
Ethyl Pyruvate Attenuates Microglial NLRP3 Inflammasome Activation via Inhibition of HMGB1/NF-κB/miR-223 Signaling

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Cell Viability Assessment with Presto Blue
2.3. Propidium Iodide Staining
2.4. ASC Speck Staining
2.5. ELISA
2.6. Detection of Intracellular ROS
2.7. Detection of Mitochondrial ROS
2.8. Analysis of Mitochondrial Membrane Potential
2.9. Western Blotting
2.10. Quantitative PCR
2.11. Transfection of miRNA Inhibitors
2.12. Statistical Analysis
3. Results
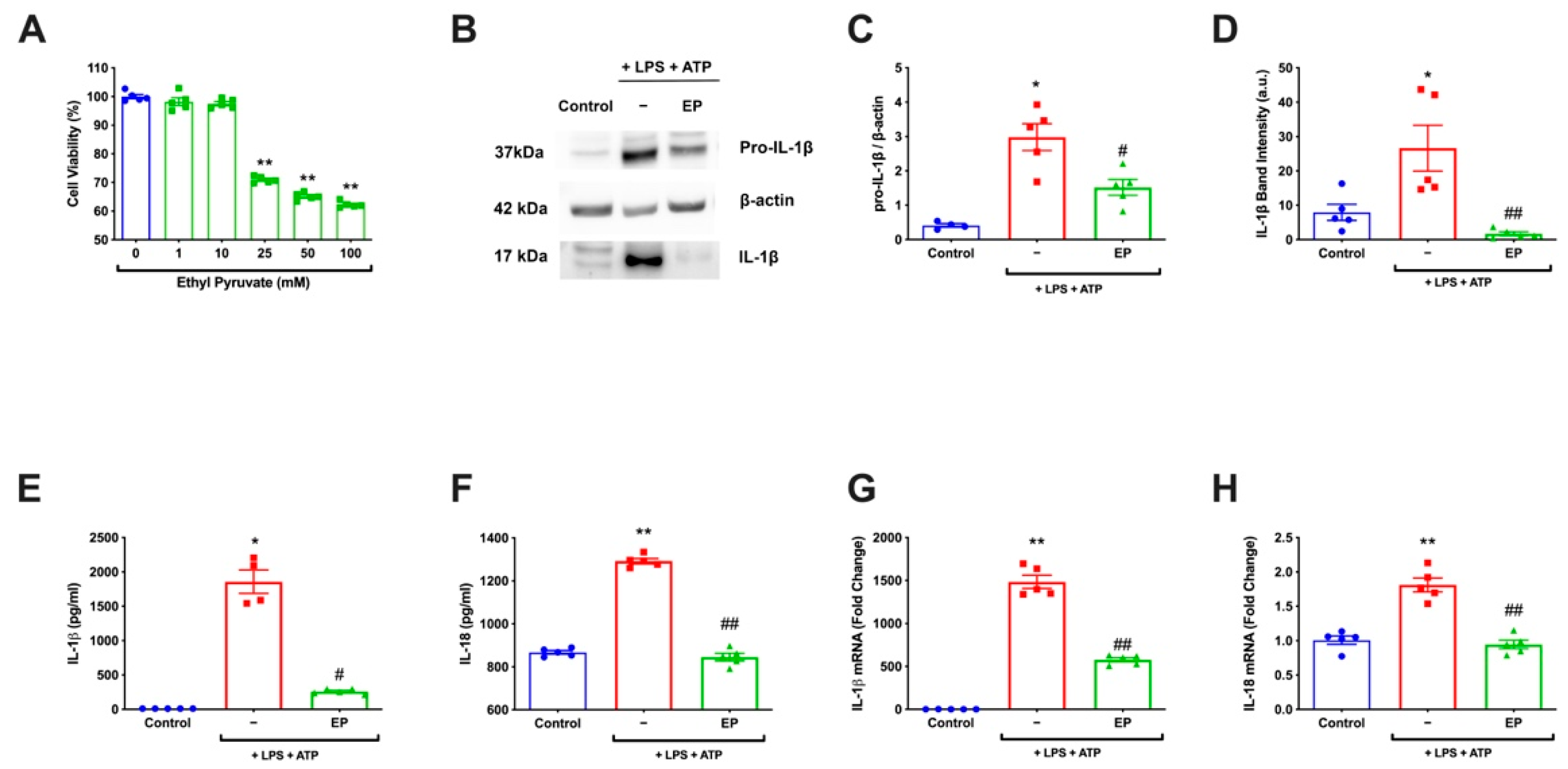
3.1. EP Decreased IL-1β and IL-18 on Both mRNA and Protein Levels
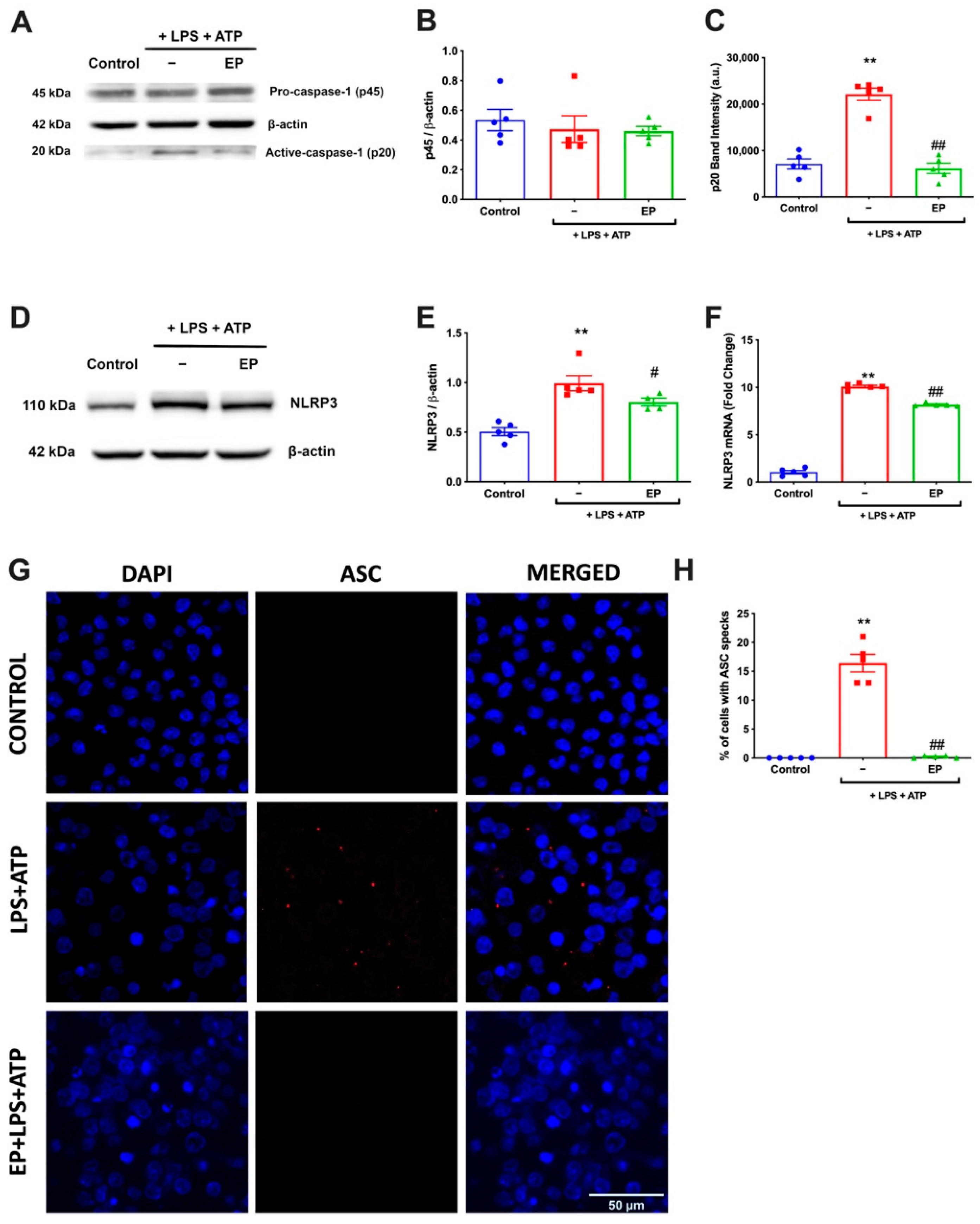
3.2. EP Diminished NLRP3 Protein Complex Formation by Reducing NLRP3, Caspase-1, and ASC Specks
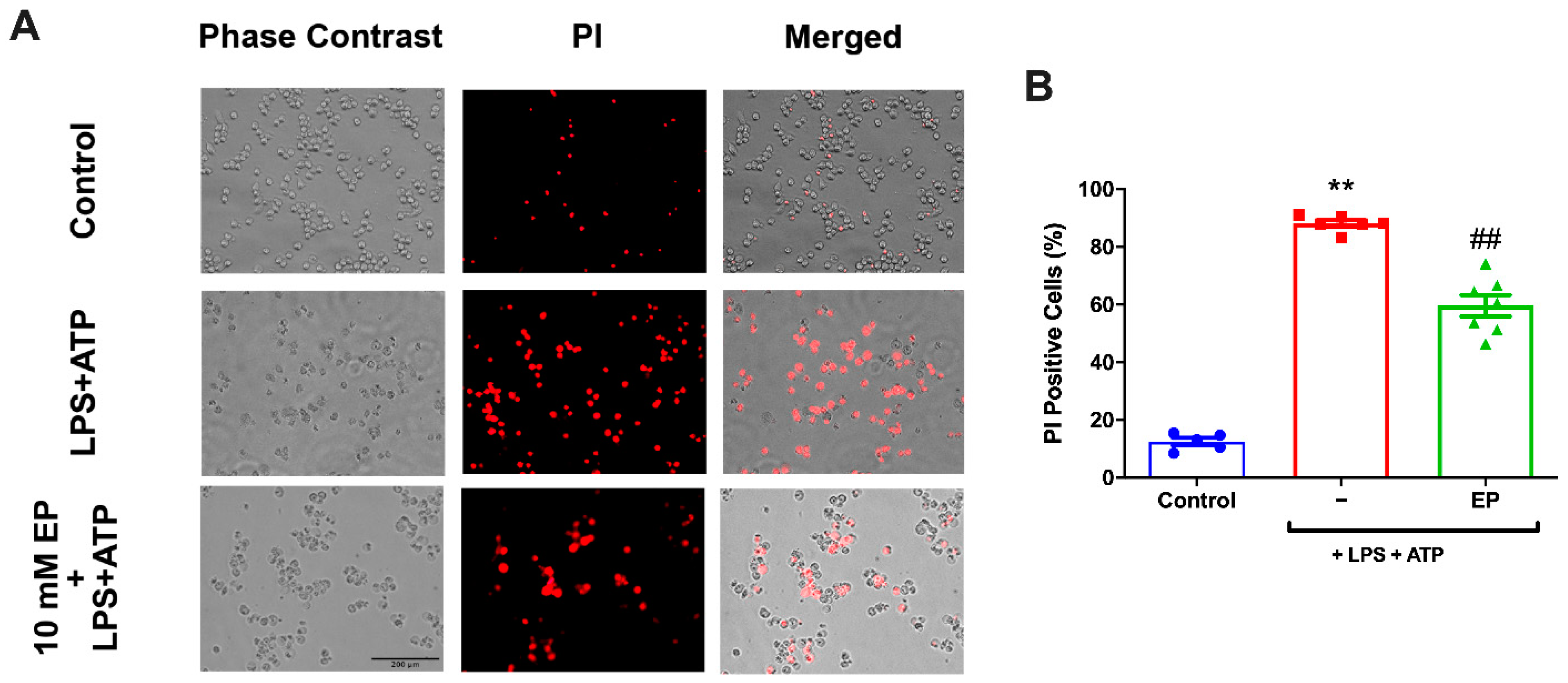
3.3. EP Protected N9 Microglial Cells Against Pyroptotic Cell Death
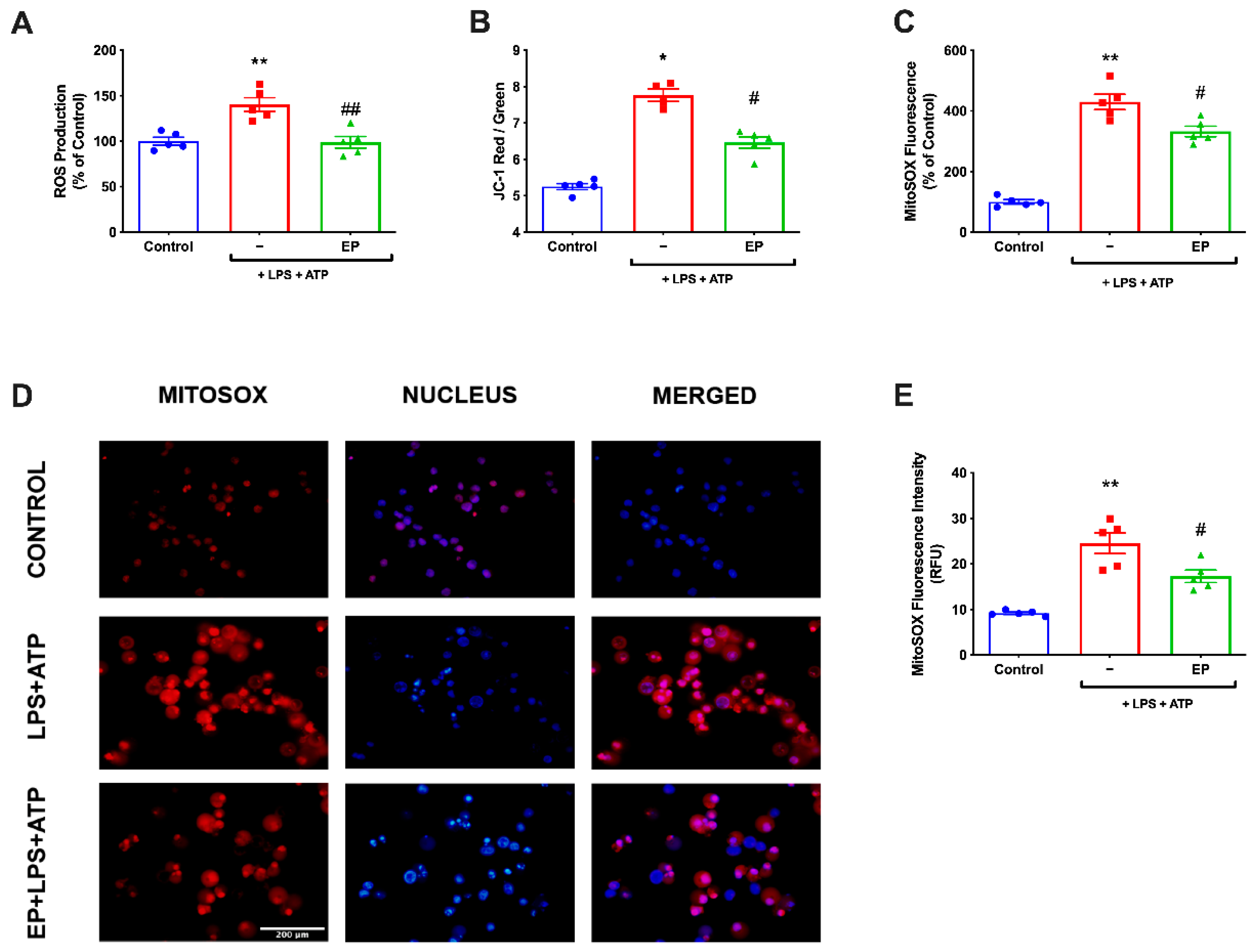
3.4. EP Ameliorated ROS Production and Restored Mitochondrial Membrane Potential
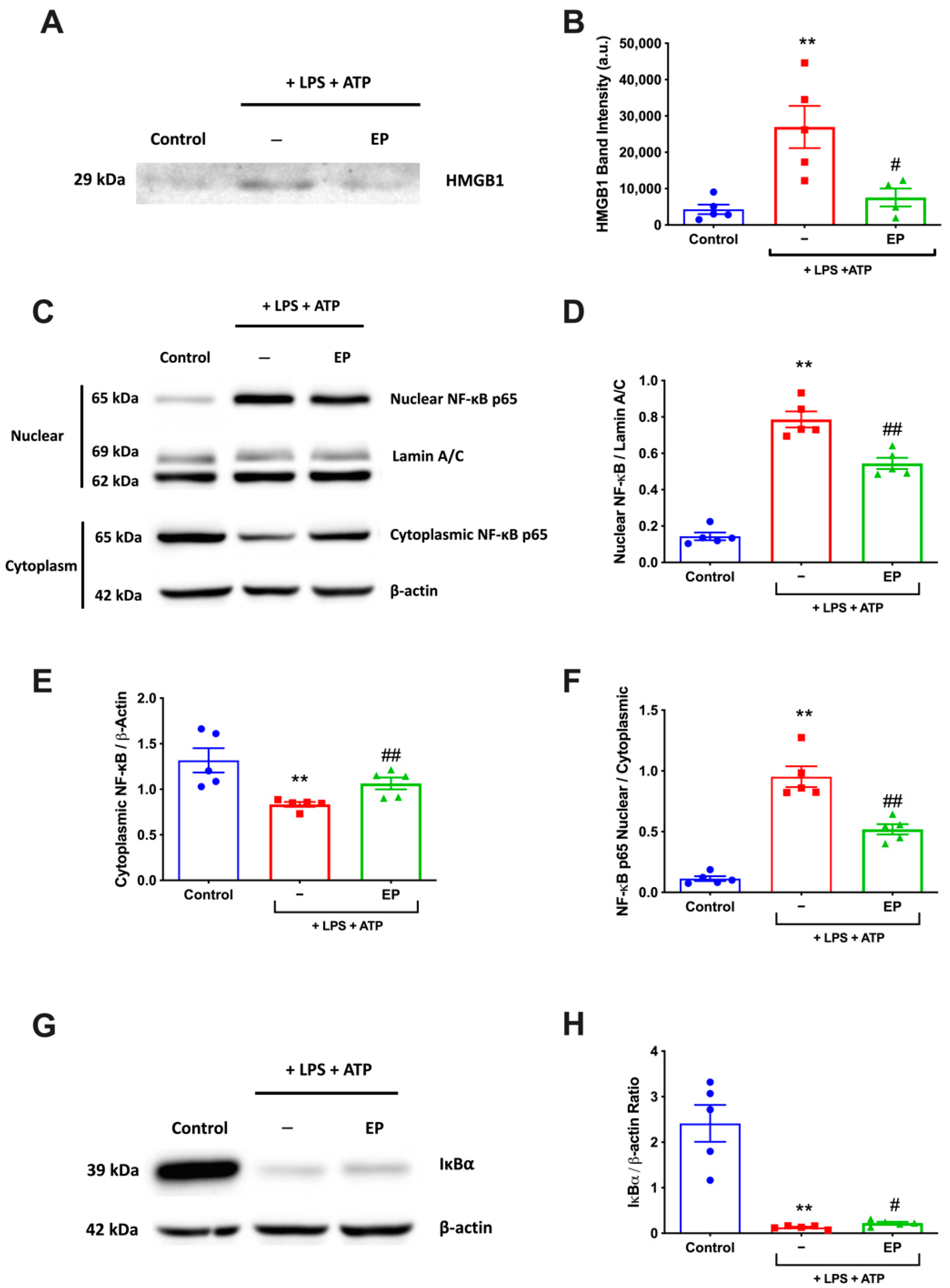
3.5. EP Reduced NF-κB Activation and HMGB1 Expression Level
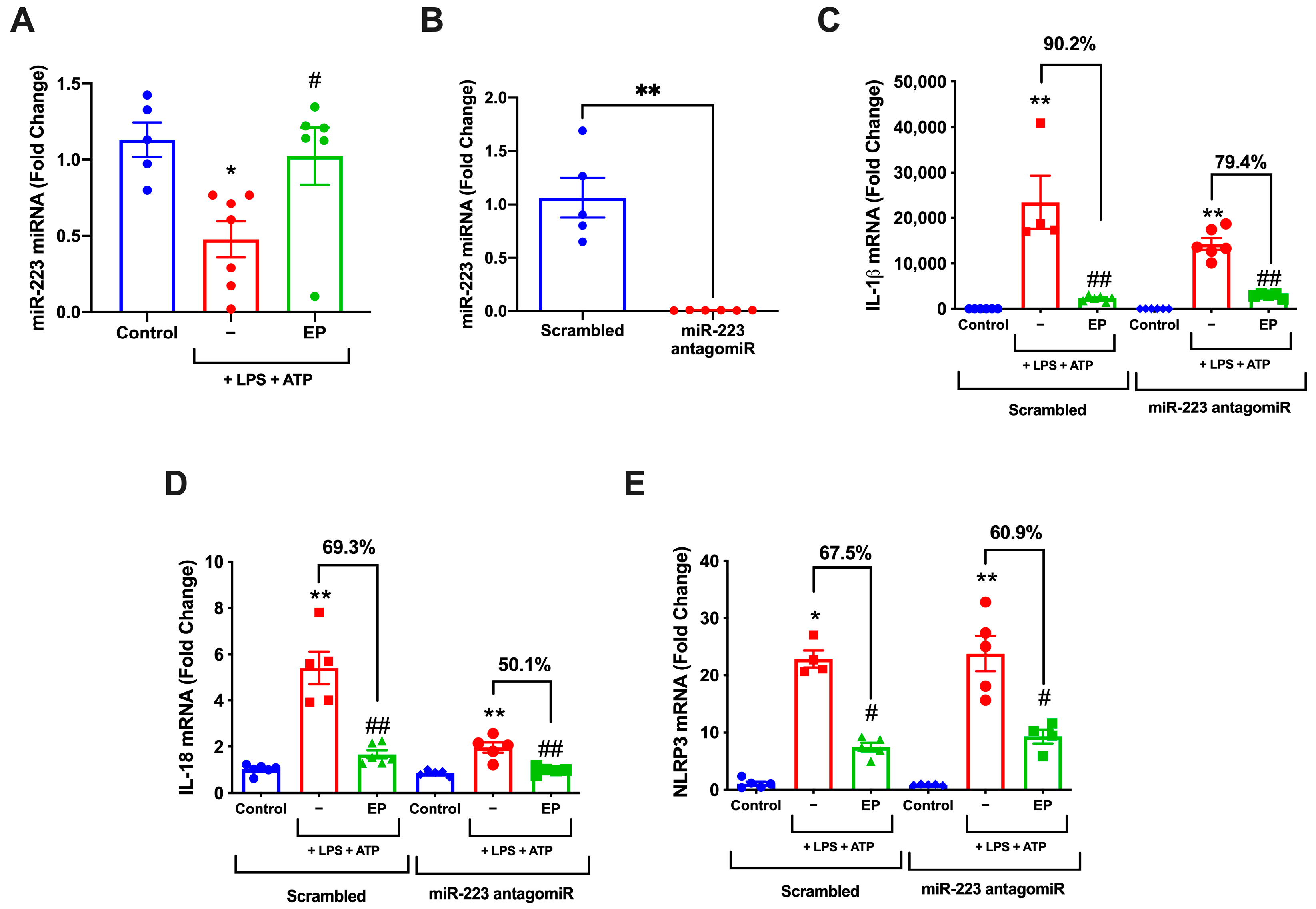
3.6. miR-223 Inhibition Attenuates Protective Roles of EP
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Voet, S.; Prinz, M.; van Loo, G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef]
- Hoffmann, S.; Beyer, C. A fatal alliance between microglia, inflammasomes, and central pain. Int. J. Mol. Sci. 2020, 21, 3764. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolivar, B.E.; Vogel, T.P.; Bouchier-Hayes, L. Inflammatory caspase regulation: Maintaining balance between inflammation and cell death in health and disease. FEBS J. 2019, 286, 2628–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Cheng, X.; Zhang, J.; Yuan, C.; Zhao, M.; Yang, X. Ethyl pyruvate confers protection against endotoxemia and sepsis by inhibiting caspase-11-dependent cell pyroptosis. Int. Immunopharmacol. 2020, 78, 106016. [Google Scholar] [CrossRef]
- Ifergan, I.; Chen, S.; Zhang, B.; Miller, S.D. Cutting edge: MicroRNA-223 regulates myeloid dendritic cell-driven Th17 responses in experimental autoimmune encephalomyelitis. J. Immunol. 2016, 196, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef] [Green Version]
- Sims, C.A.; Wattanasirichaigoon, S.; Menconi, M.J.; Ajami, A.M.; Fink, M.P. Ringer’s ethyl pyruvate solution ameliorates ischemia/reperfusion-induced intestinal mucosal injury in rats. Crit. Care Med. 2001, 29, 1513–1518. [Google Scholar] [CrossRef]
- Jang, I.S.; Park, M.Y.; Shin, I.W.; Sohn, J.T.; Lee, H.K.; Chung, Y.K. Ethyl pyruvate has anti-inflammatory and delayed myocardial protective effects after regional ischemia/reperfusion injury. Yonsei Med. J. 2010, 51, 838–844. [Google Scholar] [CrossRef]
- Yu, Y.M.; Kim, J.B.; Lee, K.W.; Kim, S.Y.; Han, P.L.; Lee, J.K. Inhibition of the cerebral ischemic injury by ethyl pyruvate with a wide therapeutic window. Stroke 2005, 36, 2238–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Wang, H.; Zhao, J.; Pan, H.; Mao, L. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-kappaB pathway after traumatic brain injury in the rat. Mediat. Inflamm. 2011, 2011, 807142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, M.P. Ethyl pyruvate. Curr. Opin. Anaesthesiol. 2008, 21, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Kim, I.D.; Kim, S.W.; Lee, H.; Park, J.Y.; Yoon, S.H.; Lee, J.K. Anti-inflammatory and anti-excitoxic effects of diethyl oxopropanamide, an ethyl pyruvate bioisoster, exert robust neuroprotective effects in the postischemic brain. Sci. Rep. 2017, 7, 42891. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Yu, Y.M.; Kim, S.W.; Lee, J.K. Anti-inflammatory mechanism is involved in ethyl pyruvate-mediated efficacious neuroprotection in the postischemic brain. Brain Res. 2005, 1060, 188–192. [Google Scholar] [CrossRef]
- Dave, S.H.; Tilstra, J.S.; Matsuoka, K.; Li, F.; DeMarco, R.A.; Beer-Stolz, D.; Sepulveda, A.R.; Fink, M.P.; Lotze, M.T.; Plevy, S.E. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J. Leukoc. Biol. 2009, 86, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Kim, H.S. Inhibitory mechanism of MMP-9 gene expression by ethyl pyruvate in lipopolysaccharide-stimulated BV2 microglial cells. Neurosci. Lett. 2011, 493, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Righi, M.; Mori, L.; de Libero, G.; Sironi, M.; Biondi, A.; Mantovani, A.; Donini, S.D.; Ricciardi-Castagnoli, P. Monokine production by microglial cell clones. Eur. J. Immunol. 1989, 19, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin attenuates LPS-induced acute depressive-like behaviors and microglial NLRP3 inflammasome activation through the SIRT1/Nrf2 pathway. Front. Immunol. 2019, 10, 1511. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Herman, F.J.; Pasinetti, G.M. Principles of inflammasome priming and inhibition: Implications for psychiatric disorders. Brain Behav. Immun. 2018, 73, 66–84. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Sharma, A.; Tate, M.; Mathew, G.; Vince, J.E.; Ritchie, R.H.; de Haan, J.B. Oxidative stress and NLRP3-inflammasome activity as significant drivers of diabetic cardiovascular complications: Therapeutic implications. Front. Physiol. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Xie, L.; Yang, X.; Liang, F.; Yang, Y.; Tong, J.; Zhong, Y.; Zhao, K.; Tang, Y.; Yuan, C. Ethyl pyruvate protects against sepsis-associated encephalopathy through inhibiting the NLRP3 inflammasome. Mol. Med. 2020, 26, 55. [Google Scholar] [CrossRef]
- Jun, J.H.; Shim, J.K.; Oh, J.E.; Shin, E.J.; Shin, E.; Kwak, Y.L. Protective effect of ethyl pyruvate against myocardial ischemia reperfusion injury through regulations of ROS-Related NLRP3 inflammasome activation. Oxid. Med. Cell. Longev. 2019, 2019, 4264580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Englert, J.A.; Yang, R.; Delude, R.L.; Fink, M.P. Ethyl pyruvate inhibits nuclear factor-kappaB-dependent signaling by directly targeting p65. J. Pharmacol. Exp. Ther. 2005, 312, 1097–1105. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Teng, J.; Miao, W. HMGB1 mediates microglia activation via the TLR4/NF-kappaB pathway in coriaria lactone induced epilepsy. Mol. Med. Rep. 2018, 17, 5125–5131. [Google Scholar] [PubMed] [Green Version]
- Li, S.; Liang, F.; Kwan, K.; Tang, Y.; Wang, X.; Tang, Y.; Li, J.; Yang, H.; Chavan, S.S.; Wang, H.; et al. Identification of ethyl pyruvate as a NLRP3 inflammasome inhibitor that preserves mitochondrial integrity. Mol. Med. 2018, 24, 8. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Sarkar, A.; Walle, L.V.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, M.A. MicroRNAs and the immune response. Trends Immunol. 2008, 29, 343–351. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, L.; Xian, R.; Yuan, B. MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol. Immunol. 2015, 65, 267–276. [Google Scholar] [CrossRef]
- Dang, C.P.; Leelahavanichkul, A. Over-expression of miR-223 induces M2 macrophage through glycolysis alteration and attenuates LPS-induced sepsis mouse model, the cell-based therapy in sepsis. PLoS ONE 2020, 15, e0236038. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, Y.; Ma, Y.; Yang, J. MiR-223 plays a protecting role in neutrophilic asthmatic mice through the inhibition of NLRP3 inflammasome. Respir. Res. 2020, 21, 116. [Google Scholar] [CrossRef]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, S.; Xue, Y.; Qiu, J.; Ye, T.; Zhang, R.; Song, B.; He, W.; Zhang, Y.; Jiang, W. MicroRNA-223 negatively regulates LPS-induced inflammatory responses by targeting NLRP3 in human dental pulp fibroblasts. Int. Endod. J. 2021, 54, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D.; Boldin, M.P.; O’Connell, R.M.; Rao, D.S.; Taganov, K.D. MicroRNAs: New regulators of immune cell development and function. Nat. Immunol. 2008, 9, 839–845. [Google Scholar] [CrossRef]
- Haneklaus, M.; Gerlic, M.; O’Neill, L.A.; Masters, S.L. MiR-223: Infection, inflammation and cancer. J. Intern. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; de Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.L.; Zhang, S.X.; Zheng, C.F.; Li, Y.F.; Zhang, L.H.; Su, Q.Y.; Hao, Y.F.; Wang, S.; Li, X.W. Long non-coding RNA MEG3 inhibits M2 macrophage polarization by activating TRAF6 via microRNA-223 down-regulation in viral myocarditis. J. Cell. Mol. Med. 2020, 24, 12341–12354. [Google Scholar] [CrossRef]
- Li, G.; Du, P.; Qiang, X.; Jin, D.; Liu, H.; Li, B.; Guo, J. Low-expressed GAS5 injure myocardial cells and progression of chronic heart failure via regulation of miR-223-3P. Exp. Mol. Pathol. 2020, 117, 104529. [Google Scholar] [CrossRef]
- Tan, J.; Fan, J.; He, J.; Zhao, L.; Tang, H. Knockdown of LncRNA DLX6-AS1 inhibits HK-2 cell pyroptosis via regulating miR-223-3p/NLRP3 pathway in lipopolysaccharide-induced acute kidney injury. J. Bioenerg. Biomembr. 2020, 52, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.M.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J.; Rushworth, S.A. NRF2-driven miR-125B1 and miR-29B1 transcriptional regulation controls a novel anti-apoptotic miRNA regulatory network for AML survival. Cell Death Differ. 2015, 22, 654–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Kim, S.W.; Jin, Y.; Kim, I.D.; Lee, J.K. Ethyl pyruvate-mediated Nrf2 activation and hemeoxygenase 1 induction in astrocytes confer protective effects via autocrine and paracrine mechanisms. Neurochem. Int. 2012, 61, 89–99. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Provider | Catalog Number | Dilution |
|---|---|---|---|
| Anti-IL-1β | Abcam | ab9722 | 1:1000 |
| Anti-Caspase-1 | Abcam | ab1872 | 1:1000 |
| Anti-NLRP3 | Adipogen | AG-20B-0014 | 1:1000 |
| Anti-HMGB1 | Cell Signaling | 3935S | 1:1000 |
| Anti-NF-κB p65 | Santa Cruz | sc-372 | 1:1000 |
| Anti- IκBα | Cell Signaling | 9242S | 1:1000 |
| Anti-β-actin | Abcam | ab8227 | 1:1000 |
| Anti-Lamin A/C | Santa Cruz | sc-20681 | 1:1000 |
| Anti-Rabbit HRP Secondary | Cell Signaling | 7074 | 1:2000 |
| Anti-Mouse HRP Secondary | Cell Signaling | 7076 | 1:2000 |
| mRNA | Sequence (5′–3′) | Product Length | Accession | |
|---|---|---|---|---|
| IL-1β | Forward | TTCTTTTCCTTCATCTTTGAAGAAG | 365 bp | NM_008361.4 |
| Reverse | TCCATCTTCTTCTTTGGGTATTGTT | |||
| IL-18 | Forward | CTTTGGAAGCCTGCTATAATCC | 363 bp | NM_008360.2 |
| Reverse | GGTCAAGAGGAAGTGATTTGGA | |||
| NLRP3 | Forward | TGCCTGTTCTTCCAGACTGGTGA | 143 bp | NM_145827.4 |
| Reverse | CACAGCACCCTCATGCCCGG | |||
| GAPDH | Forward | ACCACAGTCCATGCCATCAC | 452 bp | NM_001289726.1 |
| Reverse | TCCACCACCCTGTTGCTGTA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olcum, M.; Tufekci, K.U.; Durur, D.Y.; Tastan, B.; Gokbayrak, I.N.; Genc, K.; Genc, S. Ethyl Pyruvate Attenuates Microglial NLRP3 Inflammasome Activation via Inhibition of HMGB1/NF-κB/miR-223 Signaling. Antioxidants 2021, 10, 745. https://doi.org/10.3390/antiox10050745
Olcum M, Tufekci KU, Durur DY, Tastan B, Gokbayrak IN, Genc K, Genc S. Ethyl Pyruvate Attenuates Microglial NLRP3 Inflammasome Activation via Inhibition of HMGB1/NF-κB/miR-223 Signaling. Antioxidants. 2021; 10(5):745. https://doi.org/10.3390/antiox10050745
Chicago/Turabian StyleOlcum, Melis, Kemal Ugur Tufekci, Devrim Yagmur Durur, Bora Tastan, Irem Nur Gokbayrak, Kursad Genc, and Sermin Genc. 2021. "Ethyl Pyruvate Attenuates Microglial NLRP3 Inflammasome Activation via Inhibition of HMGB1/NF-κB/miR-223 Signaling" Antioxidants 10, no. 5: 745. https://doi.org/10.3390/antiox10050745