
NLRP3 Inflammasome in Vascular Disease: A Recurrent Villain to Combat Pharmacologically

 ,
,  , ,
, ,
Abstract
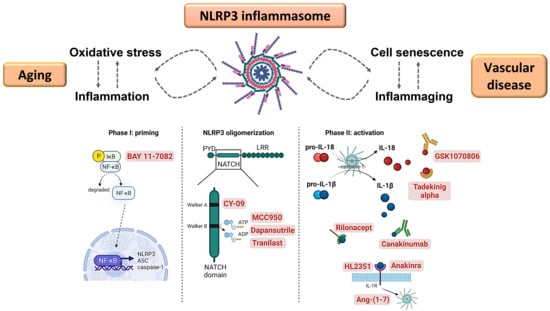
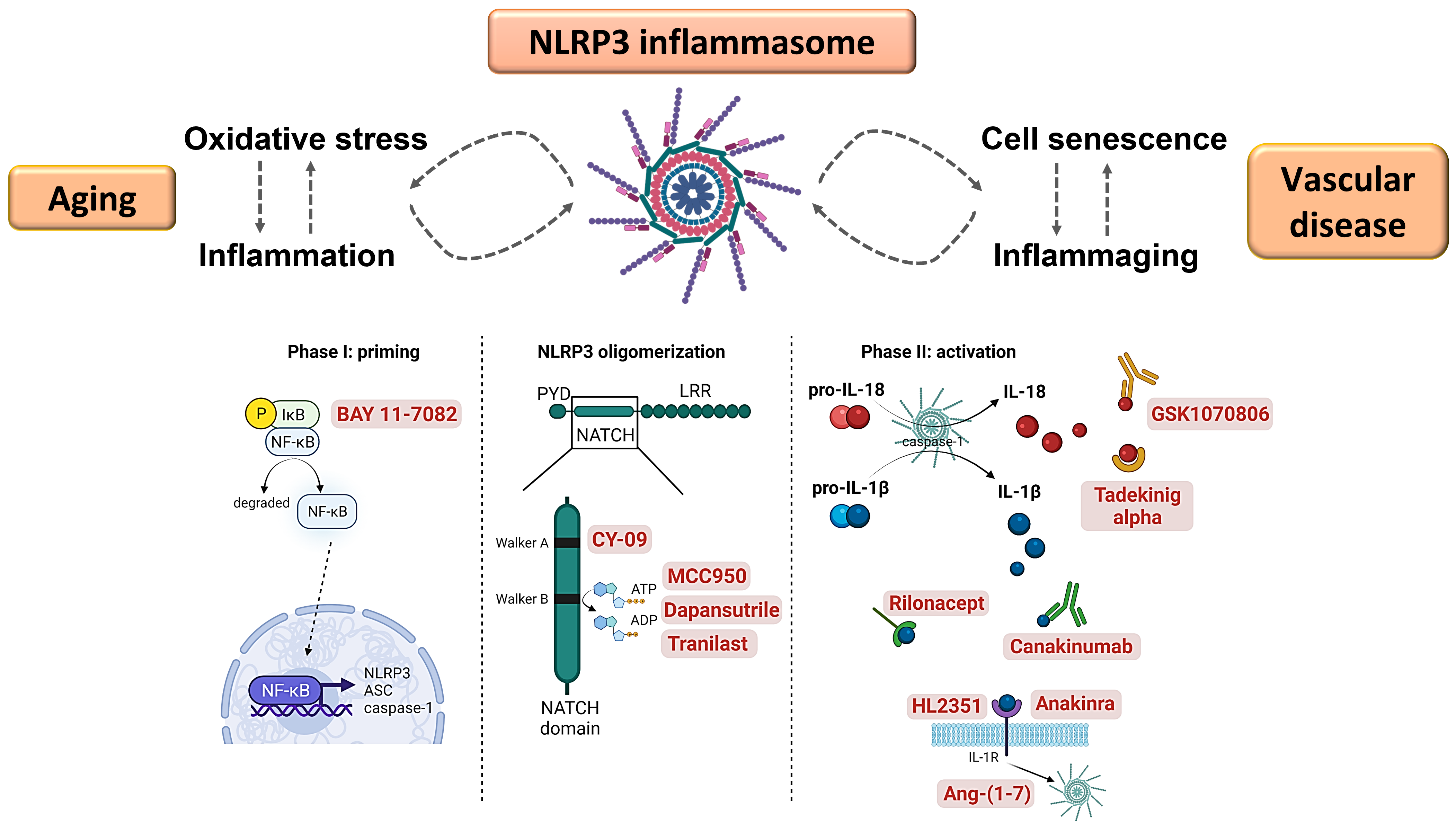
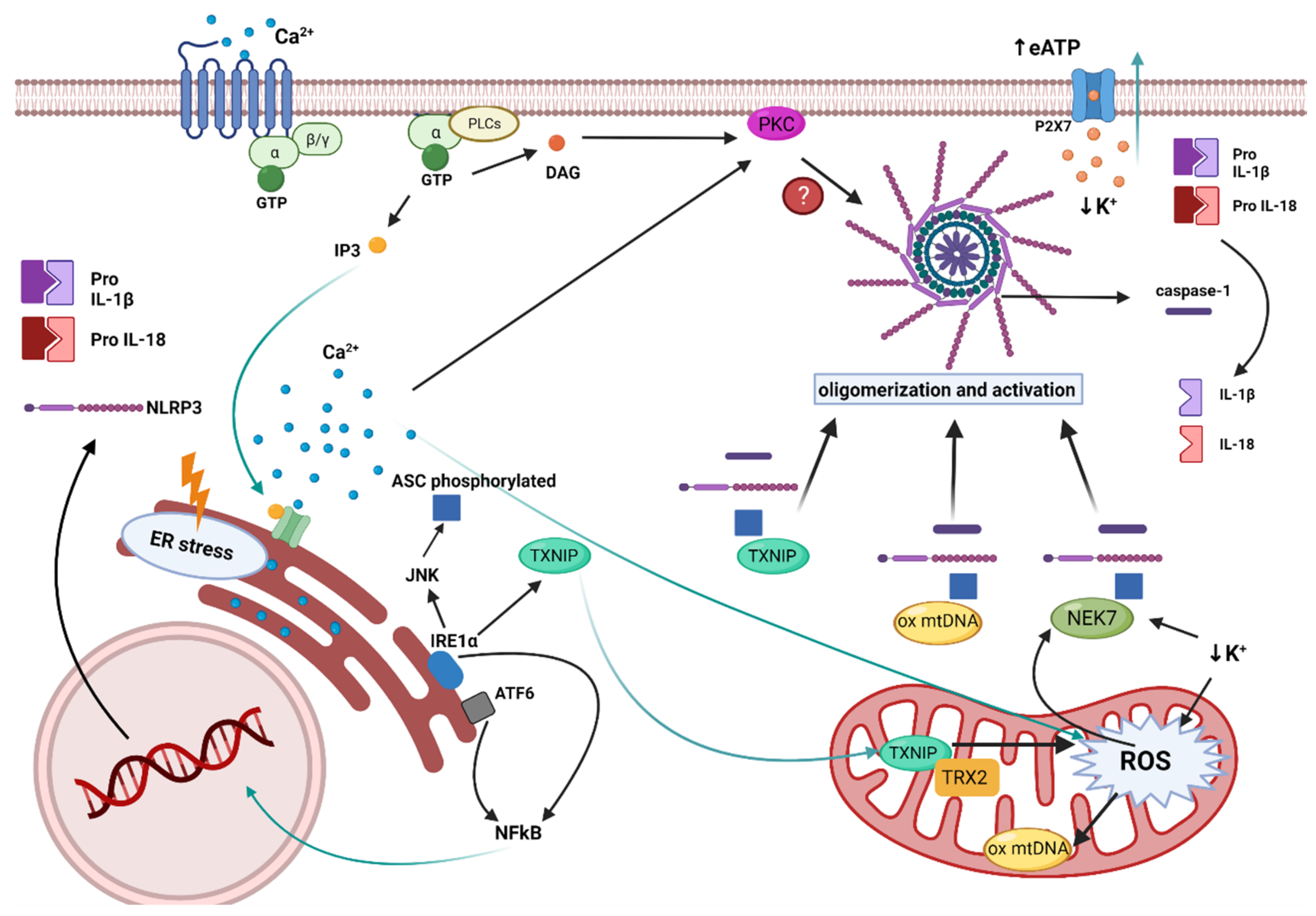
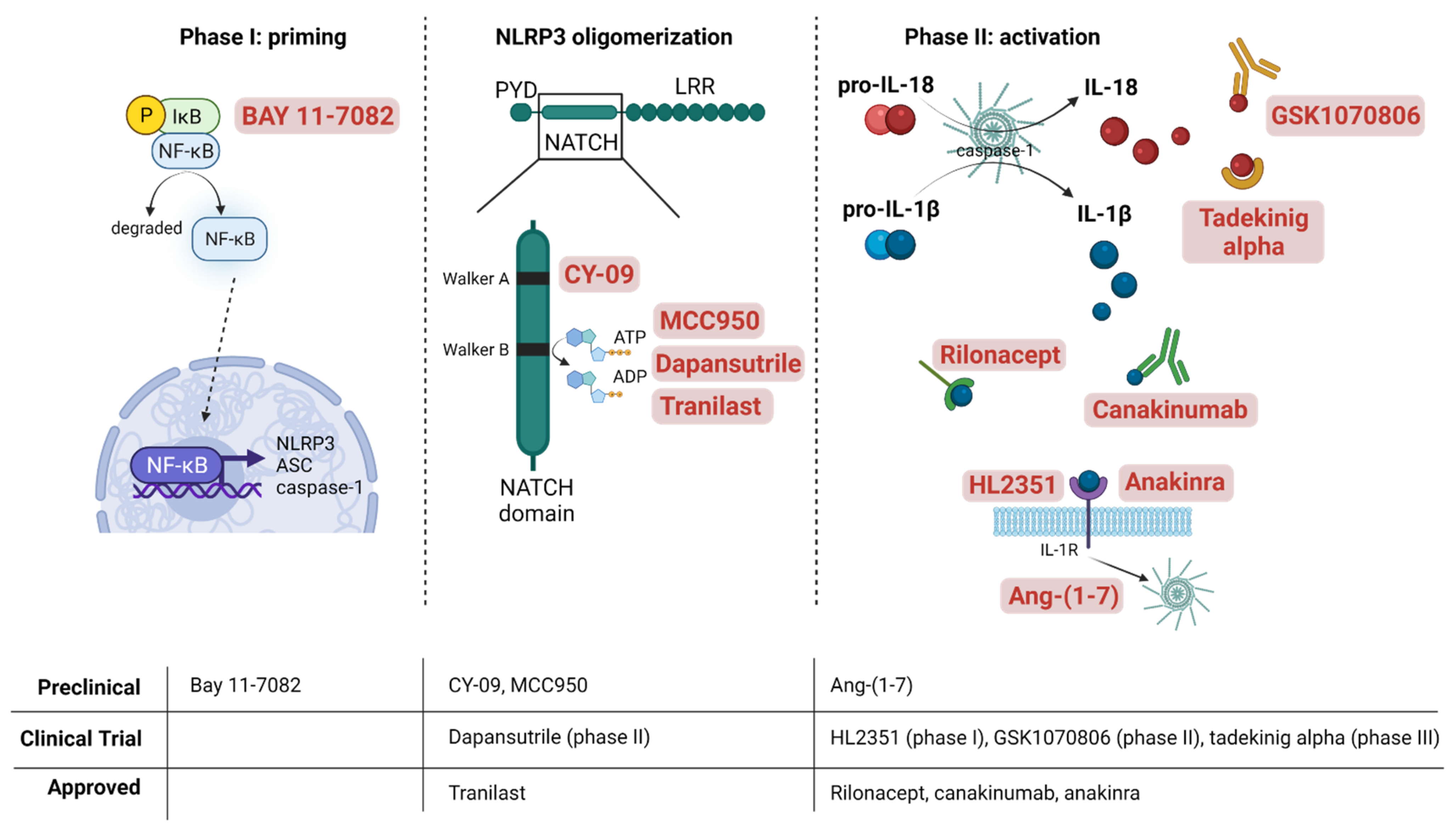
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. NLRP3 Inflammasome: Mechanisms and Physiological Function
3. NLRP3 Inflammasome in Inflammation and Vascular Disease
4. NLRP3 Inflammasome and Vascular Cell Senescence
5. Oxidative Stress and the NLRP3 Inflammasome
6. Pharmacological Interventions to Mitigate NLRP3 Inflammasome Activation in Vascular Disease
Author Contributions
Funding
Conflicts of Interest
References
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Gritsenko, A.; Green, J.P.; Brough, D.; Lopez-Castejon, G. Mechanisms of NLRP3 priming in inflammaging and age related diseases. Cytokine Growth Factor Rev. 2020, 55, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Meyers, A.K.; Zhu, X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 2020, 9, 1808. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Vitale, M.; Pugliese, G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells 2020, 9, 1812. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.-F.; Yu, T.; Chu, X.-M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinra, M.; Nampoothiri, M.; Arora, D.; Mudgal, J. Reviewing the importance of TLR-NLRP3-pyroptosis pathway and mechanism of experimental NLRP3 inflammasome inhibitors. Scand. J. Immunol. 2021, 95, e13124. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, J.A.; Bergstralh, D.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P.-Y. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Wu, Z.; Wu, H.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed NLRP3 inflammasome activation. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Akther, M.; Haque, E.; Park, J.; Kang, T.-B.; Lee, K.-H. NLRP3 Ubiquitination—A New Approach to Target NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2021, 22, 8780. [Google Scholar] [CrossRef]
- Liang, Z.; Damianou, A.; Di Daniel, E.; Kessler, B.M. Inflammasome activation controlled by the interplay between post-translational modifications: Emerging drug target opportunities. Cell Commun. Signal. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.; Holley, C.L.; Bierschenk, D.; Stacey, K.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Strandberg, T.E.; Nieminen, T. Future Perspectives on the Role of Frailty in Cardiovascular Diseases. Adv Exp Med Biol. 2020, 1216, 149–152. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Dembic, Z. Cytokines of the Immune System, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2013, 1842, 446–462. [Google Scholar] [CrossRef] [Green Version]
- De Miguel, C.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. Emerging role of the inflammasome and pyroptosis in hypertension. Int. J. Mol. Sci. 2021, 22, 1064. [Google Scholar] [CrossRef]
- He, X.; Fan, X.; Bai, B.; Lu, N.; Zhang, S.; Zhang, L. Pyroptosis is a critical immune-inflammatory response involved in atherosclerosis. Pharmacol. Res. 2021, 165, 105447. [Google Scholar] [CrossRef]
- Frostegard, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Chen, C.; Chen, Z.; Liu, L.; Jiang, J.; Wu, Z.; Zhao, M.; Chen, Y. NLRP3: A Novel Mediator in Cardiovascular Disease. J. Immunol. Res. 2018, 2018, 5702103. [Google Scholar] [CrossRef]
- Rajamäki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: A Novel Link between Cholesterol Metabolism and Inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, Y.; Mu, N.; Lou, X.; Li, W.; Chen, Y.; Fan, D.; Tan, H. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab. Investig. 2017, 97, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab. Syndr. Obes. 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikama, Y.; Aki, N.; Hata, A.; Nishimura, M.; Oyadomari, S.; Funaki, M. Palmitate-Stimulated Monocytes Induce Adhesion Molecule Expression in Endothelial Cells via IL-1 Signaling Pathway. J. Cell. Physiol. 2014, 230, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, N.; Sukhova, G.K.; Libby, P.; Reynolds, R.S.; Young, J.L.; Schönbeck, U. Expression of Interleukin (IL)-18 and Functional IL-18 Receptor on Human Vascular Endothelial Cells, Smooth Muscle Cells, and Macrophages. J. Exp. Med. 2002, 195, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.-G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Gomez, D.; Baylis, R.A.; Durgin, B.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; Hilaire, C.S.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Burger, F.; Baptista, D.; Roth, A.; da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. Int. J. Mol. Sci. 2021, 23, 340. [Google Scholar] [CrossRef]
- Clément, N.; Gueguen, M.; Glorian, M.; Blaise, R.; Andréani, M.; Brou, C.; Limon, I.; Bausero, P. Notch3 and IL-1beta exert opposing effects on a vascular smooth muscle cell inflammatory pathway in which NF-kappaB drives crosstalk. J. Cell Sci. 2007, 120, 3352–3361. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, M.; Li, Z.; Zheng, G.; Chen, A.; Zhao, L.; Danyang, L.; Wei, L.; Chen, Y.; Ruan, X.Z. Sterol-resistant SCAP Overexpression in Vascular Smooth Muscle Cells Accelerates Atherosclerosis by Increasing Local Vascular Inflammation through Activation of the NLRP3 Inflammasome in Mice. Aging Dis. 2021, 12, 747–763. [Google Scholar] [CrossRef]
- Clarke, M.C.H.; Talib, S.; Figg, N.L.; Bennett, M.R. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ. Res. 2010, 106, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Afrasyab, A.; Qu, P.; Zhao, Y.; Peng, K.; Wang, H.; Lou, D.; Niu, N.; Yuan, D. Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients. Hear. Vessel. 2015, 31, 1218–1229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Gong, Z.; Xing, S.; Xing, Q. Overexpression of Caspase-1 in Aorta of Patients with Coronary Atherosclerosis. Hear. Lung Circ. 2014, 23, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Erhart, P.; Cakmak, S.; Grond-Ginsbach, C.; Hakimi, M.; Dihlmann, S. Inflammasome activity in leucocytes decreases with abdominal aortic aneurysm progression. Int. J. Mol. Med. 2019, 44, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira-Llopis, S.; Apostolova, N.; Bañuls, C.; Muntané, J.; Rocha, M.; Victor, V.M. Mitochondria, the NLRP3 Inflammasome, and Sirtuins in Type 2 Diabetes: New Therapeutic Targets. Antioxid. Redox Signal. 2018, 29, 749–791. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 Inflammasome Activation in Patients with Type 2 Diabetes. Diabetes 2012, 62, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 Gene Silencing Ameliorates Diabetic Cardiomyopathy in a Type 2 Diabetes Rat Model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Stojanović, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Hear. J. 2020, 41, 2983–2996. [Google Scholar] [CrossRef] [Green Version]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennet, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis: Effects of Telomerase and Oxidative Stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomeres in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Zhou, Z.; Liu, W.; Chang, Q.; Sun, G.; Dai, Y. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int. J. Biochem. Cell Biol. 2017, 84, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Dongil, P.I.V.; Vallejo, S.; San Hipólito-Luengo, Á.; Díaz-Araya, G.; Peiró, C.; Bartha, J.L.; González-Arlanzón, M.M.; Rivilla, F.; de la Cuesta, F.; et al. Pharmacological Blockade of NLRP3 Inflammasome/IL- 1β-Positive Loop Mitigates Endothelial Cell Senescence and Dysfunction. Aging Dis. 2021, 13, 1–14. [Google Scholar] [CrossRef]
- Romero, A.; San Hipólito-Luengo, Á.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Peiró, C.; Romacho, T.; Leon, R.; Bartha, J.L.; et al. The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Fan, S.; Wang, X.; Lu, J.; Zhang, Z.; Wu, D.; Shan, Q.; Zheng, Y. Purple sweet potato color inhibits endothelial premature senescence by blocking the NLRP3 inflammasome. J. Nutr. Biochem. 2015, 26, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, S.; Qu, G.; Hua, J.; Zong, J.; Li, X.; Xu, F. Genistein alleviates H2O2-induced senescence of human umbilical vein endothelial cells via regulating the TXNIP/NLRP3 axis. Pharm. Biol. 2021, 59, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2017, 233, 2116–2132. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Chen, X.; Wang, Q.; Yuan, L. NEK7: A potential therapy target for NLRP3-related diseases. Biosci. Trends 2020, 14, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Solini, A. P2X receptor-ion channels in the inflammatory response in adipose tissue and pancreas—Potential triggers in onset of type 2 diabetes? Curr. Opin. Immunol. 2018, 52, 1–7. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2015, 17, 250–258. [Google Scholar] [CrossRef]
- Cai, H.; Wang, P.; Zhang, B.; Dong, X. Expression of the NEK7/NLRP3 inflammasome pathway in patients with diabetic lower extremity arterial disease. BMJ Open Diabetes Res. Care 2020, 8, e001808. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Barter, P. Antioxidant Effects of Statins in the Management of Cardiometabolic Disorders. J. Atheroscler. Thromb. 2014, 21, 997–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Zhang, Y.; Wu, B.; Wu, S.; You, S.; Lu, S.; Liu, J.; Huang, X.; Xu, J.; Cao, L.; et al. Deacetylation-dependent regulation of PARP1 by SIRT2 dictates ubiquitination of PARP1 in oxidative stress-induced vascular injury. Redox Biol. 2021, 47, 102141. [Google Scholar] [CrossRef] [PubMed]
- Sadia, K.; Ashraf, M.Z.; Mishra, A. Therapeutic Role of Sirtuins Targeting Unfolded Protein Response, Coagulation, and Inflammation in Hypoxia-Induced Thrombosis. Front. Physiol. 2021, 12, 3453. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Müller-Calleja, N.; Manukyan, D.; Canisius, A.; Strand, D.; Lackner, K.J. Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann. Rheum. Dis. 2016, 76, 891–897. [Google Scholar] [CrossRef]
- Sharma, A.; Tate, M.; Mathew, G.; Vince, J.E.; Ritchie, R.H.; de Haan, J.B. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: Therapeutic Implications. Front. Physiol. 2018, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Xi, H.; Zhang, Y.; Xu, Y.; Yang, W.Y.; Jiang, X.; Sha, X.; Cheng, X.; Wang, J.; Qin, X.; Yu, J.; et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ. Res. 2016, 118, 1525–1539. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Nie, Q.; Wang, F.; Han, Y.; Yang, B.; Sun, M.; Fan, X.; Ye, Z.; Liu, P.; Wen, J. Role of pyroptosis in atherosclerosis and its therapeutic implications. J. Cell. Physiol. 2021, 236, 7159–7175. [Google Scholar] [CrossRef]
- Cau, S.B.; Bruder-Nascimento, A.; Silva, M.B.; Ramalho, F.N.; Mestriner, F.; Alves-Lopes, R.; Ferreira, N.; Tostes, R.C.; Bruder-Nascimento, T. Angiotensin-II activates vascular inflammasome and induces vascular damage. Vasc. Pharmacol. 2021, 139, 106881. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.Y.; Hu, M.M.; Xin, Y.F.; Gang, C. Resveratrol alleviates vascular inflammatory injury by inhibiting inflammasome activation in rats with hypercholesterolemia and vitamin D2 treatment. Inflamm. Res. 2015, 64, 321–332. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. J. Cereb. Blood Flow Metab. 2016, 174, 1633–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byon, C.H.; Han, T.; Wu, J.; Hui, S.T. Txnip ablation reduces vascular smooth muscle cell inflammation and ameliorates atherosclerosis in apolipoprotein E knockout mice. Atherosclerosis 2015, 241, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Thielen, L.A.; Chen, J.; Jing, G.; Moukha-Chafiq, O.; Xu, G.; Jo, S.; Grayson, T.B.; Lu, B.; Li, P.; Augelli-Szafran, C.E.; et al. Identification of an Anti-diabetic, Orally Available Small Molecule that Regulates TXNIP Expression and Glucagon Action. Cell Metab. 2020, 32, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, D.; Wang, J.; Guo, J.; Li, Y.; Cao, Y.; Zhang, N.; Fu, Y. Melatonin inhibits endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation in lipopolysaccharide-induced endometritis in mice. Int. Immunopharmacol. 2018, 64, 101–109. [Google Scholar] [CrossRef]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [Green Version]
- Irrera, N.; Vaccaro, M.; Bitto, A.; Pallio, G.; Pizzino, G.; Lentini, M.; Altavilla, D.; Anastasi, G.P.; Ettari, R.; Squadrito, F.; et al. BAY 11-7082 inhibits the NF-κB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin. Sci. 2017, 131, 487–498. [Google Scholar] [CrossRef]
- Li, J.; Teng, X.; Jin, S.; Dong, J.; Guo, Q.; Tian, D.; Wu, Y. Hydrogen sulfide improves endothelial dysfunction by inhibiting the vicious cycle of NLRP3 inflammasome and oxidative stress in spontaneously hypertensive rats. J. Hypertens. 2019, 37, 1633–1643. [Google Scholar] [CrossRef]
- Cui, J.; Xu, G.; Bian, F. H2S alleviates aortic aneurysm and dissection: Crosstalk between transforming growth factor 1 signaling and NLRP3 inflammasome. Int. J. Cardiol. 2021, 338, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Abellán, A.; Angosto, D.; Banaclocha, H.M.; de Torre, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; Halai, R.; Cooper, M.A. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol. Rev. 2021, 73, 968–1000. [Google Scholar] [CrossRef]
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 2017, 8, 99740–99756. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [Green Version]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhuang, L.; Luo, X.; Liang, J.; Sun, E.; He, Y. Protection of MCC950 against Alzheimer’s disease via inhibiting neuronal pyroptosis in SAMP8 mice. Exp. Brain Res. 2020, 238, 2603–2614. [Google Scholar] [CrossRef]
- Beyer, M.M.S.; Lonnemann, N.; Remus, A.; Latz, E.; Heneka, M.T.; Korte, M. Enduring Changes in Neuronal Function upon Systemic Inflammation Are NLRP3 Inflammasome Dependent. J. Neurosci. 2020, 40, 5480–5494. [Google Scholar] [CrossRef]
- Van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Van Hout, G.P.J.; Bosch, L.; Ellenbroek, G.H.J.M.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.A.; Robertson, A.A.B.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Xia, L.; Zhang, F.; Zhu, D.; Xin, C.; Wang, H.; Zhang, F.; Guo, X.; Lee, Y.; Zhang, L.; et al. A novel mechanism of diabetic vascular endothelial dysfunction: Hypoadiponectinemia-induced NLRP3 inflammasome activation. Biochim. Biophys. Acta 2017, 1863, 1556–1567. [Google Scholar] [CrossRef]
- Romacho, T.; Valencia, I.; Ramos, M.; Vallejo, S.; López-Esteban, M.; Lorenzo, O.; Cannata, P.; Romero, A.; Hipólito-Luengo, A.S.; Gómez-Cerezo, J.F.; et al. Visfatin/eNampt induces endothelial dysfunction in vivo: A role for Toll-Like Receptor 4 and NLRP3 inflammasome. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klück, V.; Jansen, T.L.T.A.; Janssen, M.; Comarniceanu, A.; Efdé, M.; Tengesdal, I.W.; Joosten, L.A.B.; Marchetti, C.; Scribner, C.L.; Dinarello, C.A.; et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: An open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020, 2, e270. [Google Scholar] [CrossRef]
- Wohlford, G.F.; Van Tassell, B.W.; Billingsley, H.E.; Kadariya, D.; Canada, J.M.; Carbone, S.; Ho, A.-C.; Markley, R.; Thomas, G.; Abbate, A.; et al. Phase 1B, Randomized, Double-Blinded, Dose Escalation, Single-Center, Repeat Dose Safety and Pharmacodynamics Study of the Oral NLRP3 Inhibitor Dapansutrile in Subjects with NYHA II-III Systolic Heart Failure. J. Cardiovasc. Pharmacol. 2020, 77, 49–60. [Google Scholar] [CrossRef]
- Darakhshan, S.; Pour, A.B. Tranilast: A review of its therapeutic applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP 3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Miyachi, Y.; Imamura, S.; Niwa, Y. The effect of tranilast of the generation of reactive oxygen species. J. Pharm. Dyn. 1987, 10, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.M.; Zhang, Y.; Cox, A.J.; Kelly, D.J.; Qi, W. Tranilast attenuates the up-regulation of thioredoxin-interacting protein and oxidative stress in an experimental model of diabetic nephropathy. Nephrol. Dial. Transplant. 2010, 26, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, U.A.; Tilson, H.H.; Hawkins, P.N.; van der Poll, T.; Noviello, S.; Levy, J.; Vritzali, E.; Hoffman, H.M.; Kuemmerle-Deschner, J.B. Long-term safety and effectiveness of canakinumab therapy in patients with cryopyrin-associated periodic syndrome: Results from the β-Confident Registry. RMD Open 2021, 7, e001663. [Google Scholar] [CrossRef]
- Karabulut, Y.; Gezer, H.H.; Duruöz, M.T. Canakinumab is effective in patients with familial Mediterranean fever resistant and intolerant to the colchicine and/or anakinra treatment. Rheumatol. Int. 2021, 42, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell. Physiol. 2018, 234, 10018–10031. [Google Scholar] [CrossRef]
- Klein, A.L.; Imazio, M.; Cremer, P.; Brucato, A.; Abbate, A.; Fang, F.; Insalaco, A.; LeWinter, M.; Lewis, B.S.; Lin, D.; et al. Phase 3 Trial of Interleukin-1 Trap Rilonacept in Recurrent Pericarditis. N. Engl. J. Med. 2021, 384, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Pariano, M.; Pieroni, S.; De Luca, A.; Iannitti, R.; Borghi, M.; Puccetti, M.; Giovagnoli, S.; Renga, G.; D’Onofrio, F.; Bellet, M.; et al. Anakinra Activates Superoxide Dismutase 2 to Mitigate Inflammasome Activity. Int. J. Mol. Sci. 2021, 22, 6531. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Chonchol, M.; Ikizler, T.; Farmer-Bailey, H.; Salas, N.; Chaudhry, R.; Wang, W.; Smits, G.; Tengesdal, I.; Dinarello, C.A.; et al. IL-1 Inhibition and Vascular Function in CKD. J. Am. Soc. Nephrol. 2016, 28, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Vallejo, S.; Palacios, E.; Romacho, T.; Villalobos, L.; Peiró, C.; Sanchez-Ferrer, C.F. The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2014, 13, 158. [Google Scholar] [CrossRef] [Green Version]
- Ngo, L.; Oh, J.; Kim, A.; Back, H.; Kang, W.; Chae, J.; Yun, H.-Y.; Lee, H. Development of a Pharmacokinetic Model Describing Neonatal Fc Receptor-Mediated Recycling of HL2351, a Novel Hybrid Fc-Fused Interleukin-1 Receptor Antagonist, to Optimize Dosage Regimen. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 584. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalobos, L.A.; San Hipólito-Luengo, Á.; Ramos-González, M.; Cercas, E.; Vallejo, S.; Romero, A.; Romacho, T.; Carraro, R.; Peiro, C.; Sánchez-Ferrer, C.F. The Angiotensin-(1-7)/Mas Axis Counteracts Angiotensin II-Dependent and -Independent Pro-inflammatory Signaling in Human Vascular Smooth Muscle Cells. Front. Pharmacol. 2016, 7, 482. [Google Scholar] [CrossRef] [PubMed]
- Jhang, J.-J.; Yen, G.-C. The role of Nrf2 in NLRP3 inflammasome activation. Cell. Mol. Immunol. 2017, 14, 1011–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gillette, D.D.; Li, X.; Zhang, Z.; Wen, H. Nuclear Factor E2-related Factor-2 (Nrf2) Is Required for NLRP3 and AIM2 Inflammasome Activation. J. Biol. Chem. 2014, 289, 17020–17029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Hear. J. 2019, 41, 2153–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walle, L.V.; Stowe, I.B.; Šácha, P.; Lee, B.L.; Demon, D.; Fossoul, A.; Van Hauwermeiren, F.; Saavedra, P.H.V.; Šimon, P.; Šubrt, V.; et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease- associated mutants for inflammasome inhibition. PLoS Biol. 2019, 17, e3000354. [Google Scholar] [CrossRef]
- Mullard, A. NLRP3 inhibitors stoke anti-inflammatory ambitions. Nat. Rev. Drug Discov. 2019, 18, 405–407. [Google Scholar] [CrossRef]
- Cordani, M.; Somoza, Á. Targeting autophagy using metallic nanoparticles: A promising strategy for cancer treatment. Cell. Mol. Life Sci. 2018, 76, 1215–1242. [Google Scholar] [CrossRef] [Green Version]
- Behravan, N.; Zahedipour, F.; Jaafari, M.R.; Johnston, T.P.; Sahebkar, A. Lipid-based nanoparticulate delivery systems for HER2-positive breast cancer immunotherapy. Life Sci. 2022, 291, 120294. [Google Scholar] [CrossRef]
- Gutiérrez-Fernández, M.; de la Cuesta, F.; Tallón, A.; Cuesta, I.; Fernández-Fournier, M.; Laso-García, F.; Frutos, M.C.G.-D.; Díez-Tejedor, E.; Otero-Ortega, L. Potential Roles of Extracellular Vesicles as Biomarkers and a Novel Treatment Approach in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 9011. [Google Scholar] [CrossRef]
- Gabay, C.; Fautrel, B.; Rech, J.; Spertini, F.; Feist, E.; Kötter, I.; Morel, J.; Martin, T.; Hellmich, B.; Lamprecht, P.; et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann. Rheum. Dis. 2018, 77, 840–847. [Google Scholar] [CrossRef] [PubMed]
- McKie, E.A.; Reid, J.L.; Mistry, P.C.; DeWall, S.L.; Abberley, L.; Ambery, P.D.; Gil-Extremera, B. A Study to Investigate the Efficacy and Safety of an Anti-Interleukin-18 Monoclonal Antibody in the Treatment of Type 2 Diabetes Mellitus. PLoS ONE 2016, 11, e0150018. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Stewart, I.; Fabbri, L.; Moss, S.; Robinson, K.; Smyth, A.R.; Jenkins, G. Systematic review and meta-analysis of anakinra, sarilumab, siltuximab and tocilizumab for COVID-19. Thorax 2021, 76, 907–919. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Moro, A.; Valencia, I.; Shamoon, L.; Sánchez-Ferrer, C.F.; Peiró, C.; de la Cuesta, F. NLRP3 Inflammasome in Vascular Disease: A Recurrent Villain to Combat Pharmacologically. Antioxidants 2022, 11, 269. https://doi.org/10.3390/antiox11020269
González-Moro A, Valencia I, Shamoon L, Sánchez-Ferrer CF, Peiró C, de la Cuesta F. NLRP3 Inflammasome in Vascular Disease: A Recurrent Villain to Combat Pharmacologically. Antioxidants. 2022; 11(2):269. https://doi.org/10.3390/antiox11020269
Chicago/Turabian StyleGonzález-Moro, Ainara, Inés Valencia, Licia Shamoon, Carlos Félix Sánchez-Ferrer, Concepción Peiró, and Fernando de la Cuesta. 2022. "NLRP3 Inflammasome in Vascular Disease: A Recurrent Villain to Combat Pharmacologically" Antioxidants 11, no. 2: 269. https://doi.org/10.3390/antiox11020269