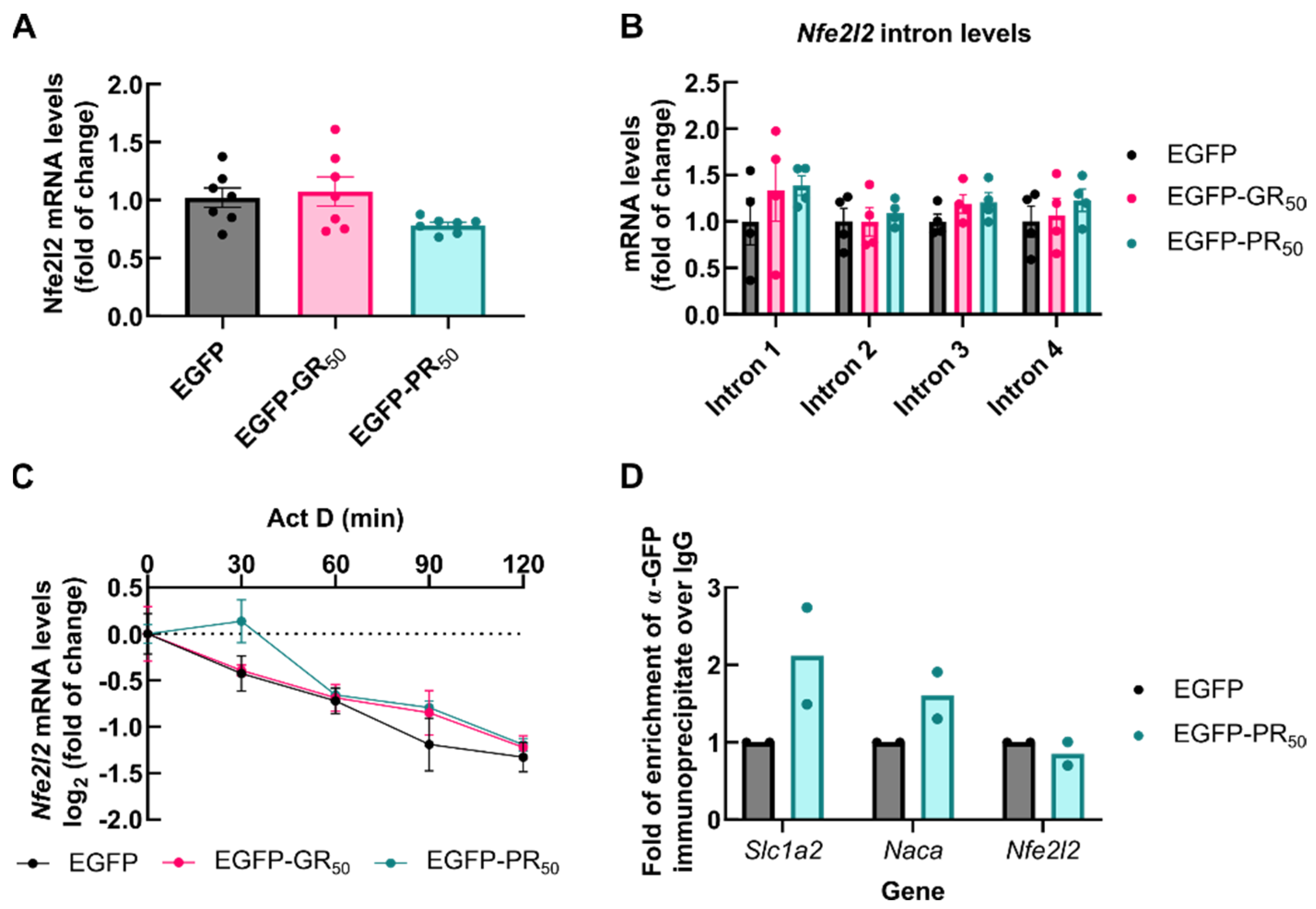
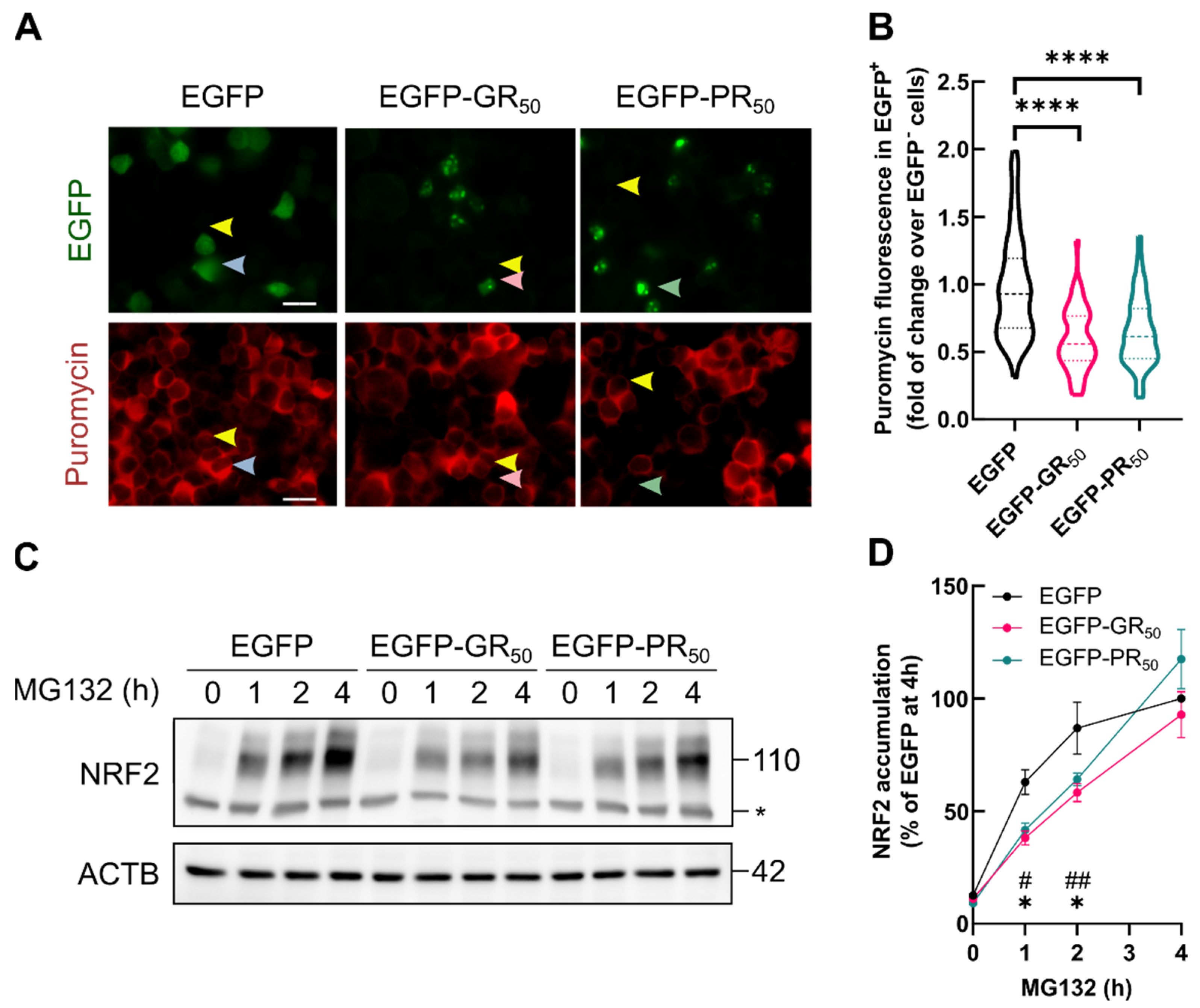
3.5. Accumulation of NRF2 Transcription Factor Is Impaired by DPRs
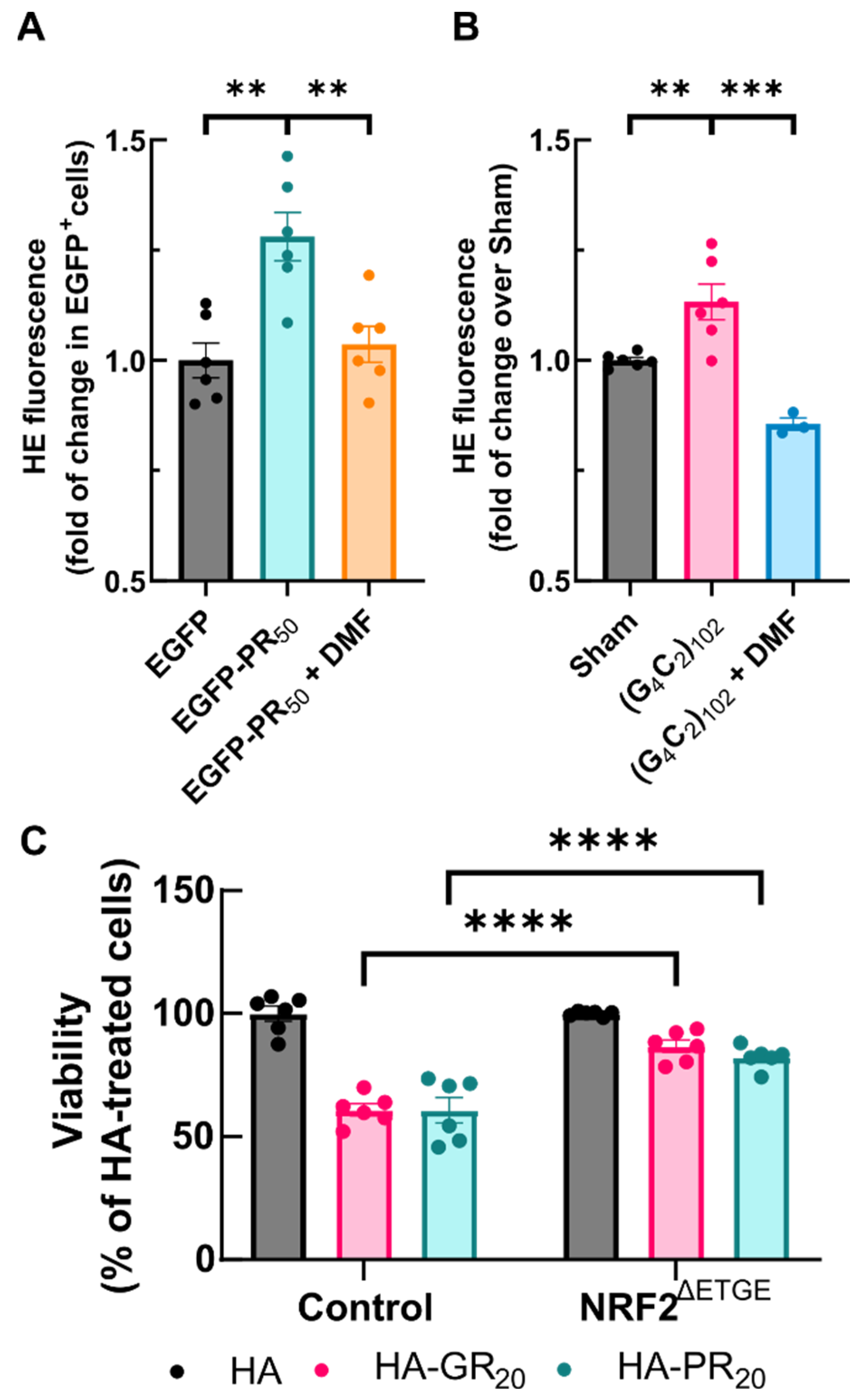
3.6. NRF2 Activation Protects against Arg-DPR Toxicity in NSC34 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic Lateral Sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Riviere, M.; Meininger, V.; Zeisser, P.; Munsat, T. An Analysis of Extended Survival in Patients with Amyotrophic Lateral Sclerosis Treated with Riluzole. Arch. Neurol. 1998, 55, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Kimura, A. Investigation of the Therapeutic Effects of Edaravone, a Free Radical Scavenger, on Amyotrophic Lateral Sclerosis (Phase II Study). Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2006, 7, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Zinman, L.; Moreno, D.; Schymick, J.; Liang, Y.; Sato, C.; Zheng, Y.; Ghani, M.; Dib, S.; Keith, J.; et al. Hypermethylation of the CpG Island near the G4C2 Repeat in ALS with a C9orf72 Expansion. Am. J. Hum. Genet. 2013, 92, 981–989. [Google Scholar] [CrossRef]
- Sudria-Lopez, E.; Koppers, M.; de Wit, M.; van der Meer, C.; Westeneng, H.-J.; Zundel, C.A.C.; Youssef, S.A.; Harkema, L.; de Bruin, A.; Veldink, J.H.; et al. Full Ablation of C9orf72 in Mice Causes Immune System-Related Pathology and Neoplastic Events but No Motor Neuron Defects. Acta Neuropathol. 2016, 132, 145–147. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.-M.; Schludi, M.H.; van der Zee, J.; Cruts, M.; et al. Bidirectional Transcripts of the Expanded C9orf72 Hexanucleotide Repeat Are Translated into Aggregating Dipeptide Repeat Proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014, 3451192–3451194. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.; Ang, C.-S.; Ormsby, A.R.; Cox, D.; Daly, J.C.; Reid, G.E.; Hatters, D.M. Arginine in C9ORF72 Dipolypeptides Mediates Promiscuous Proteome Binding and Multiple Modes of Toxicity. Mol. Cell. Proteom. MCP 2020, 19, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative Stress in ALS: Key Role in Motor Neuron Injury and Therapeutic Target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and Functional Characterization of Nrf2 Degradation by the Glycogen Synthase Kinase 3/β-TrCP Axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-Regulated Turnover of Nrf2 Is Determined by at Least Two Separate Protein Domains, the Redox-Sensitive Neh2 Degron and the Redox-Insensitive Neh6 Degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef]
- Eggler, A.L.; Small, E.; Hannink, M.; Mesecar, A.D. Cul3-Mediated Nrf2 Ubiquitination and Antioxidant Response Element (ARE) Activation Are Dependent on the Partial Molar Volume at Position 151 of Keap1. Biochem. J. 2009, 422, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Shepheard, S.R.; Parker, M.D.; Cooper-Knock, J.; Verber, N.S.; Tuddenham, L.; Heath, P.; Beauchamp, N.; Place, E.; Sollars, E.S.A.; Turner, M.R.; et al. Value of Systematic Genetic Screening of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Stopford, M.J.; Higginbottom, A.; Hautbergue, G.M.; Cooper-Knock, J.; Mulcahy, P.J.; De Vos, K.J.; Renton, A.E.; Pliner, H.; Calvo, A.; Chio, A.; et al. C9ORF72 Hexanucleotide Repeat Exerts Toxicity in a Stable, Inducible Motor Neuronal Cell Model, Which Is Rescued by Partial Depletion of Pten. Hum. Mol. Genet. 2017, 26, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, X. Antioxidant Therapies for Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2012, 2012, 472932. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear Factor-Kappa β as a Therapeutic Target for Alzheimer’s Disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-Dipeptides Encoded by the C9orf72 Repeats Bind Nucleoli, Impede RNA Biogenesis, and Kill Cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef]
- Kanekura, K.; Harada, Y.; Fujimoto, M.; Yagi, T.; Hayamizu, Y.; Nagaoka, K.; Kuroda, M. Characterization of Membrane Penetration and Cytotoxicity of C9orf72-Encoding Arginine-Rich Dipeptides. Sci. Rep. 2018, 8, 12740. [Google Scholar] [CrossRef]
- Kanekura, K.; Yagi, T.; Cammack, A.J.; Mahadevan, J.; Kuroda, M.; Harms, M.B.; Miller, T.M.; Urano, F. Poly-Dipeptides Encoded by the C9ORF72 Repeats Block Global Protein Translation. Hum. Mol. Genet. 2016, 25, 1803–1813. [Google Scholar] [CrossRef]
- Gill, A.L.; Wang, M.Z.; Levine, B.; Premasiri, A.; Vieira, F.G. Primary Neurons and Differentiated NSC-34 Cells Are More Susceptible to Arginine-Rich ALS Dipeptide Repeat Protein-Associated Toxicity than Non-Differentiated NSC-34 and CHO Cells. Int. J. Mol. Sci. 2019, 20, 6238. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of Transcription Factor Nrf2 via the Ubiquitin-Proteasome Pathway and Stabilization by Cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.S.; Rollinson, S.; Thompson, J.C.; Harris, J.M.; Stopford, C.L.; Richardson, A.M.T.; Jones, M.; Gerhard, A.; Davidson, Y.S.; Robinson, A.; et al. Distinct Clinical and Pathological Characteristics of Frontotemporal Dementia Associated with C9ORF72 Mutations. Brain J. Neurol. 2012, 135, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Llères, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory Flexibility in the Nrf2-Mediated Stress Response Is Conferred by Conformational Cycling of the Keap1-Nrf2 Protein Complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [PubMed]
- Poganik, J.R.; Long, M.J.C.; Disare, M.T.; Liu, X.; Chang, S.-H.; Hla, T.; Aye, Y. Post-Transcriptional Regulation of Nrf2-MRNA by the MRNA-Binding Proteins HuR and AUF1. FASEB J. 2019, 33, 14636–14652. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a Nonradioactive Method to Monitor Protein Synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.; Sarkar, M.; Freibaum, B.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Waggoner, D.; Subramaniam, J.R.; Tessarollo, L.; Bartnikas, T.B.; Culotta, V.C.; Price, D.L.; Rothstein, J.; Gitlin, J.D. Copper Chaperone for Superoxide Dismutase Is Essential to Activate Mammalian Cu/Zn Superoxide Dismutase. Proc. Natl. Acad. Sci. USA 2000, 97, 2886–2891. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress and Mitochondrial Damage in Neurodegenerative Diseases: From Molecular Mechanisms to Targeted Therapies. Oxid. Med. Cell. Longev. 2020, 2020, 1270256. [Google Scholar] [CrossRef]
- Riemslagh, F.W.; Verhagen, R.F.M.; van der Toorn, E.C.; Smits, D.J.; Quint, W.H.; van der Linde, H.C.; van Ham, T.J.; Willemsen, R. Reduction of Oxidative Stress Suppresses Poly-GR-Mediated Toxicity in Zebrafish Embryos. Dis. Model. Mech. 2021, 14, dmm049092. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.-B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in IPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.-D.; Almeida, S.; Gao, F.-B. C9ORF72-ALS/FTD-Associated Poly(GR) Binds Atp5a1 and Compromises Mitochondrial Function in Vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human IPSC-Derived Astrocytes from ALS Patients with Mutated C9ORF72 Show Increased Oxidative Stress and Neurotoxicity. eBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-Related Cellular Pathology in Skeletal Myocytes Derived from ALS-Patient Induced Pluripotent Stem Cells. Dis. Model. Mech. 2019, 12, dmm039552. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; von Dengler, R.; Petri, S. Nuclear Erythroid 2-Related Factor 2-Antioxidative Response Element Signaling Pathway in Motor Cortex and Spinal Cord in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-Linked Mutations of SQSTM1/P62 Disrupt Selective Autophagy and NFE2L2/NRF2 Anti-Oxidative Stress Pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lu, Y.; Qi, F.; Su, Q.; Wang, L.; You, C.; Che, F.; Yu, J. Effect of the Human SOD1-G93A Gene on the Nrf2/ARE Signaling Pathway in NSC-34 Cells. Mol. Med. Rep. 2014, 9, 2453–2458. [Google Scholar] [CrossRef]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 Mutations Causing Amyotrophic Lateral Sclerosis Are Associated with Altered Expression of RNA-Binding Protein HnRNP K and Affect the Nrf2 Antioxidant Pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.B.; Piantadosi, C.A.; et al. Heme Oxygenase-1 Regulates Mitochondrial Quality Control in the Heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 Expansion within Astrocytes Reduces Metabolic Flexibility in Amyotrophic Lateral Sclerosis. Brain J. Neurol. 2019, 142, 3771–3790. [Google Scholar] [CrossRef]
- Vucic, S.; Henderson, R.D.; Mathers, S.; Needham, M.; Schultz, D.; Kiernan, M.C. Safety and Efficacy of Dimethyl Fumarate in ALS: Randomised Controlled Study. Ann. Clin. Transl. Neurol. 2021, 8, 1991–1999. [Google Scholar] [CrossRef]
- Harder, B.; Tian, W.; La Clair, J.J.; Tan, A.-C.; Ooi, A.; Chapman, E.; Zhang, D.D. Brusatol Overcomes Chemoresistance through Inhibition of Protein Translation. Mol. Carcinog. 2017, 56, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Vercruysse, T.; Boeynaems, S.; Sicart, A.; Van Damme, P.; Daelemans, D.; Van Den Bosch, L. C9orf72-Generated Poly-GR and Poly-PR Do Not Directly Interfere with Nucleocytoplasmic Transport. Sci. Rep. 2019, 9, 15728. [Google Scholar] [CrossRef] [PubMed]
- Moens, T.G.; Niccoli, T.; Wilson, K.M.; Atilano, M.L.; Birsa, N.; Gittings, L.M.; Holbling, B.V.; Dyson, M.C.; Thoeng, A.; Neeves, J.; et al. C9orf72 Arginine-Rich Dipeptide Proteins Interact with Ribosomal Proteins in Vivo to Induce a Toxic Translational Arrest That Is Rescued by EIF1A. Acta Neuropathol. 2019, 137, 487–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Years) | Gender | Site of Onset | RIN 1 Value | FVC 2 (% Predicted) | ALSFRS-R 3 | Progression Rate | Riluzole | |
|---|---|---|---|---|---|---|---|---|
| Healthy controls | 61 | Male | - | 9.6 | - | - | - | - |
| 56 | Male | - | 9.5 | - | - | - | - | |
| 68 | Male | - | 9 | - | - | - | - | |
| 71 | Female | - | 8.6 | - | - | - | - | |
| 69 | Female | - | 8.4 | - | - | - | - | |
| 57 | Female | - | 8 | - | - | - | - | |
| 63 | Female | - | 7.3 | - | - | - | - | |
| Mean ± SD | 63.6 ± 5.9 | - | - | 8.6 ± 0.8 | - | - | - | - |
| C9orf72 patients | 53 | Male | Bulbar | 9.4 | 117 | 38 | 3.3 | no |
| 73 | Female | Bulbar | 9.1 | 51 | 32 | 4.0 | no | |
| 65 | Female | Limb | 9 | 110 | 41 | 0.3 | no | |
| 67 | Female | Limb | 8.9 | 93 | 42 | 1.2 | no | |
| 58 | Female | Limb | 8.7 | 99 | n/a | n/a | yes | |
| 61 | Male | Limb | 8.5 | 92 | 41 | 1.0 | no | |
| 70 | Female | Limb | 8 | 70 | 40 | 0.4 | no | |
| Mean ± SD | 63.9 ± 7.0 | - | - | 8.8 ± 0.5 | 90.3 ± 22.9 | 39.0 ± 3.7 | 1.7 ± 1.6 | - |
| Epitope | Dilution | Species | Source | Reference |
|---|---|---|---|---|
| NRF2 | 1:5000 | Rabbit | Homemade | - |
| HO1 | 1:2000 | Rabbit | Homemade | - |
| GFP | 1:1000 | Mouse | Sigma Aldrich | G1546 |
| ACTB | 1:4000 | Goat | Santa Cruz Biotech. | sc-1616 |
| GAPDH | 1:30000 | Mouse | Calbiochem | CB1001 |
| HA | 1:500 | Mouse | Abcam | ab9110 |
| Puromycin | 1:1000 | Mouse | Sigma Aldrich | MABE343 |
| Gene | Forward Primer (5′—3′) | Reverse Primer (5′—3′) | |
|---|---|---|---|
| Nfe2l2 | Mature | CCCGAAGCACGCTGAAGGCA | CCAGGCGGTGGGTCTCCGTA |
| Intron 1 | TTTATGACCAAATACCGAGCACA | GGCTCAATGTCTGGTAACATCC | |
| Intron 2 | AGCACAGGGTCACAACGAG | CGCTGTTCGTTAAAGGGGAG | |
| Intron 3 | CCAAGGCATGAAGAACAGACA | TAATATGGAGTCACTGGGAGGA | |
| Intron 4 | TGTGTGTTAAGTGAGTCTGGG | TAGGAGTGTGCTGTTTTCTGC | |
| Actb | CACCCAGCACATTTAGCTAGCTGA | TTCAGAGCAACTGCCCTGAAAGCA | |
| Gapdh | CGACTTCAACAGCAACTCCCACTCTTCC | TGGGTGGTCCAGGGTTTCTTACTCCTT | |
| Tbp | TGCACAGGAGCCAAGAGTGAA | CACATCACAGCTCCCCACCA | |
| Slc1a2 | ACAATATGCCCAAGCAGGTAGA | CTTTGGCTCATCGGAGCTGA | |
| Naca | GACAGTGATGAGTCAGTACCAGA | TGCTTGGCTTTACTAACAGGTTC | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Villegas, J.; Kirby, J.; Mata, A.; Cadenas, S.; Turner, M.R.; Malaspina, A.; Shaw, P.J.; Cuadrado, A.; Rojo, A.I. Dipeptide Repeat Pathology in C9orf72-ALS Is Associated with Redox, Mitochondrial and NRF2 Pathway Imbalance. Antioxidants 2022, 11, 1897. https://doi.org/10.3390/antiox11101897
Jiménez-Villegas J, Kirby J, Mata A, Cadenas S, Turner MR, Malaspina A, Shaw PJ, Cuadrado A, Rojo AI. Dipeptide Repeat Pathology in C9orf72-ALS Is Associated with Redox, Mitochondrial and NRF2 Pathway Imbalance. Antioxidants. 2022; 11(10):1897. https://doi.org/10.3390/antiox11101897
Chicago/Turabian StyleJiménez-Villegas, José, Janine Kirby, Ana Mata, Susana Cadenas, Martin R. Turner, Andrea Malaspina, Pamela J. Shaw, Antonio Cuadrado, and Ana I. Rojo. 2022. "Dipeptide Repeat Pathology in C9orf72-ALS Is Associated with Redox, Mitochondrial and NRF2 Pathway Imbalance" Antioxidants 11, no. 10: 1897. https://doi.org/10.3390/antiox11101897