
Hyperphosphatemia-Induced Oxidant/Antioxidant Imbalance Impairs Vascular Relaxation and Induces Inflammation and Fibrosis in Old Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Experimental Designs
2.4. Animal Studies
2.5. Vascular Reactivity of Mice Mesenteric Arteries
2.6. Sirius Red and Elastin Staining and DHE Detection
2.7. Immunohistochemistry of Nitrotyrosine and Nox4
2.8. Quantitative RT-PCR
2.9. Western Blot Assays
2.10. Immunofluorescence
2.11. Electrophoretic Mobility Shifts Assays (EMSA)
2.12. ROS Production
2.13. Statistical Analysis
3. Results
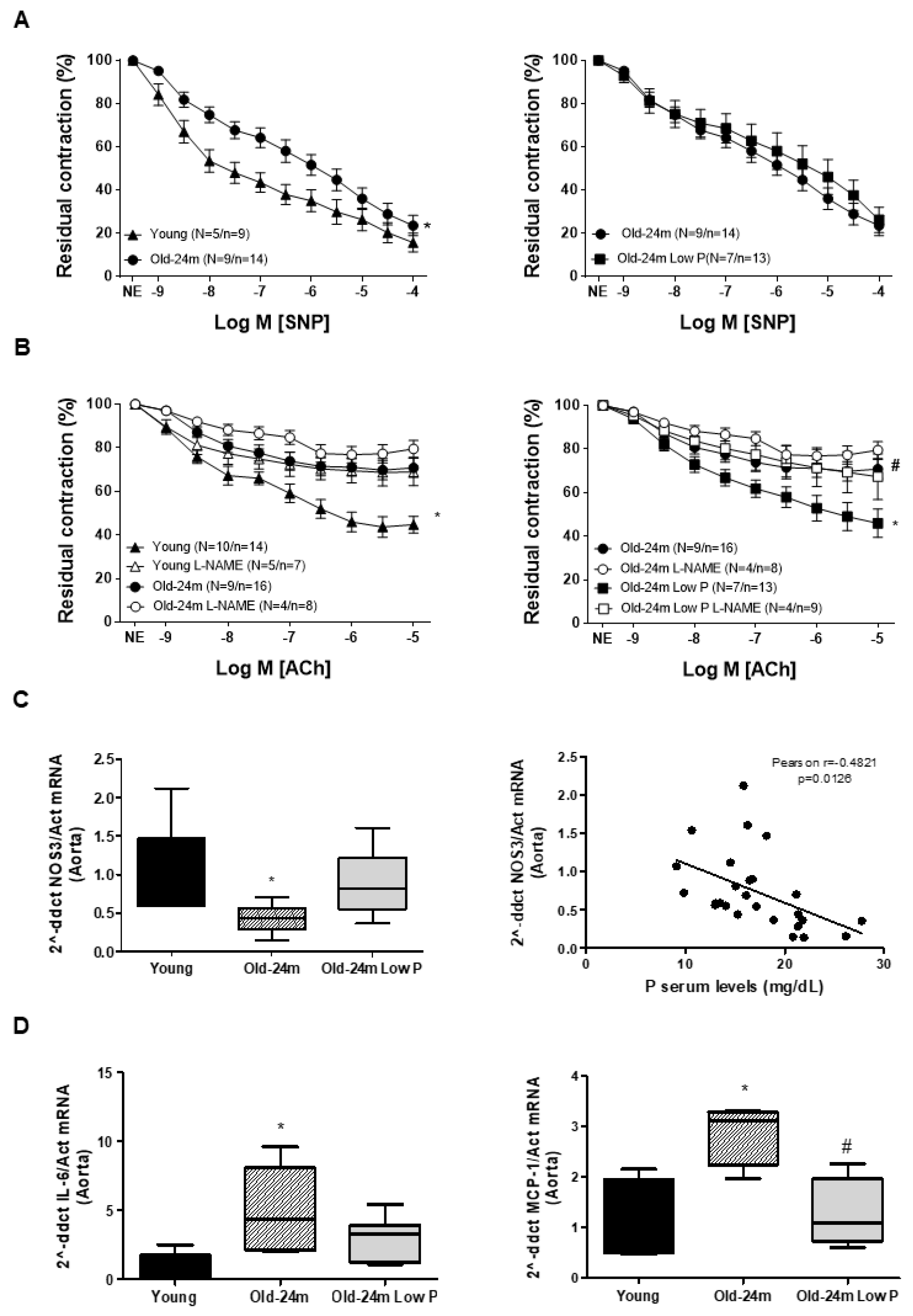
3.1. Hyperphosphatemia Induced Vascular Dysfunction in Old Mice by Reducing Endothelium-Dependent Vascular Relaxation and Increasing Inflammation and Fibrosis
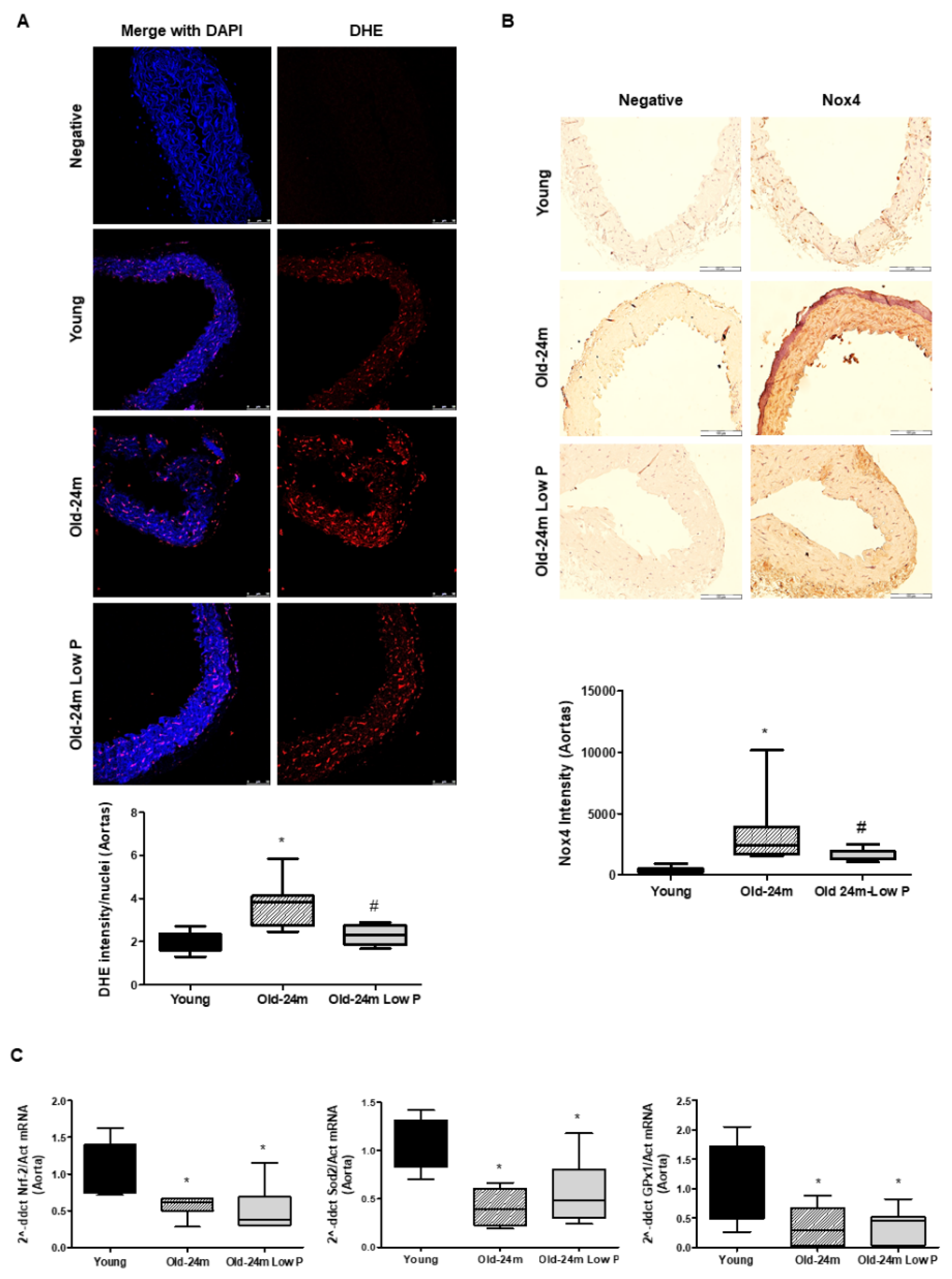
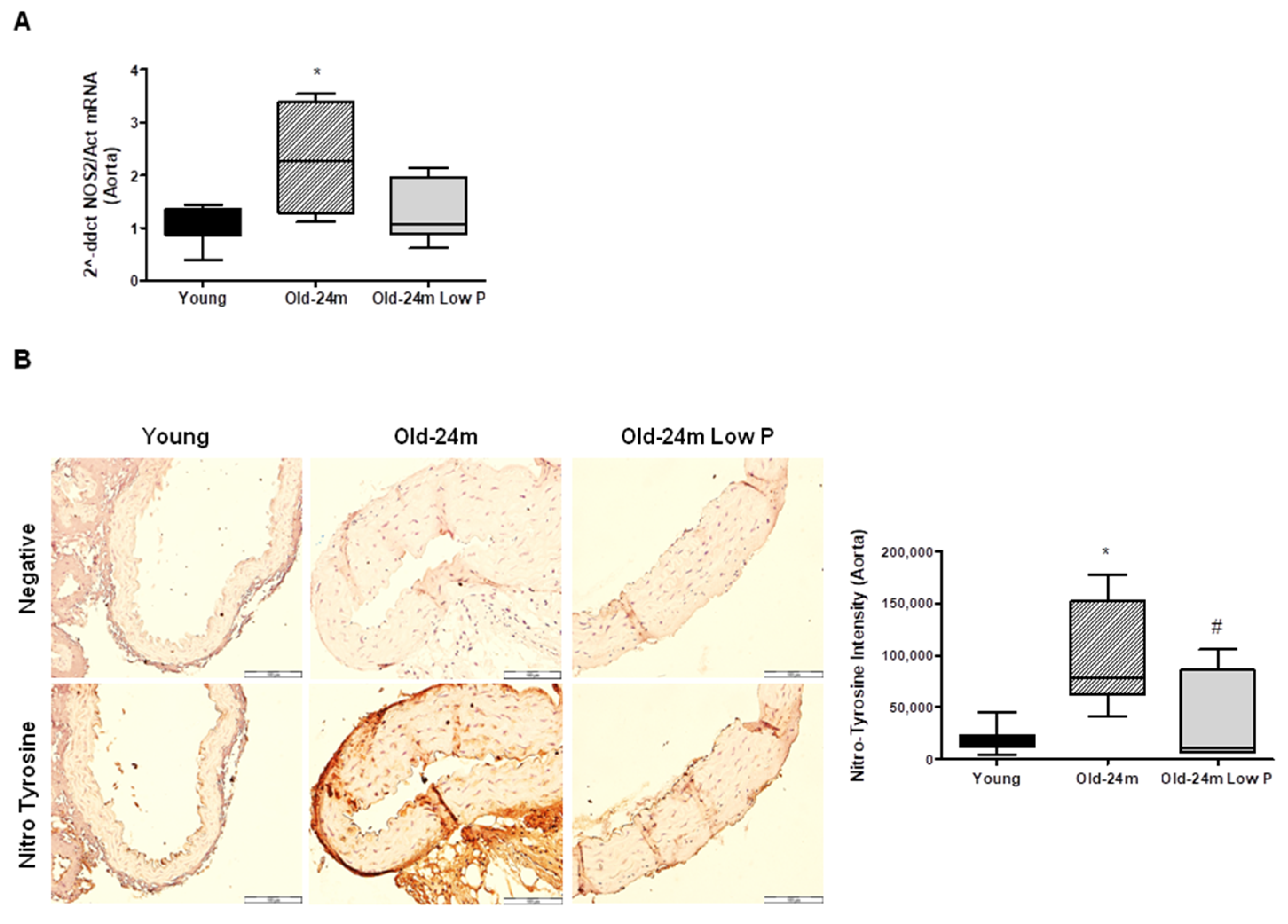
3.2. Hyperphosphatemia Impairs Oxidant/Antioxidant Balance and Induces Nitrosative Damage in Aorta from Old Mice
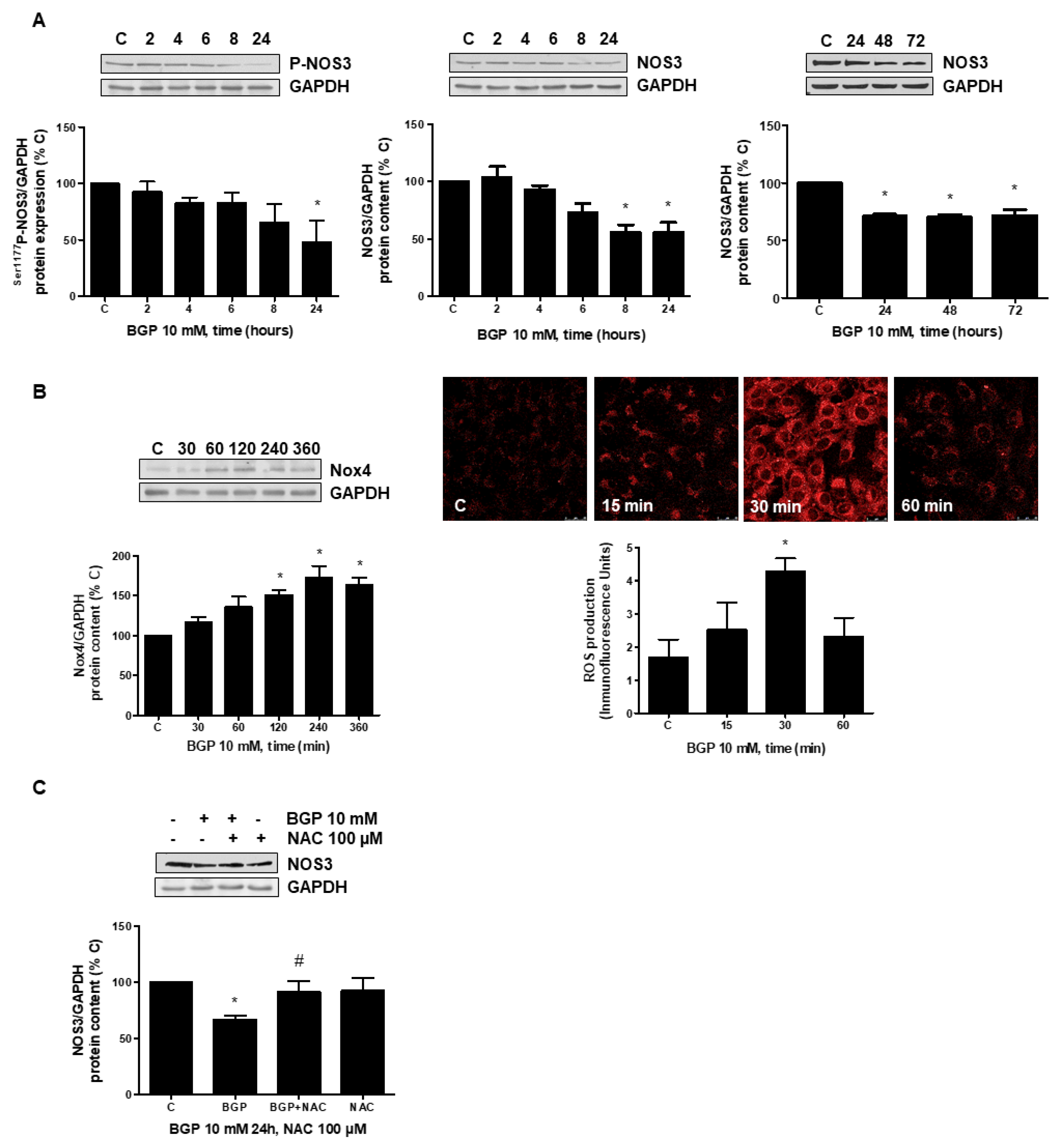
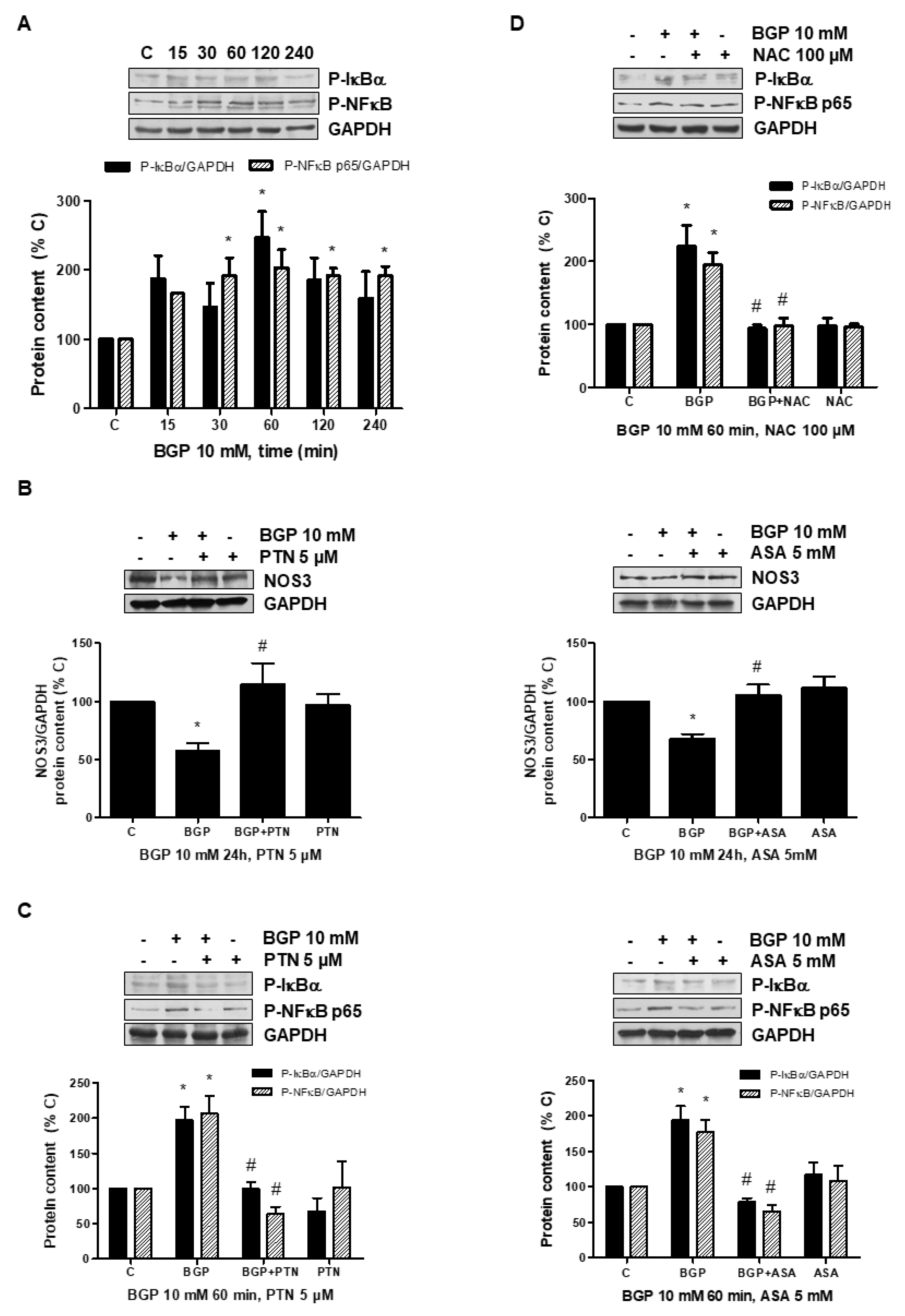
3.3. Hyperphosphatemia Downregulates NOS3 by Increasing Oxidative Stress through NFkB Activation in Endothelial Cells
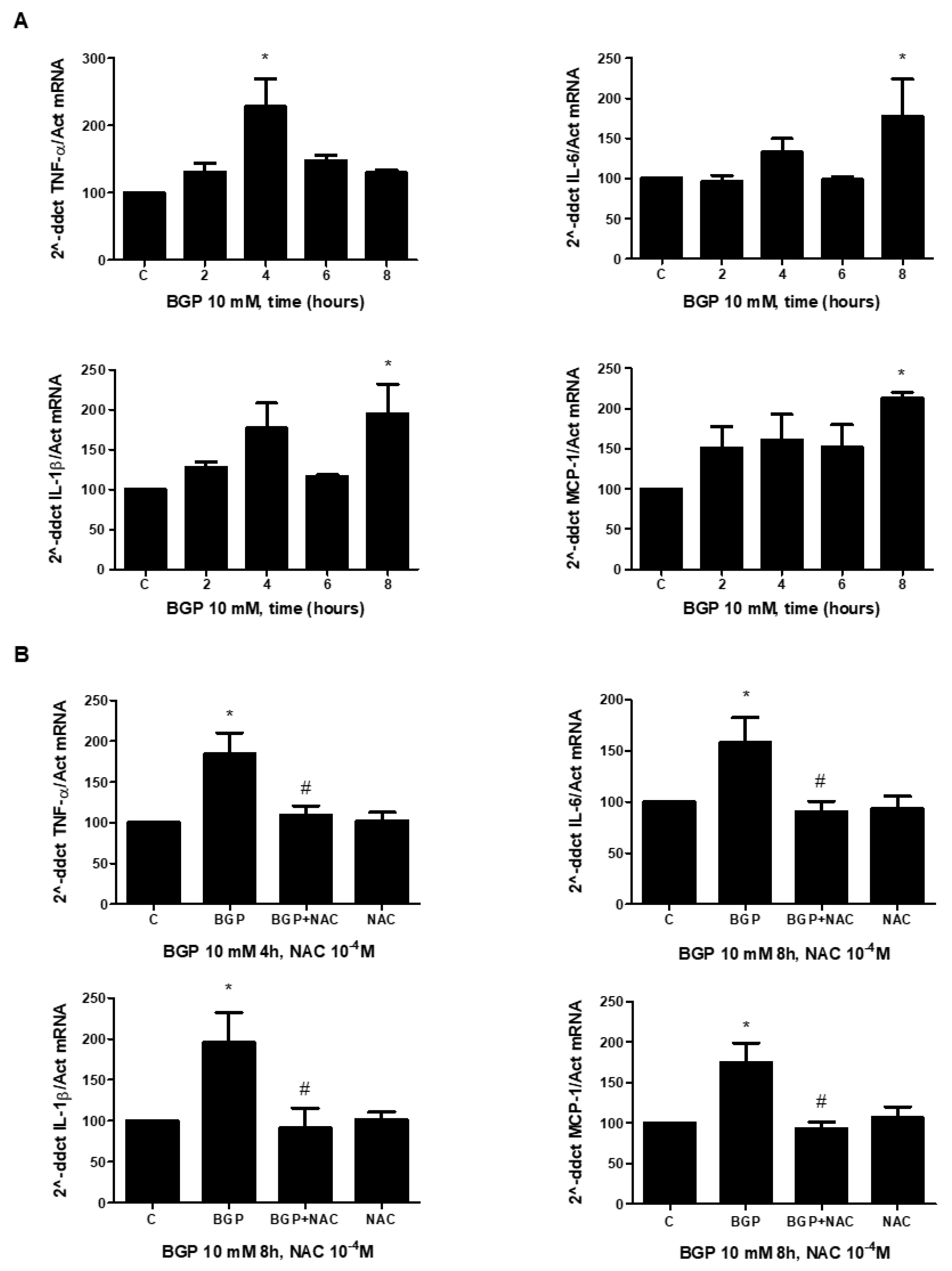
3.4. Hyperphosphatemia Induced Inflammation by Increasing Oxidative Stress in Endothelial Cells
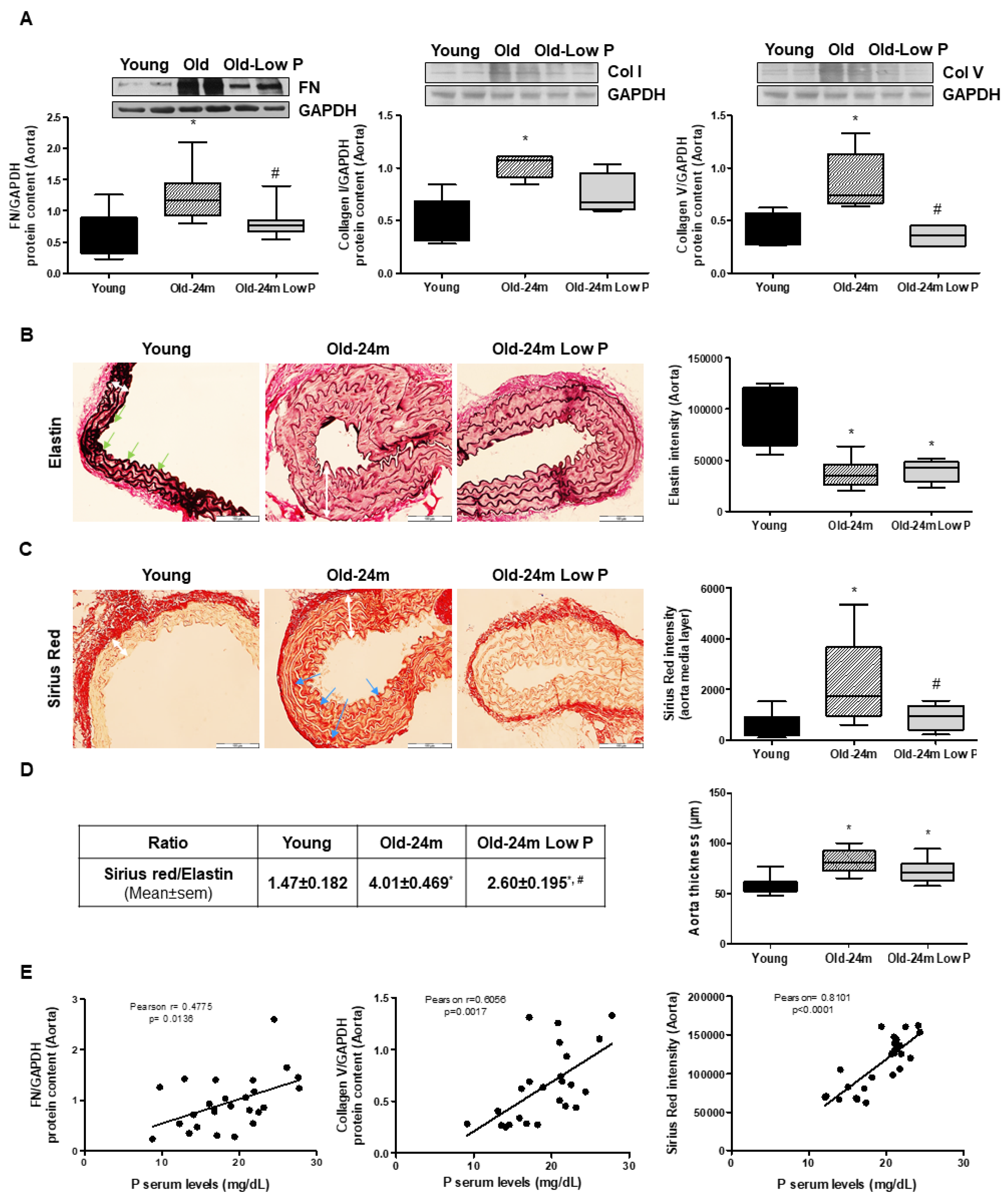
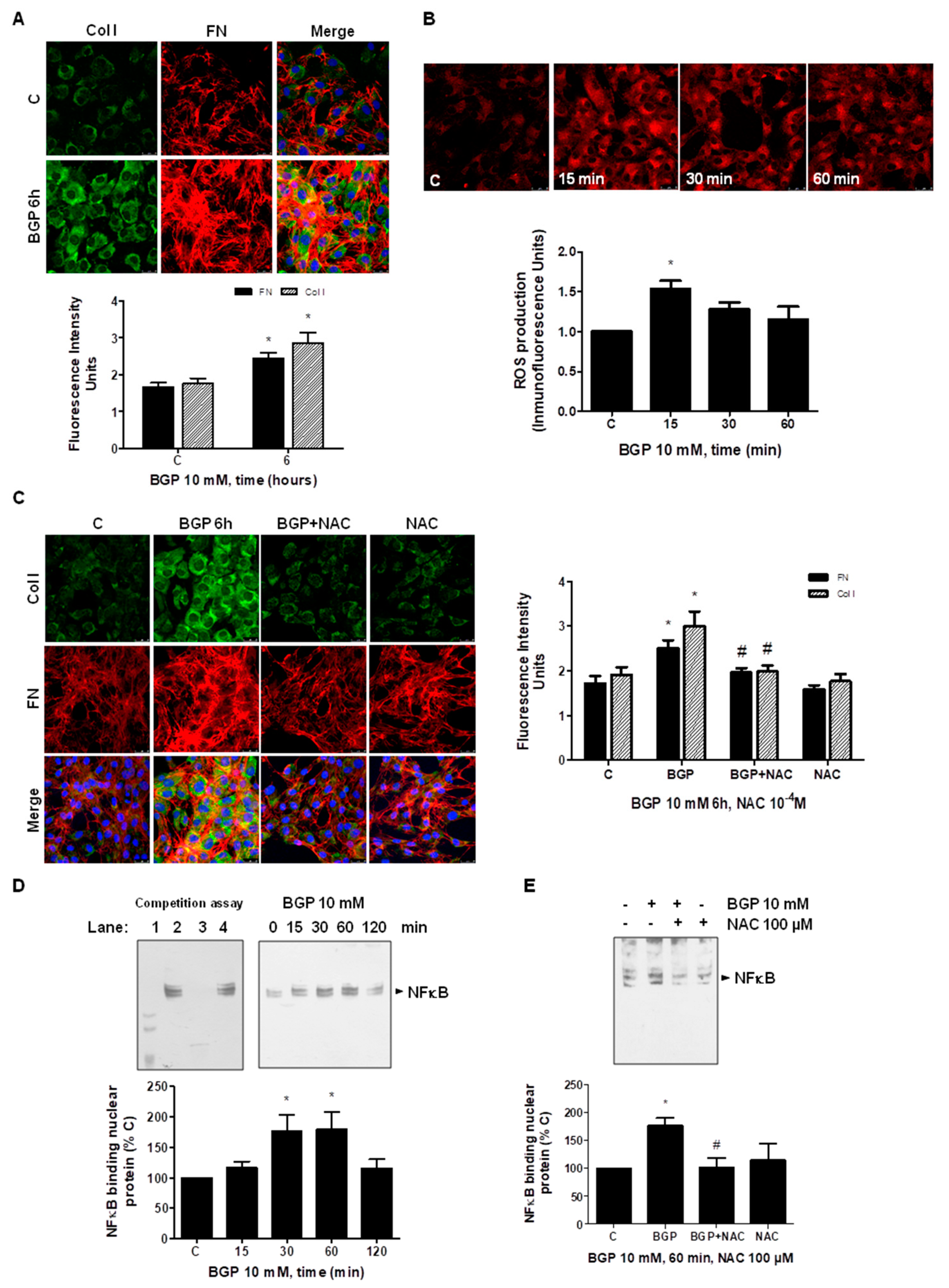
3.5. Hyperphosphatemia Induced Vascular Fibrosis by Increasing Oxidative Stress in Vascular Smooth Muscle Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Campia, U.; Tesauro, M.; Di Daniele, N.; Cardillo, C. The vascular endothelin system in obesity and type 2 diabetes: Pathophysiology and therapeutic implications. Life Sci. 2014, 118, 149–155. [Google Scholar] [CrossRef]
- Schinzari, F.; Iantorno, M.; Campia, U.; Mores, N.; Rovella, V.; Tesauro, M.; Di Daniele, N.; Cardillo, C. Vasodilator responses and endothelin-dependent vasoconstriction in metabolically healthy obesity and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E787–E792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradinaru, D.; Borsa, C.; Ionescu, C.; Prada, G.I. Oxidized LDL and NO synthesis–biomarkers of endothelial dysfunction and ageing. Mech. Ageing Dev. 2015, 151, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Vanhoutte, P.M. Cellular signaling and NO production. Pflug. Arch. 2010, 459, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.; Pickrell, A.M.; Wang, X.; Bacman, S.R.; Yu, A.; Hida, A.; Dillon, L.M.; Morton, P.D.; Malek, T.R.; Williams, S.L.; et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ. 2017, 24, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Spina, M.; Garbisa, S.; Hinnie, J.; Hunter, J.C.; Serafini-Fracassini, A. Age-related changes in composition and mechanical properties of the tunica media of the upper thoracic human aorta. Arteriosclerosis 1983, 3, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sell, D.R.; Monnier, V.M. Molecular basis of arterial stiffening: Role of glycation—A mini-review. Gerontology 2012, 58, 227–237. [Google Scholar] [CrossRef]
- Mauriello, A.; Orlandi, A.; Palmieri, G.; Spagnoli, L.G.; Oberholzer, M.; Christen, H. Age-related modification of average volume and anisotropy of vascular smooth muscle cells. Pathol. Res. Pract. 1992, 188, 630–636. [Google Scholar] [CrossRef]
- Sun, Z. Aging, arterial stiffness, and hypertension. Hypertension 2015, 65, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, P.M. Hemodynamic aging as the consequence of structural changes associated with early vascular aging (EVA). Aging Dis. 2014, 5, 109–113. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty years of saying NO: Sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smulyan, H.; Mookherjee, S.; Safar, M.E. The two faces of hypertension: Role of aortic stiffness. J. Am. Soc. Hypertens. 2016, 10, 175–183. [Google Scholar] [CrossRef]
- Noce, A.; Canale, M.P.; Capria, A.; Rovella, V.; Tesauro, M.; Splendiani, G.; Annicchiarico-Petruzzelli, M.; Manzuoli, M.; Simonetti, G.; Di Daniele, N. Coronary artery calcifications predict long term cardiovascular events in non diabetic Caucasian hemodialysis patients. Aging 2015, 7, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Kotsis, V.; Stabouli, S.; Karafillis, I.; Nilsson, P. Early vascular aging and the role of central blood pressure. J. Hypertens. 2011, 29, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Luo, R.; Tang, X.; Tang, L.; Huang, H.X.; Wen, X.; Hu, S.; Peng, B. Age-related progression of arterial stiffness and its elevated positive association with blood pressure in healthy people. Atherosclerosis 2015, 238, 147–152. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Kalluri, R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C216–C225. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.H.; Herzog, W.; MacDonald, G.Z.; Reimer, R.A.; Rios, J.L.; Smith, I.C.; Zernicke, R.F.; Hart, D.A. Obesity, metabolic syndrome, and musculoskeletal disease: Common inflammatory pathways suggest a central role for loss of muscle integrity. Front. Physiol. 2018, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodríguez-Mañas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial ageing: From endothelial dysfunction to vascular calcification. J. Intern. Med. 2017, 281, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, M.; Razzaque, M.S. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010, 24, 3562–3571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-O, M. Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nat. Rev. Nephrol. 2013, 9, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. Phosphate and klotho. Kidney Int. 2011, 79, S20–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaba, H.; Fukagawa, M. Phosphate-a poison for humans? Kidney Int. 2016, 90, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Shuto, E.; Taketani, Y.; Tanaka, R.; Harada, N.; Isshiki, M.; Sato, M.; Nashiki, K.; Amo, K.; Yamamoto, H.; Higashi, Y.; et al. Dietary phosphorus acutely impairs endothelial function. J. Am. Soc. Nephrol. 2009, 20, 1504–1512. [Google Scholar] [CrossRef] [Green Version]
- Olmos, G.; Martínez-Miguel, P.; Alcalde-Estevez, E.; Medrano, D.; Sosa, P.; Rodríguez-Mañas, L.; Naves-Diaz, M.; Rodríguez-Puyol, D.; Ruiz-Torres, M.P.; López-Ongil, S. Hyperphosphatemia induces senescence in human endothelial cells by increasing endothelin-1 production. Aging Cell 2017, 16, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Roumeiotis, S.; Mallamaci, F.; Zocalli, C. Endothelial dysfunction in chronic kidney disease, from biology to clinical outcomes: A 2020 update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef]
- Van, T.V.; Watari, E.; Taketani, Y.; Kitamura, T.; Shiota, A.; Tanaka, T.; Tanimura, A.; Harada, N.; Nakaya, Y.; Yamamoto, H.; et al. Dietary phosphate restriction ameliorates endothelial dysfunction in adenine-induced kidney disease rats. J. Clin. Biochem. Nutr. 2012, 51, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Finkel, T. Free radicals and senescence. Exp. Cell Res. 2008, 314, 1918–1922. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regina, C.; Panatta, E.; Candi, E.; Melino, G.; Amelio, I.; Balistreri, C.R.; Annicchiarico-Petruzzelli, M.; Di Daniele, N.; Ruvolo, G. Vascular ageing and endothelial cell senescence: Molecular mechanisms of physiology and diseases. Mech. Ageing Dev. 2016, 159, 14–21. [Google Scholar] [CrossRef]
- Ji, L.L.; Leeuwenburgh, C.; Leichtweis, S.; Gore, M.; Fiebig, R.; Hollander, J.; Bejma, J. Oxidative stress and aging. Role of exercise and its influences on antioxidant systems. Ann. N. Y. Acad. Sci. 1998, 854, 102–117. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Zoumakis, M.; Bixler, E.O.; Lin, H.M.; Prolo, P.; Vela-Bueno, A.; Kales, A.; Chrousos, G.P. Impaired nighttime sleep in healthy old versus young adults is associated with elevated plasma interleukin-6 and cortisol levels: Physiologic and therapeutic implications. J. Clin. Endocrinol. Metab. 2003, 88, 2087–2095. [Google Scholar] [CrossRef] [Green Version]
- Pitocco, D.; Tesauro, M.; Alessandro, R.; Ghirlanda, G.; Cardillo, C. Oxidative stress in diabetes: Implications for vascular and other complications. Int. J. Mol. Sci. 2013, 14, 21525–21550. [Google Scholar] [CrossRef] [Green Version]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzari, F.; Tesauro, M.; Rovella, V.; Di Daniele, N.; Mores, N.; Veneziani, A.; Cardillo, C. Leptin stimulates both endothelin-1 and nitric oxide activity in lean subjects but not in patients with obesity-related metabolic syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 1235–1241. [Google Scholar] [CrossRef]
- Schinzari, F.; Tesauro, M.; Rovella, V.; Galli, A.; Mores, N.; Porzio, O.; Lauro, D.; Cardillo, C. Generalized impairment of vasodilator reactivity during hyperinsulinemia in patients with obesity-related metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E947–E952. [Google Scholar] [CrossRef]
- Tesauro, M.; Schinzari, F.; Rovella, V.; Melina, D.; Mores, N.; Barini, A.; Mettimano, M.; Lauro, D.; Iantorno, M.; Quon, M.J.; et al. Tumor necrosis factor-alpha antagonism improves vasodilation during hyperinsulinemia in metabolic syndrome. Diabetes Care 2008, 31, 1439–1441. [Google Scholar] [CrossRef] [Green Version]
- Tesauro, M.; Rizza, S.; Iantorno, M.; Campia, U.; Cardillo, C.; Lauro, D.; Leo, R.; Turriziani, M.; Cocciolillo, G.C.; Fusco, A.; et al. Vascular, metabolic, and inflammatory abnormalities in normoglycemic offspring of patients with type 2 diabetes mellitus. Metabolism 2007, 56, 413–419. [Google Scholar] [CrossRef]
- Ungvari, Z.; Kaley, G.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Mechanisms of vascular aging: New perspectives. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 1028–1041. [Google Scholar] [CrossRef] [Green Version]
- Krabbe, K.S.; Pedersen, M.; Bruunsgaard, H. Inflammatory mediators in the elderly. Exp. Gerontol. 2004, 39, 687–699. [Google Scholar] [CrossRef]
- Golomb, L.; Sagiv, A.; Pateras, I.S.; Maly, A.; Krizhanovsky, V.; Gorgoulis, V.G.; Oren, M.; Ben-Yehuda, A. Age-associated inflammation connects RAS-induced senescence to stem cell dysfunction and epidermal malignancy. Cell Death Differ. 2015, 22, 1764–1774. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Fernández, A.; Sánchez-Ferrer, A.; Angulo, J.; Rodríguez-Mañas, L. Multivessel analysis of progressive vascular aging in the rat: Asynchronous vulnerability among vascular territories. Mech. Ageing Dev. 2018, 173, 39–49. [Google Scholar] [CrossRef]
- El Assar, M.; Sánchez-Puelles, J.M.; Royo, I.; López-Hernández, E.; Sánchez-Ferrer, A.; Aceña, J.L.; Rodríguez-Mañas, L.; Angulo, J. FM19G11 reverses endothelial dysfunction in rat and human arteries through stimulation of the PI3K/Akt/eNOS pathway, independently of mTOR/HIF-1α activation. Br. J. Pharm. 2015, 172, 1277–1291. [Google Scholar] [CrossRef] [Green Version]
- El Assar, M.; Angulo, J.; Santos-Ruiz, M.; Ruiz de Adana, J.C.; Pindado, M.L.; Sánchez-Ferrer, A.; Hernández, A.; Rodríguez-Mañas, L. Asymmetric dimethylarginine (ADMA) elevation and arginase up-regulation contribute to endothelial dysfunction related to insulin resistance in rats and morbidly obese humans. J. Physiol. 2016, 594, 3045–3060. [Google Scholar] [CrossRef]
- Alcalde-Estévez, E.; Asenjo-Bueno, A.; Sosa, P.; Olmos, G.; Plaza, P.; Caballero-Mora, M.A.; Rodríguez-Puyol, D.; Ruíz-Torres, M.P.; López-Ongil, S. Endothelin-1 induces cellular senescence and fibrosis in cultured myoblasts. A potential mechanism of aging-related sarcopenia. Aging 2020, 12, 11200–11223. [Google Scholar] [CrossRef] [PubMed]
- Sible, J.C.; Eriksson, E.; Oliver, N. DNA binding proteins from keloid fibroblasts form unique complexes with the human fibronectin promoter. Gene Expr. J. Liver Res. 1996, 5, 269–283. [Google Scholar]
- Lee, K.S.; Kim, J.; Kwak, S.N.; Lee, K.S.; Lee, D.K.; Ha, K.S.; Wonb, M.H.; Jeoung, D.; Lee, H.; Kwon, Y.G.; et al. Functional role of NF-κB in expression of human endothelial nitric oxide synthase. Biochem. Biophys. Res. Commun. 2014, 448, 101–107. [Google Scholar] [CrossRef]
- Kim, S.; Lee, K.S.; Choi, S.; Kim, J.; Lee, D.K.; Park, M.; Park, W.; Kim, T.H.; Hwang, J.Y.; Won, M.H.; et al. NF-κB–responsive miRNA-31-5p elicits endothelial dysfunction associated with preeclampsia via downregulation of endothelial nitric-oxide synthase. J. Biol. Chem. 2018, 293, 18989–19000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, K.S.; Kim, J.H.; Lee, D.K.; Park, M.; Choi, S.; Park, W.; Kim, S.; Choi, Y.K.; Hwang, J.Y.; et al. Aspirin prevents TNF-α-induced endothelial cell dysfunction by regulating the NF-κB-dependent miR-155/eNOS pathway: Role of a miR-155/eNOS axis in preeclampsia. Free Radic. Biol. Med. 2017, 104, 185–198. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I (kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Troyano, N.; del Nogal, M.; Mora, I.; Naves-Díaz, M.; Carrillo-López, N.; Sosa, P.; Rodriguez-Puyol, D.; Olmos, G.; Ruiz-Torres, M.P. Hyperphosphatemia induces cellular senescence in human aorta smooth muscle cells through integrin linked kinase (ILK) up-regulation. Mech. Ageing Dev. 2015, 152, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Cernadas, M.R.; Sanchez de Miguel, L.; Garcia-Duran, M.; Gonzalez-Fernandez, F.; Millas, I.; Monton, M.; Rodrigo, J.; Rico, L.; Fernandez, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Ma, Y.; Ruan, Y.; Fu, G.; Wu, S. Long-term atorvastatin improves age-related endothelial dysfunction by ameliorating oxidative stress and normalizing eNOS/iNOS imbalance in rat aorta. Exp. Gerontol. 2014, 52, 9–17. [Google Scholar] [CrossRef]
- Toda, N. Age-related changes in endothelial function and blood flow regulation. Pharmacol. Ther. 2012, 133, 159–176. [Google Scholar] [CrossRef]
- DeSouza, C.A.; Shaoiro, L.F.; Clevenger, C.M.; Dinenno, F.A.; Monahan, K.D.; Tanaka, H.; Seals, D.R. Regular aerobic exercise prevents and restores age-related declines in endothelium-dependent vasodilation healthy men. Circulation 2000, 102, 1351–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Salvetti, G.; Bernini, G.; Magagna, A.; Salvetti, A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension 2001, 38, 274–279. [Google Scholar] [CrossRef]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H425–H432. [Google Scholar] [CrossRef] [Green Version]
- Soucy, K.G.; Ryoo, S.; Benjo, A.; Lim, H.K.; Gupta, G.; Sohi, J.S.; Elser, J.; Aon, M.A.; Nyhan, D.; Shoukas, A.A.; et al. Impaired shear stress induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J. Appl. Physiol. 2006, 101, 1751–1759. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Bollrathm, J.; Greten, F.R. IKK/NF-kappaB and STAT3 pathways: Central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009, 10, 1314–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branton, M.H.; Kopp, J.B. TGF-β and fibrosis. Microbes Infect. 1999, 1, 1349–1365. [Google Scholar] [CrossRef]
- Liu, B.; Rong, Y.; Sun, D.; Li, W.; Chen, H.; Cao, B.; Wang, T. Costunolide inhibits pulmonary fibrosis via regulating NF-kB and TGF-b1/Smad2/Nrf2-NOX4 signalling pathways. Biochem. Biophys. Res. Commun. 2019, 510, 329–333. [Google Scholar] [CrossRef]
- Poli, G. Pathogenesis of liver fibrosis: Role of oxidative stress. Mol. Asp. Med. 2000, 21, 49–98. [Google Scholar] [CrossRef]
- Chiarpotto, E.; Castello, L.; Leonarduzzi, G.; Biasi, F.; Poli, G. Role of 4-hydroxy-2,3-nonenal in the pathogenesis of fibrosis. Biofactors 2005, 24, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007, 21, 3244–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef]
- Nasto, L.A.; Seo, H.Y.; Robinson, A.R.; Tilstra, J.S.; Clauson, C.L.; Sowa, G.A.; Ngo, K.; Dong, Q.; Pola, E.; Lee, J.Y.; et al. ISSLS prize winner: Inhibition of NF-κB activity ameliorates age-associated disc degeneration in a mouse model of accelerated aging. Spine 2012, 37, 1819–1825. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The role of oxidative stress and inflammation in cardiovascular aging. Biomed. Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef]
- Meyer, M.; Schreck, R.; Baeuerle, P.A. H2O2 and antioxidants, have opposite effects on activation of NF-kB and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993, 12, 2005–2015. [Google Scholar] [CrossRef]
- Kim, D.; Cheon, J.; Yoon, H.; Jun, H.S. Cudrania tricuspidata Root Extract Prevents Methylglyoxal-Induced Inflammation and Oxidative Stress via Regulation of the PKC-NOX4 Pathway in Human Kidney Cells. Oxid. Med. Cell. Longev. 2021, 2021. [Google Scholar] [CrossRef]
- Papadimitriou, A.; Peixoto, E.B.M.I.; Silva, K.C.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Inactivation of AMPK mediates high phosphate-induced extracellular matrix accumulation via NOX4/TGFß-1 signaling in human mesangial cells. Cell. Physiol. Biochem. 2014, 34, 1260–1272. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asenjo-Bueno, A.; Alcalde-Estévez, E.; El Assar, M.; Olmos, G.; Plaza, P.; Sosa, P.; Martínez-Miguel, P.; Ruiz-Torres, M.P.; López-Ongil, S. Hyperphosphatemia-Induced Oxidant/Antioxidant Imbalance Impairs Vascular Relaxation and Induces Inflammation and Fibrosis in Old Mice. Antioxidants 2021, 10, 1308. https://doi.org/10.3390/antiox10081308
Asenjo-Bueno A, Alcalde-Estévez E, El Assar M, Olmos G, Plaza P, Sosa P, Martínez-Miguel P, Ruiz-Torres MP, López-Ongil S. Hyperphosphatemia-Induced Oxidant/Antioxidant Imbalance Impairs Vascular Relaxation and Induces Inflammation and Fibrosis in Old Mice. Antioxidants. 2021; 10(8):1308. https://doi.org/10.3390/antiox10081308
Chicago/Turabian StyleAsenjo-Bueno, Ana, Elena Alcalde-Estévez, Mariam El Assar, Gemma Olmos, Patricia Plaza, Patricia Sosa, Patricia Martínez-Miguel, María Piedad Ruiz-Torres, and Susana López-Ongil. 2021. "Hyperphosphatemia-Induced Oxidant/Antioxidant Imbalance Impairs Vascular Relaxation and Induces Inflammation and Fibrosis in Old Mice" Antioxidants 10, no. 8: 1308. https://doi.org/10.3390/antiox10081308