
TNF-α Receptor Inhibitor Alleviates Metabolic and Inflammatory Changes in a Rat Model of Ischemic Stroke

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cerebral Ischemia and Treatments
2.2. Neurological Evaluation
2.3. Quantification of Ischemic Infarction
2.4. Brain Edema
2.5. Measurement of Oxidative Stress
2.6. Caspase 3 Activity Assay
2.7. Glucose Tolerance Test
2.8. Blood Sample Analyses
2.9. Measurement of Tissue Cytokines
2.10. Western Blot Analysis
2.11. RNA Isolation and Quantitative Real-Time Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
2.12. Cell Cultures
2.13. Measurement of Endothelial Barrier Integrity
2.14. Immunofluorescence Staining
2.15. Statistical Analysis
3. Results
3.1. R-7050 Alleviated Postischemic Brain Injury
3.2. R-7050 Alleviated Postischemic Inflammation
3.3. R-7050 Improved Postischemic Hyperglycemia
3.4. Cerebral Ischemia’s Impairment of Insulin Action in the Gastrocnemius and the Reversal Effect of R-7050
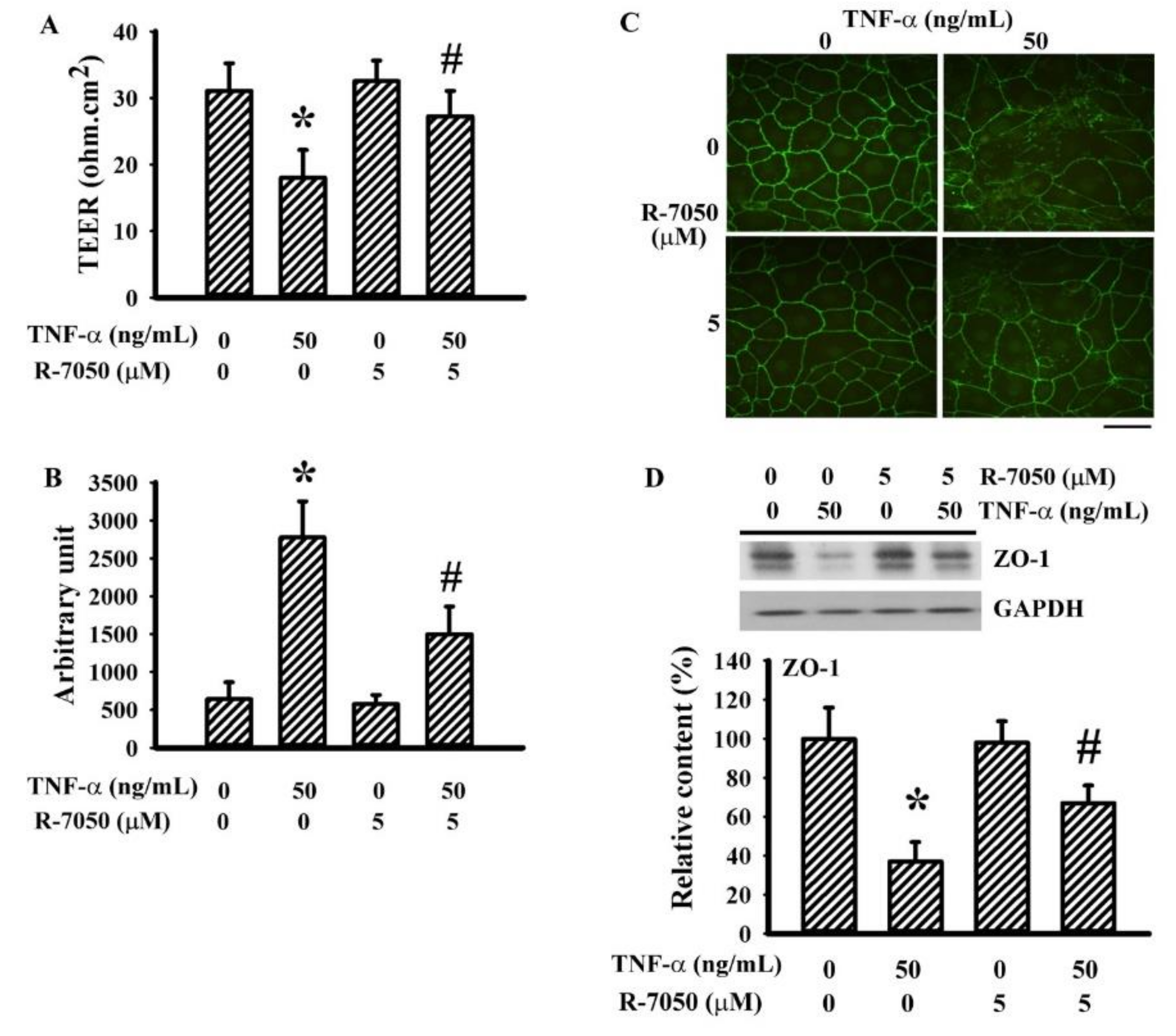
3.5. R-7050 Alleviated TNF-α-Induced Endothelial Barrier Disruption
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Navis, A.; Garcia-Santibanez, R.; Skliut, M. Epidemiology and outcomes of ischemic stroke and transient ischemic attack in the adult and geriatric population. J. Stroke Cerebrovasc. Dis. 2019, 28, 84–89. [Google Scholar] [CrossRef]
- Rasmussen, R.S.; Østergaard, A.; Kjær, P.; Skerris, A.; Skou, C.; Christoffersen, J.; Seest, L.S.; Poulsen, M.B.; Rønholt, F.; Overgaard, K. Stroke rehabilitation at home before and after discharge reduced disability and improved quality of life: A randomised controlled trial. Clin. Rehabil. 2016, 30, 225–236. [Google Scholar] [CrossRef]
- Saver, J.L.; Fonarow, G.C.; Smith, E.E.; Reeves, M.J.; Grau-Sepulveda, M.V.; Pan, W.; Olson, D.M.; Hernandez, A.F.; Peterson, E.D.; Schwamm, L.H. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013, 309, 2480–2488. [Google Scholar] [CrossRef]
- Mitchell, A.J.; Sheth, B.; Gill, J.; Yadegarfar, M.; Stubbs, B.; Yadegarfar, M.; Meader, N. Prevalence and predictors of post-stroke mood disorders: A meta-analysis and meta-regression of depression, anxiety and adjustment disorder. Gen. Hosp. Psychiatry 2017, 47, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Portegies, M.L.; Wolters, F.J.; Hofman, A.; Ikram, M.K.; Koudstaal, P.J.; Ikram, M.A. Prestroke vascular pathology and the risk of recurrent stroke and poststroke dementia. Stroke 2016, 47, 2119–2122. [Google Scholar] [CrossRef] [Green Version]
- Khoshnazar, M.; Bigdeli, M.R.; Parvardeh, S.; Pouriran, R. Attenuating effect of alpha-pinene on neurobehavioural deficit, oxidative damage and inflammatory response following focal ischaemic stroke in rat. J. Pharm. Pharmacol. 2019, 71, 1725–1733. [Google Scholar] [CrossRef]
- Qin, Y.Y.; Li, M.; Feng, X.; Wang, J.; Cao, L.; Shen, X.K.; Chen, J.; Sun, M.; Sheng, R.; Han, F.; et al. Combined NADPH and the NOX inhibitor apocynin provides greater anti-inflammatory and neuroprotective effects in a mouse model of stroke. Free Radic. Biol. Med. 2017, 104, 333–345. [Google Scholar] [CrossRef]
- Cheng, Y.; Ying, A.; Lin, Y.; Yu, J.; Luo, J.; Zeng, Y.; Lin, Y. Neutrophil-to-lymphocyte ratio, hyperglycemia, and outcomes in ischemic stroke patients treated with intravenous thrombolysis. Brain Behav. 2020, 10, e01741. [Google Scholar] [CrossRef]
- Lasek-Bal, A.; Jedrzejowska-Szypulka, H.; Student, S.; Warsz-Wianecka, A.; Zareba, K.; Puz, P.; Bal, W.; Pawletko, K.; Lewin-Kowalik, J. The importance of selected markers of inflammation and blood-brain barrier damage for short-term ischemic stroke prognosis. J. Physiol. Pharmacol. 2019, 70, 2. [Google Scholar]
- Zhou, J.; Wu, J.; Zhang, J.; Xu, T.; Zhang, H.; Zhang, Y.; Zhang, S. Association of stroke clinical outcomes with coexistence of hyperglycemia and biomarkers of inflammation. J. Stroke Cerebrovasc. Dis. 2015, 24, 1250–1255. [Google Scholar] [CrossRef]
- Chen, W.Y.; Mao, F.C.; Liu, C.H.; Kuan, Y.H.; Lai, N.W.; Wu, C.C.; Chen, C.J. Chromium supplementation improved post-stroke brain infarction and hyperglycemia. Metab. Brain Dis. 2016, 31, 289–297. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, F.; Li, J.; Wang, G. Galuteolin attenuates cerebral ischemia/reperfusion injury in rats via anti-apoptotic, anti-oxidant, and anti-inflammatory mechanisms. Neuropsychiatr. Dis. Treat. 2019, 15, 2671–2680. [Google Scholar] [CrossRef] [Green Version]
- Sá-Nakanishi, A.B.; de Oliveira, M.C.; Pateis, V.O.; Silva, L.A.P.; Pereira-Maróstica, H.V.; Gonçalves, G.A.; Oliveira, M.A.S.; Godinho, J.; Bracht, L.; Milani, H.; et al. Glycemic homeostasis and hepatic metabolism are modified in rats with global cerebral ischemia. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165934. [Google Scholar] [CrossRef]
- Bu, L.; Cao, X.; Zhang, Z.; Wu, H.; Guo, R.; Ma, M. Decreased secretion of tumor necrosis factor-alpha attenuates macrophages-induced insulin resistance in skeletal muscle. Life Sci. 2020, 244, 117304. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Biber, K.; Finsen, B. Inflammatory cytokines in experimental and human stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1677–1698. [Google Scholar] [CrossRef] [Green Version]
- Sairanen, T.; Carpén, O.; Karjalainen-Lindsberg, M.L.; Paetau, A.; Turpeinen, U.; Kaste, M.; Lindsberg, P.J. Evolution of cerebral tumor necrosis factor-alpha production during human ischemic stroke. Stroke 2001, 32, 1750–1758. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; McClure, L.A.; Zhang, Y.; Luna, J.M.; Del Brutto, O.H.; Benavente, O.R.; Elkind, M.S. Inflammatory markers and outcomes after lacunar stroke: Levels of inflammatory markers in treatment of stroke study. Stroke 2016, 47, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Tobinick, E.; Kim, N.M.; Reyzin, G.; Rodriguez-Romanacce, H.; DePuy, V. Selective TNF inhibition for chronic stroke and traumatic brain injury: An observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs 2012, 26, 1051–1070. [Google Scholar] [CrossRef]
- Arango-Dávila, C.A.; Vera, A.; Londoño, A.C.; Echeverri, A.F.; Cañas, F.; Cardozo, C.F.; Orozco, J.L.; Rengifo, J.; Cañas, C.A. Soluble or soluble/membrane TNF-alpha inhibitors protect the brain from focal ischemic injury in rats. Int. J. Neurosci. 2015, 125, 936–940. [Google Scholar] [CrossRef]
- Clausen, B.H.; Degn, M.; Martin, N.A.; Couch, Y.; Karimi, L.; Ormhøj, M.; Mortensen, M.L.; Gredal, H.B.; Gardiner, C.; Sargent, I.I.; et al. Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J. Neuroinflammation. 2014, 11, 203. [Google Scholar] [CrossRef]
- King, M.D.; Alleyne, C.H., Jr.; Dhandapani, K.M. TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice. Neurosci. Lett. 2013, 542, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Wang, Y.Y.; Chang, C.Y.; Wu, C.C.; Chen, W.Y.; Kuan, Y.H.; Liao, S.L.; Chen, C.J. Effects of β-adrenergic blockade on metabolic and inflammatory responses in a rat model of ischemic stroke. Cells 2020, 9, 1373. [Google Scholar] [CrossRef]
- Wu, M.H.; Huang, C.C.; Chio, C.C.; Tsai, K.J.; Chang, C.P.; Lin, N.K.; Lin, M.T. Inhibition of peripheral TNF-alpha and downregulation of microglial activation by alpha-lipoic acid and etanercept protect rat brain against ischemic stroke. Mol. Neurobiol. 2016, 53, 4961–4971. [Google Scholar] [CrossRef]
- Barone, F.C.; Arvin, B.; White, R.F.; Miller, A.; Webb, C.L.; Willette, R.N.; Lysko, P.G.; Feuerstein, G.Z. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 1997, 28, 1233–1244. [Google Scholar] [CrossRef]
- Vakili, A.; Mojarrad, S.; Akhavan, M.M.; Rashidy-Pour, A. Pentoxifylline attenuates TNF-alpha protein levels and brain edema following temporary focal cerebral ischemia in rats. Brain Res. 2011, 1377, 119–125. [Google Scholar] [CrossRef]
- Liao, K.Y.; Chen, C.J.; Hsieh, S.K.; Pan, P.H.; Chen, W.Y. Interleukin-13 ameliorates postischemic hepatic gluconeogenesis and hyperglycemia in rat model of stroke. Metab. Brain Dis. 2020, 35, 1201–1210. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Lin, S.Y.; Chuang, Y.H.; Chen, C.J.; Tung, K.C.; Sheu, W.H. Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E155–E163. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Chen, C.J.; Lin, S.Y.; Chuang, Y.H.; Sheu, W.H.; Tung, K.C. Hyperglycemia is associated with enhanced gluconeogenesis in a rat model of permanent cerebral ischemia. Mol. Cell. Endocrinol. 2013, 367, 50–56. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Lin, S.Y.; Chuang, Y.H.; Sheu, W.H.; Tung, K.C.; Chen, C.J. Activation of hepatic inflammatory pathways by catecholamines is associated with hepatic insulin resistance in male ischemic stroke rats. Endocrinology 2014, 155, 1235–1246. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Y.; Li, J.R.; Chen, W.Y.; Ou, Y.C.; Lai, C.Y.; Hu, Y.H.; Wu, C.C.; Chang, C.J.; Chen, C.J. Disruption of in vitro endothelial barrier integrity by Japanese encephalitis virus-Infected astrocytes. Glia 2015, 63, 1915–1932. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Fei, Y.X.; Zhu, J.P.; Zhao, B.; Yin, Q.Y.; Fang, W.R.; Li, Y.M. XQ-1H regulates Wnt/GSK3beta/beta-catenin pathway and ameliorates the integrity of blood brain barrier in mice with acute ischemic stroke. Brain Res. Bull. 2020, 164, 269–288. [Google Scholar] [CrossRef]
- McCarty, M.F.; Lerner, A. Nutraceutical induction and mimicry of heme oxygenase activity as a strategy for controlling excitotoxicity in brain trauma and ischemic stroke: Focus on oxidative stress. Expert Rev. Neurother. 2021, 21, 157–168. [Google Scholar] [CrossRef]
- Kolosowska, N.; Keuters, M.H.; Wojciechowski, S.; Keksa-Goldsteine, V.; Laine, M.; Malm, T.; Goldsteins, G.; Koistinaho, J.; Dhungana, H. Peripheral administration of IL-13 induces anti-inflammatory microglial/macrophage responses and provides neuroprotection in ischemic stroke. Neurotherapeutics 2019, 16, 1304–1319. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Bettermann, K.; Willing, L.; Sinha, K.; Simpson, I.A. The role of neutrophils in mediating stroke injury in the diabetic db/db mouse brain following hypoxia-ischemia. Neurochem. Int. 2020, 139, 104790. [Google Scholar] [CrossRef]
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Gieryng, A.; Zawadzka, M.; Kaminska, B. Dissecting functional phenotypes of microglia and macrophages in the rat brain after transient cerebral ischemia. Glia 2019, 67, 232–245. [Google Scholar] [CrossRef]
- Jin, W.N.; Shi, S.X.; Li, Z.; Li, M.; Wood, K.; Gonzales, R.J.; Liu, Q. Depletion of microglia exacerbates postischemic inflammation and brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2224–2236. [Google Scholar] [CrossRef] [Green Version]
- Otxoa-de-Amezaga, A.; Miró-Mur, F.; Pedragosa, J.; Gallizioli, M.; Justicia, C.; Gaja-Capdevila, N.; Ruíz-Jaen, F.; Salas-Perdomo, A.; Bosch, A.; Calvo, M.; et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta. Neuropathol. 2019, 137, 321–341. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.J.; Ran, Y.Y.; Qie, S.Y.; Gong, W.J.; Gao, F.H.; Ding, Z.T.; Xi, J.N. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Wium-Andersen, I.K.; Wium-Andersen, M.K.; Jørgensen, M.B.; Osler, M. Anti-inflammatory treatment and risk for depression after first-time stroke in a cohort of 147487 Danish patients. J. Psychiatry Neurosci. 2017, 42, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Chang, C.Y.; Shih, K.C.; Hung, C.J.; Wang, Y.Y.; Lin, S.Y.; Chen, W.Y.; Kuan, Y.H.; Liao, S.L.; Wang, W.Y.; et al. β-Funaltrexamine displayed anti-inflammatory and neuroprotective effects in cells and rat model of stroke. Int. J. Mol. Sci. 2020, 21, 3866. [Google Scholar] [CrossRef]
- Ma, Y.; Cheng, Q.; Wang, E.; Li, L.; Zhang, X. Inhibiting tumor necrosis factor-α signaling attenuates postoperative cognitive dysfunction in aged rats. Mol. Med. Rep. 2015, 12, 3095–3100. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Liang, Y.; Yang, P.; Wang, W.; Zhang, X.; Wang, J. TNF-α receptor antagonist attenuates isoflurane-induced cognitive impairment in aged rats. Exp. Ther. Med. 2016, 12, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Kochumon, S.; Chandy, B.; Shenouda, S.; Koshy, M.; Hasan, A.; Arefanian, H.; Al-Mulla, F.; Sindhu, S. TNF-alpha drives the CCL4 expression in human monocytic cells: Involvement of the SAPK/JNK and NF-kappaB signaling pathways. Cell Physiol. Biochem. 2019, 52, 908–921. [Google Scholar]
- Chang, Y.L.; Chen, T.H.; Wu, Y.H.; Chen, G.A.; Weng, T.H.; Tseng, P.H.; Hsieh, S.L.; Fu, S.L.; Lin, C.H.; Chen, C.J.; et al. A novel TLR2-triggered signalling crosstalk synergistically intensifies TNF-mediated IL-6 induction. J. Cell. Mol. Med. 2014, 18, 1344–1357. [Google Scholar] [CrossRef]
- Chen, L.; Cao, J.; Cao, D.; Wang, M.; Xiang, H.; Yang, Y.; Ying, T.; Cong, H. Protective effect of dexmedetomidine against diabetic hyperglycemia-exacerbated cerebral ischemia/reperfusion injury: An in vivo and in vitro study. Life Sci. 2019, 235, 116553. [Google Scholar] [CrossRef]
- He, J.; Zhou, D.; Yan, B. Eriocitrin alleviates oxidative stress and inflammatory response in cerebral ischemia reperfusion rats by regulating phosphorylation levels of Nrf2/NQO-1/HO-1/NF-kappaB p65 proteins. Ann. Transl. Med. 2020, 8, 757. [Google Scholar] [CrossRef]
- Fang, M.; Zhong, W.H.; Song, W.L.; Deng, Y.Y.; Yang, D.M.; Xiong, B.; Zeng, H.K.; Wang, H.D. Ulinastatin ameliorates pulmonary capillary endothelial permeability induced by sepsis through protection of tight junctions via inhibition of TNF-alpha and related pathways. Front. Pharmacol. 2018, 9, 823. [Google Scholar] [CrossRef]
- Pan, J.; Qu, M.; Li, Y.; Wang, L.; Zhang, L.; Wang, Y.; Tang, Y.; Tian, H.L.; Zhang, Z.; Yang, G.Y. MicroRNA-126-3p/-5p overexpression attenuates blood-brain barrier disruption in a mouse model of middle cerebral artery occlusion. Stroke 2020, 51, 619–627. [Google Scholar] [CrossRef]
- Kim, M.; Song, K.; Kim, Y.S. Alantolactone improves prolonged exposure of interleukin-6-induced skeletal muscle inflammation associated glucose intolerance and insulin resistance. Front. Pharmacol. 2017, 8, 405. [Google Scholar] [CrossRef]
- García-Eguren, G.; Sala-Vila, A.; Giró, O.; Vega-Beyhart, A.; Hanzu, F.A. Long-term hypercortisolism induces lipogenesis promoting palmitic acid accumulation and inflammation in visceral adipose tissue compared with HFD-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E995–E1003. [Google Scholar] [CrossRef]
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21, and an FGFR1/beta-Klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell. Metab. 2017, 26, 709–718.e3. [Google Scholar] [CrossRef]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef]
- Wang, D.; Liu, F.; Zhu, L.; Lin, P.; Han, F.; Wang, X.; Tan, X.; Lin, L.; Xiong, Y. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J. Neuroinflamm. 2020, 17, 257. [Google Scholar] [CrossRef]
- Lv, N.; Li, C.; Liu, X.; Qi, C.; Wang, Z. miR-34b alleviates high glucose-induced inflammation and apoptosis in human HK-2 cells via IL-6R/JAK2/STAT3 signaling pathway. Med. Sci. Monit. 2019, 25, 8142–8151. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-Y.; Wang, Y.-Y.; Chang, C.-Y.; Wu, C.-C.; Chen, W.-Y.; Liao, S.-L.; Chen, C.-J. TNF-α Receptor Inhibitor Alleviates Metabolic and Inflammatory Changes in a Rat Model of Ischemic Stroke. Antioxidants 2021, 10, 851. https://doi.org/10.3390/antiox10060851
Lin S-Y, Wang Y-Y, Chang C-Y, Wu C-C, Chen W-Y, Liao S-L, Chen C-J. TNF-α Receptor Inhibitor Alleviates Metabolic and Inflammatory Changes in a Rat Model of Ischemic Stroke. Antioxidants. 2021; 10(6):851. https://doi.org/10.3390/antiox10060851
Chicago/Turabian StyleLin, Shih-Yi, Ya-Yu Wang, Cheng-Yi Chang, Chih-Cheng Wu, Wen-Ying Chen, Su-Lan Liao, and Chun-Jung Chen. 2021. "TNF-α Receptor Inhibitor Alleviates Metabolic and Inflammatory Changes in a Rat Model of Ischemic Stroke" Antioxidants 10, no. 6: 851. https://doi.org/10.3390/antiox10060851