
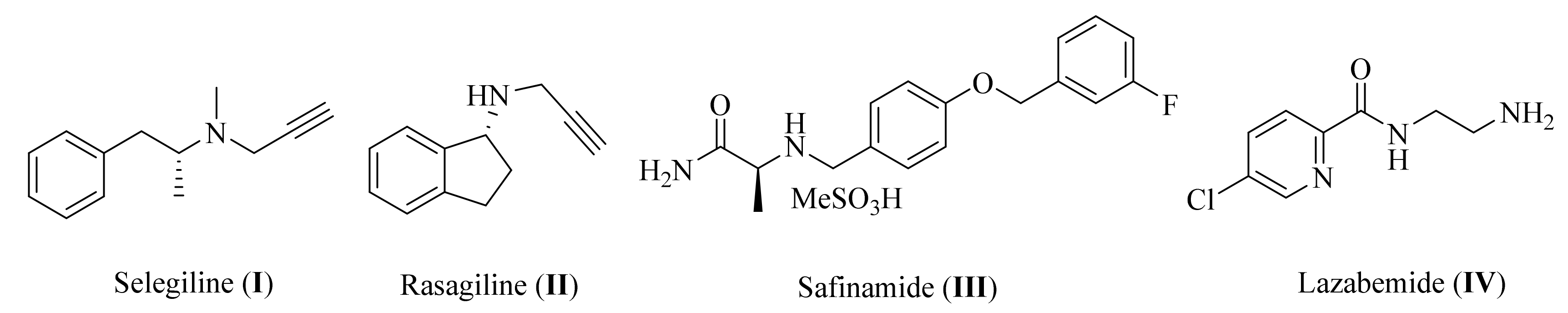
Melatonin Analogues Potently Inhibit MAO-B and Protect PC12 Cells against Oxidative Stress

 ,
,  , ,
, ,
Abstract
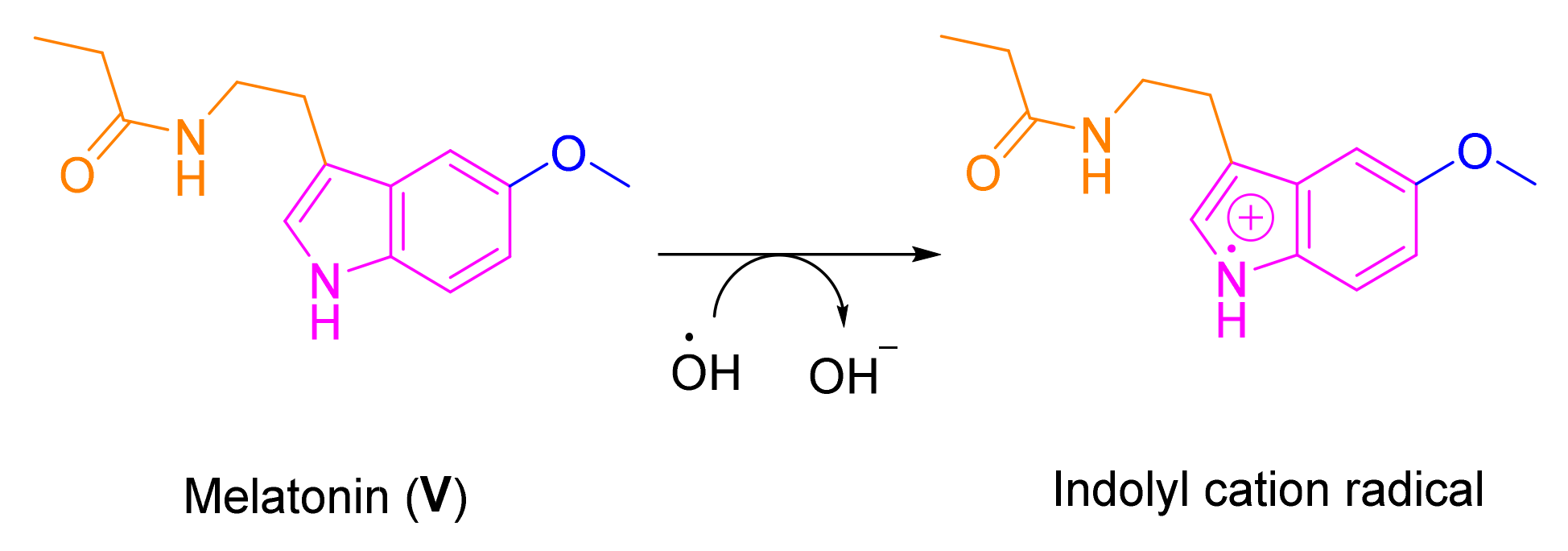
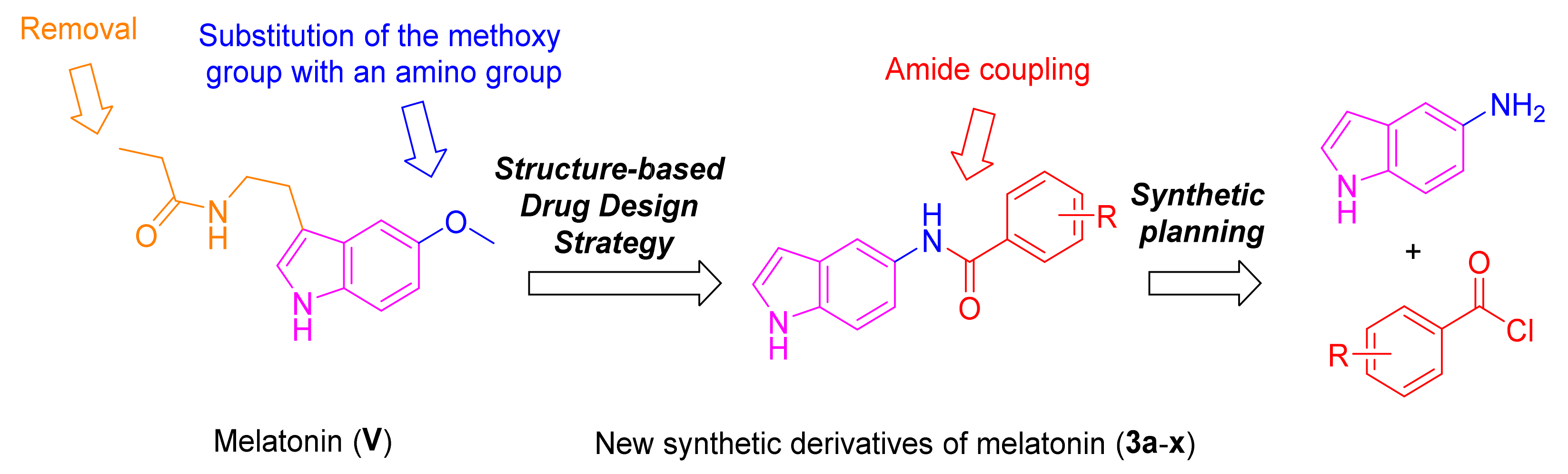
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents, Purification, and Instrumentation
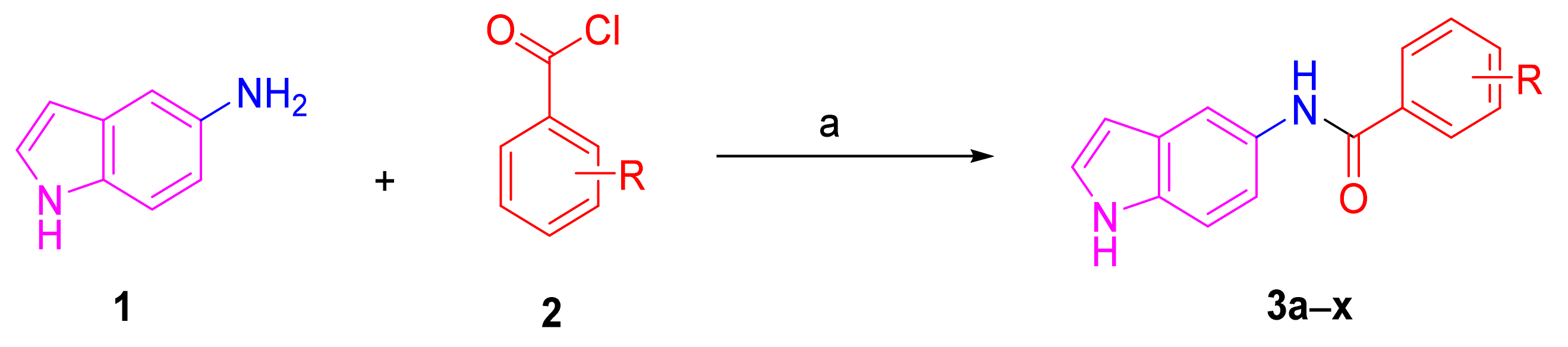
2.2. Synthesis of Melatonin Analogues 3a–x
2.2.1. N-(1H-indol-5-yl)benzamide (3a)
2.2.2. 2-Bromo-N-(1H-indol-5-yl)benzamide (3b)
2.2.3. 4-Bromo-N-(1H-indol-5-yl)benzamide (3c)
2.2.4. 2,4-Dichloro-N-(1H-indol-5-yl)benzamide (3d)
2.2.5. N-(1H-indol-5-yl)-3,5-dinitrobenzamide (3e)
2.2.6. N-(1H-indol-5-yl)-3-nitrobenzamide (3f)
2.2.7. N-(1H-indol-5-yl)-4-nitrobenzamide (3g)
2.2.8. 2,6-Difluoro-N-(1H-indol-5-yl)benzamide (3h)
2.2.9. 3-Fluoro-N-(1H-indol-5-yl)benzamide (3i)
2.2.10. 4-Fluoro-N-(1H-indol-5-yl)benzamide (3j)
2.2.11. 2-Fluoro-N-(1H-indol-5-yl)benzamide (3k)
2.2.12. 4-Fluoro-N-(1H-indol-5-yl)-2-(trifluoromethyl)benzamide (3l)
2.2.13. 3-Cyano-N-(1H-indol-5-yl)benzamide (3m)
2.2.14. 3-Chloro-N-(1H-indol-5-yl)benzamide (3n)
2.2.15. 2-Chloro-N-(1H-indol-5-yl)benzamide (3o)
2.2.16. 4-Chloro-N-(1H-indol-5-yl)benzamide (3p)
2.2.17. 4-(Tert-butyl)-N-(1H-indol-5-yl)benzamide (3q)
2.2.18. 3-Bromo-N-(1H-indol-5-yl)benzamide (3r)
2.2.19. N-(1H-indol-5-yl)-3-(trifluoromethoxy)benzamide (3s)
2.2.20. N-(1H-indol-5-yl)-4-(trifluoromethoxy)benzamide (3t)
2.2.21. N-(1H-indol-5-yl)-3-(trifluoromethyl)benzamide (3u)
2.2.22. N-(1H-indol-5-yl)-4-(trifluoromethyl)benzamide (3v)
2.2.23. 3,5-Dichloro-N-(1H-indol-5-yl)benzamide (3w)
2.2.24. 2,6-Dichloro-N-(1H-indol-5-yl)benzamide (3x)
2.3. Monoamine Oxidase (MAO) Enzyme Assay
2.4. Molecular Modeling
2.5. In Vitro Cellular and Cell-Free Bio-Assays
2.5.1. Materials
2.5.2. PC12 Cell Culture
2.5.3. Drug Treatment
2.5.4. Cell Viability Measurements
2.5.5. Western Blotting
2.5.6. Assessment of Lipid Peroxidation (LPO) in Rat Brain Homogenates
2.5.7. Statistical Analysis
3. Results and Discussion
3.1. Chemical Synthesis
3.2. In Silico Druggability Studies of the Synthesized Melatonin Analogues 3a–x
3.3. MAO Assays
3.3.1. Primary Screening of Melatonin Analogues 3a–x over MAO-B
3.3.2. Dose-Dependent Assay of the Most Active Melatonin Analogues over MAO-B
3.3.3. Selectivity Assay of Compounds 3r, 3n, and 3u–w
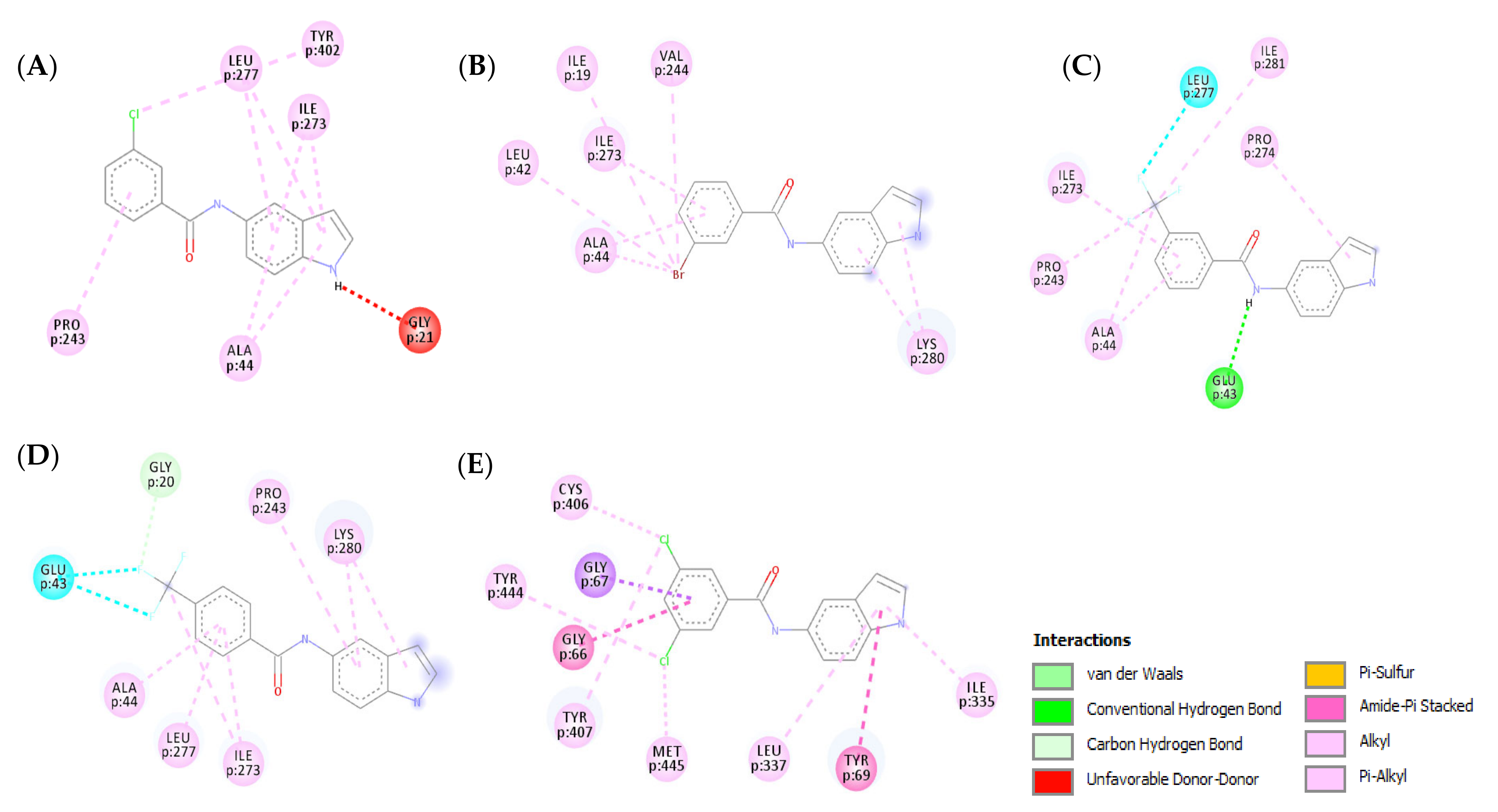
3.4. Molecular Modeling
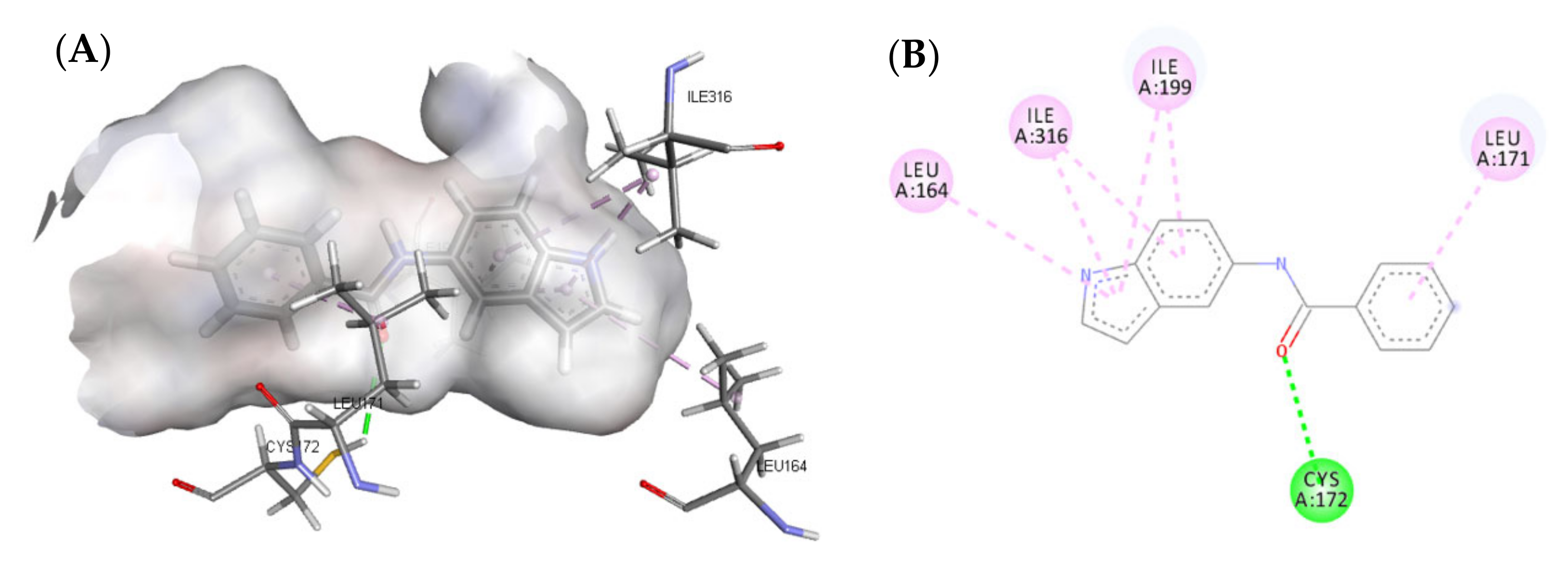
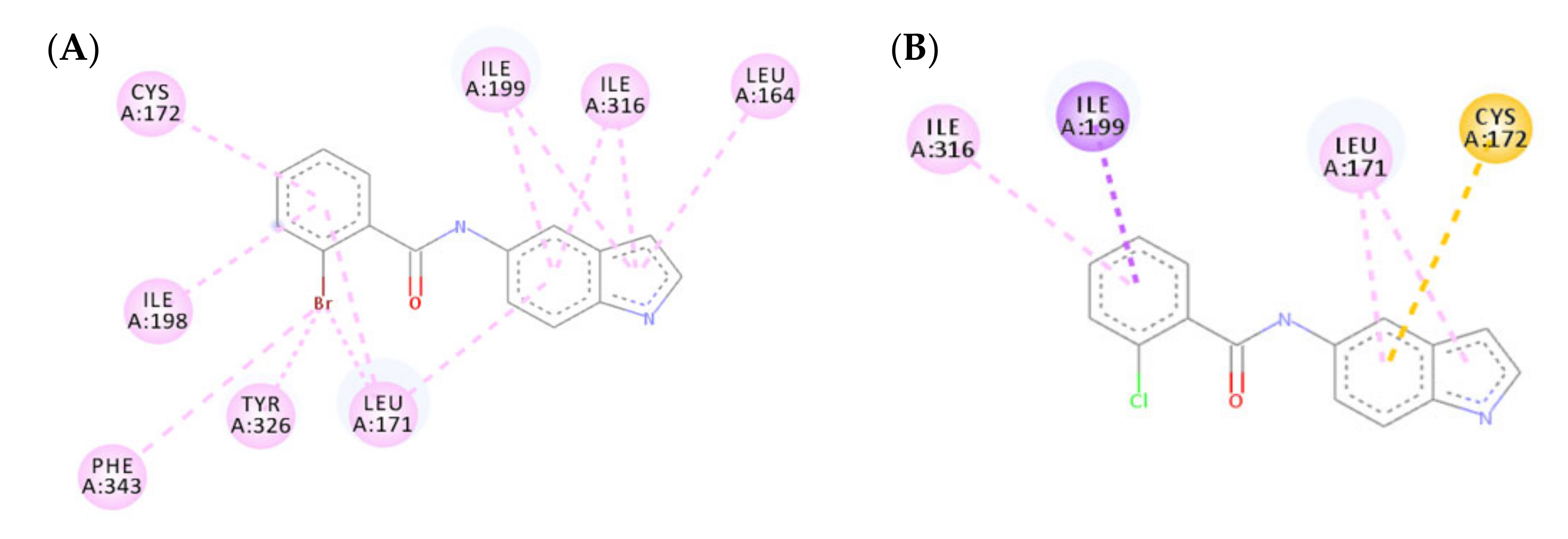
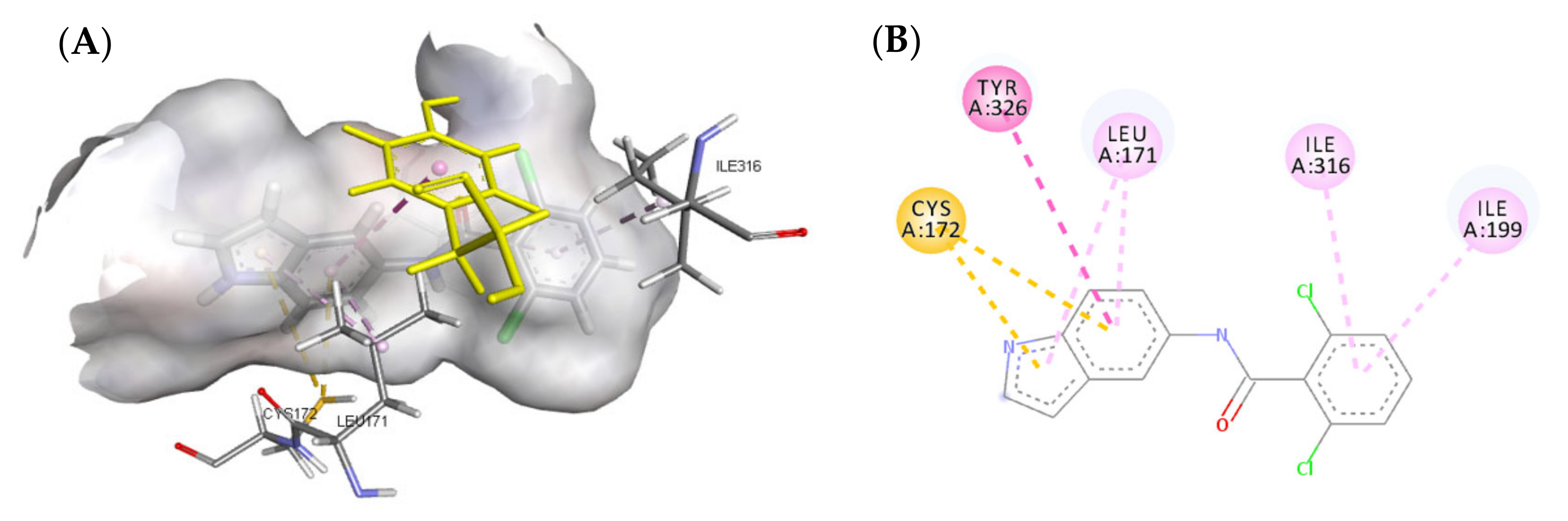
3.4.1. Molecular Docking Study over MAO-B
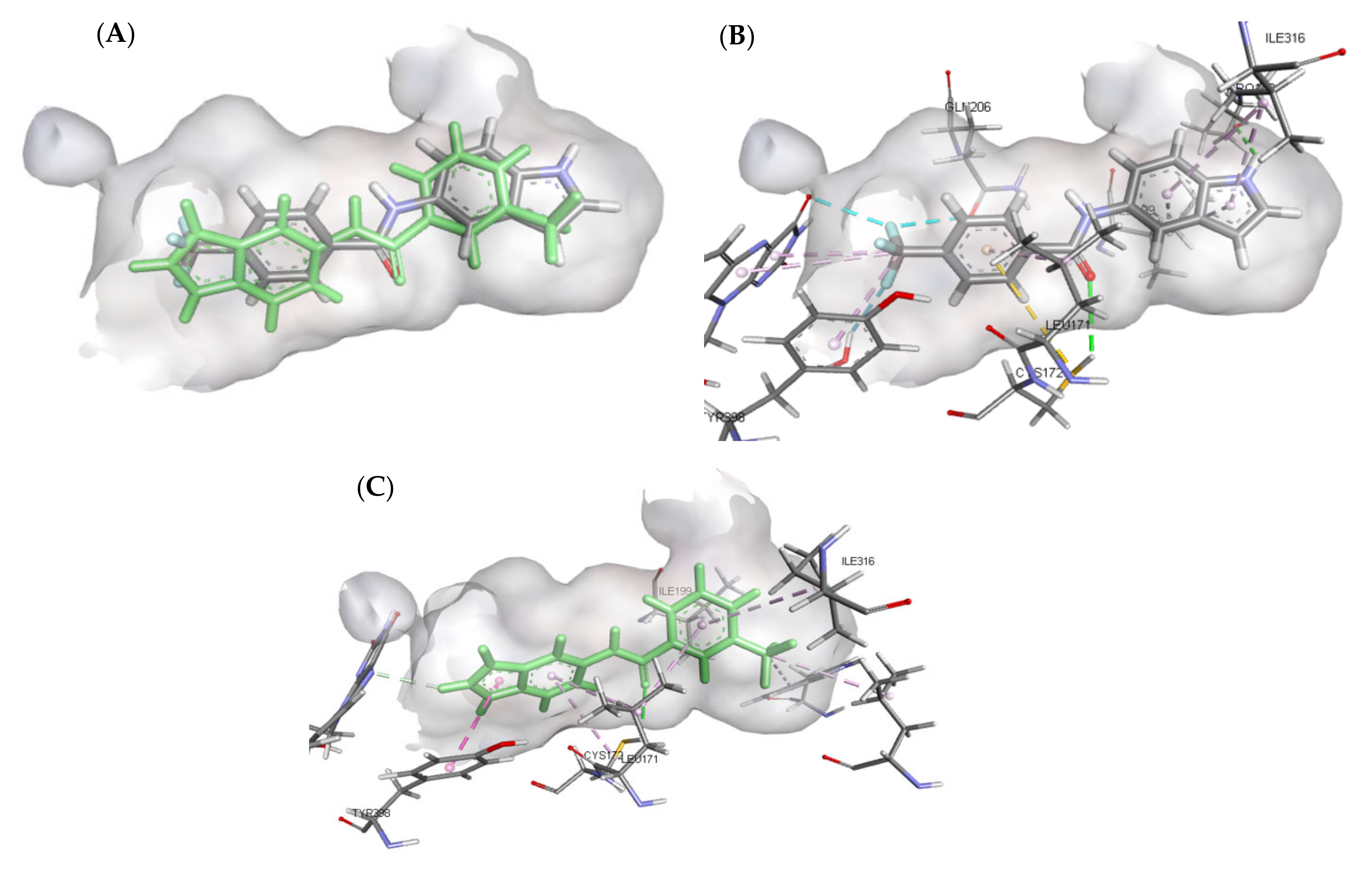
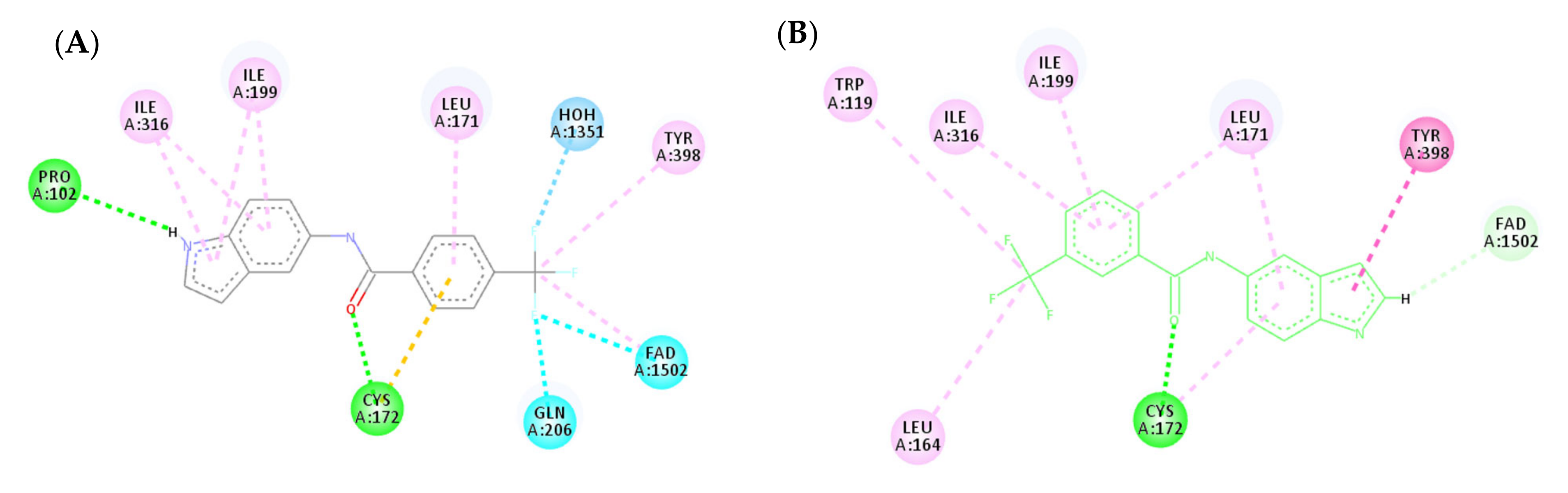
3.4.2. Molecular Docking Study over MAO-A
3.5. Biological Evaluation over PC12 Cells
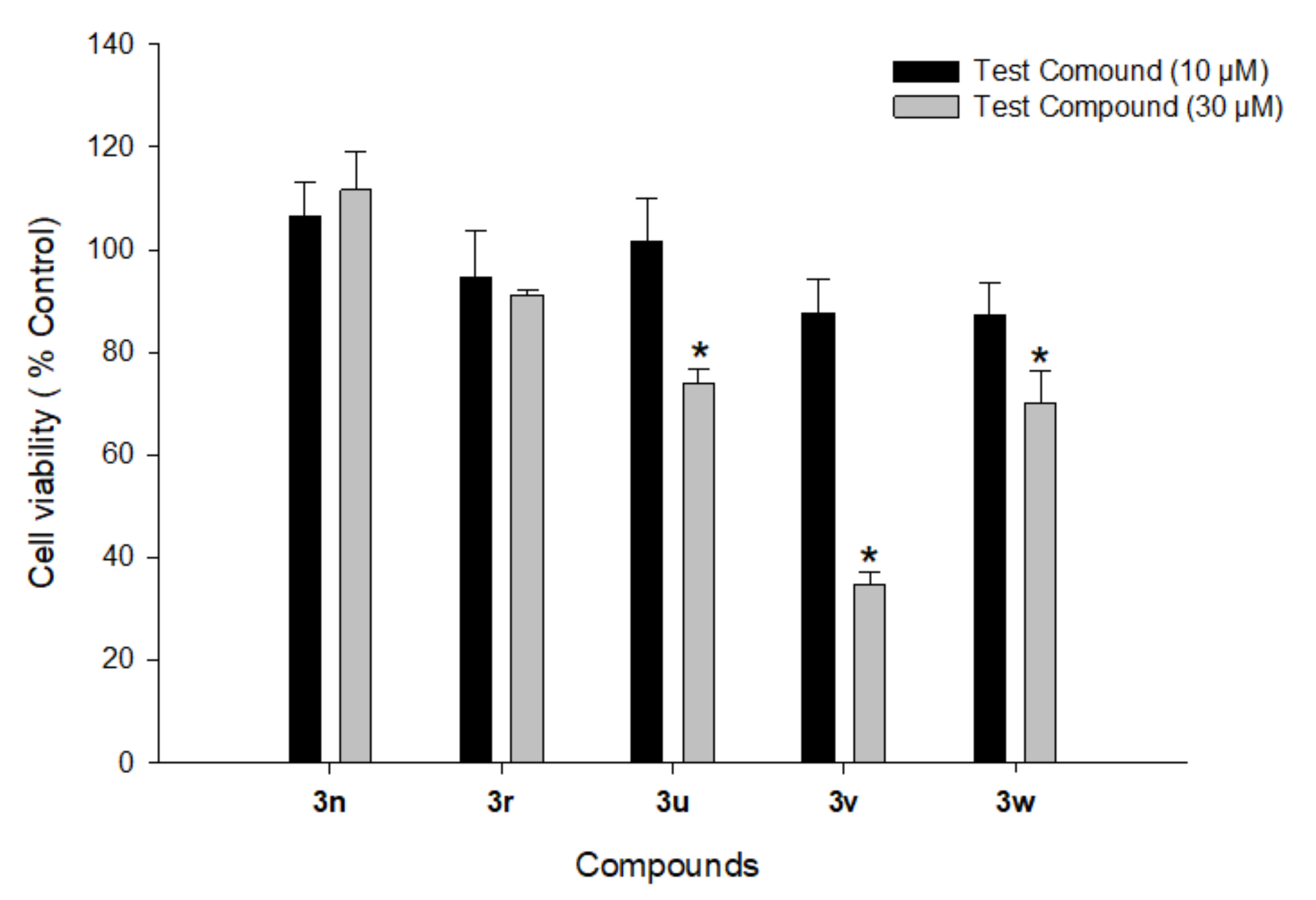
3.5.1. Cytotoxicity Profiles of Compounds 3n, 3r, and 3u–w in PC12 Cells
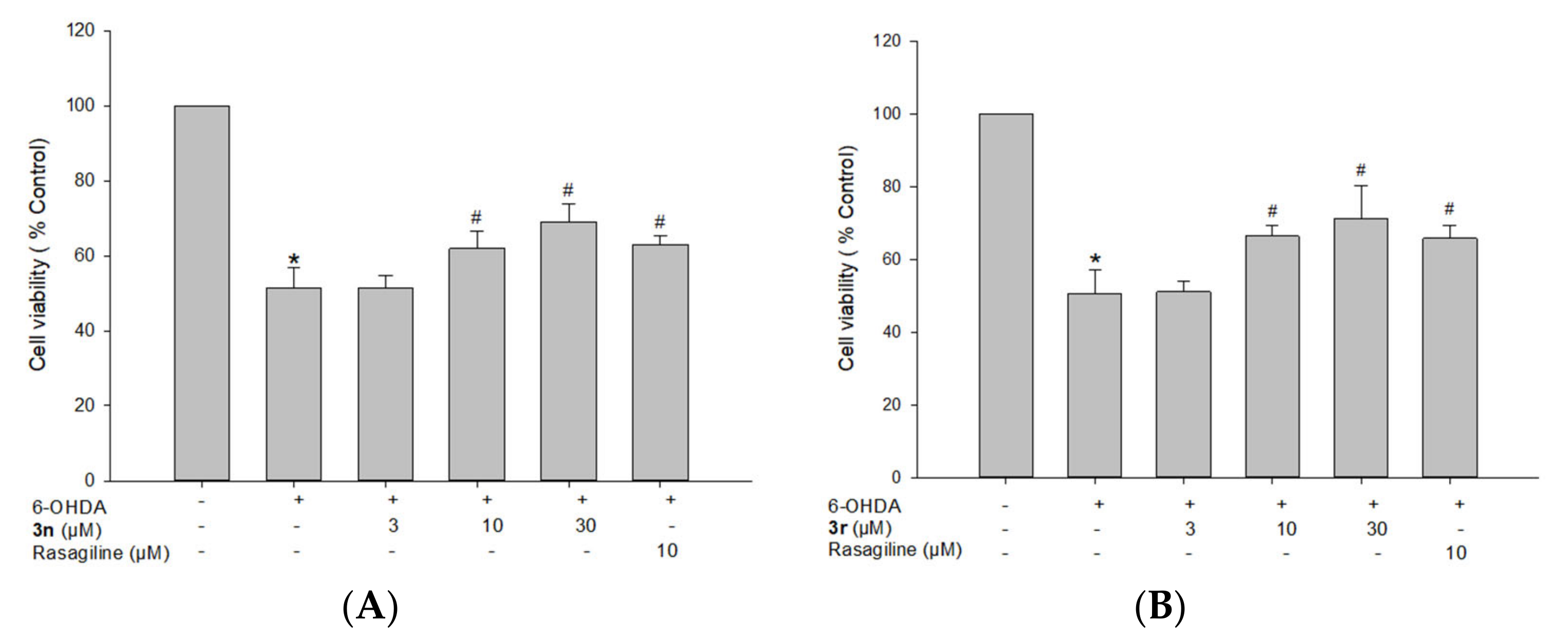
3.5.2. Neuroprotective Effects of Compounds 3n and 3r on 6-OHDA-Induced Oxidative Toxicity in PC12 Cells
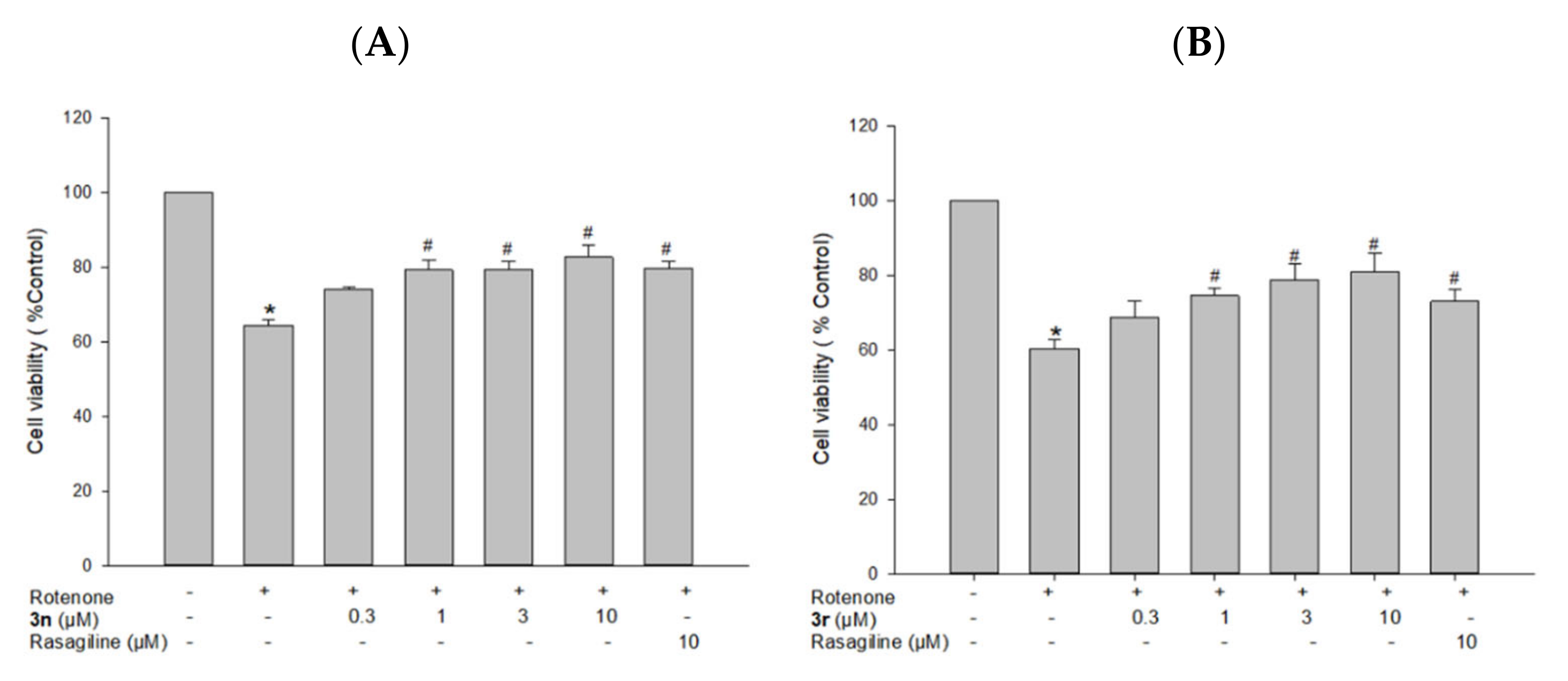
3.5.3. Neuroprotective Effects of Compounds 3n and 3r on Rotenone-Induced Toxicity in PC12 Cells
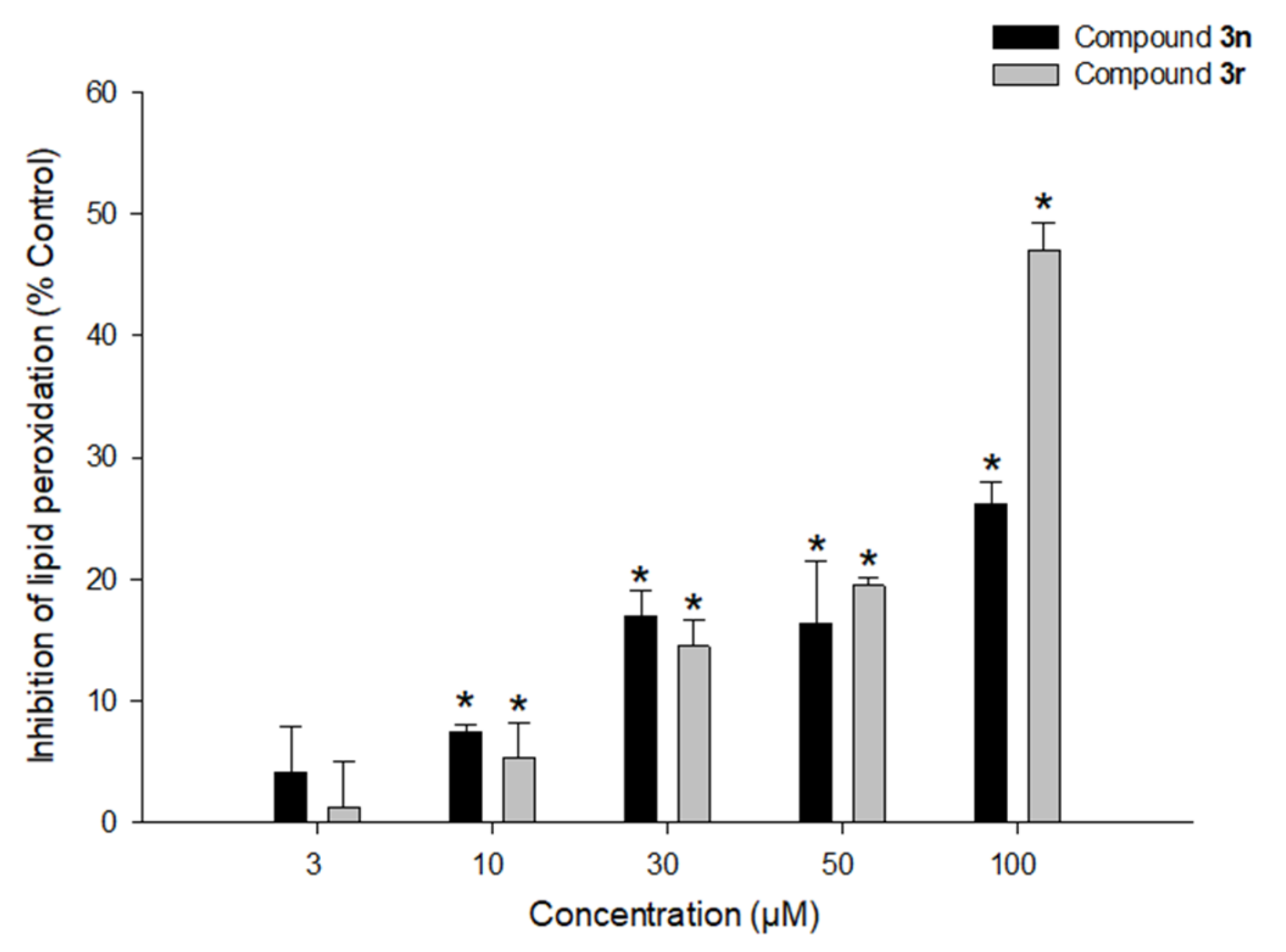
3.5.4. Effects of Compounds 3n and 3r on LPO
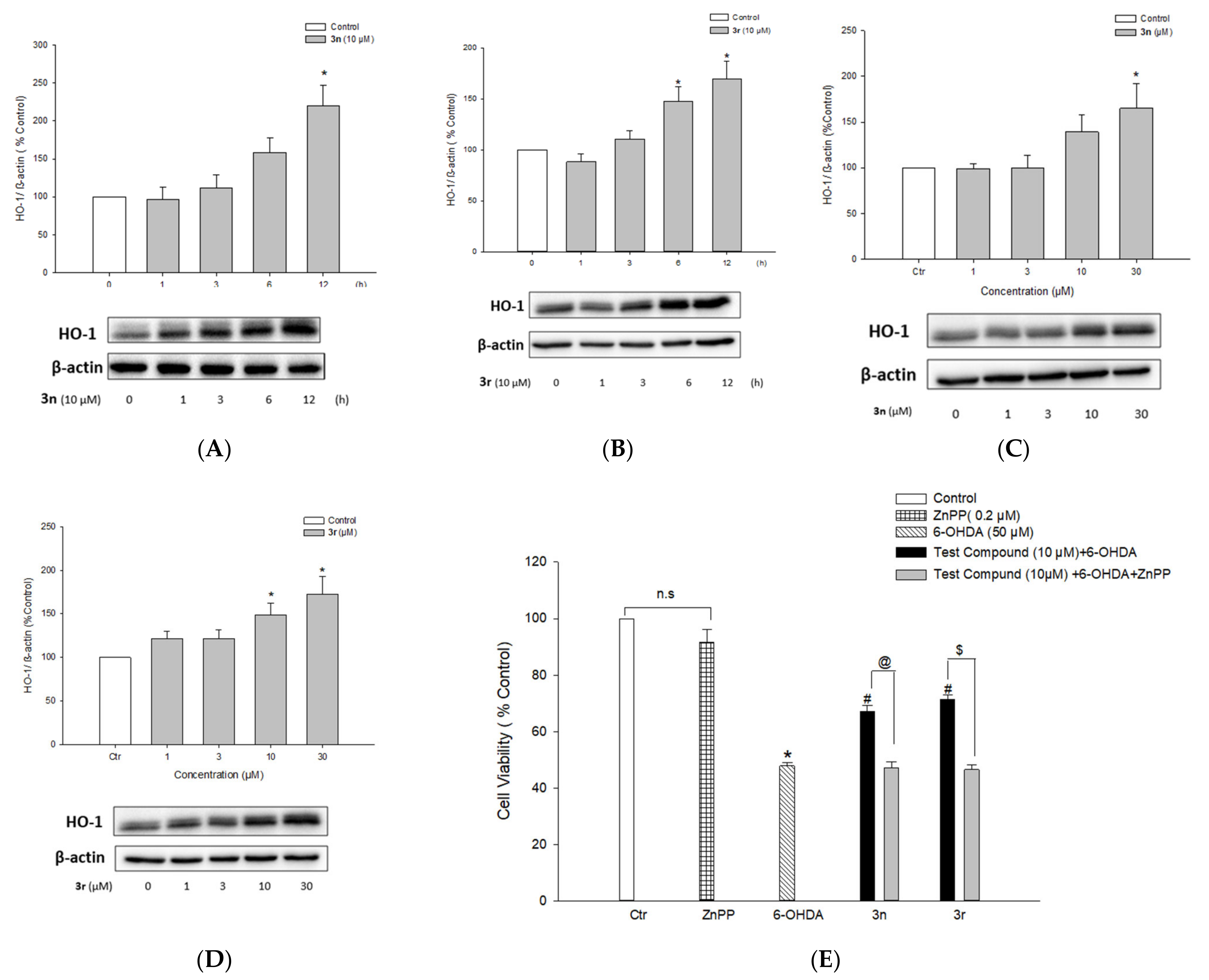
3.5.5. Up-Regulation of HO-1 Expression by Compounds 3n and 3r and Its Role in Neuroprotection
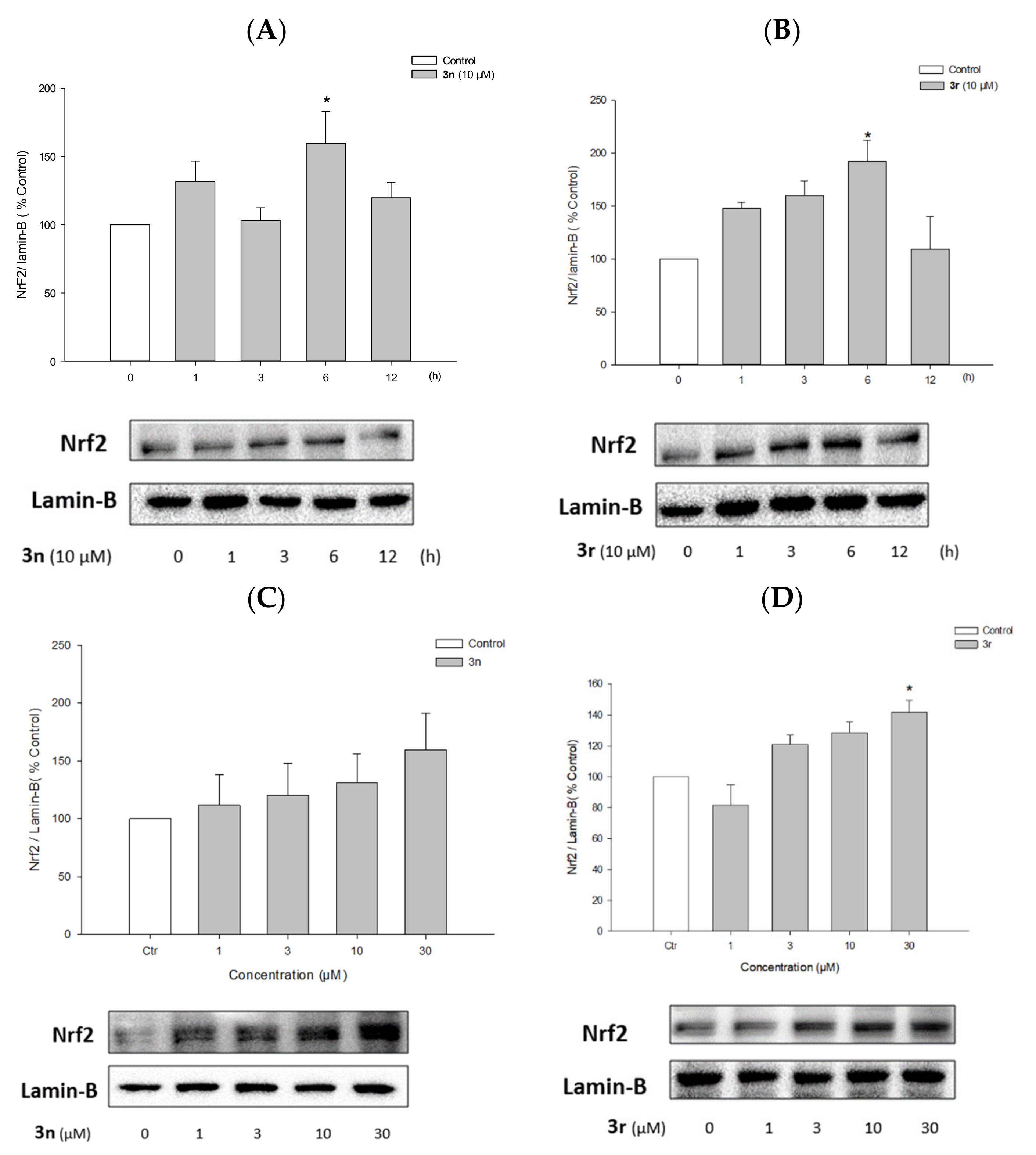
3.5.6. Activation of Nrf2 Signaling by Compounds 3n and 3r
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Fahn, S.; Sulzer, D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx J. Am. Soc. Exp. Neurother. 2004, 1, 139–154. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.H.; Chen, C.M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Hemmati-Dinarvand, M.; Saedi, S.; Valilo, M.; Kalantary-Charvadeh, A.; Alizadeh Sani, M.; Kargar, R.; Safari, H.; Samadi, N. Oxidative stress and Parkinson’s disease: Conflict of oxidant-antioxidant systems. Neurosci. Lett. 2019, 709, 134296. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative stress factors in Parkinson’s disease. Neural Regen. Res. 2021, 16, 1383–1391. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Jenner, P.; Langston, J.W. Explaining ADAGIO: A critical review of the biological basis for the clinical effects of rasagiline. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 2316–2323. [Google Scholar] [CrossRef]
- Riederer, P.; Konradi, C.; Schay, V.; Kienzl, E.; Birkmayer, G.; Danielczyk, W.; Sofic, E.; Youdim, M.B. Localization of MAO-A and MAO-B in human brain: A step in understanding the therapeutic action of L-deprenyl. Adv. Neurol. 1987, 45, 111–118. [Google Scholar]
- Saura, J.; Andrés, N.; Andrade, C.; Ojuel, J.; Eriksson, K.; Mahy, N. Biphasic and region-specific MAO-B response to aging in normal human brain. Neurobiol. Aging 1997, 18, 497–507. [Google Scholar] [CrossRef]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol. Aging 1997, 18, 431–435. [Google Scholar] [CrossRef]
- Inazu, M.; Takeda, H.; Ikoshi, H.; Uchida, Y.; Kubota, N.; Kiuchi, Y.; Oguchi, K.; Matsumiya, T. Regulation of dopamine uptake by basic fibroblast growth factor and epidermal growth factor in cultured rat astrocytes. Neurosci. Res. 1999, 34, 235–244. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Katz, D.M. Regional differences in 5-hydroxytryptamine and catecholamine uptake in primary astrocyte cultures. J. Neurochem. 1986, 47, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Sawada, M. Molecular mechanism of the relation of monoamine oxidase B and its inhibitors to Parkinson’s disease: Possible implications of glial cells. J. Neural Transm. 2006. [Google Scholar] [CrossRef]
- Kumar, M.J.; Andersen, J.K. Perspectives on MAO-B in aging and neurological disease: Where do we go from here? Mol. Neurobiol. 2004, 30, 77–89. [Google Scholar] [CrossRef]
- Vila, M.; Przedborski, S. Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Finberg, J.P.M. Inhibitors of MAO-B and COMT: Their effects on brain dopamine levels and uses in Parkinson’s disease. J. Neural Transm. 2019, 126, 433–448. [Google Scholar] [CrossRef]
- Dezsi, L.; Vecsei, L. Monoamine Oxidase B Inhibitors in Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2017, 16, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Parambi, D.G.T. Treatment of Parkinson’s Disease by MAO-B Inhibitors, New Therapies and Future Challenges—A Mini-Review. Comb. Chem. High Throughput Screen. 2020, 23, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Löhle, M.; Reichmann, H. Controversies in Neurology: Why monoamine oxidase B inhibitors could be a good choice for the initial treatment of Parkinson’s disease. BMC Neurol. 2011, 11, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkamhawy, A.; Kim, H.J.; Elsherbeny, M.H.; Paik, S.; Park, J.-H.; Gotina, L.; Abdellattif, M.H.; Gouda, N.A.; Cho, J.; Lee, K.; et al. Discovery of 3,4-dichloro-N-(1H-indol-5-yl)benzamide: A highly potent, selective, and competitive hMAO-B inhibitor with high BBB permeability profile and neuroprotective action. Bioorganic Chem. 2021, 116, 105352. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Paik, S.; Park, J.-H.; Kim, H.J.; Hassan, A.H.E.; Lee, K.; Park, K.D.; Roh, E.J. Discovery of novel and potent safinamide-based derivatives as highly selective hMAO-B inhibitors for treatment of Parkinson’s disease (PD): Design, synthesis, in vitro, in vivo and in silico biological studies. Bioorganic Chem. 2021, 115, 105233. [Google Scholar] [CrossRef]
- Reis, J.; Encarnação, I.; Gaspar, A.; Morales, A.; Milhazes, N.; Borges, F. Parkinson’s disease management. Part II- discovery of MAO-B inhibitors based on nitrogen heterocycles and analogues. Curr. Top. Med. Chem. 2012, 12, 2116–2130. [Google Scholar] [CrossRef]
- Moore, J.J.; Saadabadi, A. Selegiline. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Chang, Y.; Wang, L.B.; Li, D.; Lei, K.; Liu, S.Y. Efficacy of rasagiline for the treatment of Parkinson’s disease: An updated meta-analysis. Ann. Med. 2017, 49, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Dhillon, S. Safinamide: A Review in Parkinson’s Disease. CNS Drugs 2017, 31, 169–176. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Chapter 18—Neuroprotection in Parkinson’s Disease. In Blue Books of Neurology; Schapira, A.H.V., Lang, A.E.T., Fahn, S., Eds.; Butterworth-Heinemann: Oxford, UK, 2010; Volume 34, pp. 301–320. [Google Scholar]
- Wu, R.M.; Chen, R.C.; Chiueh, C.C. Effect of MAO-B inhibitors on MPP+ toxicity in Vivo. Ann. N. Y. Acad. Sci. 2000, 899, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Szökő, É.; Tábi, T.; Riederer, P.; Vécsei, L.; Magyar, K. Pharmacological aspects of the neuroprotective effects of irreversible MAO-B inhibitors, selegiline and rasagiline, in Parkinson’s disease. J. Neural Transm. 2018, 125, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Robakis, D.; Fahn, S. Defining the Role of the Monoamine Oxidase-B Inhibitors for Parkinson’s Disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P. Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: Focus on modulation of CNS monoamine neurotransmitter release. Pharmacol. Ther. 2014, 143, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Teo, K.C.; Ho, S.-L. Monoamine oxidase-B (MAO-B) inhibitors: Implications for disease-modification in Parkinson’s disease. Transl. Neurodegener. 2013, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Youdim, M.B.H. Why do we need multifunctional neuroprotective and neurorestorative drugs for Parkinson’s and Alzheimer’s diseases as disease modifying agents. Exp. Neurobiol. 2010, 19, 1–14. [Google Scholar] [CrossRef]
- Md Sahab, U.; Md Tanvir, K.; Md Habibur, R.; Md Abdul, A.; Md Motiar, R.; Anurag, K.; Abdullah Al, M.; Abdur, R.; Bijo, M.; Ghulam Md, A. Exploring the Multifunctional Neuroprotective Promise of Rasagiline Derivatives for Multi-Dysfunctional Alzheimer’s Disease. Curr. Pharm. Des. 2020, 26, 4690–4698. [Google Scholar]
- Sofic, E.; Rimpapa, Z.; Kundurovic, Z.; Sapcanin, A.; Tahirovic, I.; Rustembegovic, A.; Cao, G.J.J.o.n.t. Antioxidant capacity of the neurohormone melatonin. J. Neural Transm. 2005, 112, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Cipolla-Neto, J.; Amaral, F.G.D. Melatonin as a Hormone: New Physiological and Clinical Insights. Endocr. Rev. 2018, 39, 990–1028. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V. Melatonin oxidative stress and neurodegenerative diseases. Indian J. Exp. Biol. 2002, 40, 668–679. [Google Scholar] [PubMed]
- Sun, T.C.; Liu, X.C.; Yang, S.H.; Song, L.L.; Zhou, S.J.; Deng, S.L.; Tian, L.; Cheng, L.Y. Melatonin Inhibits Oxidative Stress and Apoptosis in Cryopreserved Ovarian Tissues via Nrf2/HO-1 Signaling Pathway. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of melatonin in the reduction of oxidative stress. A review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Cabrera, J.; D’Arpa, D. Melatonin and tryptophan derivatives as free radical scavengers and antioxidants. Adv. Exp. Med. Biol. 1999, 467, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Zhou, X.J.; Xu, B. Mitochondria: Central Organelles for Melatonin’s Antioxidant and Anti-Aging Actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. CMLS 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Velkov, Z.A.; Velkov, Y.Z.; Galunska, B.T.; Paskalev, D.N.; Tadjer, A.V. Melatonin: Quantum-chemical and biochemical investigation of antioxidant activity. Eur. J. Med. Chem. 2009, 44, 2834–2839. [Google Scholar] [CrossRef] [PubMed]
- Estevão, M.S.; Carvalho, L.C.; Ribeiro, D.; Couto, D.; Freitas, M.; Gomes, A.; Ferreira, L.M.; Fernandes, E.; Marques, M.M.B. Antioxidant activity of unexplored indole derivatives: Synthesis and screening. Eur. J. Med. Chem. 2010, 45, 4869–4878. [Google Scholar] [CrossRef]
- Esposito, E.; Cuzzocrea, S.J.C.n. Antiinflammatory activity of melatonin in central nervous system. Curr. Neuropharmacol. 2010, 8, 228–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Sun, C.; Laborda, P.; He, Y.; Zhao, Y.; Li, C.; Liu, F.J.P.P. Melatonin treatments reduce the pathogenicity and inhibit the growth of Xanthomonas oryzae pv. oryzicola. Plant Pathol. 2019, 68, 288–296. [Google Scholar] [CrossRef]
- Chen, X.; Sun, C.; Laborda, P.; Zhao, Y.; Palmer, I.; Fu, Z.Q.; Qiu, J.; Liu, F.J.F.i.m. Melatonin treatment inhibits the growth of Xanthomonas oryzae pv. oryzae. Front. Microbiol. 2018, 9, 2280. [Google Scholar] [CrossRef] [Green Version]
- Di Bella, G.; Mascia, F.; Gualano, L.; Di Bella, L.J.I.j.o.m.s. Melatonin anticancer effects. Int. J. Mol. Sci. 2013, 14, 2410–2430. [Google Scholar] [CrossRef] [Green Version]
- de Zanette, S.A.; Vercelino, R.; Laste, G.; Rozisky, J.R.; Schwertner, A.; Machado, C.B.; Xavier, F.; de Souza, I.C.C.; Deitos, A.; Torres, I.L.J.B.P.; et al. Melatonin analgesia is associated with improvement of the descending endogenous pain-modulating system in fibromyalgia: A phase II, randomized, double-dummy, controlled trial. BMC Pharm. Toxicol. 2014, 15, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lochner, A.; Marais, E.; Huisamen, B.J.J.o.p.r. Melatonin and cardioprotection against ischaemia/reperfusion injury: What’s new? A review. J. Pineal. Res. 2018, 65, e12490. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xia, H.; Zhang, L.; Zhang, H.; Wang, D.; Tao, X.J.B. Pharmacotherapy. Protective effects of melatonin on sepsis-induced liver injury and dysregulation of gluconeogenesis in rats through activating SIRT1/STAT3 pathway. Biomed Pharmacother. 2019, 117, 109150. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Hasan, A.U.; Kobori, H.J.H.R. Melatonin in chronic kidney disease: A promising chronotherapy targeting the intrarenal renin–angiotensin system. Hypertens. Res. 2019, 42, 920–923. [Google Scholar] [CrossRef]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Chitimus, D.M.; Popescu, M.R.; Voiculescu, S.E.; Panaitescu, A.M.; Pavel, B.; Zagrean, L.; Zagrean, A.M. Melatonin’s Impact on Antioxidative and Anti-Inflammatory Reprogramming in Homeostasis and Disease. Biomolecules 2020, 10, 1211. [Google Scholar] [CrossRef]
- Salehi, B.; Sharopov, F.; Fokou, P.V.T.; Kobylinska, A.; Jonge, L.; Tadio, K.; Sharifi-Rad, J.; Posmyk, M.M.; Martorell, M.; Martins, N.; et al. Melatonin in Medicinal and Food Plants: Occurrence, Bioavailability, and Health Potential for Humans. Cells 2019, 8, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Franco, M.I.; Fernández-Bachiller, M.I.; Pérez, C.; Hernández-Ledesma, B.; Bartolomé, B.J.J.o.m.c. Novel tacrine− melatonin hybrids as dual-acting drugs for Alzheimer disease, with improved acetylcholinesterase inhibitory and antioxidant properties. J. Med. Chem. 2006, 49, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-T.; Wang, C.-M.; Liu, Y.; Huang, Z.-G.J.E.j.o.m.c. New multifunctional melatonin-derived benzylpyridinium bromides with potent cholinergic, antioxidant, and neuroprotective properties as innovative drugs for Alzheimer’s disease. Eur. J. Med. Chem. 2015, 103, 302–311. [Google Scholar] [CrossRef]
- Bautista-Aguilera, O.M.; Esteban, G.; Bolea, I.; Nikolic, K.; Agbaba, D.; Moraleda, I.; Iriepa, I.; Samadi, A.; Soriano, E.; Unzeta, M.; et al. Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 75, 82–95. [Google Scholar] [CrossRef]
- Rivara, S.; Pala, D.; Bedini, A.; Spadoni, G. Therapeutic uses of melatonin and melatonin derivatives: A patent review (2012–2014). Expert Opin. Ther. Pat. 2015, 25, 425–441. [Google Scholar] [CrossRef]
- Wang, S.Y.; Shi, X.C.; Laborda, P. Indole-based melatonin analogues: Synthetic approaches and biological activity. Eur. J. Med. Chem. 2020, 185, 111847. [Google Scholar] [CrossRef] [PubMed]
- Mor, M.; Rivara, S.; Silva, C.; Bordi, F.; Plazzi, P.V.; Spadoni, G.; Diamantini, G.; Balsamini, C.; Tarzia, G.; Fraschini, F.; et al. Melatonin receptor ligands: Synthesis of new melatonin derivatives and comprehensive comparative molecular field analysis (CoMFA) study. J. Med. Chem. 1998, 41, 3831–3844. [Google Scholar] [CrossRef] [PubMed]
- Landagaray, E.; Ettaoussi, M.; Leclerc, V.; Traoré, B.; Perez, V.; Nosjean, O.; Boutin, J.A.; Caignard, D.H.; Delagrange, P.; Berthelot, P.; et al. New melatonin (MT1/MT2) ligands: Design and synthesis of (8,9-dihydro-7H-furo [3,2-f]chromen-1-yl) derivatives. Bioorganic Med. Chem. 2014, 22, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Angelova, V.T.; Rangelov, M.; Todorova, N.; Dangalov, M.; Andreeva-Gateva, P.; Kondeva-Burdina, M.; Karabeliov, V.; Shivachev, B.; Tchekalarova, J. Discovery of novel indole-based aroylhydrazones as anticonvulsants: Pharmacophore-based design. Bioorg. Chem. 2019, 90, 103028. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Reiter, R.J.; Manchester, L.C.; Yan, M.T.; El-Sawi, M.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Allegra, M.; Hardeland, R. Chemical and physical properties and potential mechanisms: Melatonin as a broad spectrum antioxidant and free radical scavenger. Curr. Top. Med. Chem. 2002, 2, 181–197. [Google Scholar] [CrossRef] [Green Version]
- Stasica, P.; Ulanski, P.; Rosiak, J.M. Melatonin as a hydroxyl radical scavenger. J. Pineal Res. 1998, 25, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Cytoprotective properties of melatonin: Presumed association with oxidative damage and aging. Nutrition 1998, 14, 691–696. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Paik, S.; Kim, H.J.; Park, J.-H.; Londhe, A.M.; Lee, K.; Pae, A.N.; Park, K.D.; Roh, E.J. Discovery of N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide: A novel, selective, and competitive indole-based lead inhibitor for human monoamine oxidase B. J. Enzym. Inhib. Med. Chem. 2020, 35, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Yeon, S.K.; Choi, J.W.; Park, J.-H.; Lee, Y.R.; Kim, H.J.; Shin, S.J.; Jang, B.K.; Kim, S.; Bahn, Y.-S.; Han, G.; et al. Synthesis and evaluation of biaryl derivatives for structural characterization of selective monoamine oxidase B inhibitors toward Parkinson’s disease therapy. Bioorganic Med. Chem. 2018, 26, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Cores, Á.; Abril, S.; Michalska, P.; Duarte, P.; Olives, A.I.; Martín, M.A.; Villacampa, M.; León, R.; Menéndez, J.C. Bisavenathramide Analogues as Nrf2 Inductors and Neuroprotectors in In Vitro Models of Oxidative Stress and Hyperphosphorylation. Antioxidants 2021, 10, 941. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Park, J.E.; Hassan, A.H.E.; Pae, A.N.; Lee, J.; Park, B.G.; Roh, E.J. Synthesis and evaluation of 2-(3-arylureido)pyridines and 2-(3-arylureido)pyrazines as potential modulators of Aβ-induced mitochondrial dysfunction in Alzheimer’s disease. Eur. J. Med. Chem. 2018, 144, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Viswanath, A.N.I.; Pae, A.N.; Kim, H.Y.; Heo, J.C.; Park, W.K.; Lee, C.O.; Yang, H.; Kim, K.H.; Nam, D.H.; et al. Discovery of potent and selective cytotoxic activity of new quinazoline-ureas against TMZ-resistant glioblastoma multiforme (GBM). Eur. J. Med. Chem. 2015, 103, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Hassan, A.H.E.; Paik, S.; Sup Lee, Y.; Lee, H.H.; Shin, J.S.; Lee, K.T.; Roh, E.J. EGFR inhibitors from cancer to inflammation: Discovery of 4-fluoro-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide as a novel anti-inflammatory EGFR inhibitor. Bioorganic Chem. 2019, 86, 112–118. [Google Scholar] [CrossRef]
- Park, J.E.; Elkamhawy, A.; Hassan, A.H.E.; Pae, A.N.; Lee, J.; Paik, S.; Park, B.G.; Roh, E.J. Synthesis and evaluation of new pyridyl/pyrazinyl thiourea derivatives: Neuroprotection against amyloid-β-induced toxicity. Eur. J. Med. Chem. 2017, 141, 322–334. [Google Scholar] [CrossRef]
- Al -Sanea, M.M.; Elkamhawy, A.; Zakaria, A.; Park, B.S.; Kwon, Y.; Lee, S.H.; Lee, S.W.; Kim, I.T. Synthesis and in vitro screening of phenylbipyridinylpyrazole derivatives as potential antiproliferative agents. Molecules 2015, 20, 1031–1045. [Google Scholar] [CrossRef] [Green Version]
- Musella, S.; di Sarno, V.; Ciaglia, T.; Sala, M.; Spensiero, A.; Scala, M.C.; Ostacolo, C.; Andrei, G.; Balzarini, J.; Snoeck, R.; et al. Identification of an indol-based derivative as potent and selective varicella zoster virus (VZV) inhibitor. Eur. J. Med. Chem. 2016, 124, 773–781. [Google Scholar] [CrossRef]
- Mahaney, P.E.; Heffernan, G.D.; Coghlan, R.D.; Cohn, S.T.; Kim, C.Y.; Jenkins, D.J.; Marella, M.A.; McComas, C.C.; Sabatucci, J.P.; Terefenko, E.A.; et al. Phenylaminopropanol Derivatives as Monoamine Reuptake Inhibitors and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Disease Ameliorated by Monoamine Reuptake. US20070072897 2007. [Google Scholar]
- Wang, P.-F.; Zhang, Y.-J.; Wang, D.; Hu, H.-M.; Wang, Z.-C.; Xu, C.; Qiu, H.-Y.; Zhu, H.-L. Design, synthesis, and biological evaluation of new B-RafV600E kinase inhibitors. Bioorg. Med. Chem. 2018, 26, 2372–2380. [Google Scholar] [CrossRef]
- Johnson, K.W.; Phebus, L.A. Preparation of Piperidinylindoles and Related Compounds as Serotonin 5-HT1F Agonists. WO9811895 1998. [Google Scholar]
- Frederickson, M.; Gill, A.L.; Padova, A.; Congreve, M.S. Preparation of Indoles as p38 MAP Kinase Inhibitors. WO2003087087 2003. [Google Scholar]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739. [Google Scholar] [CrossRef] [Green Version]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Cao, Q.; Sun, Z.; Chen, J.; Zheng, Q.; Xiao, F. A novel method of neural differentiation of PC12 cells by using Opti-MEM as a basic induction medium. Int. J. Mol. Med. 2018, 41, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, J.; Yang, X.; Cai, P.; Liu, Q.; Wang, K.D.G.; Kong, L.; Wang, X. Neuroprotective effects of benzyloxy substituted small molecule monoamine oxidase B inhibitors in Parkinson’s disease. Bioorganic Med. Chem. 2016, 24, 5929–5940. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Li, E.; Park, S. Insulin-like growth factor-1 inhibits 6-hydroxydopamine-mediated endoplasmic reticulum stress-induced apoptosis via regulation of heme oxygenase-1 and Nrf2 expression in PC12 cells. Int. J. Neurosci. 2012, 122, 641–649. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, W.; Wang, H.; Bao, L.; Li, G.; Li, T.; Song, S.; Li, H.; Hao, J.; Sun, J. Resveratrol Protects PC12 Cell against 6-OHDA Damage via CXCR4 Signaling Pathway. Evid. Based Complement. Altern. Med. 2015, 2015, 730121. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Liu, Y.; Sun, A.; Du, Y.; Ye, M.; Pu, X.; Qi, X. Intestinal absorption and neuroprotective effects of kaempferol-3-O-rutinoside. RSC Adv. 2017, 7, 31408–31416. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Xue, Y.Q.; Yang, C.; Yang, W.H.; Chen, L.; Zhang, Q.J.; Qu, T.Y.; Huang, S.; Zhao, L.R.; Wang, X.M.; et al. Human albumin prevents 6-hydroxydopamine-induced loss of tyrosine hydroxylase in in vitro and in vivo. PLoS ONE 2012, 7, e41226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Biswas, J.; Gupta, P.; Singh, A.; Tiwari, S.; Mishra, A.; Singh, S. Salubrinal attenuates nitric oxide mediated PERK:IRE1α: ATF-6 signaling and DNA damage in neuronal cells. Neurochem. Int. 2019, 131, 104581. [Google Scholar] [CrossRef] [PubMed]
- Buratta, S.; Chiaradia, E.; Tognoloni, A.; Gambelunghe, A.; Meschini, C.; Palmieri, L.; Muzi, G.; Urbanelli, L.; Emiliani, C.; Tancini, B. Effect of Curcumin on Protein Damage Induced by Rotenone in Dopaminergic PC12 Cells. Int. J. Mol. Sci. 2020, 21, 2761. [Google Scholar] [CrossRef]
- Park, S.-H.; Jang, J.-H.; Chen, C.-Y.; Na, H.-K.; Surh, Y.-J. A formulated red ginseng extract rescues PC12 cells from PCB-induced oxidative cell death through Nrf2-mediated upregulation of heme oxygenase-1 and glutamate cysteine ligase. Toxicology 2010, 278, 131–139. [Google Scholar] [CrossRef]
- Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.K.; Lee, H.; Cho, J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 319. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.L.; Bui, B.P.; Lee, H.; Cho, J. A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 2527. [Google Scholar] [CrossRef]
- Oh, Y.; Do, H.T.T.; Kim, S.; Kim, Y.M.; Chin, Y.W.; Cho, J. Memory-Enhancing Effects of Mangosteen Pericarp Water Extract through Antioxidative Neuroprotection and Anti-Apoptotic Action. Antioxidants 2020, 10, 34. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx J. Am. Soc. Exp. Neurother. 2005, 2, 554–571. [Google Scholar] [CrossRef] [Green Version]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx J. Am. Soc. Exp. Neurother. 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkinshaw, G.; Waters, C.M. Neurotoxin-induced cell death in neuronal PC12 cells is mediated by induction of apoptosis. Neuroscience 1994, 63, 975–987. [Google Scholar] [CrossRef]
- Zhang, G.; Buchler, I.P.; DePasquale, M.; Wormald, M.; Liao, G.; Wei, H.; Barrow, J.C.; Carr, G.V. Development of a PC12 Cell Based Assay for Screening Catechol-O-methyltransferase Inhibitors. ACS Chem. Neurosci. 2019, 10, 4221–4226. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative damage in neurodegenerative disease. Lancet 1994, 344, 796–798. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Sanchez-Pernaute, R.; Oiwa, Y.; Kohutnicka, M.; Cummins, A.; Eberling, J. Preclinical models of Parkinson’s disease. Curr. Protoc. Neurosci. 2001. [Google Scholar] [CrossRef]
- Jismy, B.; El Qami, A.; Pišlar, A.; Frlan, R.; Kos, J.; Gobec, S.; Knez, D.; Abarbri, M. Pyrimido[1,2-b]indazole derivatives: Selective inhibitors of human monoamine oxidase B with neuroprotective activity. Eur. J. Med. Chem. 2021, 209, 112911. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.N.; Singh, P. Advancement in the modelling and therapeutics of Parkinson’s disease. J. Chem. Neuroanat. 2020, 104, 101752. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Neurotoxicant-induced animal models of Parkinson’s disease: Understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res. 2004, 318, 225–241. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Johnson, J.A.J.F.R.B. Medicine. Nrf2—a therapeutic target for the treatment of neurodegenerative diseases. Free. Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Comp. | R | Isolated Yield (%) |
|---|---|---|---|
| 1 | 3a | H | 94 |
| 2 | 3b | 2-bromo | 31 |
| 3 | 3c | 4-bromo | 84 |
| 4 | 3d | 2,4-dichloro | 72 |
| 5 | 3e | 3,5-dinitro | 87 |
| 6 | 3f | 3-nitro | 85 |
| 7 | 3g | 4-nitro | 85 |
| 8 | 3h | 2,6-difluoro | 49 |
| 9 | 3i | 3-fluoro | 75 |
| 10 | 3j | 4-fluoro | 21 |
| 11 | 3k | 2-fluoro | 62 |
| 12 | 3l | 2-trifluoromethyl, 4-fluoro | 50 |
| 13 | 3m | 3-cyano | 79 |
| 14 | 3n | 3-chloro | 88 |
| 15 | 3o | 2-chloro | 100 |
| 16 | 3p | 4-chloro | 94 |
| 17 | 3q | 4-tert-butyl | 99 |
| 18 | 3r | 3-bromo | 34 |
| 19 | 3s | 3-trifluoromethoxy | 91 |
| 20 | 3t | 4-trifluoromethoxy | 86 |
| 21 | 3u | 3-trifluoromethyl | 66 |
| 22 | 3v | 4-trifluoromethyl | 86 |
| 23 | 3w | 3,5-dichloro | 52 |
| 24 | 3x | 2,6-dichloro | 78 |
| Comp. | MW | TPSA | GTI Absorption | BBB Permeability | Lipinski #Violations | ESOL Log S | ESOL Class |
|---|---|---|---|---|---|---|---|
| 3a | 236.27 | 44.89 | High | Yes | 0 | −3.12 | Soluble |
| 3b | 315.16 | 44.89 | High | Yes | 0 | −4.43 | Moderately soluble |
| 3c | 315.16 | 44.89 | High | Yes | 0 | −4.01 | Moderately soluble |
| 3d | 305.16 | 44.89 | High | Yes | 0 | −4.70 | Moderately soluble |
| 3e | 326.26 | 136.53 | Low | No | 0 | −3.17 | Soluble |
| 3f | 281.27 | 90.71 | High | No | 0 | −3.56 | Soluble |
| 3g | 281.27 | 90.71 | High | No | 0 | −3.56 | Soluble |
| 3h | 272.25 | 44.89 | High | Yes | 0 | −3.83 | Soluble |
| 3i | 254.26 | 44.89 | High | Yes | 0 | −3.68 | Soluble |
| 3j | 254.26 | 44.89 | High | Yes | 0 | −3.26 | Soluble |
| 3k | 254.26 | 44.89 | High | Yes | 0 | −3.68 | Soluble |
| 3l | 322.26 | 44.89 | High | No | 0 | −4.49 | Moderately soluble |
| 3m | 261.28 | 68.68 | High | Yes | 0 | −3.45 | Soluble |
| 3n | 270.71 | 44.89 | High | Yes | 0 | −4.12 | Moderately soluble |
| 3o | 270.71 | 44.89 | High | Yes | 0 | −4.12 | Moderately soluble |
| 3p | 270.71 | 44.89 | High | Yes | 0 | −3.69 | Soluble |
| 3q | 292.37 | 44.89 | High | Yes | 0 | −4.77 | Moderately soluble |
| 3r | 315.16 | 44.89 | High | Yes | 0 | −4.43 | Moderately soluble |
| 3s | 320.27 | 54.12 | High | Yes | 0 | −4.54 | Moderately soluble |
| 3t | 320.27 | 54.12 | High | Yes | 0 | −4.54 | Moderately soluble |
| 3u | 304.27 | 44.89 | High | Yes | 0 | −4.34 | Moderately soluble |
| 3v | 304.27 | 44.89 | High | Yes | 0 | −4.34 | Moderately soluble |
| 3w | 305.16 | 44.89 | High | Yes | 0 | −4.28 | Moderately soluble |
| 3x | 305.16 | 44.89 | High | Yes | 0 | −4.70 | Moderately soluble |
| Entry | Comp. | % Inhibition of MAO-B at 10 µM | MAO-B IC50 (µM) | MAO-A IC50 (µM) | Selectivity Index (SI) a |
|---|---|---|---|---|---|
| 1 | 3a | 14.86 ± 0.80 | - | - | - |
| 2 | 3b | 0.27 ± 0.81 | - | - | - |
| 3 | 3c | 37.09 ± 0.70 | - | - | - |
| 4 | 3d | 22.92 ± 0.70 | - | - | - |
| 5 | 3e | 2.75 ± 0.20 | - | - | - |
| 6 | 3f | 54.25 ± 0.37 | - | - | - |
| 7 | 3g | 60.98 ± 0.69 | - | - | - |
| 8 | 3h | −0.02 ± 0.26 | - | - | - |
| 9 | 3i | 35.29 ± 0.76 | - | - | - |
| 10 | 3j | 58.33 ± 1.02 | - | - | - |
| 11 | 3k | 37.86 ± 0.33 | - | - | - |
| 12 | 3l | 1.49 ± 0.26 | - | - | - |
| 13 | 3m | 8.33 ± 0.59 | - | - | - |
| 14 | 3n | 76.79 ± 0.27 | 1.41 | >100 | >71 |
| 15 | 3o | 2.39 ± 0.31 | - | - | - |
| 16 | 3p | 57.98 ± 0.07 | - | - | - |
| 17 | 3q | 50.01 ± 0.04 | - | - | - |
| 18 | 3r | 87.00 ± 0.03 | 0.91 | >100 | >109 |
| 19 | 3s | 68.34 ± 0.25 | - | - | - |
| 20 | 3t | 63.23 ± 0.45 | - | - | - |
| 21 | 3u | 85.12 ± 0.22 | 1.20 | >100 | >83 |
| 22 | 3v | 91.22 ± 0.11 | 0.66 | >100 | >151 |
| 23 | 3w | 72.63 ± 0.32 | 2.41 | >100 | >41 |
| 24 | 3x | 0.89 ± 0.31 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkamhawy, A.; Woo, J.; Gouda, N.A.; Kim, J.; Nada, H.; Roh, E.J.; Park, K.D.; Cho, J.; Lee, K. Melatonin Analogues Potently Inhibit MAO-B and Protect PC12 Cells against Oxidative Stress. Antioxidants 2021, 10, 1604. https://doi.org/10.3390/antiox10101604
Elkamhawy A, Woo J, Gouda NA, Kim J, Nada H, Roh EJ, Park KD, Cho J, Lee K. Melatonin Analogues Potently Inhibit MAO-B and Protect PC12 Cells against Oxidative Stress. Antioxidants. 2021; 10(10):1604. https://doi.org/10.3390/antiox10101604
Chicago/Turabian StyleElkamhawy, Ahmed, Jiyu Woo, Noha A. Gouda, Jushin Kim, Hossam Nada, Eun Joo Roh, Ki Duk Park, Jungsook Cho, and Kyeong Lee. 2021. "Melatonin Analogues Potently Inhibit MAO-B and Protect PC12 Cells against Oxidative Stress" Antioxidants 10, no. 10: 1604. https://doi.org/10.3390/antiox10101604